DFT studies on the mechanism of veratryl alcohol oxidation catalyzed by Cu–phen complexes†
Lisha Maa,
Qiancheng Zhanga,
Lin Cheng*a,
Zhijian Wu*b and
Jucai Yanga
aKey Laboratory of Industrial Catalysis of the Inner Mongolia Autonomous Region, Inner Mongolia University of Technology, Huhehot 010051, China. E-mail: lcheng1983@aliyun.com
bState Key Laboratory of Rare Earth Resource Utilization, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Changchun 130022, China. E-mail: zjwu@ciac.ac.cn; Fax: +86 431 85698041
First published on 26th June 2014
Abstract
Density functional theory (DFT) calculations have been performed to investigate the catalytic mechanism for the oxidation of veratryl alcohol to veratraldehyde by Cu–phen (phen = 1,10-phenanthroline) catalyst. The catalytic cycle consists of alcohol oxidation and O2 reduction. For the alcohol oxidation, both mononuclear mechanism (path A) and binuclear mechanism (path B) are proposed. Our calculations show that path B is preferred over path A. Namely, for the Cu–phen (phen = 1,10-phenanthroline) catalytic system, the mechanism is the binuclear mechanism, which is consistent with the experimental suggestion. For the O2 reduction, two possible paths are proposed as well, which are (1) “path I” in which CuI is oxidized by O2 via a binuclear mechanism, and (2) “path II” in which CuI is oxidized by O2 via a mononuclear mechanism. According to our calculations, path I is favored both in thermodynamics and kinetics.
1 Introduction
Selective oxidation of alcohols to the corresponding aldehydes and ketones plays an important role in the organic synthesis of large-scale chemical industry.1 It is known that traditional alcohol oxidation employs stoichiometric amounts of oxidizing reagents. Unfortunately, most of them suffer from the harsh conditions and toxic stoichiometric oxidations.2 Thus, the development of catalytic aerobic oxidation is necessary. Galactose oxidase (GOase) is a mononuclear copper enzyme that converts a variety of primary alcohols to aldehydes in the presence of oxygen.3 A great number of active catalysts have been synthesized as the functional models of GOase. In this aspect, the most studied systems are catalytic system Cu(II)–TEMPO (TEMPO = 2,2,6,6-tetramethyl-piperidinyloxyl).4–13 For instance, Semmelhack and co-workers reported an aerobic oxidation of benzylic and allylic alcohols with 10% CuCl/TEMPO in DMF (N,N-dimethyllformamide) as the solvent.9 Sheldon and coworkers reported a series of CuII/diimine-TEMPO systems.4,7 At room temperature and under atmospheric air, CuII/diimine-TEMPO systems could oxidize benzyl alcohol to benzaldehyde. However, the alcohol oxidation has to carry out by the high catalyst loadings (5 mol%), with tBuOK as the catalytic base and CH3CN–H2O as the mixture solvent. Reedijk and coworkers studied an efficient catalytic system TEMPO/Cu-pyrazole.10 However, this catalytic