
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Highly active iron catalysts for olefin hydrogenation enable para-hydrogen induced hyperpolarisation of 1H and 19F NMR resonances at 1.4 Tesla†
Julianna S.
Doll‡
a,
Jan
Kergassner‡
b,
Bingyu
Zhang
b,
Christina M.
Thiele
 c,
Gerd
Buntkowsky
b,
Markus
Enders
a,
Torsten
Gutmann
*b and
Dragoş-Adrian
Roşca
*ad
c,
Gerd
Buntkowsky
b,
Markus
Enders
a,
Torsten
Gutmann
*b and
Dragoş-Adrian
Roşca
*ad
aAnorganisch-Chemisches Institut, Im Neuenheimer Feld 276, Universität Heidelberg, D-69120 Heidelberg, Germany. E-mail: dragos.rosca@uni-heidelberg.de; dragos-adrian.rosca@univ-rennes.fr
bTechnische Universität Darmstadt, Eduard-Zintl-Institut für Anorganische und Physikalische Chemie, Peter-Grünberg-Straße 8, D-64287 Darmstadt, Germany. E-mail: gutmann@chemie.tu-darmstadt.de
cTechnische Universität Darmstadt, Clemens-Schöpf-Institut für Organische Chemie und Biochemie, Peter-Grünberg-Straße 16, D-64287 Darmstadt, Germany
dInstitut des Sciences Chimiques de Rennes, Université de Rennes, F-35000, Rennes, France
First published on 18th June 2025
Abstract
para-Hydrogen induced polarisation (PHIP) is an excellent tool for extracting mechanistic information in catalysis since it circumvents the intrinsic low sensitivity of nuclear magnetic resonance (NMR) spectroscopy. We report a class of iron complexes that are highly active in olefin hydrogenation catalysis and act as PHIP catalysts at 1.4 Tesla. Moreover, hyperpolarisation transfer to 19F is observed.
para-Hydrogen induced polarisation (PHIP) represents a powerful mechanistic tool, which enables the use of NMR spectroscopy to obtain local chemical environment information through a boost in sensitivity by several orders of magnitude.1 Within the field of PHIP, special approaches such as the hydrogenation of the side arm (PHIP-SAH) have been established to broaden the variety of accessible substrate systems and to optimise signal enhancement.2 The PHIP approach is currently used successfully in catalysis3 and biomolecular NMR,4 and relies on magnetic hyperpolarisation, usually through the incorporation of para-hydrogen (p-H2) in an organic substrate, through catalytic hydrogenation. In homogeneous catalysis, PHIP has enabled the detection of very low concentrations of metal-based intermediates under catalytic conditions, which would have been impossible to detect otherwise by NMR spectroscopy.5 Currently, PHIP catalysts based on noble metals (i.e. Rh, Ir, Pd, and Pt) dominate the field. Nevertheless, for the application of PHIP in bioimaging studies, reliance on noble metals has been seen as a hindrance to its implementation.6 In an effort to design catalysts based on more biocompatible and non-geopolitically geopolitically-source base metals, the last 20 years have seen rapid progress in the development of earth-abundant systems that exhibit hydrogenation activities comparable to those of noble metals. In iron chemistry, systems based on redox-active ligands are amongst the most active catalysts known for olefin functionalisation, such as hydroelementation or [2+2]-cycloaddition.7 In the direct catalytic hydrogenation of olefins, such systems can promote turnover at loadings as low as 0.1 mol%,8,9 which is comparable to and even surpasses the performance of established catalysts based on rhodium and iridium.10
This success, however, has not yet been mirrored in the development of PHIP catalysts based on iron. Unlike noble-metal catalysts, which typically operate via 2e− redox steps, in the case of iron, 1e− steps tend to be favourable. The resulting paramagnetic species involved are known to catalyse the conversion of p-H2 back to the equilibrium mixture of ortho-hydrogen (o-H2) and p-H2 (a 75![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :25 ratio at RT).11 Thus, until now, there have been very few examples employing iron-based catalysts for reactions involving p-H2.12 Recently, Fout et al. reported a rigid, redox-innocent pincer system based on two flanking NHCs as strong σ-donors and a phenyl ring, which can stabilize an iron hydride species (Fig. 1).13 This system is an active catalyst for olefin hydrogenation (down to 1 mol% catalyst loading), and remarkably, PHIP of the hydrogenation products was observed. We herein report another family of iron-based PHIP catalysts based on a potentially redox-active ligand, capable of hydrogenating olefins at catalyst loadings as low as 0.1 mol%. In addition to the PHIP effect observed at 1.4 T on