DOI:
10.1039/D5RA01985K
(Paper)
RSC Adv., 2025,
15, 16219-16227
Structural engineering of MXenes for enhanced magnesium ion diffusion: a computational study†
Received
21st March 2025
, Accepted 30th April 2025
First published on 15th May 2025
Abstract
The unique layered structure and tunable surface terminations of MXenes play a critical role in Mg2+ storage and diffusion dynamics. This study systematically investigates the behavior of Mg2+ in Ti3C2O2 and its nitrogen-doped derivatives through theoretical calculations. In Ti3C2O2 monolayers, Mg2+ exhibits a high diffusion barrier of 0.81 eV due to strong electrostatic interactions. However, AA-stacking reduces this barrier to 0.32 eV by introducing staggered active sites. The instability caused by interlayer O–O repulsion is mitigated by modulating the N/O ratio (Ti3C2O1.78N0.22), resulting in a diffusion barrier of 0.27 eV. Transition metal substitution further optimizes performance, as exemplified by Nb3C2N2, which achieves an ultralow barrier of 0.23 eV through weakened N–N covalency and enhanced metal-N interactions. Voltage analysis reveals that Nb3C2N2 possesses dual functionality as both cathode (4.00 V) and anode (0.64 V), contrasting with the anode-specific behavior observed in Ti-based MXenes.
Introduction
With the rapid depletion of fossil fuels, sustainable energy storage solutions grow increasingly vital.1 In the past few decades, rechargeable lithium-ion batteries (LIBs) have dominated portable electronics and energy storage systems through their high energy density and cycling stability.2,3 However, natural scarcity and dendritic growth risks of LIBs4–6 necessitate alternative solutions.7 Rechargeable magnesium ion batteries (MIBs) emerge as a promising alternative to LIB, offering high energy density and superior electrochemical stability.8 Mg is the fifth most abundant element in the Earth's crust,9 enabling substantial cost savings compared to Li. The melting temperature of Mg is measured at 923 K, much higher than that of Li (453.7 K), making MIBs the safer option for working at high temperatures like in aviation. Divalent Mg2+ delivers doubled volumetric capacity (3833 vs. 2046 mA h cm−3) and dendrite-free operation with low reduction potential (−2.37 vs. SHE), and is expected to achieve reversible deposition in multiple electrolytes.8,10–12
However, Mg2+ (0.72 Å) has a similar ionic radius to Li+ (0.76 Å).13The large charge/radius ratio of Mg2+ leads to strong electrostatic interaction with the electrode framework, resulting in sluggish ion diffusion.14,15 Previous research on MIB electrode materials mainly focused on the transition metal–oxygen/sulfide, polyanionic compounds, and other two-dimensional materials. In transition metal oxides/sulfides systems (e.g., α-V2O5,16 α-MoO3,17 Ti2S3/TiS2 (ref. 18)), strong Mg2+-framework electrostatic interactions induce structural transformations that elevate Mg2+ diffusion barriers to 0.9–1.30 eV. By contrast, Chevrel phases and polyanionic compounds exhibit reversible Mg2+ (de)intercalation with mitigated electrostatic interactions.19 But even for polyanionic materials with good ion mobility, their energy barrier in MIBs is usually higher than 0.6 eV, such as Mg0.25FePO4 (1.03 eV) and NaV2O2(PO4)2F (0.78 eV).20 Moreover, for the olivine-type MgxMnSiS4, the strong Mn–S covalent interaction weakens the electrostatic interactions between Mg2+ and the host material ions, thereby enhancing Mg2+ diffusion compared to its oxide counterpart MgxMnSiO4. However, the diffusion barrier for Mg2+ in MgxMnSiS4 remains as high as 0.76 eV.
Transition metals-based layered materials have stable structures and excellent electrical conductivity.21 More importantly, the layered space provides a broad diffusion channel for Mg2+. Thus, 2D materials possess a lower diffusion barrier than the above materials, such as Si2BN (0.08–0.35 eV), TiSe2 (0.88 eV), VSe2 (0.346 eV) and VS2 (0.593 eV).22,23 As a layered material, MXenes have garnered significant attention in electrochemical energy storage due to their electrical conductivity, large specific surface area, and stable layered architecture for the guest ions.16,24,25 Lukatskaya et al. showed that Ti3C2Tx can provide ample accommodation to accommodate cations, such as Li+, Na+, K+, NH4+, Mg2+ and Al3+, which produce large volumetric capacitance.26 Xie et al.15 confirm that MXene monolayer cation storage occurs in a comparatively low voltage window, which could be used as anodes for rechargeable batteries. Currently, about 40 MXenes have been synthesized experimentally and hundreds of new MXenes are simulated theoretically.27–30 It is crucial for MIB to investigate and enhance the diffusion performance of Mg ions within MXene materials.31,32 Moreover, traditional research on MXene batteries typically focuses on ion storage mechanisms under the default stacking configuration. In fact, the existence of different stacking types is an important feature of layered materials. The most typical example is the layered oxide cathode of potassium-ion batteries. Their different stacking structures can form different potassium-ion storage environments, such as edge-sharing octahedral (O3) or face-sharing prismatic (P2). O3-type materials have high structural stability and can effectively inhibit the migration of transition metals into the K layer, thereby reducing structural degradation during the cycling process. P2-type materials exhibit excellent rate performance due to their unique prismatic coordination environment, which provides shorter K ion migration paths and lower energy barriers. Obviously, stacking type has a significant impact on the storage and migration of ions.33 However, research on the impact of stacking configurations on the MXene electrochemical properties and the corresponding modulation mechanisms is still lacking.
This work investigates the Mg2+ storage and diffusion mechanisms in MXene-based materials, emphasizing the interplay between structural configurations and electrochemical performance. Based on integrating stacking engineering, functional group modulation, and transition metal substitution strategies, we elucidate the fundamental principles governing Mg2+ behavior, such as mitigating electrostatic interactions, optimizing interlayer covalent bonding, and modulating electronic structures. These findings demonstrate that hierarchical structural design-stacking control, doping, and metal substitution-significantly enhances Mg2+ kinetics and stability, advancing high-performance MIB electrodes.
Method
All first-principles density functional theory (DFT) calculations are performed using the Vienna Ab initio Simulation Package (VASP)34–37 based on Perdew–Burke–Ernzerhof's (PBE) generalized gradient approximation (GGA-PBE).38,39 The plane wave truncation energy is set to 550 eV. The valence electron selection for pseudopotential elements is as follows: Mg-3s23p0, O-2s22p4, C-2s22p2, N-2s22p3, Cl-3s23p5, S-3s23p4, F-2s22p5, Ti-3d24s2, V-3d34s2, Zr-4d25s2, Nb-4d45s1. Considering the strong correlation system of transition metal D-orbital electrons, GGA + U method is adopted in the calculation, and the U values of Ti, Nb, V and Zr are set as 2.5 eV, 2.0 eV, 2.5 eV and 2.0 eV respectively, which are obtained from the reference of element U values given by Materials Studio software. Conjugate gradient method was used to optimize the lattice structure. The energy convergence standard was set to 10–6 eV per atom and the force convergence standard was set to 0.01 eV Å−1. In addition, this work adopts the CI-NEB method40–42 to simulate the diffusion performance of Mg2+ in single/multilayer M3C2Tx (M = Ti, V, Zr, Nb, T = O, N) in a 3 × 3 × 1 supercell model. Including the beginning and end positions before and after diffusion, a total of 7 insertion points are set, and the convergence standard is that the force of each insertion point is less than 0.03 eV Å−1.
