
Spatially resolved and quantitatively revealed charge transfer between single atoms and catalyst supports
 *a
*a
Abstract
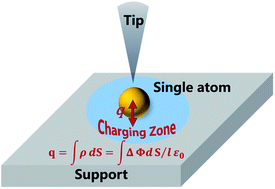
The charge state of supported single atoms is one of the most significant aspects determining the catalytic performance of single atom catalysts (SACs) which have drawn tremendous attention in recent years. In this perspective, mainly based on our previous studies and new data inputs, charge transfer between single atoms and their supports in several model systems is explored by the measurement of local work functions (LWFs). Two types of additives to tune the electronic properties of model catalysts, alkali metals and halogens, are described. The transferred charge is spatially resolved and quantitatively revealed based on LWF mapping via the Helmholtz equation. On average, Cs transfers more electrons than K does, echoing its lower first ionization energy. In contrast, Au and bromine atoms draw electrons from supports of metals like Cu and oxides like CuO. These insights into charge transfer at the atomic level are vital to understand their catalytic and promoting effects.
- This article is part of the themed collection: Single-Atom Catalysis
 Please wait while we load your content...
Please wait while we load your content...

