DOI:
10.1039/D5RA01063B
(Paper)
RSC Adv., 2025,
15, 24650-24667
Design, synthesis and anticancer evaluation of novel hydrazide-2-oxindole analogues as GSK-3β kinase inhibitors†
Received
13th February 2025
, Accepted 5th July 2025
First published on 14th July 2025
Abstract
GSK-3β plays an essential role in cancer progression, making it a promising target for therapeutic intervention with glycogen synthase kinase (GSK-3β) inhibitors. The designed compounds were discovered for their potential role against the proliferation of cancer via targeting GSK-3β. In the present study, hydrazide-2-oxindole analogues were designed, synthesised, and evaluated for anticancer efficacy against GSK-3β. Compounds were screened against Capan-1, HCT-116, LN-229, MCI-4460, DND-41, HL-60, K-562, MOLT4, Z-138 cells and normal cell line HEK 293. Among the compounds, 6Eb and 6Ec showed the highest selective cytotoxicity against prostate cancer (Capan-1) with CC50 values of 9.40 μM, and 8.25 μM, respectively. Furthermore, the mechanism of the anticancer effect was evidenced by 6Eb and 6Ec against GSK-3β kinase with IC50s of 11.02 μM and 59.81 μM, respectively. In addition, elevation of β-catenin and downregulation of NF-kB and STAT3 in the western blot by 6Eb evidenced inhibition of GSK-3β kinase as the cause of cytotoxicity. Furthermore, in vitro results were supported by docking scores of −10.5 kcal mol−1 and −8.8 kcal mol−1, respectively, as compared to bio-acetoxime (−7.7 kcal mol−1). Furthermore, the higher stability in molecular dynamics simulations validated the docking approach and indicated anti-cancer effects by inhibiting GSK-3β. In addition, the density functional theory analysis identified electronic distribution in compounds, which was correlated to the findings of docking and molecular dynamic simulation on their participation in polar and lipophilic interactions with kinase. Moreover, the compounds meet the drug-likeness criteria, suggesting they could be candidates for the development of drugs against cancer that target GSK-3β.
1. Introduction
Many human disorders, including cancer, have been linked to abnormal GSK-3β activity, which suggests that it could be a therapeutic target for anticancer treatment. In around 25 distinct cancer types, GSK-3β has been recognized as a possible therapeutic target in recent investigations, with many of these studies being published in the last decade. In addition, there is growing evidence that blocking GSK-3β activity shields healthy cells and tissues from the negative consequences linked to traditional cancer treatments.1–4 Although only active GSK-3β is expressed in cancer cells, abnormal nuclear accumulation of GSK-3β has been recognized as a characteristic of cancer cells in malignant tumors of various origins.5–8 Recent findings have highlighted the critical involvement of GSK-3β in the anticancer immune response.9–13
GSK-3, a serine/threonine kinase, exists in two isoforms: GSK-3α and GSK-3β. Multiple processes, such as phosphorylation, protein complex formation, and subcellular distribution, contribute to GSK-3, activity regulation.14,15 So far, GSK-3β has been linked to several illnesses, including diabetes, cancer, bipolar disorder, and neurodegenerative disorders. GSK-3β regulates apoptosis, proliferation, and the cell cycle.16 In contrast; GSK-3β has been described as a tumour suppressor in malignancies such as skin, breast, oral cavity, and lung cancer. There are two types of GSK-3β inhibitors: non-ATP competitive inhibitors and ATP competitive inhibitors. Non-ATP competitive inhibitors form a weak binding association with the enzyme and do not compete with ATP concentration, providing a distinct pharmacological advantage. The bulk of known GSK-3β inhibitors are ATP-competitive, targeting the ATP-binding region of GSK-3β. As illustrated in Fig. 1, there are multiple inhibitors from various chemical classes, including bisindolylmaleimide, indolyl-maleimide, pyrimidine, maleimide, indirubine, paullone, pyrazolamide, oxadiazole, thiazole, and thiadiazole.17,18
 |
| | Fig. 1 Pharmacophores of GSK-3β inhibitors. The key structural components involved in the interaction are highlighted. | |
Staurosporine, a natural substance from Streptomyces staurosporeus, inhibits GSK-3β with an IC50 of 15 nM.19 Compound A is a 7-azaindazolyl-indolyl-maleimide analogue with a morpholine side chain that fills the expanded pocket via H-bonds with Lys183 and Asp200.19 The ketone and amine of indolyl-maleimide inhibitor, B, interacted via polar contacts (Fig. 1). Furthermore, the side chain of aliphatic amino alcohol stabilized the complex from the end via an H-bond with Gln185.20 The nitrogen in the pyrimidine and anilino rings of compound C was connected to Tyr134 and Val135 via H-bonds. While in the kinase's back pocket, amide and hydroxyl groups protruded and established additional hydrogen bond with Asp200. Furthermore, their oral bioavailability was comparable in rats (F = 65%; t1/2 = 3 h) and mice (F = 67%; t1/2 = 4.4 h).21 In in vivo research on zebrafish embryos, maleimide analogue D demonstrated a higher affinity and IC50 of 92 μM. An SAR investigation found that substituents at the C-4 or C-5 locations fit into the front area of the ATP site. Larger substitutions had more hydrophobic interactions and a stronger binding affinity.22 Likewise, indirubin analogue E showed higher selectivity against GSK-3β with an IC50 of 22 nM compared to other kinases. The isatin ring's amino and ketone groups created strong hydrogen bonds and lipophilic contacts to stabilize the complex.23,24 Paullone analogue F, containing a cyclic amide and a nitro group that enhanced molecular interactions with the target, demonstrated potent kinase inhibition with an IC50 of 4 nM.25 Likewise, pyrazolamide analogue G exhibited strong kinase binding through a combination of polar and nonpolar interactions, mediated by its amide linkage and phenyl ring, also resulting in an IC50 of 4 nM.26 The compound ‘H’ interacts with the ATP binding site, where the O1 oxygen atom and hydrogen atom on the C2-carbon of the benzodioxole form hydrogen bonds with the amide NH hydrogen and the carbonyl oxygen of Val135 located in the hinge region, respectively. The 4-methoxy-3-fluorobenzyl group occupies the hydrophobic pocket, while both the N3 and N4 nitrogen atoms of the oxadiazole are involved in a distinctive hydrogen bond network connecting Lys85–Glu97–Asp200 through two water molecules.27 Moreover, the urea linkage of compound I formed a key interaction with Gln137 at the centre of the binding site. The methoxyphenyl ring further stabilized the complex by occupying the rear pocket and forming close contact with Ile62, Lys183, and Gln185.28 Additionally, the thiadiazolidinone ring ketones in TDZD-8 (J) established hydrogen connections with Lys206 and Arg96.29 Pharmacophore searches revealed common pharmacophoric components such as amide and urea linkages (open and cyclic), oxime, and a fused ring involved in polar contact. In addition, the core ATP binding site was occupied by the parent heterocyclic ring, which included pyrrole, pyrazole, oxadiazole, thiazole, thiadiazole, indole, and pyrimidines (Fig. 1). Furthermore, substituent's like nitro, methoxy, bromo, fluoro, methyl, and ethyl boosted activity by interacting with the backside of the pocket. Inspired by these findings, we decided to include heteryl rings, such as triazole and indole rings, as well as the linkage of hydrazide in our new molecules to preserve similar types of interactions.
1.1. Rationale and design
Several indolylmaleimides, such as A and B, have been developed to be effective GSK-3β inhibitors based on staurosporine, a microbial alkaloid discovered as an initial GSK-3β inhibitor (Fig. 1). The majority of these indolylmaleimides exhibit toxicity, low solubility, and poor selectivity, making them inappropriate for treating disorders such as diabetes, Alzheimer's disease, and cancer.19,30 As a result, we chose smaller thiadiazolidinone synthetic analogues TDZD-8 and 10, the first non-competitive GSK-3β inhibitors that lack all of the aforementioned toxicities and exhibit drug-like characteristics. TDZD-8 and 10 inhibited GSK-3β at 2 and 10 μM, respectively. They trigger apoptosis, causing extremely rapid cell death and loss of membrane integrity.29 Furthermore, indirubin-3-monoxime analogues such as bio-acetoxime, and 44 were more selective against GSK-3β than other kinases, with affinities of 22 nM and 3 nM, respectively.31 In SY5Y-MYCN cells, bio-acetoxime significantly lowers c-MYC expression and p-SMAD3 levels. It also reduces the viability of KCN, KCNR, SY5Y, Kelly, and IMR32 cells by inducing apoptosis.32 The increased potency of these analogues prompted us to investigate them as potential leads for the development of design compounds via molecular hybridization approach. The thiadiazolidinone ring's ketones in TDZD-8 formed two hydrogen connections with Lys206 and Arg96. To keep the binding pattern consistent, we bioisosterically substituted the thiadiazolidinones with a triazole ring in our proposed molecule. Furthermore, the amino and ketone groups of the indole ring in bio-acetoxime and compound 44 formed strong H-bonds with Asp133, Val135, and Asp200. Additionally, the indole ring increased binding affinity through numerous lipophilic interactions with Ala83, Tyr134, and Leu188. Because of it's significant contribution, we bioisosterically replaced the benzyl ring of TDZD-8 in our developed compound with indole moiety to retain a similar interaction. Furthermore, the monoxime group in the bio-acetoxime stabilized protein–ligand complex by filling the back side of the pocket. To preserve the same stability and binding connection in our developed compounds, we added different substituted hydrazides to the indole ring (Fig. 2).
 |
| | Fig. 2 Modification strategies of designed compounds. | |
In the present study, a series of indole-linked triazoles (6Ab–Ed) were designed, synthesized, and evaluated for their anti-proliferative activity. Their mechanism of action was elucidated through a combination of GSK-3β kinase inhibition assays, western blot analysis, and in silico studies.