system also requires organic solvent and basic co-catalyst. Recently, S. S. Stahl and coworkers reported a series (bpy)CuI/TEMPO systems.11–13 Among the aerobic alcohol oxidation reported to date, CH3CN has been used as solvent. Since this will cause safety issues associated with the flammability of organic solvent and oxygen mixtures,14 it would be highly desirable to use water as the reaction solvent. In this sense, CuII/1,10-phenanthroline catalytic system for aerobic oxidation of veratryl alcohol using water as the only solvent would be interesting.15 In experimental study, binuclear reaction mechanism has been proposed,15 similar to the mechanism in GOase functional complex.16 However, previous studies17,18 suggest that besides binuclear reaction mechanism, the mononuclear reaction mechanism is also possible for Cu/bipy-TEMPO systems.4,7,19 Therefore, based on the density functional theory, the reaction mechanism of aerobic veratryl alcohol oxidation catalyzed by CuII/1,10-phenanthroline has been studied by considering both mechanisms. We hope the obtained results could provide useful insights for the reaction process and are useful for future design of more efficient catalysts.2 Computational methods
The calculations were performed by use of Gaussian 03 suite of programs.20 In the model reaction, the substrate veratryl alcohol is simplified to benzyl alcohol. Geometry optimizations and thermal corrections are performed with B3LYP21 functional and the standard 6-31G(d) basis set. Relative free energies are obtained by single point calculations at B3LYP/6-311+G(d,p) and by including thermal corrections to free energies at B3LYP/6-31G(d). This includes entropy contributions by taking into account the vibrational, rotational and translational motions of the species at 298.15 K. Open-shell calculations for anti-ferromagnetic coupled singlet state usually result in a spin contamination. Therefore, an energy correction was estimated from the Heisenberg spin–Hamiltonian formalism.22 Similar energy correction approach was used in the previous studies.23,24 The spin densities (ρ) were generated by Mulliken population analysis with a larger basis set (6-311+G(d,p)). The intrinsic reaction coordinate (IRC) approach was used to confirm that the transition state connects the two relevant minima.25 The solvent effect on the potential energy surface was investigated by single-point calculations at the B3LYP/6-311+G(d,p) level with the conductor-like polarized continuum solvent model (CPCM)26 using water (ε = 78.39) as solvent. All the energies values are obtained at solvent (and in kcal mol−1).3 Result and discussion