Results and discussion
Structure and properties of bulk phase Ti3C2O2
The Ti3C2 structure is stacked by the Ti–C–Ti–C–Ti layer. In each layer, the atoms are arranged in hexagons. During the preparation of MXene, the –OH in the etched aqueous solution will spontaneously adsorb on the surface, forming –O terminated groups and water. Based on the type of molten salt and subsequent treatment conditions, other terminated groups, such as –Cl, –I, –Br, –S, –Se and –N could also be adsorbed on the surface of MXene layers. Among these function groups, previous theoretical studies mainly considered –O adsorbed in MXene electrode materials. Because divalent O2− with a larger chemical activity has more affinity for the cation exposed framework. The adsorbed atoms inherit the hexagonal arrangement of the Ti–C–Ti–C–Ti layer, which distributes above the middle Ti atoms (Fig. 1).
 |
| | Fig. 1 (a) The top and side views of the three Mg2+ adsorption sites, namely, H1, H2 and T site on monolayer Ti3C2O2. (b) Binding energy and valence states of Mg2+ at H1, H2 and T sites. | |
Generally, the synthesized MXene materials are bulk rather than monolayer. According to the relative position of adjacent layers, the Mxene structure has three possible stacking types, including AA (Table S1†), ABC1 (Table S2†) and ABC2 (Table S3†) types (Fig. 2a). For ABC1 and ABC2 types, terminal O is directly opposite to the outer Ti and C in the adjacent Ti3C2O2 layer, respectively. The AA-stacking structure is characterized by the alignment of the terminal O groups of adjacent Ti3C2O2 layers. According to the calculated phonon spectra, the phonon band for AA-stacking appears at −0.40 THz, which indicates a slight kinetic instability (Fig. 2b). By contrast, the phonon band structures without imaginary frequencies suggest that ABC1 and ABC2-stacking configurations are more likely to exist in nature. Moreover, our calculated relative energies show that the Ti3C2O2 bulk with ABC1-stacking is the most stable configuration (Fig. 2c). In comparison, the AA and ABC2 types have relatively higher energy of 0.30 and 0.09 eV, respectively. Such relative stability of ABC1-stacking is also applies consistently to the case of –Cl and –F terminate.
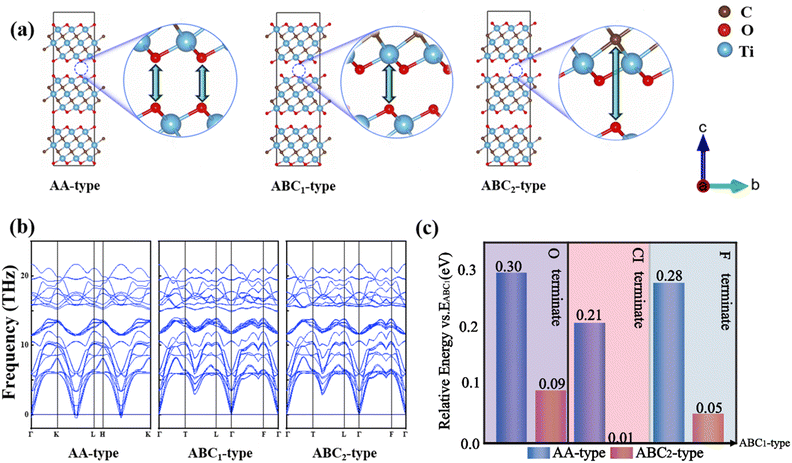 |
| | Fig. 2 (a) The side-views of the three stacked Ti3C2O2, namely ABC1 type, AA type and ABC2 type. (b) The phonon band structures of AA-, ABC1- and ABC2-stacked configuration. (c) The relative energy of ABC1- and ABC2-type vs. the energy of AA-type for Ti3C2O2, Ti3C2Cl2 and Ti3C2F2, respectively, in left to right order. | |
Adsorption of Mg2+ in Ti3C2O2
Each surface of the Ti3C2O2 layer has three possible Mg2+ active sites named H1, H2 and T, which are above the C, Ti and O atoms, respectively (Fig. 1a). H1 and H2 sites in two triangular hollow regions formed by three O and three Ti atoms, respectively. While the T site has only one coordinated O atom. To determine the most favorable Mg2+ active site, the Mg2+ binding energy Eb on the 2 × 2 × 1 Ti3C2O2 monolayer was calculated through the following formula:| | |
Eb = ETi3C2N2Mgx − ETi3C2N2 − xEMg
| (1) |
where EMgxTi3C2O2, ETi3C2O2 and EMg are the total energy of Ti3C2O2Mgx, Ti3C2O2 and one atom of metal Mg. The calculation results show that the H1 site exhibited a more negative Eb (−2.16 eV) than H2 (−1.36 eV) site. While the Mg2+ on T site migrates to the adjacent H1 site after structural optimization. In the case of fixed Mg2+ at a specific xy coordinate, the binding energy at the T site is only −0.68 eV. This indicates that a low coordination degree is not favorable for Mg2+ storage. According to the Bader charge analysis, the valence states of Mg2+ on H1, H2 and T site was +1.55, +1.41 and +0.84, respectively (Fig. 1b). Obviously, Mg2+ at the H1 site is closer to its theoretical standard valence state, which is more conducive to its binding. When the Ti3C2O2 layers are stacked to form the bulk, the Mg2+ active site is provided by both upper and lower Ti3C2O2 layers. All possible adsorption sites of Mg2+ in the three 42 configurations are taken into account. For the AA-stacking type, the two Ti3C2O2 layers form H1, H12 and T active sites for Mg2+, where the “H12” site means the combination of the H1 and H2 sites from two adjacent Ti3C2O2 layers, respectively. ABC1-Ti3C2O2 forms H1, H2 and H12 sites. ABC2-Ti3C2O2 forms TH1, H1 and H2 sites. For all these three stacking configurations, the H1 site exhibited stronger binding energy than the other sites (Fig. 3), which is consistent with the Mg2+ adsorption behavior for Ti3C2O2 monolayer. Among the three configurations, the H1 site in AA-stacked structure exhibited the most negative adsorption energy, which indicated that the AA-stacking is more favorable adsorptive configuration for Mg2+. According to the optimized crystal, the lower the binding energy, the smaller the lattice parameter c. This suggests that the strong coulomb attraction between the O atoms and the adsorbed Mg2+ can mitigate volume expansion during ion storage. Moreover, it is evident that the structural energy difference between the ABC1- and ABC2-Ti3C2O2 is very small (Fig. 2c and 4). Furthermore, during structural optimized, Mg2+ adsorbed ABC2-Ti3C2O2 will relax into ABC1 type through the sliding of adjacent Ti3C2O2 layers. Therefore, the ABC2 configuration is improbable to form under practical conditions and will hence not be considered in subsequent studies.