2. Materials and methods
2.1. Chemistry
All chemicals and reagents were procured from BLD Pharmatech, Hyderabad, and Spectrochem Bengaluru, India. The progress of reactions was monitored by thin layer chromatography (TLC) on pre-coated silica gel 60 F254 plates (Merck, Germany) with chloroform and ethanol as the mobile phase. Melting points (MP) were determined using a digital melting point apparatus (DBK 50 Hz, 220 V) and reported uncorrected. Proton and carbon nuclear magnetic resonance (1H/13C NMR) spectra were recorded on a 400/100 MHz Bruker instrument in CDCl3/DMSO-d6 and chemical shift values (J) were reported in hertz (Hz). Fourier-transform infrared (FT-IR) spectra were obtained on JASCO460+ FTIR spectrophotometer. High-resolution mass spectrometry (HRMS) data were collected by electrospray ionization (ESI) technique using Waters Synapt G2 QTOF mass spectrometry. The 1-(prop-2-yn-1-yl)indoline-2,3-dione (3) was synthesized by reacting indole-2,3-dione (1) with 3-bromoprop-1-yne (2) in presence of K2CO3 in dimethylformamide (DMF).33 The respective aryl azides (4A–E) were obtained by reacting aryl bromides with sodium azide as per literature.33 Other intermediate derivatives (5A–D) were prepared according to literature (Scheme 1).34
 |
| | Scheme 1 Synthesis of 3-substituted-1-((1-benzyl-1H-1,2,3-triazol-4-yl) methyl)indoline-2-one (6Ab–Ed). | |
2.1.1. Synthesis of 1-((1-(4-methoxybenzyl)-1H-1,2,3-triazol-4-yl)methyl)indoline-2,3-dione (5E). 1-(Prop-2-yn-1-yl)indoline-2,3-dione (3) 3.12 mmol and respective azides (4A–E) were dissolved in 20 mL of tertiary butanol. To the resulting solution, sodium ascorbate 1.24 mmol and copper sulphate pentahydrate (20 mL distilled water) 0.62 mmol were added and stirred for 5 h at 35 °C. Progress of reaction was monitored by TLC. After completion, the reaction mixture was extracted with ethyl acetate. Organic layer was separated and dried over anhydrous Na2SO4. Excess solvent was removed under vacuum and recrystallized from ethanol. MP: 163–165 °C; yield (%) 85; FT-IR KBr (v cm−1): 3149, 3079, 2959, 1739, 1613, 1470, 1248; 1H-NMR (400 MHz, DMSO-d6 δ/ppm): 8.13 (1H, s), 7.62–7.58 (1H, t, J = 15.6 Hz), 7.54 (1H, d, J = 7.6 Hz), 7.26 (2H, d, J = 11.2 Hz), 7.14 (2H, t, J = 15.2 Hz), 6.89 (2H, d, J = 8.8 Hz), 5.44 (2H, s, –CH2), 4.92 (2H, s, –CH2), 3.70 (3H, s, –OCH3).
2.2. General procedure for synthesis of 1-((1-benzyl-1H-1,2,3-triazol-4-yl)methyl)-3-hydrazonoindolin-2-one (6A–E)
1-((1-Benzyl-1H-1,2,3-triazole-4-yl)methyl)indoline-2,3-dione (5A–E) 1.0 mmol, and respective hydrazines (R1NHNH2) were refluxed in 50 mL absolute ethanol and 0.5 mL glacial acetic acid. The reaction mixture was cooled and, the precipitated mass of 6 was re-crystallized from the ethanol-chloroform mixture in appropriate ratio.
2.2.1. 2-(1-((1-Benzyl-1H-1,2,3-triazol-4-yl)methyl)-2-oxoindolin-3-ylidene)hydrazine carbothioamide (6Ab). MP: 254–256 °C; yield (%) 65; FT-IR KBr (ν cm−1): 3410, 3150, 3055, 1692, 1613, 1469 & 1219; 1H-NMR (400 MHz, DMSO-d6 δ/ppm): 12.37 (1H, s, –NH), 9.10 (1H, s), 8.75 (1H, s), 8.21 (1H, s), 7.72 (1H, d, J = 7.2 Hz), 7.42–7.27 (6H, m), 7.18–7.12 (2H, m), 5.54 (2H, s, –N–CH2), 5.02 (2H, s, –N–CH2); 13C NMR (100 MHz, DMSO-d6 δ/ppm): 179.15, 160.89, 142.83, 142.26, 136.33, 131.57, 131.43, 129.23, 128.65, 128.36, 124.28, 123.54, 121.26, 119.83, 110.84, 53.33, 35.05; HRMS (ESI) M + H for C19H17N7OS, calculated: 391.1215, observed: 392.1290.
2.2.2. 1-((1-Benzyl-1H-1,2,3-triazol-4-yl)methyl)-3-(2-phenylhydrazono)indolin-2-one (6Ac). MP: 224–226 °C; yield (%) 55; FT-IR KBr (ν cm−1): 3437, 3181–3064, 2966, 2944, 1665, 1612, 1466 & 1245; 1H-NMR (400 MHz, DMSO-d6 δ/ppm): 12.66 (1H, s, –NH), 8.18 (1H, s), 7.62 (1H, d, J = 6.8 Hz), 7.47–7.26 (10H, m), 7.17–7.05 (3H, m), 5.54 (2H, s, –N–CH2), 5.07 (2H, s, –N–CH2); 13C NMR (100 MHz, DMSO-d6 δ/ppm): 161.24, 142.83, 142.74, 140.33, 136.37, 130.00, 129.22, 128.82, 128.63, 128.37, 127.00, 124.07, 123.68, 123.03, 120.96, 118.93, 114.73, 110.29, 53.29, 34.78; HRMS (ESI) M + H for C24H20N6O, calculated: 408.1699, observed: 409.1780.
2.2.3. 1-((1-Benzyl-1H-1,2,3-triazol-4-yl)methyl)-3-(2-(5-chloro-6-oxo-1,6-dihydropyrida-zin-4-yl)hydrazono)indolin-2-one (6Ad). MP: >300 °C; yield (%) 65; FT-IR KBr (ν cm−1): 3477, 3054, 2875, 1683, 1608, 1470 & 1270; 1H-NMR (400 MHz, DMSO-d6 δ/ppm): 13.26 (1H, s, –NH), 12.92 (1H, s, –NH), 8.56 (1H, s), 8.24 (1H, s), 7.76 (1H, d, J = 10.4 Hz), 7.43 (1H, t, J = 14.4 Hz), 7.38–7.32 (3H, m), 7.29–7.27 (2H, m), 7.25 (1H, d J = 8.4 Hz), 7.18 (1H, t, J = 14.4 Hz), 5.56 (2H, s, –N–CH2), 5.09 (2H, s, –N–CH2); 13C NMR (100 MHz, DMSO-d6 δ/ppm): 161.50, 158.16, 142.29, 142.21, 140.97, 136.33, 134.55, 131.47, 129.23, 128.65, 128.38, 127.42, 124.29. 123.68, 119.48, 110.99, 109.84, 53.33, 35.03; HRMS (ESI) M + H for C22H17ClN8O2, calculated: 460.1163, observed: 461.1238.
2.2.4. 2-(1-((1-(4-Chlorobenzyl)-1H-1,2,3-triazol-4-yl)methyl)-2-oxoindolin-3-ylidene) hydrazinecarbothioamide (6Bb). MP: 247–248 °C; yield (%) 50; FT-IR KBr (ν cm−1): 3420, 3085, 2989, 2934, 1679, 1607, 1490 & 1224; 1H NMR (400 MHz, DMSO-d6 δ/ppm): 12.37 (1H, s, –NH), 9.11 (1H, s), 8.76 (1H, s), 8.22 (1H, s), 7.72 (1H, d, J = 7.2 Hz), 7.44–7.38 (3H, m), 7.32 (2H, d, J = 8.4 Hz), 7.18–7.11 (2H, m), 5.56 (2H, s, –N–CH2), 5.03 (2H, s, –N–CH2); 13C NMR (100 MHz, DMSO-d6 δ/ppm): 179.15, 160.88, 142.82, 142.32, 135.30, 133.36, 131.57, 131.41, 130.35, 129.21, 124.34, 123.54, 121.26, 119.82, 110.83, 52.52, 35.03; HRMS (ESI) M + H for C19H16ClN7OS, calculated: 425.0826, observed: 426.0902.
2.2.5. 1-((1-(4-Chlorobenzyl)-1H-1,2,3-triazol-4-yl)methyl)-3-(2-phenylhydrazono)indolin-2-one (6Bc). MP: 236–238–240 °C; yield (%) 65; FT-IR KBr (ν cm−1): 3310, 3069, 2983, 2938, 1661, 1594, 1469 & 1221; 1H-NMR (400 MHz, DMSO-d6 δ/ppm): 12.68 (1H, s, –NH), 8.19 (1H, s), 7.62 (1H, d, J = 6.8 Hz), 7.49 (2H, d, J = 8.8 Hz), 7.44–7.38 (3H, m), 7.32–7.28 (3H, m), 7.18–7.06 (4H, m), 5.56 (2H, s, –N–CH2), 5.09 (2H, s, –N–CH2); 13C NMR (100 MHz, DMSO-d6 δ/ppm): 161.24, 142.80, 140.32, 135.34, 133.35, 130.36, 130.01, 129.21, 128.84, 124.12, 123.69, 123.04, 120.95, 118.94, 114.73, 110.29, 52.49, 35.06, 34.06; HRMS (ESI) M + H for C24H19ClN6O, calculated: 442.1309, observed: 443.1386.
2.2.6. 3-(2-(5-Chloro-6-oxo-1,6-dihydropyridazin-4-yl)hydrazono)-1-((1-(4-chlorobenzyl)-1H-1,2,3-triazol-4-yl)methyl)indolin-2-one (6Bd). MP: 241–242 °C; yield (%) 65; FT-IR KBr (ν cm−1): 3423, 3067, 3032, 2983–2923, 1685, 1607, 1470 & 1226; 1H-NMR (400 MHz, DMSO-d6 δ/ppm): 13.25 (1H, s, –NH), 12.91 (1H, s, NH), 8.56 (1H, s), 8.24 (1H, s), 7.76 (1H, d, J = 6.8 Hz), 7.46–7.42 (3H, m), 7.32 (2H, d, J = 8 Hz), 7.23–7.17 (3H, dt, J = 8.4, 15.2 Hz), 5.57 (2H, s, –N–CH2), 5.09 (2H, s, –N–CH2); 13C NMR (100 MHz, DMSO-d6 δ/ppm): 161.50, 158.17, 142.27, 140.97, 135.30, 134.54, 131.49, 130.37, 129.21, 127.42, 124.34, 123.70, 120.95, 119.48, 110.98, 109.84, 52.52, 35.02; HRMS (ESI) M + H for C22H16Cl2N8O2, calculated: 494.0733, observed: 495.0855.
2.2.7. 2-(1-((1-(4-Methylbenzyl)-1H-1,2,3-triazol-4-yl)methyl)-2-oxoindolin-3-ylidene) hydrazinecarbothioamide (6Cb). MP: 263–264 °C; yield (%) 60; FT-IR KBr (ν cm−1): 3413, 3060, 2920, 1685, 1612, 1489 & 1220; 1H-NMR (400 MHz, DMSO-d6 δ/ppm): 12.37 (1H, s, –NH), 9.11 (1H, s), 8.76 (1H, s), 8.17 (1H, s), 7.71 (1H, d, J = 6.8 Hz), 7.40 (1H, t, J = 15.2 Hz), 7.19–7.09 (6H, m), 5.49 (2H, s, –N–CH2), 5.02 (2H, s, –N–CH2), 2.26 (3H, s, –CH3); 13C NMR (100 MHz, DMSO-d6 δ/ppm): 179.16, 160.88, 142.84, 142.22, 138.03, 133.30, 131.56, 131.42, 129.75, 128.43, 124.09, 123.53, 121.25, 119.83, 110.84, 53.14, 35.05, 21.13; HRMS (ESI) M + H for C20H19N7OS, calculated: 405.1372, observed: 406.1465.
