Cu(I)-catalyzed tandem decarboxylative/C–H activation coupling of cyclic diketones, proline and alkynes: synthesis of α-alkynylated pyrrolidine-oxyindoles†
Atul Kumar*,
Mukesh Kumar,
Lalit Prakash Gupta and
Maneesh Kumar Gupta
Medicinal and Process Chemistry Division, Central Drug Research Institute, CSIR, Lucknow, India. E-mail: dratulsax@gmail.com; Fax: +91-522-26234051; Tel: +91-522-2612411
First published on 15th January 2014
Abstract
An efficient ligand/base and oxidant-free copper(I) catalyzed intermolecular direct alkynylation (IDA) strategy has been developed for the synthesis of α-alkynylated pyrrolidine-oxindole derivatives using cyclic diketones, amino acids and alkynes via tandem decarboxylative/C–H activation and reductive-amination strategy.
The carbon–carbon bond formation by transition-metal-catalyzed coupling has reinvigorateded the science of synthesis due to its inventive contribution to the synthetic organic chemistry. Generally, these synthetic methodologies involve expensive metal catalysts, and preformed “organo-metallic reagents” that produce unwanted, often toxic, stoichiometric side-products.1
With increasing environmental concerns and waste management, especially in the area of pharmaceuticals, development of methods that avoid preformed “organo-metallic reagents” and organic solvents are very important because the removal of metallic impurities from final pharmaceutical entities increases the cost considerably. Construction of new C–C bond by decarboxylative coupling reaction is also a powerful and attractive alternative synthetic method because of their high efficiency, selectivity and convenience.2 Further, extrusion of CO2 as the waste product during the reaction is considered to be environmentally benign and requires no special separation procedures.
Transition-metal catalyzed decarboxylative coupling reaction of amino acids provides new synthetic route for the construction of C–C or C–N bond. For examples, Cohen et al. reported a decarboxylative reaction of proline with sterically congested 2-hydroxyacetophenones in 1979.3 In 2008, Seidel et al.described the synthesis of aminals by the coupling reaction of proline with 2-aminobenzaldehyde.4 Recently, Li's and co-worker reported C–C bond-forming reaction using copper- or iron-catalyzed oxidative decarboxylative coupling of sp3-hybridized carbons with N-benzylproline.5a Concurrently, Seidel6 and Li's group5b,c also reported new methods involving aldehyde-induced intermolecular tandam decarboxylative coupling reaction of amino acids and alkynes to afford propargylic amino acids derivatives (Fig. 1). These new reactions increases the scope and synthetic utility of the catalytic decarboxylative coupling reactions. However, these decarboxylative coupling protocols requiring complex ligands, long reaction time, harsh reaction conditions, and superstoichiometric amount of oxidants or transition metals besides copper to facilitate the reaction. However, to the best of our knowledge, there are no literature examples describing the synthesis of α-alkynylated pyrrolidine-oxyindole via tandam decarboxylative coupling reaction using isatin and cyclic aromatic diketones.