The catalytic cycle of Cu–phen system consists of two parts, namely, alcohol oxidation and O2 reduction (Scheme 1). The corresponding cartesian coordinates of all structures provided in the ESI.† For the alcohol oxidation, two paths were proposed, i.e., mononuclear mechanism (path A) and binuclear mechanism (path B). For the O2 reduction, two paths were proposed as well, i.e., path I and path II. The corresponding energy profiles and the optimized geometries for the alcohol oxidation and O2 reduction are given in Fig. 1–3, respectively. | ||
| Scheme 1 Proposed catalytic mechanisms of the title complexes. The green, purple colors represent path A and path B. The red, blue colours represent path I and path II, respectively. (The superscript “r” represents closed shell singlet state.) | ||
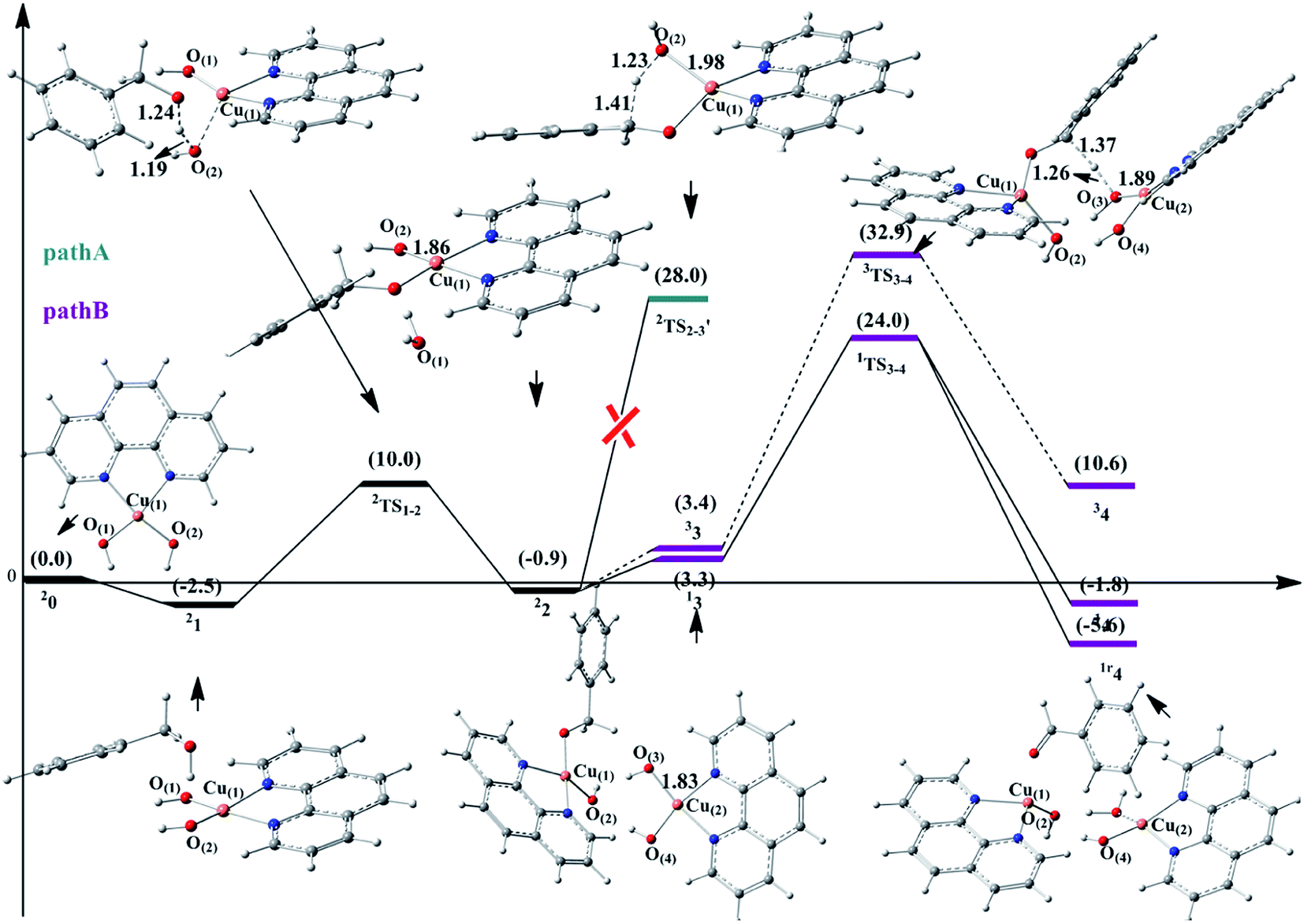 | ||
| Fig. 1 The optimized geometries and energy profile of the reactant, intermediates, transition states and product for substrate oxidation in path A and path B. The dashed line represents triplet spin state; solid line represents open-shell state. The energy values are in kcal mol−1 and bond lengths are in Å. (The superscript “r” represents closed shell singlet state. The orange, red, white balls represent Cu atoms, O atoms and H atoms, respectively. The blue, gray balls represent N atoms and C atoms, respectively.) | ||
 | ||
| Fig. 2 The optimized geometries and energy profile of the reactant, intermediates, transition states and product for O2 reduction in path I. The dashed line represents triplet spin state; solid line represents singlet state. The energy values are in kcal mol−1 and bond lengths are in Å. (The orange, red, white balls represent Cu atoms, O atoms and H atoms, respectively. The blue, gray balls represent N atoms and C atoms, respectively.) | ||
 | ||
| Fig. 3 The optimized geometries and energy profile of the reactant, intermediates, transition states and product for O2 reduction in path II. The dashed line represents triplet spin state; solid line represents singlet state. The energy values are in kcal mol−1 and bond lengths are in Å. (The orange, red, white balls represent Cu atoms, O atoms and H atoms, respectively. The blue, gray balls represent N atoms and C atoms, respectively.). | ||
3.1 Substrate oxidation
For 13, the positive spin density of ρ = 0.59 on Cu(1), ρ = 0.13 on phen(1), ρ = 0.11 on O(2)H, and ρ = 0.13 on the substrate benzyl alcohol are accompanied by a spin density of opposite sign of ρ = −0.56 on Cu(2), ρ = −0.13 on phen(2), ρ = −0.15 on O(3)H, and ρ = −0.14 on O(4)H. According to the spin density distribution, the complex 13 possesses{[Cu(1)II(phen)(1)(O(2)H)(OCH2Ph)][Cu(2)II(phen)(2)(O(3)H)(O(4)H)]}character. For the product of the H atom abstraction (1r4), all the spin densities of the atoms in 1r4 are zero. 1r4 is best formulated as {[Cu(1)I(phen)(1)(O(2)H)(OCHPh)][Cu(2)I(phen)(2)(O(4)H)]}. In comparison with 13 and 14, the above change indicates that the oxidation state of Cu for both Cu(1) and Cu(2) center is reduced from CuII to CuI, accompanied by the formation of H2O and the product PhCHO, which is consistent with the experimental observation.