2.2.8. 1-((1-(4-Methylbenzyl)-1H-1,2,3-triazol-4yl)methyl)-3-(2-phenylhydrazono)indolin-2-one (6Cc). MP: 216–217 °C; yield (%) 70; FT-IR KBr (ν cm−1): 3423, 3068, 2923, 1662, 1612, 1469 & 1221; 1H-NMR (400 MHz, DMSO-d6 δ/ppm): 12.67 (1H, s, –NH), 8.14 (1H, s), 7.61 (1H, d, J = 7.2 Hz), 7.54 (2H, d, J = 9.6 Hz), 7.39 (2H, t, J = 15.2 Hz), 7.30 (1H, t, J = 16.8 Hz), 7.19–7.05 (7H, m), 5.49 (2H, s, –N–CH2), 5.07 (2H, s, –N–CH2), 2.26 (3H, s,–CH3); 13C NMR (100 MHz, DMSO-d6 δ/ppm): 161.23, 142.83, 142.71, 140.33, 138.02, 133.35, 130.00, 129.74, 128.83, 128.44, 126.99, 123.88, 123.68, 123.03, 120.94, 118.93, 114.73, 110.29, 53.10, 34.77, 21.12; HRMS (ESI) M + H for C25H22N6O, calculated: 422.1855, observed: 423.1956.
2.2.9. 3-(2-(5-Chloro-6-oxo-1,6-dihydropyridazin-4-yl)hydrazono)-1-((1-(4-methylbenzyl)-1H-1,2,3-triazol-4-yl)methyl)indolin-2-one (6Cd). MP: >300 °C; yield (%) 60; FT-IR KBr (ν cm−1): 3298, 3045, 2869, 1687, 1609, 1469 & 1270; 1H-NMR (400 MHz, DMSO-d6 δ/ppm): 13.28 (1H, s, –NH), 12.90 (1H, s, –NH), 8.55 (1H, s), 8.19 (1H, s), 7.75 (1H, d, J = 6.0 Hz), 7.42 (1H, t, J = 15.2 Hz), 7.23–7.14 (6H, m), 5.49 (2H, s, –N–CH2), 5.07 (2H, s, –N–CH2), 2.26 (3H, s, –CH3); 13C NMR (100 MHz, DMSO-d6 δ/ppm): 161.50, 158.17, 142.30, 142.17, 140.98, 138.04, 133.31, 131.48, 129.75, 128.45, 127.43, 124.10, 123.69, 120.94, 119.48, 109.83, 53.14, 35.03, 21.12; HRMS (ESI) M + H for C23H19ClN8O2, calculated: 474.1319, observed: 475.1425.
2.2.10. 2-(1-((1-(4-Nitrobenzyl)-1H-1,2,3-triazol-4-yl)methyl)-2-oxoindolin-3-ylidene) hydrazinecarbothioamide (6Db). MP: 277–278 °C; yield (%) 60; FT-IR KBr (ν cm−1): 3466, 3111, 3079, 2996, 2953, 1684, 1598, 1490, 1347 & 1222; 1H-NMR (400 MHz, DMSO-d6 δ/ppm): 12.36 (1H, s, –NH), 9.13 (1H, s), 8.77 (1H, s), 8.29 (1H, s), 8.23 (2H, d, J = 9.2 Hz), 7.72 (1H, d, J = 8.0 Hz), 7.51 (2H, d, J = 8.4 Hz), 7.41 (1H, t, J = 15.2 Hz), 7.20–7.13 (2H, m), 5.74 (2H, s, –N–CH2), 5.05 (2H, s, –N–CH2); 13C NMR (100 MHz, DMSO-d6 δ/ppm): 179.16, 160.90, 147.71, 143.72, 142.82, 142.46, 131.58, 131.37, 129.51, 124.73, 124.37, 123.55, 121.25, 119.86, 110.85, 52.42, 35.06; HRMS (ESI) M + H for C19H16N8O3S, calculated: 436.1066, observed: 437.1143.
2.2.11. 1-((1-(4-Nitrobenzyl)-1H-1,2,3-triazol-4-yl)methyl)-3-(2-phenylhydrazono)indolin-2-one (6Dc). MP: 216–218 °C; yield (%) 65; FT-IR KBr (ν cm−1): 3433, 3074, 2978, 2942, 1664, 1597, 1467, 1520, 1350 & 1245; 1H-NMR (400 MHz, DMSO-d6 δ/ppm): 12.68 (1H, s, –NH), 8.26 (1H, s), 8.23 (2H, d, J = 8.4 Hz), 7.62 (1H, d, J = 6.8 Hz), 7.54–7.46 (4H, m), 7.39 (2H, t, J = 16.4 Hz), 7.31 (1H, t, J = 16.0 Hz), 7.19–7.03 (3H, ddm, J = 7.2, 8 Hz), 5.74 (2H, s, –N–CH2), 5.10 (2H, s, –N–CH2); 13C NMR (100 MHz, DMSO-d6 δ/ppm): 161.25, 147.70, 143.72, 142.93, 142.81, 140.30, 130.00, 129.50, 128.84, 126.97, 124.52, 124.35, 123.69, 123.04, 120.96, 118.94, 114.72, 110.28, 52.40, 34.77; HRMS (ESI) M − H for C24H19N7O3, calculated: 453.1549, observed: 452.1864.
2.2.12. 3-(2-(5-Chloro-6-oxo-1,6-dihydropyridazin-4-yl)hydrazono)-1-((1-(4-nitrobenzyl)-1H-1,2,3-triazol-4-yl)methyl)indolin-2-one (6Dd). MP: >300 °C; yield (%) 60; FT-IR KBr (ν cm−1): 3130, 3046, 2974, 2841, 1685, 1609, 1470, 1349 & 1272; 1H-NMR (400 MHz, DMSO-d6 δ/ppm): 13.26 (1H, s, –NH), 12.91(1H, s, –NH), 8.55 (1H, s), 8.31 (1H, s), 8.23 (2H, d, J = 8.0 Hz), 7.76 (1H, d, J = 8.0 Hz), 7.51 (1H, d, J = 8.0 Hz), 7.44 (2H, t, J = 15.2 Hz), 7.26 (1H, d, J = 8.0 Hz), 7.18 (1H, t, J = 18.4 Hz), 5.75 (2H, s, –N–CH2), 5.10 (2H, s, –N–CH2); 13C NMR (100 MHz, DMSO-d6 δ/ppm): 162.89, 161.51, 158.16, 147.71, 143.69, 142.39, 142.27, 140.96, 134.52, 131.50, 129.52, 127.41, 124.74, 124.36, 123.71, 120.95, 119.49, 110.98, 109.86, 52.42, 35.03; HRMS (ESI) M − H for C22H16ClN9O4, observed: 505.1014, calculated: 504.0933.
2.2.13. 2-(1-((1-(4-Methoxybenzyl)-1H-1,2,3-triazol-4-yl)methyl)-2-oxoindolin-3-ylidene) hydrazinecarbothioamide (6Eb). MP: 256–258 °C; yield (%) 60; FT-IR KBr (ν cm−1): 3419, 3127, 3067, 2999, 2962, 1676, 1613, 1467 & 1251; 1H-NMR (400 MHz, DMSO-d6 δ/ppm): 12.36 (1H, s, –NH), 9.12 (1H, s), 8.77 (1H, s), 8.15 (1H, s), 7.71 (1H, d, J = 9.2 Hz), 7.39 (1H, t, J = 15.2 Hz), 7.27 (2H, d, J = 9.2 Hz), 7.18–7.13 (2H, m), 6.91 (2H, d, J = 8.4 Hz), 5.45 (2H, s, –N–CH2), 5.01 (2H, s, –N–CH2), 3.72 (3H, s, –OCH3); 13C NMR (100 MHz, DMSO-d6 δ/ppm): 179.16, 160.88, 159.60, 142.84, 142.20, 131.57, 131.43, 130.06, 128.22, 123.91, 123.53, 121.25, 119.82, 114.57, 110.84, 55.59, 52.89, 35.05; HRMS (ESI) M + H forC20H19N7O2S, calculated: 421.1321, observed: 422.1401.
2.2.14. 1-((1-(4-Methoxybenzyl)-1H-1,2,3-triazol-4-yl)methyl)-3-(2-phenylhydrazono) indolin-2-one (6Ec). MP: 198–200 °C; yield (%) 65; FT-IR (v cm−1): 3202, 3064, 3021, 2971, 2956, 1669, 1613, 1469 & 1246; 1H-NMR (400 MHz, DMSO-d6 δ/ppm): 12.67 (1H, s, –NH), 8.12 (1H, s), 7.61 (1H, d, J = 7.6 Hz), 7.48 (2H, d, J = 7.2 Hz), 7.39 (2H, t, J = 15.2 Hz), 7.31–7.25 (3H, m), 7.18–7.05 (3H, m), 6.91 (2H, d, J = 8.0 Hz), 5.45 (2H, s, –N–CH2), 5.06 (2H, s, –N–CH2), 3.72 (3H, s, –OCH3); 13C NMR (100 MHz, DMSO-d6 δ/ppm): 161.23, 159.59, 142.83, 142.69, 140.34, 130.07, 130.00, 128.83, 128.26, 126.99, 123.03, 120.94, 118.93, 114.73, 114.56, 110.29, 55.58, 52.85, 34.77; HRMS (ESI) M + H for C25H22N6O2, calculated: 438.1804, observed: 439.1880.
2.2.15. 3-(2-(5-Chloro-6-oxo-1,6-dihydropyridazin-4-yl)hydrazono)-1-((1-(4-methoxy-benzyl)-1H-1,2,3-triazol-4-yl)methyl)indolin-2-one (6Ed). MP: >300 °C; yield (%) 70; FT-IR (v cm−1): 3298, 3210, 2954, 1678, 1609, 1468 & 1255; 1H-NMR (400 MHz, DMSO-d6 δ/ppm): 13.26 (1H, s, –NH), 12.90 (1H, s, –NH), 8.55 (1H, s), 8.18 (1H, s), 7.75 (1H, d, J = 7.6 Hz), 7.44 (1H, t, J = 16.0 Hz), 7.30–7.22 (3H, m), 7.17 (1H, t, J = 15.2 Hz), 6.91 (2H, d, J = 8.8 Hz), 5.46 (2H, s, –N–CH2), 5.06 (2H, s, –N–CH2), 3.72 (3H, s, –OCH3); 13C NMR (100 MHz, DMSO-d6 δ/ppm): 161.49, 159.60, 158.17, 142.31, 142.16, 140.98, 134.55, 131.48, 130.08, 128.22, 127.43, 123.92, 123.68, 120.94, 119.48, 114.57, 110.98, 109.83, 55.59, 52.89, 35.03; HRMS (ESI) M + H for C23H19ClN8O3, calculated: 490.1269, observed: 491.1142.
2.3. Biological investigation
2.3.1. Cancer cell lines. The human cancer cell lines Capan-1, HCT-116, NCI-H460, LN-229, HL-60, K-562, MOLT4, Z-138 and HEK293 were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA). The DND-41 cell line was sourced from the Deutsche Sammlung von Mikroorganismen und Zellkulturen (DSMZ Leibniz-Institut, Braunschweig, Germany). All cell lines were cultured according to the suppliers' recommendations. Culture media were purchased from Gibco (Gibco Life Technologies, Merelbeke, Belgium) and supplemented with 10% fetal bovine serum (HyClone, Cytiva, MA, USA). For all biological assays, the test compounds and reference drugs were dissolved in DMSO at a concentration of 100 μM.