 | ||
| Fig. 1 Comparison of different intermolecular decarboxylative cross coupling protocols. | ||
The oxindole skeleton has been widely known as a privileged scaffold in naturally occurring alkaloids and biologically active molecules.7 Furthermore, Pyrrolidine-oxyindoles also forms the core unit of several medicinal and natural spiropyrrolidine-oxindole alkaloids like nelivaptan, spirotyrpstatin and compound I etc.8 (Fig. 2)
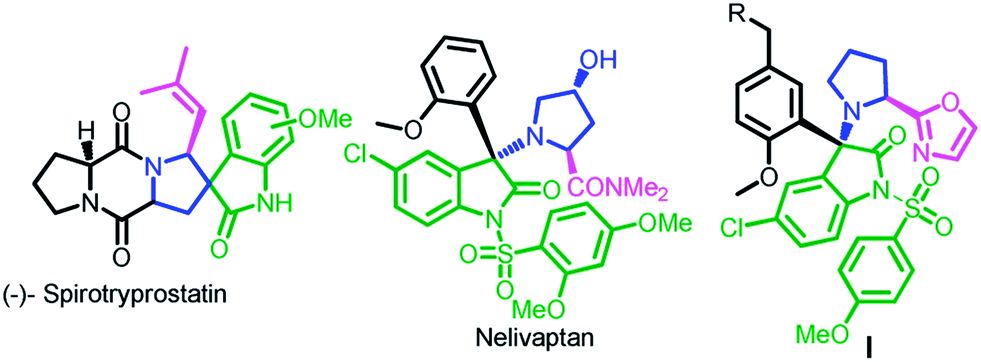 | ||
| Fig. 2 Important biologically active compounds possessing pyrrolidine-oxindoles motif. | ||
The direct and straightforward approach for the synthesis of α-phenyl-ethynyl pyrrolidine-oxyindole via tandem decarboxylative/C–H activation in one-pot remains a challenging task. Herein, we wish to describe, the highly efficient Cu(I)-catalyzed intermolecular direct alkynylation (IDA) of in situ generated oxindoles-pyrrolidine ylide via tandem decarboxylative/Csp3-Csp C–H activation coupling reaction using aromatic cyclic ketones, α-amino acid and alkyne for the synthesis of α-alkynylated pyrrolidine-oxyindole without use of any ligant and oxidants outlined in Scheme 1.
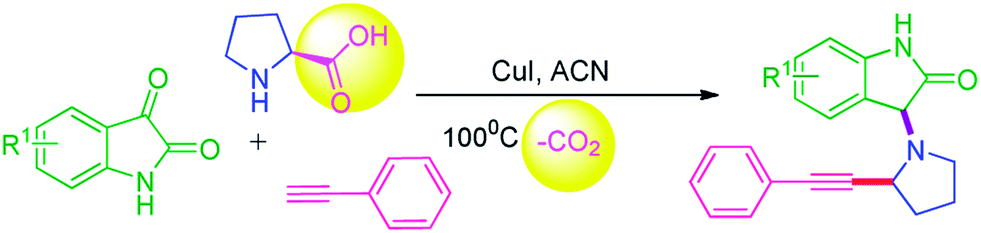 | ||
| Scheme 1 Synthesis of α-alkynylated pyrrolidine-oxyindole. | ||
As illustrated in Table 1, our initial investigation were initiated employing isatin (1a), proline (2a) and phenylacetylene (3a) as model substrate to find the optimal reaction conditions. Our first attempts was focused on to carry out the reaction in the absence of any catalysts in 5 mL of acetonitrile at 100 °C for 8 hours, unfortunately the desired coupling product 4a was not observed (Table 1, entry 1). The desired 4a was obtained in 20% yield when reaction was carried out in 15 mol% of CuBr (Table 1, entry 2). Delightfully, 65% yield of 4a was achieved when 15 mol% of CuI was employed as catalyst (Table 1, entry 3). To further improve the yield, we used different amount of CuI, and the best yield (82%) of coupling product 4a was obtained with 20 mol% of CuI in 6 hours (Table 1, entries 4, 5, and 6). Among the copper catalysts examined, CuI was the most successful catalyst for this reaction compared to CuCl2, Cu(OTf)2, Cu(OAc)2, Cu2O, and CuBr2 (Table 1, entries 14–18). We attempts to use other catalyst like PdCl2 and Pd(OAc)2, but the trials were also unsuccessful (Table 1, entries 19 and 20). The reaction was explored in different solvents as well. Preeminent result was found when reaction was performed in acetinitrile as reaction medium, the coupling product 4a could be isolated in 82% yield, whereas no observable improvement was achieved in other solvents like toluene, DMSO, DMF, THF, dioxane, ethanol, and methanol (Table 1, entries 4, and 7–13).
