The synthesis of a hierarchically porous BEA zeolitevia pseudomorphic crystallization†
Minkee
Choi
,
Kyungsu
Na
and
Ryong
Ryoo
*
Center for Functional Nanomaterials, Department of Chemistry, Graduate School of Nanoscience and Technology (WCU), KAIST, Daejeon 305-701, Korea. E-mail: rryoo@kaist.ac.kr; Fax: (+82)42-350-8130
First published on 22nd April 2009
Abstract
A synthesis route to a hierarchically meso-/microporous BEA zeolite has been developed with a cyclic diammonium as a structure-directing agent (SDA), which leads to pseudomorphic crystallization of the zeolite by suppressing the mobility of silicates during crystallization.
Crystalline microporous zeolites can be synthesized with a morphology that has 3-dimensionally interconnected intra- or intercrystalline mesoporosity.1–13 Such zeolites, with a micro-/mesoporous hierarchical structure, have attracted much attention in recent years due to their improved diffusion,12catalytic activity, selectivity and lifetime.6,13 Significant efforts have been devoted to the development of preparation methods for hierarchical zeolites, such as mesopore generation with a hard template,1–5 the use of organosilane surfactants or polymers as a soft template,6–8,13 packing of nanosized zeolite crystals,9 and demetallation of zeolites for the post-synthetic generation of mesopores.10–12 In the present work, we demonstrate a new approach to hierarchical zeolites, namely, pseudomorphic transformation of porous silica gel using a 3,10-diazoniabicyclo[10.2.2]hexadeca-12,14,15-triene, 3,3,10,10-tetramethyl-dichloride (designated as CD for the cyclic diammonium-type structure) as a zeolite structure-directing agent (SDA).
Zeolite crystallization can be generalized by a ‘solution-mediated’ mechanism.14 According to this mechanism, a gel-type or solid-like silica source becomes partially dissolved during the initial stage of crystallization, so that there is an equilibrium between undissolved and dissolved species. The dissolved species re-assembles into crystalline zeolite by the aid of an SDA. If the mobility of the dissolved species is very high, it can migrate to a remote growth site of developing crystals and consequently, the zeolite crystal has a very different morphology from the initial gel morphology. On the other hand, if the mobility is very low, or the dissolved silicate species is immediately captured by an SDA to form a zeolite crystal, the morphology of the initial gel is maintained in the final product. This limiting case (i.e., pseudomorphiccrystallization) was traditionally described as ‘solid-state’ crystallization. To the best of our knowledge, however, no zeolite crystallization has been free from a morphological change, even at the submicron length scale. Even steam-assisted crystallizations of dried gel have shown significant diffusion.14,15–17
Here, we report that when the cyclic diammonium in Scheme 1 (CD) was used as an SDA, BEA zeolite could be synthesized according to the pseudomorphic crystallization in nanoscale. The pseudomorphic crystallization led to the formation of BEA zeolite with mesoporosity, whereas the SDA gelated silicates into a highly mesoporous gel structure (Scheme 1). Furthermore, when a hierarchically structured silica source like diatomaceous earth was used, the initial morphology was maintained in the zeolite product.