2.3.2. Cytotoxicity assays. Adherent cell lines were seeded at densities ranging from 500 to 1500 cells per well in 384-well plates (Greiner Bio-One, Vilvoorde, Belgium). Following overnight incubation, cells were treated with seven different concentrations of the test compounds, ranging from 100 to 0.006 μM. Untreated cell lines (i.e., without compound treatment) were used as negative controls.Suspension cell lines were seeded at densities ranging from 2500 to 5000 cells per well in 384-well culture plates containing the test compounds at the same concentration points. All cell lines were incubated for 72 h with compounds and then analyzed using the CellTiter 96® AQueous One Solution Cell Proliferation Assay (MTS) reagent (Promega, Leiden, The Netherlands) according to the manufacturer's instructions. Absorbance of the samples was measured at 490 nm using a SpectraMaxPlus 384 (Molecular Devices, CA, USA), and OD values were used to calculate the 50% inhibitory concentration (IC50). Compounds were tested in two independent experiments.35
2.3.3. GSK3β kinase assay using ADP-Glo. A reaction mixture of 25 μL was prepared in a 96-well plate, consisting of 5X reaction buffer, 50 μM ATP, 50 μM DTT, 1 μg per μL GSK3β substrate, and 0.5 ng per μL GSK-3β enzyme (Catalognumber: V1991 Promega), along with increasing concentrations of the compounds 6Eb and 6Ec. A reaction mixture containing DMSO served as the vehicle control, while 12 nM laduviglusib was used as a positive control for the experiment. The reaction was incubated at room temperature for 60 min. Following incubation, 5 μL of ADP-Glo TM Reagent (Catalog number: V6930, Promega) was added and the mixture was incubated at room temperature for an additional 40 min. Subsequently, 10 μL of kinase detection reagent was added, and the mixture was incubated at room temperature for 30 min. Luminescence was measured using a plate-reader with an integration time of 0.25 to 1 second.
2.3.4. Western blotting. Compounds 6Eb, 6Ec treated and control cells were harvested and lysed in RIPA buffer (1 M Tris pH 8, 1% Triton X-100, 0.5% sodium deoxycholate, 1% sodium chloride, 0.1% SDS, 1 mM sodium orthovanadate) supplemented with a protease inhibitor cocktail from MP Biomedicals. The total protein concentration was determined using the Bradford method, and 35 μg of protein was resolved by SDS-PAGE before being transferred to a PVDF membrane. The membrane was cut and probed with the appropriate antibodies. The antibody concentrations were as follows: GAPDH (BioLegend) at 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5000, NF-kB (BioLegend) at 1:1000, PTEN (CST) at 1:1000, STAT-3 (CST) at 1:1000, β-catenin at 1:1000. Anti-rabbit IgG-HRP (CST) at 1:1000, and anti-mouse-IgG-HRP (CST) at 1:5000 and 1:1000. Protein bands were visualised using the enhanced chemiluminescence substrate from Bio-Rad. Images were captured on the Bio-Rad Doc.
:5000, NF-kB (BioLegend) at 1:1000, PTEN (CST) at 1:1000, STAT-3 (CST) at 1:1000, β-catenin at 1:1000. Anti-rabbit IgG-HRP (CST) at 1:1000, and anti-mouse-IgG-HRP (CST) at 1:5000 and 1:1000. Protein bands were visualised using the enhanced chemiluminescence substrate from Bio-Rad. Images were captured on the Bio-Rad Doc.
2.4. Computational (in silico) study
2.4.1. Docking protocol. The protein–ligand interactions were performed using the PyRx software (version 0.8) using AutoDockVina. The docking method had previously been established.36,37 In brief, GSK-3β receptor protein (PDB ID: 1Q41) was extracted from the Protein Data Bank (PDB). The ligand structures were drawn using ChemDraw Ultra 12 and modified in 3D conformers. The protein was treated before being saved in PDBQT format, which included eliminating excess metals and water, reducing energy, and introducing hydrogen to the native ligand. The ligand was altered by reducing its energy content level and introducing Gasteiger charges and polar hydrogen. The file was saved using PDBQT, with active torsion set to 6. The grid box for GSK-3β was created using centre attributes (X = 39.65, Y = 6.22, Z = 37.53) and size (X = 25, Y = 25, and Z = 25). The number of conformers was set at ten with an exhaustiveness of eight. The Discovery Studio Visualization software (version 4) was used to view the interaction. The docking scores were captured, and the 3D contact photos were stored as JPG files.
2.4.2. Molecular dynamics simulation studies. The DESMOND software (Schrodinger Release 2021-3; DE Shaw Research, New York; academic license) was used to evaluate molecular dynamics of the best docked protein–ligand combination. This study used the Berendsen thermostat and barostat procedures for 100 ns. The system was resolved, reduced, and stored in a TIP3P. The Desmond package for protein production, ligand preparation, and Epik tools all validated the correctness of molecular structures. Physiological states were mimicked by neutralizing the solvated complex system with suitable Na+/Cl− counterions at a salt concentration of 0.15 M using an optimum potential for liquid simulations (OPLS) all-atom force field (at 300 K and 1.01325 bars). To relax the system, the Broyden–Fletcher–Goldfarb–Shanno and steepest descent algorithms were used. The Nose–Hoover thermostat and Martyna–Tobias–Klein barostat algorithms were used in the dynamics simulation at 300 K (Kelvin) and 1 atm. (atmospheric) pressure. The smooth particle mesh Ewald technique was used to manage both long-range and short-range coulombic interactions, using 9 Å endpoint values. MD simulations were performed for 100 ns, with data on trajectory recorded for the remaining 2 ns. The stability of the bound complexes 6Ec-1Q41 and bioacetoxime-1Q41 was assessed using root mean square deviation (RMSD).37
2.4.3. MM-GBSA calculations. The binding free energies of the GSK-3β protein (1Q41), 6Ec, and standard (bioacetoxime) complex were examined using the molecular mechanics-generalized born surface area (MM-GBSA) methodology and the prime module's Python script called MMPBSA.py.38,39 The binding free energy was calculated using the OPLS 2005 force field, the VSGB solvent model, and the rotamer search method.40 The following eqn (1) was used to compute the binding free energy of ligands with the GSK-3β receptor:| | |
ΔGbind = Gcomplex − (Gprotein + Gligand)
| (1) |
where ΔGbind = binding free energy, Gcomplex = free energy of the complex, Gprotein = free energy of the target protein, and Gligand = free energy of the ligand.
2.4.4. Density functional theory (DFT) calculations on 6Ec molecule. The theoretical computations were performed using the DFT method to calculate properties like chemical potential (μ), hardness (η), softness (S), electrophilicity index (w), and electronegativity (c). Molecular electrostatic potential and frontier molecular orbitals were orbitals were analyzed using optimized geometry of the studied compound (6Ec). All DFT calculations were carried out using the Gaussian 16 program, taking hybrid Becke's three parameters (B3) with a Lee–Yang–Par (LYP) correlation functional (B3LYP) and a 6-31G(d) basis set. GaussView 6.1 interface program was used to visualize three-dimensional structures and to prepare input files for Gaussian.41
2.4.5. ADME/toxicity prediction. Molecular descriptors commonly used in absorption, distribution, metabolism, and elimination (ADME) analysis were calculated using the Swiss-ADME web server.42,43
3. Results and discussion
3.1. Chemistry
A total of fifteen indole linked 1,2,3-triazoles (6Ab–Ed) were prepared by reacting 1.0 mmol of 1-((1-benzyl-1H-1,2,3-triazole-4-yl)methyl)indoline-2,3-dione (5A–E), and respective hydrazines (R1NHNH2). All structures were confirmed for their structure by FTIR, 1H/13C NMR and, HRMS data. In the FT-IR spectra of synthesized derivatives (6Ab–6Ed), the indole-NH stretching peaks appeared between 3477–3130 cm−1 while, the aromatic and aliphatic –CH stretching were observed in the range of 3210–3021 and 2999–2841 cm−1 respectively. Intense peaks for carbonyl (![[double bond splayed left]](https://www.rsc.org/images/entities/char_e009.gif) C
C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) stretching appeared between 1692–1661 cm−1. In 1H-NMR spectra of 6Ab–Ed the –NH protons appeared between δ 13.88–12.26 ppm. Aromatic protons observed in the range of δ 9.38–6.84 ppm. Peaks for –CH2 protons appeared between δ 5.75–5.01 ppm. The methoxy protons (–OCH3) of 6Eb, 6Ec & 6Ed appeared at δ 3.72 ppm. The methyl protons (–CH3) of 6Cb, 6Cc & 6Cd appeared at δ 2.26 ppm. The 13C NMR spectra of all molecules had shown peak for CS at δ 179 ppm, between δ 162.89 to 160.88 ppm for CO, and between δ 159.60 to 109.83 ppm for aromatic carbons, at δ 55.89 ppm for OCH3 and between δ 53.33 to 34.77 ppm for –N–CH2, at δ 21.13 ppm for CH3 carbons. The m/z values of all molecules (6Ab–Ed) were found in close proximity with calculated mass of the molecule and confirming their structure.
O) stretching appeared between 1692–1661 cm−1. In 1H-NMR spectra of 6Ab–Ed the –NH protons appeared between δ 13.88–12.26 ppm. Aromatic protons observed in the range of δ 9.38–6.84 ppm. Peaks for –CH2 protons appeared between δ 5.75–5.01 ppm. The methoxy protons (–OCH3) of 6Eb, 6Ec & 6Ed appeared at δ 3.72 ppm. The methyl protons (–CH3) of 6Cb, 6Cc & 6Cd appeared at δ 2.26 ppm. The 13C NMR spectra of all molecules had shown peak for CS at δ 179 ppm, between δ 162.89 to 160.88 ppm for CO, and between δ 159.60 to 109.83 ppm for aromatic carbons, at δ 55.89 ppm for OCH3 and between δ 53.33 to 34.77 ppm for –N–CH2, at δ 21.13 ppm for CH3 carbons. The m/z values of all molecules (6Ab–Ed) were found in close proximity with calculated mass of the molecule and confirming their structure.
3.2. Cytotoxicity study
The cytotoxicity studies in a series of hydrazide-2-oxindole analogues (6Ab–Ed) carried out across a broad range of human cancer cell lines include Pancreatic adenocarcinoma (Capan-1), Colorectal carcinoma (HCT-116), Glioblastoma (LN229), Lung carcinoma (NCI-H460), Acute lymphoblastic leukaemia (DND-41), Acute myeloid leukaemia (HI-60), Chronic myeloid leukaemia (K562), T lymphoblast (MOLT4), Non-Hodgkin lymphoma (Z138) and normal human embryonic kidney (HEK 293) cell lines. Sunitinib and 5-Fluorouracil were used as reference standard during the study. In comparison to sunitinib (1.5 μM) and 5-fluorouracil (2.7 μM), molecules 6Eb and 6Ec exhibited moderate cytotoxicity specifically against Capan-1 cells with IC50 values in the range of 8.25–9.40 μM indicating some level of cytotoxicity over other molecules in the series. Two more compounds namely 6Cb and 6Eb showed cytotoxicity against LN229 cells (CC50: 10.20–12.55 μM). The compounds 6Eb and 6Ec displayed promising toxicity against MOLT4 cells with 2.7 and 0.9 μM respectively (ESI Fig. 1†). Rest of the molecules failed to exhibit significant cytotoxicity against tested human cancer cell lines. To test these compounds toxicity on normal cells, both 6Eb and 6Ec were tested on HEK 293 cells and found to be non-toxic with IC50 46 μM and 144 μM respectively. These results indicate that these two molecules are non-toxic to normal cells with the great selectivity to cancer cells. The study suggests that the substituent on the triazole benzyl ring (specifically –H or electron donating CH3 and 4-OCH3) and the carbothiamide and pridazinone substitutions on the hydrazide nitrogen are critical in the observed cytotoxicity by 6Cb, 6Eb and 6Ec. Based on cytotoxicity data we can conclude that specific substituents other than –H or methyl and methoxy became potent cytotoxic compounds, particularly against the Capan-1 and LN229 cells. Further, despite the moderate activity of 6Cb, 6Eb against LN229, & 6Eb, 6Ec against Capan-1 cells, none of the derivatives exhibited notable cytotoxicity against other cancer cell lines suggesting, a certain degree of selectivity for LN229 and Capan-1 cells. Overall these compounds are selectively potent against adherent type cancer cell lines tested but not to suspension type cancer cell lines (Table 1).