 |
| | Fig. 3 The binding energy of Mg2+ at each adsorption site of Ti3C2O2 and the change trend of lattice parameter c. | |
 |
| | Fig. 4 (a) Diffusion path of magnesium on single layer Ti3C2O2 surface. (b) Diffusion paths of Mg2+ in AA type and (c) ABC1 type Ti3C2O2 structure, respectively. (d) Diffusion energy barrier of Mg2+ in AA type, ABC1 type and single layer Ti3C2O2. | |
Mg2+ diffuses in AA, ABC1 and monolayer Ti3C2O2
The rate performance of anode is determined by the kinetics of electronic and ionic diffusion. To further study the Mg2+ diffusion kinetic properties for the AA- and ABC1-stacking structures, the Mg2+ diffusion barrier is evaluated in the 3 × 3 × 1 Ti3C2O2 bulk/monolayer using the CI-NEB method. In each configuration, Mg2+ is simulated to migrate between two energetically favorable active sites (Fig. 4a). On Ti3C2O2 monolayer, the diffusion of Mg2+ between two adjacent H1 sites across H2 site (described as H1 → H2 → H1) has a relatively large barrier of 0.81 eV (Fig. 4d). Such a value is close to the Mg2+ barrier (0.72 eV) on the Ti2NO2 monolayer reported by Wang et al.28 According to that work, the univalent Li+, Na+, K+ diffusion barriers are lower than 0.3 eV, which suggests that the pathway of Ti2NO2 monolayer is insufficient to provide an adequate coordination environment for the bivalent Mg2+.
In the AA-stacking Ti3C2O2, the Mg2+ barrier is significantly improved to 0.32 eV. The optimized AA-stacking structure shows that the adjacent H12 active sites provided by the upper and lower Ti3C2O2 surfaces are opposite, which can be described as 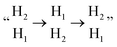 (Fig. 4b). Notably, compared with the Ti3C2O2 monolayer, Mg2+ can only move from
(Fig. 4b). Notably, compared with the Ti3C2O2 monolayer, Mg2+ can only move from 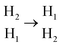 through a single step, and can thereby transfer from one H1 active site to another. It can be seen that the periodic migration path of Mg2+ has been shortened by almost half. Besides, according to the CI-NEB method, the ionic migration barrier is obtained by subtracting the energy of the stable state (SS) from that of the transition state (TS), i.e., Ebarrier = ETS − ESS.43 The contribution of metastable H2 site in AA-Ti3C2O2 increases the ESS, alleviating energy changes during Mg2+ migration. In comparison, the Mg2+ diffusion channel in the ABC1-Ti3C2O2 can be described as
through a single step, and can thereby transfer from one H1 active site to another. It can be seen that the periodic migration path of Mg2+ has been shortened by almost half. Besides, according to the CI-NEB method, the ionic migration barrier is obtained by subtracting the energy of the stable state (SS) from that of the transition state (TS), i.e., Ebarrier = ETS − ESS.43 The contribution of metastable H2 site in AA-Ti3C2O2 increases the ESS, alleviating energy changes during Mg2+ migration. In comparison, the Mg2+ diffusion channel in the ABC1-Ti3C2O2 can be described as  (Fig. 4c). The stable active site composed of two stable H1 sites lead to a low ESS value. While the metastable H2 and exceptionally unstable T site significantly increases the ETS. Such substantial energy disparity between ETS and ESS lead to a large diffusion barrier of 2.01 eV for ABC1-Ti3C2O2. To further investigate the influencing factors of Mg2+ diffusion barrier, the Bader charge of Mg2+ in stable states (eS) and transition states (eT) as well as the charge difference (Δe = |es − eT|) between them were calculated based on the CI-NEB image. The result showed that the Δe for AA and ABC1 types are 0.005 and 0.206e, which are consistent with their barrier order of 0.32, and 2.01 eV, respectively. The Mg2+ channel in ABC1-Ti3C2O2 has large fluctuation on electron capture, which is also an important factors that cause the largest diffusion barrier.
(Fig. 4c). The stable active site composed of two stable H1 sites lead to a low ESS value. While the metastable H2 and exceptionally unstable T site significantly increases the ETS. Such substantial energy disparity between ETS and ESS lead to a large diffusion barrier of 2.01 eV for ABC1-Ti3C2O2. To further investigate the influencing factors of Mg2+ diffusion barrier, the Bader charge of Mg2+ in stable states (eS) and transition states (eT) as well as the charge difference (Δe = |es − eT|) between them were calculated based on the CI-NEB image. The result showed that the Δe for AA and ABC1 types are 0.005 and 0.206e, which are consistent with their barrier order of 0.32, and 2.01 eV, respectively. The Mg2+ channel in ABC1-Ti3C2O2 has large fluctuation on electron capture, which is also an important factors that cause the largest diffusion barrier.
Diffusion of Mg2+ in AA-Ti3C2Tx (T = O, N)
According to the above discussion, the adsorption and migration behavior of Mg2+ exhibits a pronounced preference for the AA-Ti3C2O2. Regrettably, in layered stacking structures, the coulombic effect between layers significantly influence the stacking configuration. For the AA-Ti3C2O2, the O–O electrostatic repulsion from the adjacent layers leads to a preference for staggered arrangement in Ti3C2O2 bulk, thereby increasing the interlayer spacing and rendering the AA-Ti3C2O2 unstable. One effective approach to mitigate electrostatic repulsion is to substitute bivalent O with monovalent halogens such as F or Cl. However, the introduction of F/Cl functional groups does reduce the energy difference between the AA- and ABC1-Ti3C2O2, yet the AA configuration remains relatively unstable (Fig. 2c). The remaining stacking control method is to utilize higher valence N as functional group, thereby converting electrostatic repulsion into covalent bonding through the addition of electrons.
Consequently, the O function group in AA, ABC1 and ABC2 configurations are replaced by N (Fig. 5a). The calculated relative energy revealed that the AA-type Ti3C2N2 become the most stable configuration. Taking AA-Ti3C2N2 as the reference structure, the relative energies of ABC1 and ABC2 are 6.82 eV and 7.89 eV, respectively (Fig. 5b). Such large values indicate that the introduction of interlayer N–N covalent bonding significantly enhances the stability of the AA-stacking configuration. In the AA-Ti3C2N2 system, the diffusion energy barrier of Mg2+ calculated using the CI-NEB method is 1.07 eV, which is 0.94 eV lower than that in the stable ABC1 configuration for Ti3C2O2. Evidently, the electrostatic interaction between layers does not directly influence the migration of Mg2+. Instead, it indirectly facilitates a favorable migration environment for Mg2+ by inducing structural changes. Despite this reduction, the barrier of 1.07 eV still represents a poor diffusion performance. This can be attributed to the strong bonding between nitrogen atoms in adjacent Ti3C2N2 layers, which results in reduced layer spacing and hinders the migration of Mg2+. Consequently, it is crucial to determine an appropriate N doping concentration to maintain optimal layer spacing while controlling the MXene stacking type.