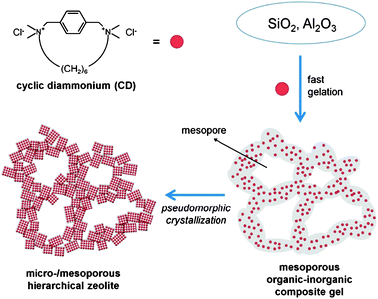 | ||
| Scheme 1 A schematic representation for the ‘pseudomorphic crystallization’ of a hierarchical BEA zeolite in the presence of cyclic diammonium (CD) as a structure-directing agent. | ||
The addition of CD immediately caused gelation, even under the highly alkaline conditions used in the present zeolite synthesis (see the ESI†). Approximately 90% of the silicate in the synthesis composition was precipitated as an organic-silicate composite gel less than 1 min after the CD mixing. As shown in the TEM images (Fig. 1), the initial gel (0 h) possessed a highly mesoporous structure with disordered connectivity. This result indicated that the CD molecules acted as a very effective gelator. This is in contrast to the formation of clear sol by tetraethylammonium (TEA+), which is a typical SDA in BEA synthesis.18–21 After 4 h crystallization with CD, small crystalline domains (<10 nm) were developed within the amorphous gel. After 24 h, the gel domains were completely converted into zeolite. N2 adsorption isotherms (see the ESI†) indicated that the zeolite had a similar mesopore volume and pore size distribution as compared to the initial gel phase. A high-resolution TEM image (Fig. 1, bottom panel) showed that the mesoporous framework was composed of interconnected zeolite nanocrystals of 10–15 nm in diameter. The crystal size was similar to the original framework thickness in the initial gel structure. Very broad XRD peak widths were in good agreement with the nanosized crystals (see the ESI†). These results support that the original mesoporous structure of amorphous gel was almost retained after the complete crystallization into BEA zeolite.
 | ||
| Fig. 1 High-resolution TEM images of solid products collected during the crystallization of hierarchical BEA zeolite at 170 °C. | ||
Elemental analysis revealed that the initial gel phase (0 h) and the final zeolite product (24 h) had about the same CD content (20 wt% based on dried weight at 130 °C), indicating that there was no significant change in the framework composition during crystallization. This is in contrast to previous studies using other SDAs, where there was a significant compositional change.14,18 In the conventional BEA synthesis, Na+ and K+ were the predominant cations in the initial gel, and these cations were replaced by TEA+ after prolonged hydrothermal crystallization.18 Besides, calcination of the initial gel generated ca. 50% of the microporosity of the fully crystalline BEA zeolite. The similar compositions and microporosity between amorphous and zeolite phases in the present work suggest that the initial amorphous phase could possess a zeolite-like locally organized silicate structure around organic SDA. Such ‘pre-organized’ phases (exhibiting similar compositions and microporosities to those of zeolite, but X-ray amorphous) were detected in previous studies, but only after prolonged hydrothermal crystallization.14,18
Conventionally, the syntheses of BEA zeolites were performed with tetraethylammonium hydroxide (TEAOH) and alkali-deficient silica sources, such as colloidal silica, fumed silica and tetraethylorthosilicate.18–20 When TEA+ was used as an SDA, the presence of Na+ above a certain concentration resulted in the formation of MFI and MOR zeolites.21 In contrast to previous results, the present CD led to the generation of a pure BEA structure with twelve-membered microporous channels, even under high concentrations of Na+ (see the ESI†). Notably, CD molecules could generate a pure BEA product, even in the presence of tetrapropylammonium (TPA+) ions as a competing SDA. This result is very surprising as TPA+ was known as the most effective SDA for MFI zeolite,22 and it generated MFI zeolite even in the presence of other organic amines and alkylammonium cations.23 Our result demonstrated the extraordinary ability of CD for BEA structure direction.
The strong interaction of CD with silicates and its effective structure-directing ability to BEA zeolite may be attributed to its unique molecular structure. Multiple electrostatic interactions between silicate anions and two ammonium sites would be advantageous for the silicate polymerization (rapid gelation) as compared to ordinary quaternary ammoniums (e.g., TEA+ and TPA+). Geometric optimization by molecular dynamics revealed that the CD molecule has approximately an ellipsoidal geometry (11.6 × 5.5 Å). The molecular dynamics simulation indicated that conformational change by thermal rotation and vibration is greatly suppressed, even at elevated temperature (200 °C), due to the rigid phenyl moiety in the cyclic geometry. Such a limited conformational freedom is known as an important prerequisite for the selective structure direction of zeolites.24 The shortest dimension of CD molecules (5.5 Å) well fits within the twelve-membered BEA zeolite pores (pore dimensions: 6.6 × 6.7 Å for [100] and 5.6 × 5.6 Å for [001]), while the longest dimension (11.6 Å) is aligned along the direction of straight channels. The molecular dimension, however, is too large for the micropores (4.5–5.5 Å in diameter) of zeolites with ten-membered apertures (e.g., MFI).