To further investigate the H atom abstraction, the electron transfer process during 13 → 1TS3–4 is discussed. In 1TS3–4, the spin density on the Cu(2) center is −0.56, indicating that Cu(2) center is in the CuII oxidation state. This means that the homolytic cleavage of the Cu(2)–O(3)H bond does not occur in 1TS3–4. This conclusion could be supported by the Cu(2)–O(3)H bond length change (13 → 1TS3–4: 1.83 Å → 1.89 Å). The bond length change for the similar homolytic cleavage of the Cu–OH bond should be about 0.26 Å.18 Therefore, the homolytic cleavage of the Cu(2)–O(3)H bond did not happen in 1TS3–4. Namely, the spin density located on O(3)H should not be affected by the Cu(2)–O(3)H bond in 1TS3–4. However, for the O(3)H group, the spin density changes from −0.15 to −0.05 in the process of 13 → 1TS3–4, indicating that a very small fraction of α-spin electron has been migrated to the O(3)H species. This fraction of the α-spin electron comes from the homolytic cleavage of Cα–H bond of –OCH2Ph. The corresponding β-spin electron generating from homolytic cleavage of Cα–H bond migrates to the substrate, which should result in the decrease of the α-spin density on the substrate. However, it is interesting to mention that the α-spin density on the substrate increases from +0.16 to +0.55, indicating some α-spin densities migrate to the substrate. In addition, the spin density on Cu(1) center decreases from +0.59 to +0.3 (13 → 1TS3–4), indicating that a small fraction of β-spin electron transferring to the Cu(1) center. The α-spin densities migrating to the substrate and the β-spin electron migrating to the Cu(1) center could trace to a fraction of the homolytic cleavage of the Cu(1)–OCHPh bond. This could give a fraction of β-spin electron to the Cu(1) atom and α-spin electron to the substrate radical, which results in the decrease of the α-spin density on Cu(1) center (+0.59 → +0.3), coupled with the increase of the α-spin density on OCHPh (+0.13 → +0.55). In a word, in the process of 13 → 1TS3–4, two bonds were partially homolytic cleavage (partially homolytic cleavage of Cu(1)–OCHPh bond and minor homolytic cleavage of the Cα–H bond of the –OCH2Ph group). This conclusion is further supported by the molecular orbitals for 13 → 1TS3–4 (Scheme 2). In the process of 13 → 1TS3–4, the increase of a small fraction of the α-spin electron on substrate and a new small fraction of the β-spin electron distributed on Cu(1) indicates the partial homolytic cleavage of the Cu(1)–OCHPh bond. Moreover, a small fraction of the α-spin electron on O(3)H and a new small fraction of the β-spin electron located on substrate implies the partial homolytic cleavage of the Cα–H bond of the –OCH2Ph group. Thus, only Cu(1)–OCHPh bond and Cα–H bond are partially homolytic cleavage in the process of 13 → 1TS3–4.