Table 1 Cytotoxic effects of the compounds on Capan-1, HCT-116, LN229, NCI-H460, DND-41, HI-60, K562 & Z138 cells. Results are presented as mean ± standard deviation of two independent experiments
| Compounds |
Cytotoxicity adherent cells (CC50) 100 μM |
Cytotoxicity suspension cells (CC50) 100 μM |
| Capan-1 |
HCT-116 |
LN229 |
NCI-H460 |
DND-41 |
Hl-60 |
K562 |
Z138 |
| 6Ab |
>100 |
>100 |
>100 |
>100 |
>100 |
>100 |
>100 |
>100 |
| 6Ac |
>100 |
>100 |
>100 |
>100 |
>100 |
>100 |
>100 |
>100 |
| 6Ad |
>100 |
>100 |
>100 |
>100 |
>100 |
>100 |
>100 |
>100 |
| 6Bb |
>100 |
54.05 ± 0.71 |
>100 |
>100 |
>100 |
>100 |
>100 |
>100 |
| 6Bc |
>100 |
>100 |
>100 |
>100 |
>100 |
>100 |
>100 |
>100 |
| 6Bd |
74.30 ± 0.28 |
>100 |
>100 |
>100 |
>100 |
>100 |
>100 |
>100 |
| 6Cb |
41.95 ± 2.33 |
70.70 ± 3.25 |
12.55 ± 0.78 |
>100 |
>100 |
>100 |
>100 |
>100 |
| 6Cc |
62.15 ± 7.28 |
>100 |
70.85 ± 10.96 |
>100 |
>100 |
>100 |
>100 |
>100 |
| 6Cd |
>100 |
>100 |
>100 |
>100 |
>100 |
>100 |
>100 |
>100 |
| 6Db |
>100 |
>100 |
23.55 ± 1.91 |
>100 |
>100 |
>100 |
>100 |
>100 |
| 6Dc |
26.10 ± 3.82 |
>100 |
>100 |
>100 |
>100 |
>100 |
>100 |
>100 |
| 6Dd |
>100 |
>100 |
>100 |
>100 |
>100 |
>100 |
>100 |
>100 |
| 6Eb |
9.40 ± 0.57 |
46.30 ± 6.22 |
10.20 ± 0.99 |
>100 |
>100 |
>100 |
>100 |
>100 |
| 6Ec |
8.25 ± 0.78 |
34.25 ± 5.16 |
67.90 ± 1.70 |
>100 |
73.80 ± 14.42 |
76.05 ± 3.75 |
88.85 ± 10.25 |
53.80 ± 2.12 |
| 6Ed |
>100 |
>100 |
>100 |
>100 |
>100 |
>100 |
>100 |
>100 |
| Sunitinib |
1.5 ± 0.01 |
4.25 ± 0.71 |
11.80 ± 1.41 |
8.85 ± 3.04 |
12.90 ± 1.98 |
5.00 ± 1.27 |
8.4 ± 2.12 |
8.15 ± 1.49 |
| 5-Fluorouracil |
2.7 ± 0.01 |
2.3 ± 0.71 |
7.35 ± 0.35 |
5.00 ± 1.70 |
65.35 ± 4.60 |
5.90 ± 0.99 |
12.90 ± 0.42 |
7.85 ± 0.71 |
3.3. Inhibition study of GSK-3β kinase
A kinase activity experiment was conducted in the presence of inhibitors to determine their specificity for GSK-3β. A reaction mixture containing 50 μM ATP, 50 μM DTT, 1 μg per μL GSK-3β substrate, and 0.5 ng per μL GSK3β enzyme, together with increasing quantities of the 6Eb and 6Ec (1 μM to 125 μM), was incubated. The reaction mixture including DMSO served as the vehicle control, whereas 12 nM laduviglusib was employed as a positive control in the experiment. At sub-micromolar concentrations, GSK-3β inhibitors reduced their efficacy by 50%. Specifically, 6Eb exhibited 50% inhibition of activity at a concentration of 11.02 ± 1.95 μM. The activity of 6Eb was 60%, 50%, and 40% at concentrations of 5 μM, 7.5 μM, and 10 μM, respectively. These observations suggest that 6Eb decreased the activity of GSK-3β in a concentration-dependent manner. While compound 6Ec showed a 50% inhibition at higher values of 59.81 ± 7.56 μM (Fig. 3A–C). These investigations suggest that our designed compounds acted as anticancer agents via inhibition of GSK-3β.
 |
| | Fig. 3 The figure illustrates the dose–response relationship of compounds 6Eb and 6Ec in inhibiting GSK-3β activity. (A) Effect of compound 6Eb, at a broad range assay. (B) Effects ofcompound 6Ec, at a broad range. (C) Effect of compound 6Eb, at a narrower range. Data are presented as mean ± SDM. Significance was plotted based on p values and denoted as * (p-value ≤ 0.05), ** (p-value ≤ 0.01), *** (p-value ≤ 0.001), **** (p-value ≤ 0.0001) and ns as not significant. | |
3.4. 6Eb regulated GSK-3β targets NF-kB and β-catenin
To check the activity of the GSK-3β inhibitors in cells, MOLT4 cells were treated with varying concentrations of 6Eb and 6Ec and the IC50 was calculated based on MTT assay (ESI Fig. 1a and b†). In MOLT4 cells, IC50 for 6Eb and 6Ec was 2.7 and 0.9 μM respectively, which was lowest among all the cell lines screened for GSK-3β inhibiton. We performed western blot for select GSK-3β targets, NFkB, β-catenin and PTEN. 6Eb showed downregulation of NF-kB, upregulation of β-catenin and no change in PTEN levels. It is very well known that phosphorylation of β-catenin by GSK-3β leads to ubiquitin mediated destruction of β-catenin, therefore inhibition led to an increase in β-catenin. Upregulation of β-catenin and downregulation of NF-kB lead to increase in cytotoxicity upon GSK-3β inhibiton. 6Ec did not show any change in the levels of any of the proteins tested (Fig. 4). To check whether 6Ec downregulated any of the GSK-3β target, we selected STAT3 (ESI Fig. 2a and b†), and found downregulation of STAT3 in 6Eb and not 6Ec, which is also evident from the kinase assay suggesting low kinase inhibitor activity of 6Ec, although higher cytotoxicity was observed with 6Ec.
 |
| | Fig. 4 GSK-3β inhibitors induced alterations in MOLT4 cells: (A) western blot analysis of GSK-3β targets 6Eb (GSKi) (A) treated MOLT4 cells lysates and 6Ec (B) and corresponding quantitation (i, ii, iii) of NF-kB, β-catenin and PTEN. Each experiment was done in duplicates, and a representative image is shown for each marker. Quantification was done for each marker (i, ii, iii) and is represented as a bar graph of mean ± SEM. One sample t-test and one way ANOVA test was performed, and the p-value were calculated between control, and compounds treated groups (*: p-value < 0.05). | |
3.5. In silico study
3.5.1. Molecular docking studies. The most potent compounds in biological screening, 6Eb and 6Ec, were picked out and docked to study how indole derivatives interact with GSK-3β. The docking protocol was validated by re-docking the co-crystal ligand back to the target. The RMSD between the co-crystal ligand and the observed pose was 0.62 Å, indicating that the docking protocol is accurate to evaluate the interaction. In the study, the docking scores of molecules 6Eb, 6Ec, and bio-acetoxime were −10.5 kcal mol−1, −8.8 kcal mol−1, and −7.7 kcal mol−1, respectively. The binding pocket contains Pro214, Ala231, Ala160, Leu194, Val147, Leu210, Lys162, Ala273, Leu215, Arg137, Gly218, Leu139, Thr217, Glu260, and Gly216. The thiourea side chain of compound 6Eb developed two strong hydrogen bonds and a pi-cation contact with Asp200. Furthermore, the p-methoxyphenyl ring developed several lipophilic bonds with Ile60, Val70, Val110, Ala83, Leu132, Leu132, and Cys199, firmly holding the enzyme from the ends and aiding in filling the expanded cavity. In addition, the triazole ring stabilized the kinase from the middle by interacting with Ile62 in a lipophilic way. The indole ring also helped to stabilize the protein–ligand complex by interacting with Cys199 in a lipophilic way.There were strong lipophilic interactions between the phenylhydrazone ring of compound 6Ec and Ala83, Val110, Leu132, and Leu188 at the end of the kinase. These numerous interactions keep the molecule in a stable shape and stabilize the ligand–protein combination. A p-methoxyphenyl ring connected the kinase with Ile62 via lipophilic contacts at its extremities. The ketone of the indole ring also improved the stability of the complex through H-bonds and lipophilic interactions with Cya199 and Val70. Furthermore, the indole ring increased binding affinity by supporting the kinase from the center via a significant multiple pi-cation interaction with Asp200. The binding and stability of the protein–ligand complex to GSK-3β were significantly influenced by many interactions, including pi-cation interactions, lipophilic contact, and H-bonds. These findings align with their biological investigation, indicating that compound 6Ec exhibited anticancer effects through GSK3β inhibition.
The replacement of a smaller linkage of hydrazinecarbothioamide with phenylhydrazide in 6Ec changed the orientation within the pocket, but it retained binding with comparable residues. These changes in 6Ec could be attributed to the presence of two bulky phenyl rings, such as p-methoxyphenyl and phenylhydrazide, on both sides of the molecule. In contrast, 6Eb had an aliphatic part of hydrazinecarbothioamide on one side and p-methoxypheyl on the other. The change in the substitution pattern causes both compounds to orientate differently within the same pocket occupied by standard.