|
||||
|---|---|---|---|---|
| Entry | Catalyst (mol%) | Solvent | t (h) | Yieldb (%) |
| a Reaction conditions: Isatin 1a (1 mmol), proline 2a (1.5 mmol), and alkyne 3a (2 mmol), in 5 mL of solvent in the presence of catalysts at refluxed condition under nitrogen atmosphere.b Yield of the isolated product.c Refluxed at 110 °C. Nr = not reacted. DMSO = dimethylsulfoxide; DMF = N,N-dimethylformamide; THF = tetrahydrofuran. | ||||
| 1 | — | CH3CN | 8 | 0 |
| 2 | CuBr (15) | CH3CN | 6 | 20 |
| 3 | CuI (15) | CH3CN | 6 | 65 |
| 4 | CuI (20) | CH3CN | 6 | 82 |
| 5 | CuI (10) | CH3CN | 7 | 47 |
| 6 | CuI (5) | CH3CN | 7 | 35 |
| 7 | CuI (20) | Toluene | 6 | 40 |
| 8c | CuI (20) | DMSO | 6 | 25 |
| 9c | CuI (20) | DMF | 6 | 27 |
| 10 | CuI (20) | THF | 6 | 30 |
| 11 | CuI (20) | Dioxane | 6 | 20 |
| 12 | CuI (20) | Ethanol | 6 | 12 |
| 13 | CuI (20) | Methanol | 6 | 10 |
| 14 | CuCl2 (20) | CH3CN | 6 | 12 |
| 15 | Cu(OTf)2(20) | CH3CN | 8 | 15 |
| 16 | Cu(OAc)2(20) | CH3CN | 8 | <10 |
| 17 | Cu2O (20) | CH3CN | 6 | <5 |
| 18 | CuBr2 (20) | CH3CN | 6 | <5 |
| 19 | PdCl2 (15) | CH3CN | 8 | n.r. |
| 20 | Pd(OAc)2 (15) | CH3CN | 8 | n.r. |
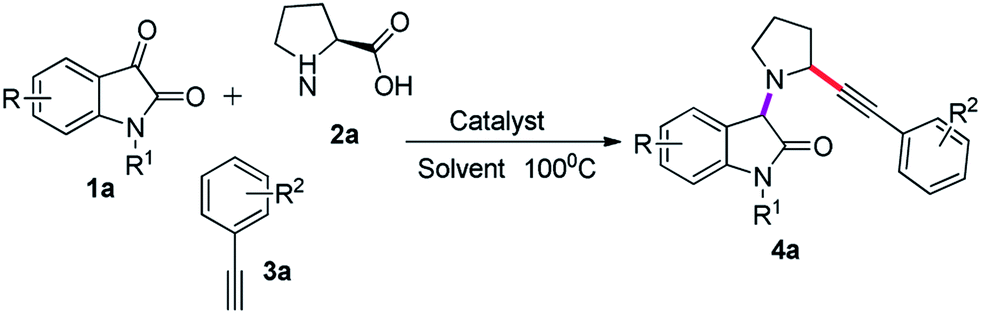
With the optimized reaction conditions established, we then examined the scope of decarboxylative coupling reaction, various isatin derivatives reacted with proline and alkyne under optimized conditions, and the results are illustrated in Fig. 3. A wide range of isatin bearing chloro, bromo, nitro, flouro, and iodo at the 5th position along with electron-neutal isatin 1a could be employed as coupling partner with proline 2a which were smoothly transformed to the corresponding α-substituted pyrrolidine-oxindole 4a with phenylacetylene 3a in good to excellent yield by NMR (Fig. 3 entries 4a–4f). N-substituted isatin like methyl, ethyl, propyl, butyl, and benzyl also furnished desired product in good to moderate yield (Fig. 3, entries 4g–4k).
 | ||
| Fig. 3 Synthesized α-alkynylated pyrollidine-oxindoles. | ||
Subsequently, we also explored the use of various alkynes reacted under the same conditions in Fig. 3. Aromatic alkynes bearing methyl group at para and meta position afforded good yields (Fig. 3, entries 4l–4m). However, aliphatic alkyne like pentyne and hexyne did not react well and gave an inseparable complex mixture (Fig. 3, entries 4o–4p), whereas methylpropiolate furnished desired product in better yields (Fig. 3, 4n). All the synthesized product of the reaction was fully characterized by 1H and 13C NMR methods and mass spectroscopic data. A proposed mechanism for the Cu(I) catalyzed decarboxylation/C–H activation for the synthesis of α-substituted pyrolidine oxindoles in one-pot is illustrated in Scheme 2. Condensation of proline with isatin in the presence of CuI to gave imine-type intermediate A which converted into reactive intermediate spiro[indoline-3,3′-pyrrolo[1,2-c]oxazole]-1′,2-dione B. B immediately undergoes decarboxylation by CuI to afforded C which subsequently gave intermediate D. Intermediate D (pyrrolidine-type ylide) will be in equilibrium with D′. D or D′ further co-ordinated with copper and reacted with 3a to generated organo-copper complex intermediate E, which undergoes intramolecular nucleophilic addition to give desired coupling products and regenerates the copper catalyst for additional reaction.