 | ||
| Scheme 2 Molecular orbitals isosurface for 13 and 1TS3–4. SOMO indicates singly occupied molecular orbital. The red, blue colors represent the occupied molecular orbital for the homolytic cleavage of Cu(1)–OCHPh bond and Cα–H bond, respectively (contour values = ±0.04). | ||
3.2 O2 reduction
For the O2 reduction step, two pathways (path I and path II) are proposed based on the experimental observation (Scheme 1). In path I, O2 initially replaces the product PhCHO to coordinate to the Cu(1) center in 4 generating 5. Then, after the H atom and a proton transfer steps, the H2O2 and PhCHO are formed (6 → 7). Finally, the release of the H2O2 and PhCHO will result in the regeneration of 20. In contrast, in path II, the H2O and product PhCHO dissociate before O2 binding, generating of two units of 8, which is formulated as [CuI(phen)(OH)]. Subsequently, another alcohol molecule coordinates to the [CuI(phen)(OH)] complex (9 → 10), followed by the O2 binding to the CuI center to give 11. Then, after the proton and H atom transfer steps, alcohol is converted to aldehyde (11 → 12). Finally, the release of the H2O2 and product PhCHO will result in the regeneration of 8.In summary, the 1r4 → 35 process is exothermic by 26.8 kcal mol−1 and the energy barrier for path I (about 3 kcal mol−1) is observed (22.3 kcal mol−1 in path II). Therefore, for the O2 oxidation reaction, path I is the preferred pathway.
3.3 Calculations of the veratryl alcohol oxidation
Because the substrate veratryl alcohol was simplified to benzyl alcohol, which could introduce extra errors in the activation barriers, we performed calculations on benzyl alcohol as the substrate for the critical reaction steps in both path A and path B. On the basis of our calculations, the geometry structures (Fig. 4) and the energy barriers for the critical reaction steps in path A and path B are quite similar for veratryl alcohol and benzyl alcohol. As an example, the energy barriers for the H abstraction step (rate-determining step in path B) for veratryl alcohol and benzyl alcohol are within 0.5 kcal mol−1. And the critical bond lengths are shown in the following Table 1. As seen in the table below, the differences are within 0.03 Å. This implies that benzyl alcohol represents a good trade-off between accuracy and computational cost in exploring the whole energetic profile. | ||
| Fig. 4 The optimized geometries for the transition states in both substrate benzyl alcohol and veratryl alcohol. The bond lengths are in Å. (The orange, red, white balls represent Cu atoms, O atoms and H atoms, respectively. The blue, gray balls represent N atoms and C atoms, respectively.). | ||
| Model | O(1)⋯H | C⋯H | Cu(1)–O(2) | Cu(2)–O(1) |
|---|---|---|---|---|
| Benzyl alcohol | 1.19 Å | 1.45 Å | 1.86 Å | 1.91 Å |
| Veratryl alcohol | 1.20 Å | 1.42 Å | 1.87 Å | 1.91 Å |
3.4 wB97XD
wB97XD is the latest functional from Head-Gordon and coworkers, which includes empirical dispersion.30 Therefore, we chose the wB97XD functional as the alternative functional to recalculate the critical reaction steps in both path A and path B. For the open shell singlet state, the large spin contamination (S2 > 3) for wB97XD was observed, implying wB97XD is not suitable to describe the open shell singlet. But for the triplet state, wB97XD functional gives the very similar geometry structures and the energy barriers compared to B3LYP. In addition, B3LYP has been tested extensively in many studies of molecular structures and is a suitable functional for describing the open shell singlet.29 Thus, we suppose the B3LYP functional give reliable results in this catalytic system.4 Conclusions
The catalytic mechanism for the oxidation of veratryl alcohol to veratraldehyde by Cu–phen (phen = 1,10-phenanthroline) catalyst have been performed by use of density functional method. The catalytic cycle consists of two parts, namely, alcohol oxidation and O2 reduction. For the alcohol oxidation, two mechanisms were proposed, i.e., mononuclear mechanism and binuclear mechanism. The calculations show that the H-atom transfer step within the alcohol oxidation is the rate-determining step for both paths. The calculated overall reaction barrier for path A is 28.9 kcal mol−1, larger than 24.9 kcal mol−1 for path B, indicating that path B is favorite. Namely, for the Cu–phen (phen = 1,10-phenanthroline) catalytic system, the mechanism is the binuclear mechanism, in agreement with experiment. In addition, for the O2 reduction, two possible paths are proposed as well (path I and path II). According to our calculations, path I is favored.Acknowledgements
The authors thank the National Natural Science Foundation of China (NSFC) (Grant no. 21343007) and the Inner Mongolia University of Technology (Grant no. ZD201207) for financial support.References
- J. M. Hoover, B. L. Ryland and S. S. Stahl, J. Am. Chem. Soc., 2013, 135, 2357 CrossRef CAS PubMed.