 |
| | Fig. 5 (a) AA-, ABC1- and ABC2-Ti3C2N2 interlayer structure. (b) The relative energy of ABC1- and ABC2-Ti3C2N2 with respect to AA-type. (c) The relative energies of AA and ABC1 configurations with different N content. (d) Diffusion barrier comparison of Mg2+ in AA-stacking Ti3C2N2 and Ti3C2N1.78N0.22 structures. | |
The calculation of the relative energies for AA and ABC1 configurations with varying N contents reveals that when the N concentration reaches 11%, the stable phase MXene bulk transitions to the AA-stacking type, corresponding to the chemical formula Ti3C2O1.78N0.22 (Fig. 5c). For this configuration, the Mg2+ diffusion barrier is decreased to 0.27 eV, which is significantly lower by 0.8 eV compared to Ti3C2N2 bulk. Notably, Mg2+ diffusion predominantly occurs near O functional groups rather than N functional groups (Fig. 5d). This can be attributed to the additional energy required for Mg2+ to overcome the interlayer N–N covalent bonds during diffusion, thereby increasing the migration energy barrier.
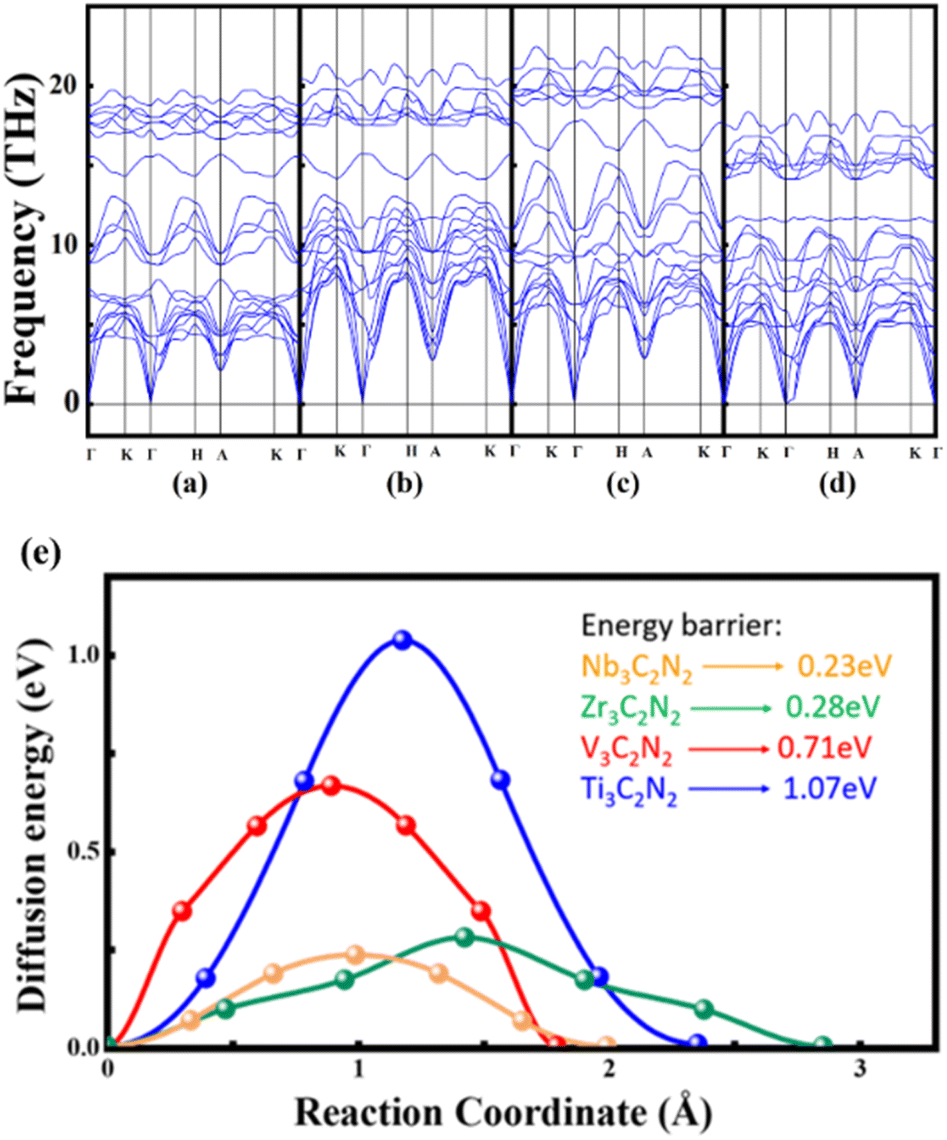
On the other hand, the strong interlayer N–N covalent bonding in the AA-Ti3C2N2 structure is primarily responsible for the poor Mg diffusion kinetic property. Based on the such bonding situation, it can be proposed that enhancing the metal–N bond strength may weaken the interlayer N–N covalency, thereby improving ionic conductivity. In the periodic table, we have selected three transition metal elements, V, Zr and Nb, to substitute for Ti in AA-Ti3C2N2. V (3d34s2), Nb (4d45s1), and Zr (4d25s2) each enhance N bonding through distinct mechanisms compared to Ti (3d24s2). V and Nb provide additional valence electrons for covalent interactions, while Zr achieves stronger binding via reduced electronegativity-driven ionic attraction. After replacing Ti atoms in AA-stacking Ti3C2N2 with V, Zr and Nb, the dynamical stability of these four conformations are checked through examining the phonon band structures (Fig. 6a–d), which indicated that they are dynamically stable configurations with no states associated with imaginary frequencies.
 |
| | Fig. 6 Phonon spectra of (a) Nb3C2N2, (b) Ti3C2N2, (c) V3C2N2 and (d) Zr3C2N2. (e) Diffusion energy barrier of Mg2+ in AA-M3C2N2 (M = Ti, V, Zr, Nb). | |
Diffusion of Mg2+ in AA-M3C2N2 (M = Nb, V, Zr)
Subsequently, to analyze the diffusion behavior of Mg2+ in AA-M3C2N2 (M = V, Zr and Nb), the Mg2+ migration pathways and corresponding energy barriers are investigated within 3 × 3 × 1 supercell. With a single Mg2+ intercalation into M3C2N2, it was relaxed to the  site after structure optimization and the corresponding the half of diffusion pathways were described as
site after structure optimization and the corresponding the half of diffusion pathways were described as 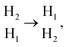 which showed that Mg2+ diffusion behavior in M3C2N2 bulks (including Ti3C2N2) is consistent. The diffusion barriers of Mg2+ in V3C2N2, Zr3C2N2 and Nb3C2N2 had decreased compared with in Ti3C2N2 by 0.37 eV, 0.79 eV and 0.84 eV, respectively. Notably, Nb3C2N2 exhibits the smallest barrier of 0.23 eV. This indicates that N–N covalent binding is weakened more effectively by replacing Ti with Nb than with V and Zr (Fig. 6e). Overall, the Nb3C2N2 obtained by using the transition metal element Nb to replace Ti in AA-Ti3C2N2 has the lowest energy barrier (Fig. S1†).
which showed that Mg2+ diffusion behavior in M3C2N2 bulks (including Ti3C2N2) is consistent. The diffusion barriers of Mg2+ in V3C2N2, Zr3C2N2 and Nb3C2N2 had decreased compared with in Ti3C2N2 by 0.37 eV, 0.79 eV and 0.84 eV, respectively. Notably, Nb3C2N2 exhibits the smallest barrier of 0.23 eV. This indicates that N–N covalent binding is weakened more effectively by replacing Ti with Nb than with V and Zr (Fig. 6e). Overall, the Nb3C2N2 obtained by using the transition metal element Nb to replace Ti in AA-Ti3C2N2 has the lowest energy barrier (Fig. S1†).