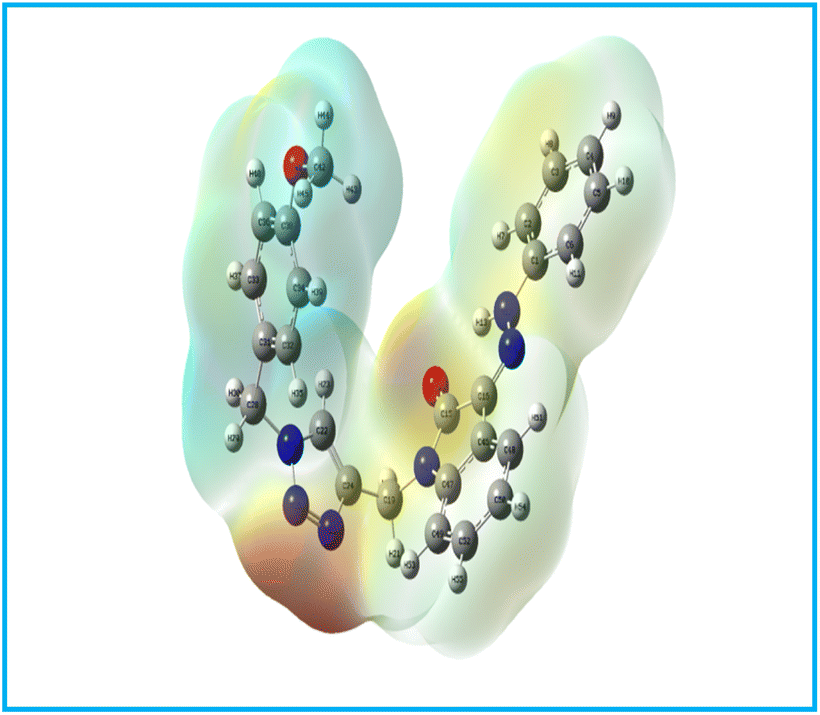
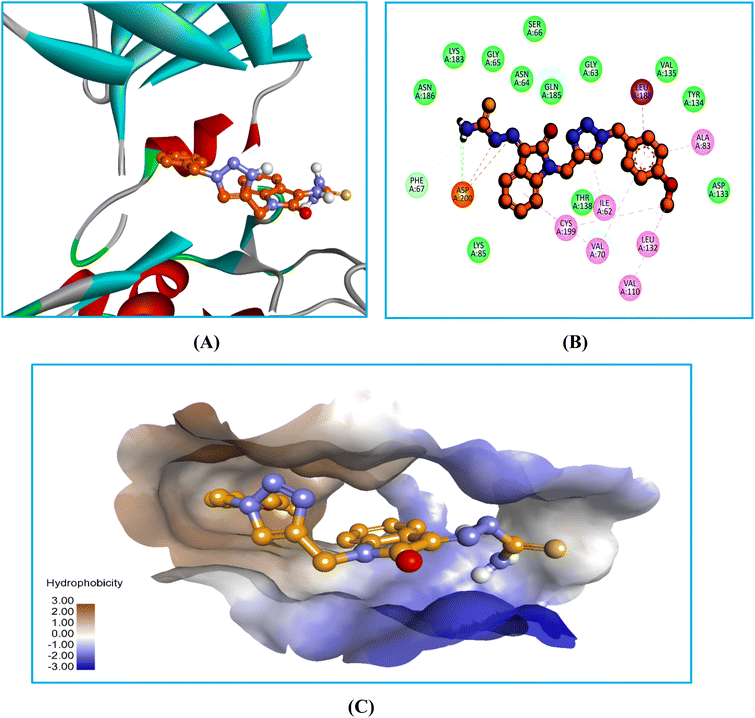
In the bio-acetoxime, the ketone group at the second position of indole formed strong hydrogen bonds with Lys85 and Asp200. The presence of bromine at the C6 position of indole increased lipophilic interactions with Ala83, Tyr134, and Leu188. Furthermore, indole showed many lipophilic and pi-cation interactions with Val70 and Leu188 and Cys199, respectively. Furthermore, strong pi-cation interactions between the indole ring and Asp200 were observed, stabilizing the protein–ligand complex from one end. The images in (Fig. 5–7) demonstrated that our developed compounds bind to residues and binding sites similar to those of the standard. Moreover, their higher docking score indicates that these compounds could act as an anticancer agent by targeting GSK-3β.
 |
| | Fig. 5 Docking interaction images of 6Eb against GSK-3β: (A) 3D view, (B) 2D view, (C) 3D hydrophobic surface view; H-bond denoted by green dashed line, pink and gray dashed line-lipophilic contacts, orange dashed line-pi-cation interaction. | |
 |
| | Fig. 6 Docking interaction images of 6Ec against GSK-3β: (A) 3D view, (B) 2D view, (C) 3D hydrophobic surface view; H-bond denoted by green dashed line, pink and gray dashed line-lipophilic contacts, orange dashed line-pi-cation interaction. | |
 |
| | Fig. 7 Docking interaction images of bio-acetoxime (standard) against GSK-3β: (A) 3D view, (B) 2D view; H-bond denoted by green dashed line, pink and gray dashed line-lipophilic contacts, orange dashed line-pi-cation interaction. | |
3.5.2. Molecular dynamic simulation study. The molecular dynamics technique was utilized to look into the stability and conformational changes of molecules in the simulated protein. Compound 6Ec had stronger cytotoxicity, a higher docking score, and a promising ADME/Tox profile. These findings suggest investigating the GSK-3β protein-bound complex of 6Ec for molecular dynamics simulation in comparison to bio-acetoxime (standard). The stability of the complexes was assessed using potential energy throughout a 100-ns simulation time with a 2-ns interval. This study examined the stability of protein–ligand complexes using RMSD graphs.Root Mean Square Deviation (RMSD) is a measure of how far atoms move from one frame to another. During simulation, the RMSD of the protein and ligand can reveal structural conformation and ligand stability in relation to the protein and binding site. Changes of 1–3 Å are suitable for small and globular proteins.37 Compound 6Ec and protein stabilized after modest alterations over the first 15 ns of MD simulation. After a brief period of drifting (17–19 ns), it returned and remained steady for the following 31 ns. Between 32–37 ns and 40–41 ns, there was another drift away from the complex with minor fluctuations. After 43 ns, it stayed connected to the protein for the remainder of the simulation session. The RMSD value for compound 6Ec and GSK-3β was 1 Å, which falls within the acceptable range of 1–3 Å, demonstrating stability (Fig. 8A and B).
 |
| | Fig. 8 RMSD and RMSF plots. (A and C) compound 6Ec-GSK-3β; (B and D) bio-acetoxime-GSK-3β. | |
The RMSD plot of the target GSK-3β with the bound ligand bio-acetoxime (standard) in (Fig. 8B) demonstrated that the ligand–protein complex was stable within the first 5 ns, with a minor fluctuation at a distance of 3.0 Å. Despite moving away from the protein for 37–68 ns, the ligand returned to its original position and remained fixed until the simulation was over. The RMSD value for target GSK-3β with standard was around 1.0 Å, which is within acceptable limits (1–3 Å).
3.5.3. The root mean square fluctuations (RMSF). The RMSF graph shows protein amino acid residue fluctuations over simulation time. Modifications of 1–3 Å are appropriate for small and globular proteins.38 Compound 6Ec (Fig. 8C and D) had lower RMSF values in the system (0.6–3 Å). Its residues 25 and 240 exhibited the highest peaks at 3 Å, indicating significant fluctuation in some regions, whereas the rest were below 2.4 Å. The standard's fluctuation ranged from 0.6 to 4.8 Å, which was higher than 3 Å. In comparison to compound 6Ec, the peaks were substantially higher and reached 4.8 Å, indicating increased variance in specific regions. Furthermore, the bio-acetoxime (standard) showed steeper and higher-frequency peaks, indicating greater variability in certain residues. The spike around residues 60 and 240 was significantly greater than the rest, indicating instability or flexibility in that region. These findings suggest that 6Ec did not significantly affect the protein's structure and organization upon binding.
3.5.4. Protein–ligand contacts. Compound 6Ec had strong to moderate protein–ligand interactions, with interaction percentages as high as 0.6 (Fig. 9A and B). It formed a strong hydrogen bond with Asn64 for 35% of the simulation duration and a weak H-bond with Val135. Moreover, 6Ec and the bio-acetoxime (standard) engaged in lipophilic interactions with similar residues such as Phe67, Val70, Ala83, and Leu188, which accounted for 60%, 30%, 50%, and 30% of the total occupancy. Furthermore, 6Ec had a strong 40% water-mediated H bridge with Ile62, whereas the standard had less than 10% time. Standard also had a strong H-bond and water bridge with Val135 50% of the time, with a fraction of 1% interaction. This demonstrates that certain protein residues may interact with the ligand multiple times using the same subtype. It also created weak hydrogen bonds with Ile62, Lys85, and Cys199. Both 6Ec and bio-acetoxime (standard) demonstrated a concentrated pattern of interaction over many residues. These findings reveal that 6Ec had a comparable binding pattern to the standard, implying that our developed compound had an affinity against the target protein.
 |
| | Fig. 9 Protein–ligand contact and timeline plot. (A and C) compound 6Ec-GSK-3β; (B and D) bio-acetoxime-GSK-3β. | |
3.5.5. Protein–ligand timeline contacts. The interaction and contact timeline (H-bonds, hydrophobic, ionic, and water bridges) are summarized in (Fig. 9C and D). The top panel displays the total number of individual interactions the protein had with the ligand 6Ec during the 100 ns MD simulation trajectory. The bottom panel displays the residues that interact with the ligand in each trajectory frame. Some residues, especially Ile62, Asn64, Phe67, Val70, Ala83, and Leu188 in 6Ec, and Phe67, Ala83, and Val135 in the bio-acetoxime (standard), demonstrated more than one unique interaction with the ligand, as indicated by a darker orange shading on the plot's scale on the right. In the docking analysis of 6Ec, the residues Ile62, Val70, Ala83, and Leu188 also contributed significantly to binding. These findings are consistent with their important role in a protein–ligand contact study.
3.5.6. MM-GBSA study. The MM-GBSA approach is often used to determine the binding free energy of a ligand to a protein molecule.38 The binding free energy of the 6Ec-GSk-3β and bio-acetoxime-GSk-3β complexes were determined, along with the impact of various non-bonded interaction energies (Table 2). The complex 6Ec GSK-3B had a binding energy of −66.019 kcal mol−1, which was 1.34 times lower than the bio-acetoxime (standard) complex (−49.204 kcal mol−1). Non-bonded interactions, including ΔGbindLipo, ΔGbindCovalent, ΔGbindHbond, ΔGbindCoulomb, ΔGbindSolvGB, and ΔGbindvdW, affect the binding free energy of ΔGbind. In all types of interactions, ΔGbindvdW and ΔGbindLipo were significantly lower in energy than standard and contributed the most to average binding energy in protein–ligand interactions (Table 2). Also, ΔGbindCoulomb made an important contribution, but its energy was 10 kcal higher than the standard. ΔGbindCovalent and ΔGbindHbond made moderate contributions. In contrast, ΔGbindSolvGB showed unfavorable energy contributions and thus opposed binding. Furthermore, the greater number of lipophilic contacts in the MD simulation and docking of 6Ec is corroborated by the higher value of ΔGbindlip. Additionally docking and MD studies revealed that, 6Ec and the bio-acetoxime (standard) showed comparable H-bonds with proteins, as evidenced by their close ΔGbindHbond values.