- (a) L. Wolfgang, T. Michael, S. Erichand, W. W. Karl, Ullman's Encyclopedia of Industrial Chemistry, V. C. H. Wiley, Weinheim, 6th edn, 2002, vol. 12, p. 609 Search PubMed; (b) Blackie Academic & Proffesional, Principles of Organic Synthesis, 3rd edn. London, 1993, p. 96 Search PubMed; (c) R. V. Stevens, K. T. Chapman and H. N. J. Weller, Org. Chem., 1980, 45, 2030 CrossRef CAS; (d) J. R. J. Holum, Org. Chem., 1961, 26, 4814 CrossRef CAS; (e) D. G. Lee and U. A. J. Spitzer, Org. Chem., 1970, 35, 3589 CrossRef CAS; (f) R. J. Highet and W. C. Wildman, J. Am. Chem. Soc., 1955, 77, 4399 CrossRef CAS; (g) F. M. Menger and C. Lee, Tetrahedron Lett., 1981, 22, 1655 CrossRef CAS; (h) L. M. Berkowitz and P. N. Rylander, J. Am. Chem. Soc., 1958, 80, 6682 CrossRef CAS.
- (a) J. W. Whittaker, H. Sigel, A. Sigel and M. Dekker, The free radical-coupled copper active site of galactose oxidase, New York, 1994, vol.30, p. 315 Search PubMed; (b) P. F. Knowles, Bio-Inorg. Chem., 1994, 2, 207 Search PubMed.
- P. Gamez, I. W. C. E. Arends, R. A. Sheldon and J. Reedijk, Adv. Synth. Catal., 2004, 346, 805 CrossRef CAS.
- Z. Lu, J. S. Costa, O. Roubeau, I. Mutikainen, U. Turpeinen, S. J. Teat, P. Gamez and J. Reedijk, Dalton Trans., 2008, 27, 3567 RSC.
- N. Mase, T. Mizumori and Y. Tatemoto, Chem. Commun., 2011, 47, 2086 RSC.
- P. Gamez, I. W. C. E. Arends, J. Reedijk and R. A. Sheldon, Chem. Commun., 2003, 2414 RSC.
- J. S. Uber, Y. Vogels, D. van den Helder, I. Mutikainen, U. Turpeinen, W. T. Fu, O. Roubeau, P. Gamez and J. Reedijk, Eur. J. Inorg. Chem., 2007, 26, 4197 CrossRef.
- M. F. Semmelhack, C. R. Schmid, D. A. Cortés and C. S. Chou, J. Am. Chem. Soc., 1984, 106, 3374 CrossRef CAS.
- J. S. Uber, Y. Vogels, D. van den Helder, I. Mutikainen, U. Turpeinen, W. T. Fu, O. Roubeau, P. Gamez and J. Reedijk, Eur. J. Inorg. Chem., 2007, 26, 4197 CrossRef.
- J. M. Hoover and S. S. Stahl, J. Am. Chem. Soc., 2011, 133, 16901 CrossRef CAS PubMed.
- J. F. Greene, J. M. Hoover, D. S. Mannel, T. W. Root and S. S. Stahl, Org. Process Res. Dev., 2013, 17, 1247 CrossRef CAS.
- N. J. Hill, J. M. Hoover and S. S. Stahl, J. Chem. Educ., 2013, 90, 102 CrossRef CAS.
- A. Sheldon, P. J. Dunn, A. S. Wells and M. T. Williams, Green Chemistry in the Pharmaceutical Industry, 2010, p. 1 Search PubMed.