Theoretical voltage value of AA-stacking Nb3C2N2
According to the above discussion, the favorite absorption site of Mg2+ is  site. Based on this, the formation energy of Nb3C2N2Mgx (0 ≤ x ≤ 1) was calculated via the following equation to explore the thermodynamically stable Mg-containing phases,
site. Based on this, the formation energy of Nb3C2N2Mgx (0 ≤ x ≤ 1) was calculated via the following equation to explore the thermodynamically stable Mg-containing phases,| |
 | (2) |
where ENb3C2N2Mgx, ENb3C2N2Mg2 and ENb3C2N2 are total energies for Nb3C2N2Mgx, Nb3C2N2Mg2 and Nb3C2N2, correspondingly, x is the Mg2+ adsorption degrees in Nb3C2N2Mgx, including 0, 0.25, 0.5, 0.75, 1, 1.25, 1.5, 1.75 and 2 (Fig. 7a). Our result suggests that the thermodynamically stable configurations are Nb3C2N2Mg0.75, Nb3C2N2Mg and Nb3C2N2Mg2, with their Ef points located at the bottom edge of the convex hull. Based on this, the open circuit voltage (OCV) of AA-stacking Nb3C2N2 with different Mg2+ adsorption degrees is calculated from the following formula,| |
 | (3) |
where  and
and  are the total energies of Nb3C2N2 with different Mg2+ adsorption degrees x1 and x2, EMg is the energy of a Mg atom in metal Mg, charge value n = 2 for Mg2+. Interestingly, the calculated voltages contain two distinct plateaus at 4.00 V and 0.64 V, corresponding to the ranges of x = 0–0.75 and x = 1–2, respectively (Fig. 7b). This results indicate that when a monolayer of Mg2+ is intercalated between the Nb3C2N2 layers, the binding affinity of Mg2+ to the Nb3C2N2 layers significantly decreases. Such a significant voltage difference indicates that the material can function as both the positive and negative electrode of the battery under varying ion concentrations. This behavior is analogous to that observed in Prussian blue materials used in potassium-ion batteries.44 By contrast, the voltage platforms presented by Ti-based MXene belong to the anode range (Fig. S2†). From a voltage stability perspective, Ti3C2O2Mgx and Ti3C2O1.78N0.22Mgx exhibit three short platforms decreasing in the concentration of 0 < x < 1. Although Ti3C2N2 possesses two stable platforms, the voltage of 0.02 V is too low for electrode, which is easy to transform the Mg2+ intercalation mechanism into electroplating.
are the total energies of Nb3C2N2 with different Mg2+ adsorption degrees x1 and x2, EMg is the energy of a Mg atom in metal Mg, charge value n = 2 for Mg2+. Interestingly, the calculated voltages contain two distinct plateaus at 4.00 V and 0.64 V, corresponding to the ranges of x = 0–0.75 and x = 1–2, respectively (Fig. 7b). This results indicate that when a monolayer of Mg2+ is intercalated between the Nb3C2N2 layers, the binding affinity of Mg2+ to the Nb3C2N2 layers significantly decreases. Such a significant voltage difference indicates that the material can function as both the positive and negative electrode of the battery under varying ion concentrations. This behavior is analogous to that observed in Prussian blue materials used in potassium-ion batteries.44 By contrast, the voltage platforms presented by Ti-based MXene belong to the anode range (Fig. S2†). From a voltage stability perspective, Ti3C2O2Mgx and Ti3C2O1.78N0.22Mgx exhibit three short platforms decreasing in the concentration of 0 < x < 1. Although Ti3C2N2 possesses two stable platforms, the voltage of 0.02 V is too low for electrode, which is easy to transform the Mg2+ intercalation mechanism into electroplating.
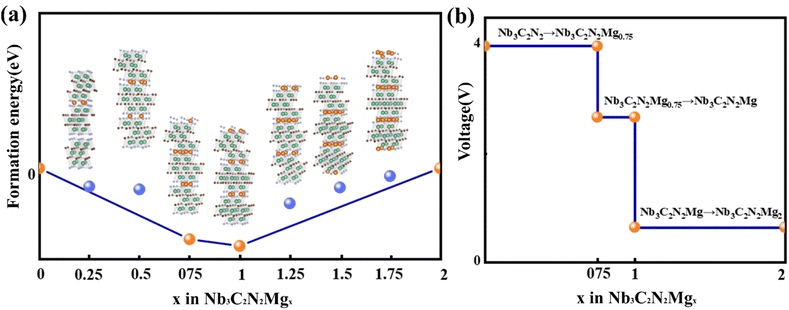 |
| | Fig. 7 (a) Formation energy and (b) theoretical voltage value of Nb3C2N2Mgx. | |
Conclusions
This study systematically investigates the Mg2+ storage and diffusion mechanisms in MXene materials, focusing on Ti3C2O2 and its nitrogen-doped derivatives. The strong electrostatic interaction between divalent Mg2+ and electrode frameworks leads to high diffusion barriers (0.81 eV in monolayers), which are mitigated by optimizing MXene stacking configurations and functional group engineering. AA-stacking Ti3C2O2 exhibits strong Mg2+ adsorption energy of −2.16 eV and low diffusion barrier of 0.32 eV due to staggered active sites shortening migration paths. However, its instability from interlayer O–O repulsion necessitates structural modifications. Replacing O with N transforms stable ABC1-Ti3C2O2 to AA-Ti3C2N2 through enhanced interlayer N–N covalent bonding. However, excessive N doping increases the energy barrier (1.07 eV) due to decreased layer spacing. One effective approach is to control the N/O ratio, which balances interlayer spacing and covalent interactions, resulting in an energy barrier of 0.27 eV in the Ti3C2O1.78N0.22 structure. Another strategy involves substituting transition metals into the MXene framework to modulate interlayer bonding. Introducing transition metals (V, Zr, Nb) into Ti3C2N2 weakens N–N covalency, with Nb3C2N2 achieving an ultralow barrier of 0.23 eV. Nb3C2N2 possesses dual voltage platforms (4.00 V and 0.64 V), enabling its dual role as cathode/anode, while Ti-based MXenes exhibit anode-specific behavior. These findings highlight that tailored MXene structures, through stacking control, nitrogen doping, and metal substitution, significantly enhance Mg2+ kinetics and stability, positioning them as promising high-performance electrodes for next-generation Mg-ion batteries.