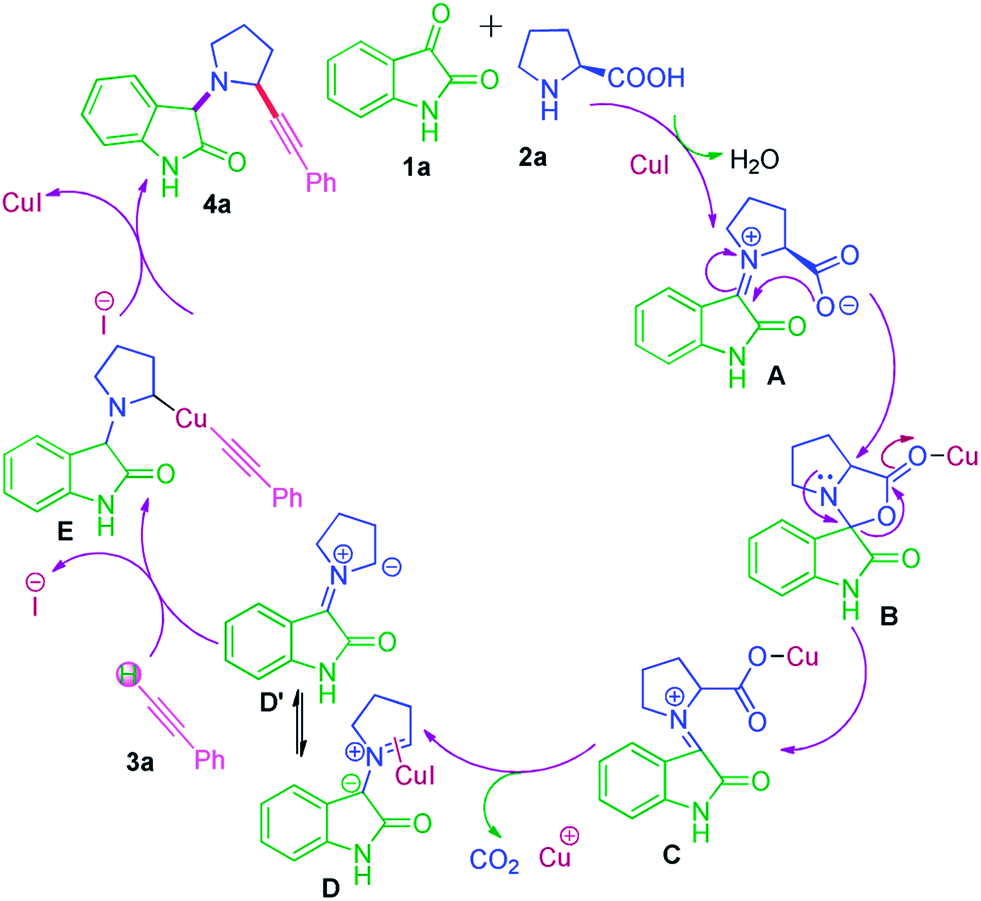 | ||
| Scheme 2 Proposed mechanism. | ||
This proposed mechanism readily illuminates the formation of the two regioisomers and suggest that the regioselectivity depends on the charge distribution in between intermediate D and D′ in addition to potential steric factor. The negative charge on intermediate D appear to stabilize with amidic oxygen of isatin that favours the protonation at 3-position of isatin and ultimately leads to the predominantly formation of regioisomer 4 whereas in case of intermediate D′ the protonation at 2-position of pyrrolidine ring is not favour since negative charge is not more stabilize at that position, resulted no formation of regioisomer 5. Steric factor also inhibit the formation of 5 Fig. 4.