Fig. 2 shows SEM images of BEA zeolite, which was synthesized with diatomaceous earth as the silica precursor. Diatomaceous earth (kieselguhr) is the fossilized remains of diatoms, a type of silica-shelled algae. When TEAOH was used, the distinct morphology of the diatomaceous earth was completely lost and bulk BEA zeolite crystals (1–5 μm) were produced (Fig. 2b). On the contrary, when the CD was used as an SDA for zeolite crystallization, the original macroporous morphology of diatomaceous earth was fully retained after complete crystallization (Fig. 2c). A high-magnification SEM image shows that the macroporous framework was composed of highly nanocrystalline BEA zeolites (Fig. 2d). The resultant zeolite possessed a structural hierarchy in three different length scales, i.e., macropores due to the original morphology of diatomaceous earth, mesopores due to the intercrystalline void space between zeolite nanocrystals, and micropores due to the zeolite framework. In previous work, Anderson et al.25 investigated the crystallization of MFI zeolite by using a diatomaceous earth as a silica source. Their results showed that the macroporous morphology was maintained at the initial stage of crystallization, but disappeared after prolonged crystallization.
 | ||
| Fig. 2 SEM images of pristine diatomaceous earth and BEA zeolites synthesized using diatomaceous earth as a silica source. a: Pristine diatomaceous earth. b: BEA zeolite synthesized using TEAOH. c and d: BEA zeolite synthesized using CD as SDA. | ||
Due to the twelve-membered micropores and strong acidity, BEA zeolites have been used as catalysts for various hydrocarbon conversions such as hydrodewaxing,26,27reforming28 and acylation/alkylation.29,30 In the present work, we investigated the catalytic properties of the micro-/mesoporous hierarchical BEA zeolite in the isopropylation of naphthalene. The alkylation product, 2,6-diisopropylnaphthalene, is an industrially important raw material for the production of advanced aromatic polymers. The result shows a dramatic difference in the catalytic lifetime between hierarchical and bulk BEA zeolites (see the ESI†). The hierarchical BEA zeolite retained 74% of its initial catalytic activity after 5 h, while the conventional zeolite completely lost its catalytic activity. This remarkable difference could be attributed to the enhanced molecular diffusion through mesopores. The presence of secondary mesopores can enhance the diffusion of the coke precursor out of micropores, inhibiting the coke formation at the micropores.13
In summary, a hierarchically micro-/mesoporous BEA zeolite was synthesized using an organic SDA with a cyclic diammonium structure. From the synthesis result, it was concluded that the cyclic diammonium SDA caused very rapid formation of a mesoporous organic-silicate gel, which was converted into the hierarchical zeolite structure according to a ‘pseudomorphic crystallization’ mechanism. The ability of the SDA for the pseudomorphic zeolite generation was also confirmed in synthesis of BEA zeolite from diatomaceous earth. The hierarchical BEA zeolite exhibited a dramatic improvement of catalytic lifetime in isopropylation of naphthalene, as compared with the conventional counterpart.
This work was supported by the National Honor Scientist Program of the Ministry of Education, Science and Technology in Korea.
Notes and references
- Y. Tao, H. Kanoh, L. Abrams and K. Kaneko, Chem. Rev., 2006, 106, 896 CrossRef CAS
.