- H. Korpi, P. J. Figiel, E. Lankinen, P. Ryan, M. Leskel and T. Repo, Eur. J. Inorg. Chem., 2007, 17, 2465 CrossRef.
- T. T. K. Esa and M. P. K. Ari, Chem.–Eur. J., 2009, 15, 10901 CrossRef PubMed.
- L. Cheng, J. P. Wang, M. Y. Wang and Z. J. Wu, Inorg. Chem., 2010, 49, 9392 CrossRef CAS PubMed.
- L. Cheng, J. P. Wang, M. Y. Wang and Z. J. Wu, Dalton Trans., 2010, 39, 5377 RSC.
- P. J. Figiel, M. Leskelä and T. Repo, Adv. Synth. Catal., 2007, 349, 1173 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery, T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. M. Nakai, X. Klene, J. E. Li, H. P. Knox, J. B. Hratchian, C. Cross, J. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, J. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, Gaussian 03, Gaussian, Inc., Pittsburgh, PA, 2003 Search PubMed.
- (a) A. J. Cohen and N. C. Handy, Mol. Phys., 2001, 99, 607 CrossRef CAS; (b) J. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS.
- (a) J. M. Mouesca, J. L. Chen, L. Noodleman, D. Bashford and D. A. Case, J. Am. Chem. Soc., 1994, 116, 11898 CrossRef CAS; (b) L. Noodleman, J. Chem. Phys., 1981, 74, 5737 CrossRef CAS PubMed; (c) L. Ciofini and C. A. Daul, Coord. Chem. Rev., 2003, 238 Search PubMed.
- V. Bachler, G. Olbrich, F. Neese and K. Wieghardt, Inorg. Chem., 2002, 41, 4179 CrossRef CAS PubMed.
- P. E. M. Siegbahn and V. Pelmenschikov, J. Am. Chem. Soc., 2006, 128, 7466 CrossRef PubMed.
- (a) K. Fukui, Acc. Chem. Res., 1981, 14, 363 CrossRef CAS; (b) K. Fukui, J. Phys. Chem., 1970, 74, 4161 CrossRef CAS.
- (a) V. Barone and M. Cossi, J. Phys. Chem. A, 1998, 102, 1995 CrossRef CAS; (b) M. Cossi, G. Scalmani, N. Rega and V. Barone, J. Comput. Chem., 2003, 24, 669 CrossRef CAS PubMed.
- (a) H. Hirao, D. Kumar, W. Thiel and S. Shaik, J. Am. Chem. Soc., 2005, 127, 13007 CrossRef CAS PubMed; (b) S. Cohen, S. Kozuch, C. Hazan and S. Shaik, J. Am. Chem. Soc., 2006, 128, 11028 CrossRef CAS PubMed; (c) E. Derat and S. Shaik, J. Am. Chem. Soc., 2006, 128, 8185 CrossRef CAS PubMed; (d) D. Kumar, S. P. de Visser and S. Shaik, Chem.–Eur. J., 2005, 11, 2825 CrossRef CAS PubMed; (e) M. Guell, J. M. Luis, P. E. M. Siegbahn and M. J. Sola, Biol. Inorg. Chem., 2009, 14, 273 CrossRef CAS PubMed.
- H. Hirao, D. Kumar, L. Que, Jr and S. Shaik, J. Am. Chem. Soc., 2006, 128, 8590 CrossRef CAS PubMed.
- G. Jürgen, K. Elfi, F. Michael and C. Dieter, Int. J. Mol. Sci., 2002, 3, 360 CrossRef PubMed.
- M. Yury, S. Åsmund and O. Giovanni, Dalton Trans., 2012, 41, 5526 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Table S1 contains the cartesian coordinates of all the structures considered in this work from the B3LYP optimized geometries. See DOI: 10.1039/c4ra02896a |
| This journal is © The Royal Society of Chemistry 2014 |