 | ||
| Fig. 4 Proposed rationalization of regioselectivity. | ||
Using same reaction conditions we have also synthesized several pharmacologically valuable 2-(2-(phenylethynyl)pyrrolidin-1-yl)acenaphthylen-1(2H)-one,2-(2-(phenylethynyl)pyrrolidin-1-yl)aceanthrylen-1(2H)-one, and 2-(2-(phenylethynyl)pyrrolidin-1-yl)-1H-indene-1,3(2H)-dione in excellent yields using polycyclic di and triketones (Schemes 3–5).
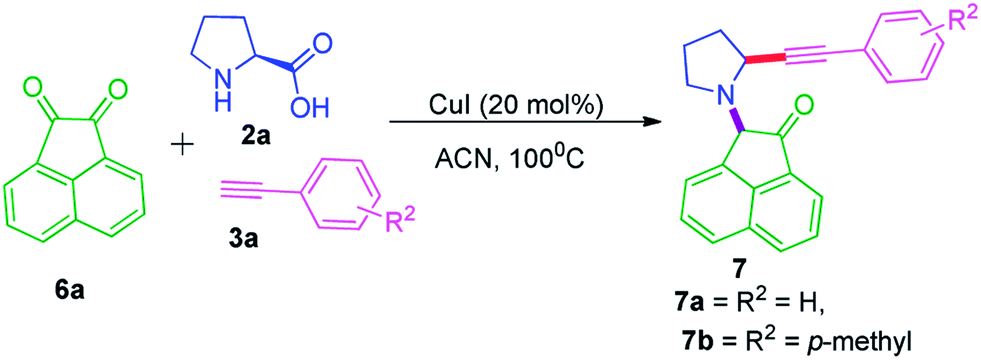 | ||
| Scheme 3 Synthesis of α-phenylethynyl pyrrolidine acenaphthylnone. | ||
 | ||
| Scheme 4 Synthesis of α-phenylethynyl pyrrolidine aceanthrylenone. | ||
 | ||
| Scheme 5 Synthesis of 2-(2-(phenylethynyl)pyrrolidin-1-yl)-1H-indene-1,3(2H)-dione. | ||
In conclusion, we have developed a novel copper(I)-mediated intermolecular tandem decarboxylative Csp3-Csp C–H activation for the direct synthesis of α-substituted pyrrolidine-oxyindoles through multi-component under mild reaction conditions. This intermolecular direct alkynylation (IDA) is important for environment as well as for providing a convenient synthetic pathway for pharmaceutically relevant pyrrolidine oxindoles, polycyclic alkynylated di and tri ketones. This methodology is also useful in the formation of important core of natural Spiro-alkaloids. Further studies on the application of this reaction are under way in our laboratory.
Acknowledgements
Authors MK, LPG and MKG are thankful to CSIR UGC New Delhi for the award of a senior research fellowship. We are also gratefully acknowledge SAIF, CDRI for providing analytical facilities. The CDRI communication no. is 8610.Notes and references
- (a) E. Negishi, L. F. Valente and M. Kobayashi, J. Am. Chem. Soc., 1980, 102, 3298–3299 CrossRef CAS; (b) E. Negishi, Acc. Chem. Res., 1982, 15, 340–348 CrossRef CAS; (c) X. Zeng, M. Quian, Q. Hu and E. Negishi, Angew. Chem., Int. Ed., 2004, 43, 2259–2263 CrossRef CAS PubMed; (d) V. Farina, V. Krishnamurthy and W. J. Scott, Org. React., 1997, 50, 1–652 CAS; (e) N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457–2483 CrossRef CAS; (f) J. P. Wolfe and S. P. Buchwald, Angew. Chem., Int. Ed., 1999, 38, 2413–2416 CrossRef CAS; (g) K. J. Sonogashira, J. Organomet. Chem., 2002, 653, 46–49 CrossRef CAS; (h) K. Tamao, K. Sumitani and M. Kumada, J. Am. Chem. Soc., 1972, 94, 4374–4376 CrossRef CAS; (i) Metal-Catalyzed Cross-Coupling Reactions, ed. F. Diederich and P. J. Stang, Wiley-VCH, New York, 1998 Search PubMed; (j) K. C. Nicolaou, P. G. Bulger and D. Sarlah, Angew. Chem., Int. Ed., 2005, 44, 4442–4489 CrossRef CAS PubMed.