- K. Egeblad, Christina H. Christensen, M. Kustova and Claus H. Christensen, Chem. Mater., 2008, 20, 946 CrossRef CAS
- C. J. H. Jacobsen, C. Madsen, J. Houzvicka, I. Schmidt and A. Carlsson, J. Am. Chem. Soc., 2000, 122, 7116 CrossRef CAS
- Y. S. Tao, H. Kanoh and K. Kaneko, J. Am. Chem. Soc., 2003, 125, 6044 CrossRef CAS
- B. T. Holland, L. Abrams, L. Abrams and A. Stein, J. Am. Chem. Soc., 1999, 121, 4308 CrossRef CAS
- M. Choi, H. S. Cho, R. Srivastava, C. Venkatesan, D.-H. Choi and R. Ryoo, Nat. Mater., 2006, 5, 718 CrossRef CAS
- D. P. Serrano, J. Aguado, J. M. Escola, J. M. Rodriguez and A. Peral, Chem. Mater., 2006, 18, 2462 CrossRef CAS
- H. Wang and T. J. Pinnavaia, Angew. Chem., Int. Ed., 2006, 45, 7603 CrossRef CAS
- L. Tosheva and V. P. Valtchev, Chem. Mater., 2005, 17, 2494 CrossRef CAS
- S. van Donk, A. H. Janssen, J. H. Bitter and K. P. de Jong, Catal. Rev. Sci. Eng., 2003, 45, 297 CrossRef CAS
- J. C. Groen, J. A. Moulijn and J. Pérez-Ramírez, J. Mater. Chem., 2006, 16, 2121 RSC
- J. C. Groen, W. Zhu, S. Brouwer, S. J. Huynink, F. Kapteijn, J. A. Moulijn and J. Pérez-Ramírez, J. Am. Chem. Soc., 2007, 129, 355 CrossRef CAS
- R. Srivastava, M. Choi and R. Ryoo, Chem. Commun., 2006, 4489 RSC
- C. S. Cundy and P. A. Cox, Microporous Mesoporous Mater., 2005, 82, 1 CrossRef CAS
- W. Xu, J. Dong, J. Li, J. Li and F. Wu, J. Chem. Soc., Chem. Commun., 1990, 755 RSC
- M. Matsukata, M. Ogura, T. Osaki, P. R. H. R. Rao, M. Nomura and E. Kikuchi, Top. Catal., 1999, 9, 77 CrossRef CAS
- S. P. Naik, A. S. T. Chiang and R. W. Thompson, J. Phys. Chem. B, 2003, 107, 7006 CrossRef CAS
- M. A. Camblor and J. Pérez-Pariente, Zeolites, 1991, 11, 202 CAS
-
R. L. Wadlinger, G. T. Kerr and E. J. Rosinski, US Patent, 3
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 308069, 1967 Search PubMed.
308069, 1967 Search PubMed. - P. R. Hari Prasad Rao and M. Matasukata, Chem. Commun., 1996, 1441 RSC
- Y. J. Lee, S. D. Kim, S. C. Byun, J. W. Park, Y. J. Jeong, Y. J. Kwon, H. O. Song and W. J. Kim, J. Cryst. Growth, 2006, 297, 138 CrossRef CAS
- V. Shen and A. T. Bell, Microporous Mesoporous Mater., 1996, 7, 187 CAS
- L. Gang, G. Xinwen, W. Xiangsheng, Z. Qi, B. Xinhe, H. Xiuwen and L. Liwu, Appl. Catal., A, 1999, 185, 11 CrossRef
- Y. Kubota, M. M. Helmkamp, S. I. Zones and M. E. Davis, Microporous Mater., 1996, 6, 213 CrossRef CAS
- M. W. Anderson, S. M. Holmes, S. M. Holmes, N. Hanif and C. S. Cundy, Angew. Chem., Int. Ed., 2000, 39, 2707 CrossRef CAS
- J. A. Martens, J. Pérez-Pariente and P. A. Jacobs, Proceedings of the International Symposium on Zeolite Catalysis, Siofok, Hungary, 1989, pp. 487 Search PubMed.
- G. Kinger and H. Vinek, Appl. Catal., A, 2001, 218, 139 CrossRef CAS
- W. Zhang and P. G. Smirniotis, Catal. Lett., 1999, 60, 223 CrossRef CAS
- M. Guidotti, C. Canaff, J.-M. Coustard, P. Magnoux and M. Guisnet, J. Catal., 2005, 230, 375 CrossRef CAS
- S.-J. Chu and Y.-W. Chen, Appl. Catal., A, 1995, 123, 51 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Experimental, XRD data, N2 adsorption isotherms and catalysis. See DOI: 10.1039/b905087f |
| This journal is © The Royal Society of Chemistry 2009 |