Table 2 Post simulation binding energy analysis (MM/GBSA) of compound 6Ec and bio-acetoxime
| Compound |
ΔGbind |
ΔGbindLipo |
ΔGbindvdW |
ΔGbindCoulomb |
ΔGbindHbond |
ΔGbindSolvGB |
ΔGbindCovalent |
| 6Ec |
−66.019 |
−27.785 |
−59.977 |
−12.463 |
−1.433 |
14.968 |
−4.068 |
| Bio-acetoxime |
−49.204 |
−15.185 |
−45.547 |
−22.258 |
−1.572 |
13.730 |
−2.151 |
3.5.7. Correlation of DFT study with docking and MD simulation of 6Ec. Compound 6Ec demonstrated the highest cytotoxicity, a greater docking score, and significant protein–ligand stability in the MD simulation study. Furthermore, DFT research was conducted to determine how the molecular and electrical characteristics of 6Ec influence biological activity.The lowest energy of optimization in the ground suggests the most stable conformation of 6Ec (obtained using the B3LYP/6-31G(d) level of theory). It had a higher dipole moment implies a greater potential to undergo polar interaction with the target protein, which was justified via pi-cation and H-bond interaction in the docking (Fig. 10 and Table 3).
 |
| | Fig. 10 Ground state optimized and labeled structures of 6Ec were calculated using DFT/B3LYP/6-31G(d) level of theory. | |
Table 3 Optimization energy and dipole moment of compound 6Ec
| Compound |
Optimisation energy (hartree) |
Dipole moment (debye) |
| 5Ec |
−1444.826 |
3.709 |
In order to understand molecular properties of a neutral system, we calculated the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO). The energy gap between EHOMO and ELUMO was found to be 0.12559 eV, which was less than 0.2 eV, indicating significant chemical reactivity and less kinetic stability of the molecules (Fig. 11). These findings were also supported by higher softness and lower hardness values. Together, these might reflect that 6Ec had a higher affinity for the target protein. Furthermore, lower values of ionization potential and chemical potential showed that the compound had a considerable inclination to accommodate the ionic state as well as greater reactivity towards the target (Table 4), which has verified the finding of approximately six types of total interaction in the MD investigation. Furthermore, increased reactivity of 6Ec was observed in docking studies via multiple types of lipophilic, pi-cation, and H-bond interactions. Furthermore, lower values of electronegativity, electron affinity, and electrophilicity index may suggest that 6Ec had a less electron-loving nature, which could be due to the presence of an electron-donating group-containing ring such as p-methoxyphenyl, as well as basic hydrazone linkage and a triazole ring. Furthermore, HOMO images revealed a greater electron density on the indole to phenylhydrazone area. In contrast, the indole ring had a higher concentration of LUMO than the phenylhydazone ring, which could be attributed to the presence of an electron-withdrawing carbonyl group. It resulted in a higher electron deficit in LUMO, which may be cause of one lipophilic contact with Val70 during the docking experiment.
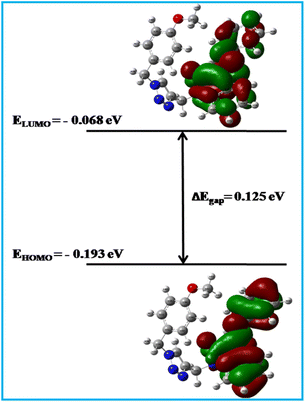 |
| | Fig. 11 Frontier molecular orbitals (HOMO and LUMO) and their energy gap (ΔEgap) of 6Ec were calculated using the DFT/B3LYP/6-31G method. | |
Table 4 Global and local reactivity descriptors of compound 6Ec
| EHOMOa (eV) |
ELUMOb (eV) |
ΔEgapc (eV) |
Id (eV) |
Ae (eV) |
μf (eV) |
χg (eV) |
ηh (eV) |
Si (eV) |
ωj (eV) |
| Energy of HOMO. Energy of LUMO. Energy gap between HOMO & LUMO. Ionization potential. Electron affinity. Chemical potential. Electronegativity. Hardness. Softness. Electrophilicity index. |
| −0.19345 |
−0.06786 |
0.12559 |
0.19345 |
0.06786 |
−0.130655 |
0.130655 |
0.062795 |
7.962417 |
0.135975 |
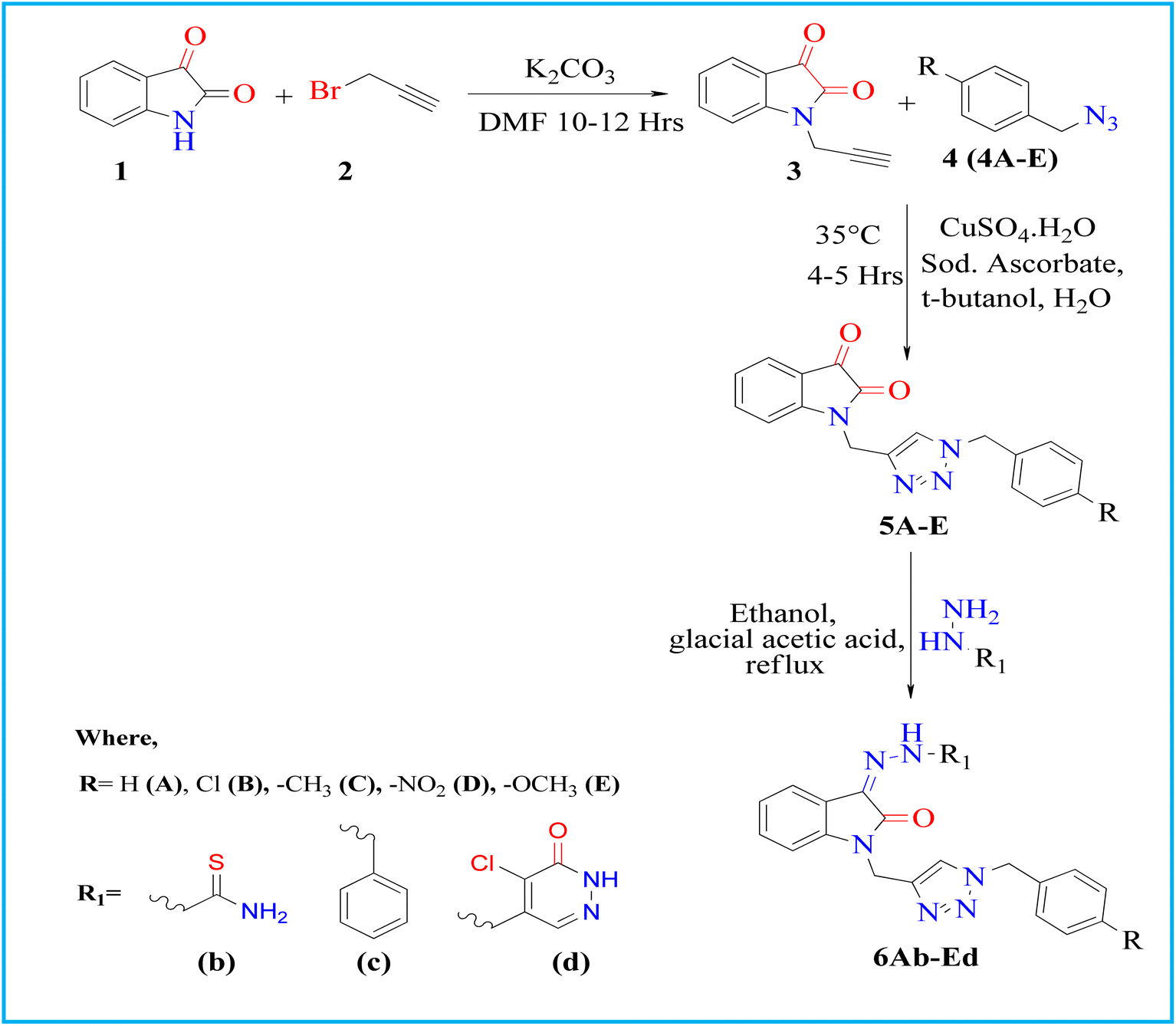
Also, the molecular electrostatic potential (MEP) map tells about reactivity regions in an organic molecule depending upon the electron density of each atom, which is the most probable region for electrophilic attack. Here, different colors represent different values of electrostatic potential, molecular size, and shape. Potential increases in order from red < orange < yellow < green < blue.39–41 High electron density is manifested by the red zone towards the triazole ring, hydrazone linkage, and oxygen of indole in the MEP map (Fig. 12). On the other hand, a phenylhydrazone ring oriented towards yellow color indicates a slightly electron-rich region, which may be due to the electron-releasing property of the hydrazone ring. These observations also supported multiple lipophilic and polar interactions of phenylhydrazone with Ala83, Val110, Leu132, Leu188, Cys199, and Asp200 in the docking and MD study. Taken together, the promising electrical and molecular features of 6Ec warrant further biological investigation.
 |
| | Fig. 12 Molecular electrostatic potential plots of 6Ec calculated using the DFT/B3LYP/6-31G(d) method. | |
3.5.8. In silico ADME determination. Synthesized compounds 6Eb and 6Ec demonstrated significant cytotoxicity and kinase inhibition as well as greater stability in the in silico work. As a result, those two compounds were examined for an in silico ADME investigation. The parameters molecular weight (MW), lipophilicity (logP), topological polar surface area (tPSA), and Lipinski's rule of five (Ro5) were computed using the Swiss-ADME web server.In silico results summarized in (Table 5) reveal that compound 6Ec had a greater drug-likeness property over 6Eb and standard among the studies. 6Ec exhibited a tPSA value (84.64 Å2) within the range of ≤140, which is ideal for improved oral absorption and intestinal permeability. Both compounds showed H-bonds within the range, indicating that aqueous solubility and binding affinity were optimized without compromising solubility. Both compounds 6Eb and 6Ec had excellent logP values (2.96 and 1.98), indicating sufficient lipophilicity to permeate cellular membranes without CNS damage. Further, 6Eb and 6Ec had optimum number of rotatable bonds, which may led to greater rigidity, higher binding affinity, and lower conformational changes during target engagement. Besides, Fsp3 indicates a saturation of sp3 hybridized carbons. A greater score of Fsp3 indicates a drug's similarity with known medicines, which often contain a larger proportion of saturated carbon atoms. Also, drug–drug interactions, metabolic interactions, and toxicity typically decrease with increasing Fsp3 values. While, 6Ec exhibited a lower Fsp3 value compared to bio-acetoxime (standard), suggesting a more rigid interaction with the target. The finding collectively reflects that 6Ec had a better ADME profile than 6Eb and bio-acetoxime (standard). Moreover, it demonstrated a balanced set of properties favouring optimal absorption, distribution, and target binding. Additionally, it met the necessary tPSA, lipophilicity, Fsp3, and binding affinity. These characteristics collectively suggest that 6Ec could be a promising candidate with better pharmacokinetics and smaller metabolic liabilities.