the 1H resonances of the hydrogenated substate, transfer of polarisation to 19F resonances is also observed. This opens an interesting pathway to iron-based PHIP catalysts capable of polarizing marker molecules with potential applications in MRI.14 We have previously reported that the PNN-ligand (PNN = phosphine α-iminopyridine) framework can stabilise dinuclear iron dinitrogen complexes (1), as well as a rare example of an anionic iron hydride complex bearing a N2 ligand (2).15 While both complexes are based on formal Fe(0) centres, exhibiting a singlet ground state, the electronic structures of these complexes are significantly different. Namely, for [PNNFe(N2)]2(μ-N2) (1), the PNN ligand is redox non-innocent with a narrow singlet–triplet separation (ΔEST = 3.7 kcal mol−1, DFT), while 2 has a closed-shell singlet ground state, featuring a redox-inactive PNN chelate and a considerably widened singlet triplet separation (ΔEST = 9.2 kcal mol−1, see the ESI†).15
:25 ratio at RT).11 Thus, until now, there have been very few examples employing iron-based catalysts for reactions involving p-H2.12 Recently, Fout et al. reported a rigid, redox-innocent pincer system based on two flanking NHCs as strong σ-donors and a phenyl ring, which can stabilize an iron hydride species (Fig. 1).13 This system is an active catalyst for olefin hydrogenation (down to 1 mol% catalyst loading), and remarkably, PHIP of the hydrogenation products was observed. We herein report another family of iron-based PHIP catalysts based on a potentially redox-active ligand, capable of hydrogenating olefins at catalyst loadings as low as 0.1 mol%. In addition to the PHIP effect observed at 1.4 T on the 1H resonances of the hydrogenated substate, transfer of polarisation to 19F resonances is also observed. This opens an interesting pathway to iron-based PHIP catalysts capable of polarizing marker molecules with potential applications in MRI.14 We have previously reported that the PNN-ligand (PNN = phosphine α-iminopyridine) framework can stabilise dinuclear iron dinitrogen complexes (1), as well as a rare example of an anionic iron hydride complex bearing a N2 ligand (2).15 While both complexes are based on formal Fe(0) centres, exhibiting a singlet ground state, the electronic structures of these complexes are significantly different. Namely, for [PNNFe(N2)]2(μ-N2) (1), the PNN ligand is redox non-innocent with a narrow singlet–triplet separation (ΔEST = 3.7 kcal mol−1, DFT), while 2 has a closed-shell singlet ground state, featuring a redox-inactive PNN chelate and a considerably widened singlet triplet separation (ΔEST = 9.2 kcal mol−1, see the ESI†).15
 | ||
| Fig. 1 State-of-the-art iron-based molecular PHIP catalysts. | ||
Since 1 and 2 possess labile N2 ligands, which can be displaced by more π-accepting olefins, they seem well-suited for olefin functionalisation reactions such as hydrogenation. Both systems are highly active in catalytic hydrogenation of olefins at low catalyst loadings (Fig. 2). Under the same reaction conditions (0.1 mol% loading per Fe centre, 7 bar H2, RT) using p-F-styrene 3 as a model substrate, hydride 2 displayed slightly higher activity (full conversion in 10 min) when compared to 1 (97% conversion after 15 min). Hydride 2 can additionally hydrogenate a wide range of mono-substituted aromatic and aliphatic olefins (3–8). Geminal disubstituted olefins (9) remained reactive, albeit with a significantly lower reaction rate. Activated internal olefins (e.g. norbornene 10) underwent hydrogenation rapidly, while less activated internal olefins (11 and 12) required significantly longer reaction times. Interestingly, conjugated olefins (e.g.12) did not resist turnover, suggesting that an η4-diene intermediate is not a viable thermodynamic sink in this methodology. In the absence of H2, styrene derivatives undergo polymerisation in the presence of 2 at higher catalyst loadings (>0.5 mol%) and higher substrate concentrations (>1.4 M), while this side reaction could mostly be suppressed at lower catalyst loadings and concentrations (<0.5 mol%, <0.7 M). An exception to this is o- and m-F-styrenes (vide infra). Substrates containing more fragile functional groups (e.g. esters, 15) required higher catalyst loadings and led to incomplete conversion. For longer chain olefins (14 and 15), in addition to olefin hydrogenation, competitive double bond isomerisation was observed (ca. 30%). Vinylic phenols and allylic amines remained unreactive (see the ESI†).