Data availability
The datasets used and/or analyzed during the current study are available from the corresponding author upon reasonable request.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the Hebei Natural Science Foundation Youth Foundation (No. 606020324086), the Advanced Talents Incubation Program of the Hebei University (No. 521000981394), the Scientific Research and Innovation Team of Hebei University (Grant No. IT2023B03) and the High-Performance Computing Center of Hebei University.
References
- Y. Shabeer, S. S. Madani, S. Panchal, M. Mousavi and M. Fowler, Different Metal–Air Batteries as Range Extenders for the Electric Vehicle Market: A Comparative Study, Batteries, 2025, 11(1), 35 Search PubMed.
- D. A. Kolosov and O. E. Glukhova, High-Capacity Ion Batteries Based on Ti2C MXene and Borophene First Principles Calculations, Inorganics, 2023, 11(3), 95 Search PubMed.
- X. Zhang, A. Wang, X. Liu and J. Luo, Dendrites in Lithium Metal Anodes: Suppression, Regulation, and Elimination, Acc. Chem. Res., 2019, 52(11), 3223–3232 Search PubMed.
- F. Sun, H. Liu, L. Ni and X. Wang, Growth Mechanisms of Li Dendrite and the Suppression Strategies, Chin. Sci. Bull., 2021, 66(9), 1046–1056 Search PubMed.
- X. Chen, B. Zhao, C. Yan and Q. Zhang, Review on Li Deposition in Working Batteries: From Nucleation to Early Growth, Adv. Mater., 2021, 33(8), 2004128 Search PubMed.
- Y.-G. Lee, S. Fujiki, C. Jung, N. Suzuki, N. Yashiro, R. Omoda, D.-S. Ko, T. Shiratsuchi, T. Sugimoto, S. Ryu, J. H. Ku, T. Watanabe, Y. Park, Y. Aihara, D. Im and I. T. Han, High-Energy Long-Cycling All-Solid-State Lithium Metal Batteries Enabled by Silver–Carbon Composite Anodes, Nat. Energy, 2020, 5(4), 299–308 Search PubMed.
- G. A. Shaikh, A. M. Satawara, S. K. Gupta and P. N. Gajjar, Unlocking the Potential of Mg-Ion Batteries: Cu2C MXene Anode with Ultrahigh Storage and Energy Density with Rapid Mg Diffusion, Chem. Eng. J., 2024, 498, 155368 Search PubMed.
- R. Dominko, J. Bitenc, R. Berthelot, M. Gauthier, G. Pagot and V. Di Noto, Magnesium Batteries: Current Picture and Missing Pieces of the Puzzle, J. Power Sources, 2020, 478, 229027 Search PubMed.
- Y. Du, Y. Chen, S. Tan, J. Chen, X. Huang, L. Cui, J. Long, Z. Wang, X. Yao, B. Shang, G. Huang, X. Zhou, L. Li, J. Wang and F. Pan, Strong Solvent Coordination Effect Inducing Gradient Solid-Electrolyte-Interphase Formation for Highly Efficient Mg Plating/Stripping, Energy Storage Mater., 2023, 62, 102939 Search PubMed.
- K. A. Papadopoulou, A. Chroneos and S.-R. G. Christopoulos, Mg-Ion Diffusion on the Surface of Ti3C2S2 MXene, J. Phys. Chem. Solids, 2022, 166, 110713 Search PubMed.
- B. Li, R. Masse, C. Liu, Y. Hu, W. Li, G. Zhang and G. Cao, Kinetic Surface Control for Improved Magnesium-Electrolyte Interfaces for Magnesium Ion Batteries, Energy Storage Mater., 2019, 22, 96–104 Search PubMed.
- M. Deng, L. Wang, B. Vaghefinazari, W. Xu, C. Feiler, S. V. Lamaka, D. Höche, M. L. Zheludkevich and D. Snihirova, High-Energy and Durable Aqueous Magnesium Batteries: Recent Advances and Perspectives, Energy Storage Mater., 2021, 43, 238–247 Search PubMed.
- J. Nai, Y. Han, S. Zhang, Y. Li, X. Chen, R. Wu, L. Wang and L. Jiang, Sulfite Activation by ZnO-Encapsulated Hydrogels for Degradation of Trimethylphenol, Nano Res., 2023, 16(10), 12345–12356 Search PubMed.
- M. Hammouri, M. Arsentev and A. Petrov, Ab Initio Studies of Ti2S3 as a New Cathode Material for Magnesium Secondary Batteries, Mater. Today Commun., 2019, 20, 100598 Search PubMed.
- Y. Xie, Y. Dall'Agnese, M. Naguib, Y. Gogotsi, M. W. Barsoum, H. L. Zhuang and P. R. C. Kent, Prediction and Characterization of MXene Nanosheet Anodes for Non-Lithium-Ion Batteries, ACS Nano, 2014, 8(9), 9606–9615 Search PubMed.
- R. Xiao, J. Xie, T. Luo, L. Huang, Y. Zhou, D. Yu, C. Chen and Y. Liu, Phase Transformation and Diffusion Kinetics of V2 O5 Electrode in Rechargeable Li and Mg Batteries: A First-Principle Study, J. Phys. Chem. C, 2018, 122(3), 1513–1521 Search PubMed.
- T. A. Barnes, L. F. Wan, P. R. C. Kent and D. Prendergast, Hybrid DFT Investigation of the Energetics of Mg Ion Diffusion in α-MoO3, Phys. Chem. Chem. Phys., 2018, 20(38), 24877–24884 Search PubMed.
- D. S. Tchitchekova, A. Ponrouch, R. Verrelli, T. Broux, C. Frontera, A. Sorrentino, F. Bardé, N. Biskup, M. E. Arroyo-de Dompablo and M. R. Palacín, Electrochemical Intercalation of Calcium and Magnesium in TiS2 : Fundamental Studies Related to Multivalent Battery Applications, Chem. Mater., 2018, 30(3), 847–856 Search PubMed.
- G. Li, Z. Yao and C. Li, In-Situ Multi-Scale Structural Engineering of Cathode and Electrolyte for High-Rate and Long-Life Mg Metal Batteries, J. Energy Chem., 2025, 105, 44–53 Search PubMed.
- J. Wang, S. Tan, G. Zhang, Y. Jiang, Y. Yin, F. Xiong, Q. Li, D. Huang, Q. Zhang, L. Gu, Q. An and L. Mai, Fast and Stable Mg2+ Intercalation in a High Voltage NaV2O2(PO4)2F/rGO Cathode Material for Magnesium-Ion Batteries, Sci. China Mater., 2020, 63(9), 1651–1662 Search PubMed.
- S. Li, Y. Sun, Y. Pang, S. Xia, T. Chen, H. Sun, S. Zheng and T. Yuan, Recent Developments of Layered Transition Metal Oxide Cathodes for Sodium-ion Batteries toward Desired High Performance, Asia-Pac. J. Chem. Eng., 2022, 17(4), e2762 Search PubMed.