Table 5 In silico ADME data for the compounds 6Eb, 6Ec and bio-acetoxime
| Compound code |
Binding affinity |
MW |
Fsp3 |
RB |
HBA |
HBD |
tPSA |
logP |
Lipinski |
ESOL logS |
ESOL class |
logKP |
F |
| 6Eb |
−8.8 |
421.48 |
0.15 |
7 |
5 |
2 |
142.75 |
1.98 |
0 |
−3.75 |
Soluble |
−7.36 |
0.55 |
| 6Ec |
−10.1 |
438.48 |
0.12 |
7 |
5 |
1 |
84.64 |
2.96 |
0 |
−5.08 |
Moderately soluble |
−6.19 |
0.55 |
| Bio-acetoxime |
−7.7 |
73.09 |
0.67 |
0 |
2 |
1 |
32.59 |
1.22 |
0 |
−0.37 |
Very soluble |
−6.66 |
0.55 |
4. Conclusions
The current work examines the cytotoxic effects of newly synthesized hydrazide-2-oxindole analogues against several cancer cell lines. The compound 6Cb exhibited cytotoxicity against the LN229 cell line (CC50: 12.22 μM). The other two compounds, 6Eb and 6Ec, demonstrated the maximum cytotoxicity against the MOLT4 cells (2.7 μM and 0.9 μM) followed by Capan-1 cell line (CC50: 9.40 μM and 8.25 μM, respectively). These compounds 6Eb and 6Ec were non-toxic to normal cells (HEK 293 IC50 46 μM and 144 μM respectively). Furthermore, the compounds 6Eb and 6Ec strongly inhibited GSK-3β, indicating an anticancer mechanism. In western blot, 6Eb enhanced the β-catenin fraction and decreased NF-kB and STAT3 expression, confirming its anticancer mechanism. Also, docking and MD tests corroborated cytotoxicity, kinase inhibition, and western blotting results, demonstrating that compound 6Eb inhibited GSK-3β activity. Moreover, DFT analysis revealed electron-rich and deficient regions on molecules, which were used to compare non-polar and polar interactions with kinase in docking and MD studies. Likewise, in silico ADMET discovered the drug-like properties of the proposed molecule, demonstrating its novelty as a potential lead. However, the study findings indicate that additional in vivo and biochemical studies are required to evaluate the efficacy and safety of 6Eb and 6Ec, strengthening their potential as therapeutic agents.
Abbreviations
| p-SMAD3 | Mothers against decape ntaplegic homolog 3 |
| c-Myc | Cellular Myc |
| DTT | Dithiothreitol |
| PDBQT | Protein data bank, partial charge, and atom type |
| OPLS | Optimised potential for liquid simulations |
| TIP3P | Transferable intermolecular potential with three point |
| MM-GBSA | Molecular mechanics/generalised born surface area |
| NF-kB | Nuclear factor kappa-light enhancer of activated B cells |
| PTEN | Phosphatase and tensin homolog |
| STAT3 | Signal transducer and activation of transcription 3 |
| RMSD | Root mean square deviation |
| RMSF | Root mean square fluctuations |
| ΔGbind | Binding free energy |
| ΔGbindLipo | Lipophilic contribution to the overall binding free energy of a ligand–protein complex |
| ΔGbindvdW | van der Waals force contribution to the overall binding free energy of a ligand–protein complexs |
| ΔGbindCoulomb | Change in Gibbs free energy of binding that is specifically attributed to electrostatic interactions (coulomb interactions) between a ligand and a protein or receptor |
| ΔGbindHbond | Binding free energy of hydrogen bonds |
| ΔGbindSolvGB | Binding free energy of solvation |
| ΔGbindCovalent | Binding free energy of covalent bond |
| MEP | Molecular electrostatic potential |
| ADME | Absorption, distribution, metabolism and elimination |
Data availability
The data are included within the article and in ESI.†
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
Thanks to Saumya K. Patel and Nandan Dixit from the Department of Botany, bioinformatics and climate change impacts management, School of Science, Gujarat University, Ahmedabad, Gujarat, India, for providing the MD simulation study.
References
- N. Embi, D. B. Rylatt and P. Cohen, Eur. J. Biochem., 1980, 107, 519–527 CrossRef CAS PubMed.
- A. J. Harwood, Cell, 2001, 105, 821–824 CrossRef CAS PubMed.
- H. Yao, P. Shaw, C. Wrong and D. C. Wan, J. Chem. Neuroanat., 2002, 23, 291–297 CrossRef CAS.
- J. R. Woodgett, EMBO J., 1990, 9, 2431–2438 CrossRef CAS.
- H. Eldar-Finkelman, Trends Mol. Med., 2002, 8, 126–132 CrossRef CAS PubMed.
- B. W. Doble and J. R. Woodgett, J. Cell Sci., 2003, 116, 1175–1186 CrossRef CAS PubMed.
- R. S. Jope, C. J. Yuskaitis and E. Beurel, Neurochem. Res., 2007, 32, 577–595 CrossRef CAS.
- S. Frame and P. Cohen, Biochem. J., 2001, 359, 1–16 CrossRef CAS PubMed.
- L. Kim and A. R. Kimmel, Curr. Opin. Genet. Dev., 2000, 10, 508–514 CrossRef CAS.
- A. S. Wagman and J. M. Nuss, Curr. Pharm. Des., 2001, 7, 417–450 CrossRef CAS.
- P. Cohen, Eur. J. Biochem., 2001, 268, 5001–5010 CrossRef CAS PubMed.
- C. Sasaki, T. Hayashi, W. R. Zhang, H. Warita, Y. Manabe, K. Sakai and K. Abe, Neurol. Res., 2001, 23, 588–592 CrossRef CAS PubMed.
- A. Castro and A. Martinez, Expert Opin. Ther. Pat., 2000, 10, 1519–1527 CrossRef CAS.
- J. A. Bertrand, S. Thieffine, A. Vulpetti, C. Cristiani, B. Valsasina, S. Knapp, H. M. Kalisz and M. Flocco, J. Mol. Biol., 2003, 333, 393–407 CrossRef CAS PubMed.
- I. Hers, J. M. Tavare and R. M. Denton, FEBS Lett., 1999, 460, 433–436 CrossRef CAS PubMed.
- H.-C. Zhang, K. B. White, H. Ye, D. F. McComsey, C. K. Derian, M. F. Addo, P. Andrade- Gordon, A. J. Eckardt, B. R. Conway, L. Westover, J. Z. Xu, R. Look, K. T. Demarest, S. Emanuel and B. E. Maryanoff, Bioorg. Med. Chem. Lett., 2003, 13, 3049–3053 CrossRef CAS PubMed.
- E. erHaar, J. T. Coll, D. A. Austen, H. M. Hsiao, L. Swenson and J. Jain, Nat. Struct. Biol., 2001, 8, 593–596 CrossRef.
- R. Dajani, E. Fraser, S. M. Roe, N. Young, V. Good, T. C. Dale and L. H. Pearl, Cell, 2001, 105, 721–732 CrossRef CAS PubMed.
- Q. Ye, Y. Shen, Y. Zhou, D. Lv, J. Gao, J. Li and Y. Hu, Eur. J. Med. Chem., 2013, 68, 361–371 CrossRef CAS.
- L. Gong, D. Hirschfeld and Y. C. Tan, et al., Bioorg. Med. Chem. Lett., 2010, 20, 1693–1696 CrossRef CAS.
- A. M. Aronov, T. Qing and G. Martinez-Botella, et al., J. Med. Chem., 2009, 52, 6362–6368 CrossRef CAS PubMed.
- H. Zou, L. Zhou and Y. Li, et al., J. Med. Chem., 2010, 53, 994–1003 CrossRef CAS PubMed.
- L. Meijer, A. L. Skaltsounis and P. Magiatis, et al., Chem. Biol., 2003, 10, 1255–1266 CrossRef CAS.
- P. Polychronopoulos, P. Magiatis and A. L. Skaltsounis, et al., J. Med. Chem., 2004, 47, 935–946 CrossRef CAS.
- J. A. Bertrand, S. Thieffine and A. Vulpetti, et al., J. Mol. Biol., 2003, 333, 393–407 CrossRef CAS.
- J. Witherington, V. Bordas and D. Haigh, et al., Bioorg. Med. Chem. Lett., 2003, 13, 1581–1584 CrossRef CAS PubMed.
- M. Saitoh, J. Kunitomo and E. Kimura, et al., Bioorg. Med. Chem., 2009, 17, 2017–2029 CrossRef CAS PubMed.
- M. L. Selenica, H. S. Jensen and A. K. Larsen, et al., Br. J. Pharmacol., 2007, 152, 959–979 CrossRef CAS.
- A. Martinez, M. Alonso, A. Castro, C. Pérez and F. J. Moreno, J. Med. Chem., 2002, 45, 1292–1299 CrossRef CAS PubMed.
- T. Kramer, B. Schmidt and F. Lo Monte, Int. J. Alzheimers Dis., 2012, 2012, 381029 Search PubMed.
- P. Polychronopoulos, P. Magiatis, A. L. Skaltsounis, V. Myrianthopoulos, E. Mikros, A. Tarricone, A. Musacchio, S. M. Roe, L. Pearl, M. Leost, P. Greengard and L. Meijer, J. Med. Chem., 2004, 47, 935–946 CrossRef CAS.
- K. Vougogiannopoulou, Y. Ferandin and K. Bettayeb, et al., J. Med. Chem., 2008, 51, 6421–6431 CrossRef CAS.
- A. Das, G. Greco, S. Kumar, E. Catanzaro, R. Morigi, A. Locatelli, D. Schols, H. Alici, H. Tahtaci, F. Ravindran, C. Fimognari and S. S. Karki, Comput. Biol. Chem., 2023, 97, 107641 CrossRef.
- K. Aman, K. Lal, M. Murtaza, S. Jaglan, Y. Rohila, P. Singh, M. B. Singh and K. Kumari, J. Biomol. Struct. Dyn., 2024, 42, 9919–9938 CrossRef.
- T. V. de Walle, A. Theppawong, C. Grootaert, S. D. Jonghe, L. Persoons, D. Daelemans, K. V. Hecke, J. V. Camp and M. D’hooghe, Monatsh. Chem., 2019, 150, 2045–2051 CrossRef.
- O. Trott and A. J. Olson, J. Comput. Chem., 2010, 31, 455–461 CrossRef CAS.
- D. Yevale, N. Teraiya, T. Lalwani, M. Dalasaniya, K. Kapadiya, R. K. Ameta, C. B. Sangani and Y. T. Duan, Bioorg. Chem., 2024, 147, 107323 CrossRef CAS PubMed.
- S. Rajamanikandan, J. Jeyakanthan and P. Srinivasan, Appl. Biochem. Biotechnol., 2017, 181, 192–218 CrossRef CAS.
- K. K. Bharadwaj, T. Sarkar, A. Ghosh, D. Baishya, B. Rabha, M. Panda, B. R. Nelson, A. John, H. I. Sheikh, B. P. Dash, H. A. Edinur and S. Pati, ChemRxiv, 2021, 193, 3371–3394 CrossRef CAS PubMed.
- L. Piao, Z. Chen, Q. Li, R. Liu, W. Song, R. Kong and S. Chang, Int. J. Mol. Sci., 2019, 20, 224 CrossRef PubMed.
- M. Kawad, S. Sangani, J. K. Parmar, R. C. Dabhi, P. S. Arya, N. Teraiya, S. Ahmed and R. K. Ameta, Polyhedron, 2024, 264, 117242 CrossRef CAS.
- A. Diana, O. Michielin and V. Zoete, Sci. Rep., 2017, 7, 42717 CrossRef.
- D. F. Veber, S. R. Johnson, H. Y. Cheng, B. R. Smith, K. W. Ward and K. D. Kopple, J. Med. Chem., 2002, 45, 2615–2623 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ab,
Sujeet Kumar
ab,
Sujeet Kumar