- For a review, see: (a) L. J. Gooßen, N. Rodriguez and K. Gooßen, Angew. Chem., 2008, 120, 3144–3164 (Angew. Chem., Int. Ed., 2008, 47, 3100–3120) CrossRef; (b) J. G. Knight, S. W. Ainge, A. M. Harm, S. J. Harwood, H. I. Maughan, D. R. Armour, D. M. Hollinshead and A. A. Jaxa-Chamiec, J. Am. Chem. Soc., 2000, 122, 2944–2945 CrossRef CAS; (c) S. H. Sim, H.-J. Park, S. I. Lee and Y. K. Chung, Org. Lett., 2008, 10, 433–436 CrossRef CAS PubMed; (d) D. Bourgeois, D. Craig, N. P. King and D. M. Mountford, Angew. Chem., 2005, 117, 624–627 (Angew. Chem., Int. Ed., 2005, 44, 618–621) CrossRef; (e) G. Lalic, A. D. Aloise and M. D. Shair, J. Am. Chem. Soc., 2003, 125, 2852–2853 CrossRef CAS PubMed; (f) B. M. Trost and J. Xu, J. Am. Chem. Soc., 2005, 127, 17180 CrossRef CAS PubMed.
- N. Cohen, J. F. Blount, R. J. Lopresti and D. P. Trullinger, J. Org. Chem., 1979, 44, 4005 CrossRef CAS.
- C. Zhang, C. K. De, R. Mal and D. Seidel, J. Am. Chem. Soc., 2008, 130, 416 CrossRef CAS PubMed.
- (a) H.-P. Bi, L. Zhao, Y.-M. Liang and C.-J. Li, Angew. Chem., Int. Ed., 2009, 48, 792 CrossRef CAS PubMed; (b) H.-P. Bi, W.-W. Chen, Y.-M. Liang and C.-J. Li, Org. Lett., 2009, 11, 3246 CrossRef CAS PubMed; (c) H.-P. Bi, Q. F. Teng, M. Guan, W.-W. Chen, Y.-M. Liang, X. J. Yao and C.-J. Li, J. Org. Chem., 2010, 75, 783 CrossRef CAS PubMed.
- (a) C. Zhang and D. Seidel, J. Am. Chem. Soc., 2010, 132, 798 CrossRef PubMed; (b) C. Zhang, D. Das and D. Seidel, Chem. Sci., 2011, 2, 233 RSC.
- Review: C. Marti and E. Carreira, Eur. J. Org. Chem., 2003, 2209 CrossRef CAS.
- (a) M. N. G. James and G. J. B. Williams, Can. J. Chem., 1972, 50, 2407 CrossRef CAS; (b) A. Jossang, P. Jossang, A. H. Hamid, T. Sevenet and B. J. Bodo, J. Org. Chem., 1991, 56, 6527 CrossRef CAS; (c) C.-B. Cui, H. Kakeya and H. J. J. Osada, J. Antibiot., 1996, 49, 832 CrossRef CAS; (d) C.-B. Cui, H. Kakeya and H. J. Osada, Tetrahedron, 1996, 52, 12651 CrossRef CAS; (e) C. Serradeil-Le Gal, J. Wagnon, B. Tonnerre, R. Roux, G. Garcia, G. Griebel and A. Aulombard, CNS Drug Rev., 2005, 11, 53–68 CAS; (f) J. B. Robert, G. Griebel, C. Farrokhi, C. Markham, M. Yang and D. C. Blanchard, Pharmacol., Biochem. Behav., 2005, 80, 189–194 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3ra47238h |
| This journal is © The Royal Society of Chemistry 2014 |