- P. Panigrahi, S. B. Mishra, T. Hussain, B. R. K. Nanda and R. Ahuja, Density Functional Theory Studies of Si2 BN Nanosheets as Anode Materials for Magnesium-Ion Batteries, ACS Appl. Nano Mater., 2020, 3(9), 9055–9063 Search PubMed.
- M. Mao, X. Ji, S. Hou, T. Gao, F. Wang, L. Chen, X. Fan, J. Chen, J. Ma and C. Wang, Tuning Anionic Chemistry To Improve Kinetics of Mg Intercalation, Chem. Mater., 2019, 31(9), 3183–3191 Search PubMed.
- Y. Zhang, Z. Yuan, L. Zhao, Y. Li, X. Qin, J. Li, W. Han and L. Wang, Review of Design Routines of MXene Materials for Magnesium-ion Energy Storage Device, Small, 2023, 19(34), 2301815 Search PubMed.
- B. Zhou, H. Shi, R. Cao, X. Zhang and Z. Jiang, Theoretical Study on the Initial Stage of a Magnesium Battery Based on a V2 O5 Cathode, Phys. Chem. Chem. Phys., 2014, 16(34), 18578–18585 Search PubMed.
- M. R. Lukatskaya, O. Mashtalir, C. E. Ren, Y. Dall'Agnese, P. Rozier, P. L. Taberna, M. Naguib, P. Simon, M. W. Barsoum and Y. Gogotsi, Cation Intercalation and High Volumetric Capacitance of Two-Dimensional Titanium Carbide, Science, 2013, 341(6153), 1502–1505 Search PubMed.
- M. Naguib, M. W. Barsoum and Y. Gogotsi, Ten Years of Progress in the Synthesis and Development of MXenes, Adv. Mater., 2021, 33(39), 2103393 Search PubMed.
- D. Wang, Y. Gao, Y. Liu, D. Jin, Y. Gogotsi, X. Meng, F. Du, G. Chen and Y. Wei, First-Principles Calculations of Ti2 N and Ti2 NT2 (T = O, F, OH) Monolayers as Potential Anode Materials for Lithium-Ion Batteries and Beyond, J. Phys. Chem. C, 2017, 121(24), 13025–13034 Search PubMed.
- D. Er, J. Li, M. Naguib, Y. Gogotsi and V. B. Shenoy, Ti3 C2 MXene as a High Capacity Electrode Material for Metal (Li, Na, K, Ca) Ion Batteries, ACS Appl. Mater. Interfaces, 2014, 6(14), 11173–11179 Search PubMed.
- M. Naguib, J. Halim, J. Lu, K. M. Cook, L. Hultman, Y. Gogotsi and M. W. Barsoum, New Two-Dimensional Niobium and Vanadium Carbides as Promising Materials for Li-Ion Batteries, J. Am. Chem. Soc., 2013, 135(43), 15966–15969 Search PubMed.
- Z. Zhang, Z. Sun, S. Sun, J. Liu, J. Jiang, W. Shang and Y. Wen, MXene Modified Anode of Magnesium Battery to Improve the Problems of Voltage Lag and Irreversibility, J. Alloys Compd., 2023, 960, 170609 Search PubMed.
- J. Zhu, R. Shi, Y. Liu, Y. Zhu, J. Zhang, X. Hu and L. Li, 3D Interwoven MXene Networks Fabricated by the Assistance of Bacterial Celluloses as High-Performance Cathode Material for Rechargeable Magnesium Battery, Appl. Surf. Sci., 2020, 528, 146985 Search PubMed.
- R. Rajagopalan, Y. Tang, X. Ji, C. Jia and H. Wang, Adv. Funct. Mater., 2020, 1909486 Search PubMed.
- G. Kresse and J. Hafner, Ab Initio Molecular Dynamics for Open-Shell Transition Metals, Phys. Rev. B:Condens. Matter Mater. Phys., 1993, 48(17), 13115–13118 Search PubMed.
- G. Kresse and J. Furthmüller, Efficient Iterative Schemes for Ab Initio Total-Energy Calculations Using a Plane-Wave Basis Set, Phys. Rev. B:Condens. Matter Mater. Phys., 1996, 54(16), 11169–11186 Search PubMed.
- P. E. Blöchl, Projector Augmented-Wave Method, Phys. Rev. B:Condens. Matter Mater. Phys., 1994, 50(24), 17953–17979 Search PubMed.
- G. Kresse and D. Joubert, From Ultrasoft Pseudopotentials to the Projector Augmented-Wave Method, Phys. Rev. B:Condens. Matter Mater. Phys., 1999, 59(3), 1758–1775 Search PubMed.
- J. P. Perdew and W. Yue, Accurate and Simple Density Functional for the Electronic Exchange Energy: Generalized Gradient Approximation, Phys. Rev. B:Condens. Matter Mater. Phys., 1986, 33(12), 8800–8802 Search PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized Gradient Approximation Made Simple, Phys. Rev. Lett., 1996, 77(18), 3865–3868 Search PubMed.
- J. Luo, T. Fan, J. Zhang, P. Qiu, X. Shi and L. Chen, Automatic Identification of Slip Pathways in Ductile Inorganic Materials by Combining the Active Learning Strategy and NEB Method, npj Comput. Mater., 2025, 11(1), 41 Search PubMed.
- G. Henkelman, B. P. Uberuaga and H. Jónsson, A Climbing Image Nudged Elastic Band Method for Finding Saddle Points and Minimum Energy Paths, J. Chem. Phys., 2000, 113(22), 9901–9904 Search PubMed.
- G. Henkelman and H. Jónsson, Improved Tangent Estimate in the Nudged Elastic Band Method for Finding Minimum Energy Paths and Saddle Points, J. Chem. Phys., 2000, 113(22), 9978–9985 Search PubMed.
- C. Teng, Y. Wang and J. L. Bao, Physical Prior Mean Function-Driven Gaussian Processes Search for Minimum-Energy Reaction Paths with a Climbing-Image Nudged Elastic Band: A General Method for Gas-Phase, Interfacial, and Bulk-Phase Reactions, J. Chem. Theory Comput., 2024, 20(10), 4308–4324 Search PubMed.
- Q. Wang, J. Li, H. Jin, S. Xin and H. Gao, Prussian-blue Materials: Revealing New Opportunities for Rechargeable Batteries, Infomat, 2022, 4(6), e12311 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ra01985k |
| ‡ Mingxiao Ma and Xiangyu Yao contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Ruining Wang
a,
Ruining Wang



 (Fig. 4b). Notably, compared with the Ti3C2O2 monolayer, Mg2+ can only move from
(Fig. 4b). Notably, compared with the Ti3C2O2 monolayer, Mg2+ can only move from  through a single step, and can thereby transfer from one H1 active site to another. It can be seen that the periodic migration path of Mg2+ has been shortened by almost half. Besides, according to the CI-NEB method, the ionic migration barrier is obtained by subtracting the energy of the stable state (SS) from that of the transition state (TS), i.e., Ebarrier = ETS − ESS.43 The contribution of metastable H2 site in AA-Ti3C2O2 increases the ESS, alleviating energy changes during Mg2+ migration. In comparison, the Mg2+ diffusion channel in the ABC1-Ti3C2O2 can be described as
through a single step, and can thereby transfer from one H1 active site to another. It can be seen that the periodic migration path of Mg2+ has been shortened by almost half. Besides, according to the CI-NEB method, the ionic migration barrier is obtained by subtracting the energy of the stable state (SS) from that of the transition state (TS), i.e., Ebarrier = ETS − ESS.43 The contribution of metastable H2 site in AA-Ti3C2O2 increases the ESS, alleviating energy changes during Mg2+ migration. In comparison, the Mg2+ diffusion channel in the ABC1-Ti3C2O2 can be described as  (Fig. 4c). The stable active site composed of two stable H1 sites lead to a low ESS value. While the metastable H2 and exceptionally unstable T site significantly increases the ETS. Such substantial energy disparity between ETS and ESS lead to a large diffusion barrier of 2.01 eV for ABC1-Ti3C2O2. To further investigate the influencing factors of Mg2+ diffusion barrier, the Bader charge of Mg2+ in stable states (eS) and transition states (eT) as well as the charge difference (Δe = |es − eT|) between them were calculated based on the CI-NEB image. The result showed that the Δe for AA and ABC1 types are 0.005 and 0.206e, which are consistent with their barrier order of 0.32, and 2.01 eV, respectively. The Mg2+ channel in ABC1-Ti3C2O2 has large fluctuation on electron capture, which is also an important factors that cause the largest diffusion barrier.
(Fig. 4c). The stable active site composed of two stable H1 sites lead to a low ESS value. While the metastable H2 and exceptionally unstable T site significantly increases the ETS. Such substantial energy disparity between ETS and ESS lead to a large diffusion barrier of 2.01 eV for ABC1-Ti3C2O2. To further investigate the influencing factors of Mg2+ diffusion barrier, the Bader charge of Mg2+ in stable states (eS) and transition states (eT) as well as the charge difference (Δe = |es − eT|) between them were calculated based on the CI-NEB image. The result showed that the Δe for AA and ABC1 types are 0.005 and 0.206e, which are consistent with their barrier order of 0.32, and 2.01 eV, respectively. The Mg2+ channel in ABC1-Ti3C2O2 has large fluctuation on electron capture, which is also an important factors that cause the largest diffusion barrier.
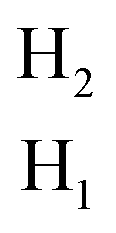 site after structure optimization and the corresponding the half of diffusion pathways were described as
site after structure optimization and the corresponding the half of diffusion pathways were described as  which showed that Mg2+ diffusion behavior in M3C2N2 bulks (including Ti3C2N2) is consistent. The diffusion barriers of Mg2+ in V3C2N2, Zr3C2N2 and Nb3C2N2 had decreased compared with in Ti3C2N2 by 0.37 eV, 0.79 eV and 0.84 eV, respectively. Notably, Nb3C2N2 exhibits the smallest barrier of 0.23 eV. This indicates that N–N covalent binding is weakened more effectively by replacing Ti with Nb than with V and Zr (Fig. 6e). Overall, the Nb3C2N2 obtained by using the transition metal element Nb to replace Ti in AA-Ti3C2N2 has the lowest energy barrier (Fig. S1†).
which showed that Mg2+ diffusion behavior in M3C2N2 bulks (including Ti3C2N2) is consistent. The diffusion barriers of Mg2+ in V3C2N2, Zr3C2N2 and Nb3C2N2 had decreased compared with in Ti3C2N2 by 0.37 eV, 0.79 eV and 0.84 eV, respectively. Notably, Nb3C2N2 exhibits the smallest barrier of 0.23 eV. This indicates that N–N covalent binding is weakened more effectively by replacing Ti with Nb than with V and Zr (Fig. 6e). Overall, the Nb3C2N2 obtained by using the transition metal element Nb to replace Ti in AA-Ti3C2N2 has the lowest energy barrier (Fig. S1†).
 site. Based on this, the formation energy of Nb3C2N2Mgx (0 ≤ x ≤ 1) was calculated via the following equation to explore the thermodynamically stable Mg-containing phases,
site. Based on this, the formation energy of Nb3C2N2Mgx (0 ≤ x ≤ 1) was calculated via the following equation to explore the thermodynamically stable Mg-containing phases,

 and
and 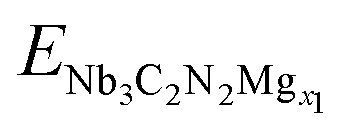 are the total energies of Nb3C2N2 with different Mg2+ adsorption degrees x1 and x2, EMg is the energy of a Mg atom in metal Mg, charge value n = 2 for Mg2+. Interestingly, the calculated voltages contain two distinct plateaus at 4.00 V and 0.64 V, corresponding to the ranges of x = 0–0.75 and x = 1–2, respectively (Fig. 7b). This results indicate that when a monolayer of Mg2+ is intercalated between the Nb3C2N2 layers, the binding affinity of Mg2+ to the Nb3C2N2 layers significantly decreases. Such a significant voltage difference indicates that the material can function as both the positive and negative electrode of the battery under varying ion concentrations. This behavior is analogous to that observed in Prussian blue materials used in potassium-ion batteries.44 By contrast, the voltage platforms presented by Ti-based MXene belong to the anode range (Fig. S2†). From a voltage stability perspective, Ti3C2O2Mgx and Ti3C2O1.78N0.22Mgx exhibit three short platforms decreasing in the concentration of 0 < x < 1. Although Ti3C2N2 possesses two stable platforms, the voltage of 0.02 V is too low for electrode, which is easy to transform the Mg2+ intercalation mechanism into electroplating.
are the total energies of Nb3C2N2 with different Mg2+ adsorption degrees x1 and x2, EMg is the energy of a Mg atom in metal Mg, charge value n = 2 for Mg2+. Interestingly, the calculated voltages contain two distinct plateaus at 4.00 V and 0.64 V, corresponding to the ranges of x = 0–0.75 and x = 1–2, respectively (Fig. 7b). This results indicate that when a monolayer of Mg2+ is intercalated between the Nb3C2N2 layers, the binding affinity of Mg2+ to the Nb3C2N2 layers significantly decreases. Such a significant voltage difference indicates that the material can function as both the positive and negative electrode of the battery under varying ion concentrations. This behavior is analogous to that observed in Prussian blue materials used in potassium-ion batteries.44 By contrast, the voltage platforms presented by Ti-based MXene belong to the anode range (Fig. S2†). From a voltage stability perspective, Ti3C2O2Mgx and Ti3C2O1.78N0.22Mgx exhibit three short platforms decreasing in the concentration of 0 < x < 1. Although Ti3C2N2 possesses two stable platforms, the voltage of 0.02 V is too low for electrode, which is easy to transform the Mg2+ intercalation mechanism into electroplating.