 | ||
| Fig. 2 Olefin hydrogenation substrate scope using 2 as a pre-catalyst. Conversions were determined by 1H NMR spectroscopy with respect to an internal standard (C6Me6). | ||
Given the high activity of 1 and 2 in olefin hydrogenation, we investigated the catalytic conversion of styrene substrates in the presence of para-hydrogen (p-H2). For polarisation transfer from p-H2 to the hydrogenated substrate to occur, a series of pre-conditions are required, including (i) the pair-wise addition of H2 to the substrate needs to occur with the breaking of symmetry of p-H2, (ii) the transfer of hydrogen atoms to the substrate needs to proceed faster than the relaxation of the nuclear spin of the product,16 and (iii) the formation of paramagnetic (or ferromagnetic) particles needs to be inhibited, as this can lead to a faster spin relaxation.
The fulfilment of pre-conditions (i) and (ii) mandates the hydrogenation of unsymmetrical olefinic substrates, mediated by highly active catalytic systems. We have chosen propene and p-F-styrene (3) as representatives of aliphatic and aromatic olefins as model substrates, in the presence of iron-based catalysts 1 and 2. Since the two catalysts are structurally similar but have very different electronic structures (vide supra), subjecting the two systems to PHIP conditions would reveal more information about ligand design in iron-based polarisation transfer methodologies. Under ALTADENA conditions,17 the hydrogenation of propene and p-F-styrene (3) was performed with catalyst 1 or 2, respectively, by treatment with 4 bar p-H2 and subsequent transfer of the appropriate sample into the probe of a 1.4 T benchtop NMR spectrometer. The signal enhancements (labelled as ε1 for catalyst 1 and ε2 for catalyst 2) and decays of polarisation obtained in 1H NMR were monitored in THF-d8 and C6D6 solutions for 1, and in only THF-d8 solution for 2 due to solubility reasons. The obtained 1H signal enhancement factors are summarized in Table S1 (ESI†) and the corresponding spectra are shown in Fig. S37–S45 (ESI†).
As shown in Fig. 3a, for propene, the highest 1H enhancement of 270 was obtained with 1 in THF-d8, while for p-F-styrene (3) the highest enhancement of 2250 was obtained with 2 in THF-d8, which corresponds to a polarisation of 1.09%, as described in the ESI.† In comparison, the enhancement reported in ref. 13 (ca. 200) corresponds to a polarisation of 0.97%, which is statistically identical. This convincingly demonstrates the feasibility of 1H PHIP at 1.4 Tesla for both gaseous and liquid substrates employing catalysts 1 and 2 in THF-d8. For the hydrogenation of 3 with catalyst 1, the influence of solvent on the enhancement factor was investigated. An impact is observed (ε1 = 415 for THF-d8, ε1 = 515 for C6D6), in line with previous reports on rhodium and iridium catalysts.18 Since various parameters influence the obtained enhancement (see the ESI†), systematic studies are required, which are beyond the scope of this communication.
 | ||
| Fig. 3 A summary of the PHIP experiments and excerpts from 1H (a) and 19F (b) NMR data (RT, 1.4 Tesla benchtop NMR spectrometer). For the full dataset, see the ESI.† | ||
Subsequently, we have studied the feasibility of PHIP polarization transfer from 1H to 19F. To measure the dependence of the enhancement factor on the position of substitution, we have explored o-F-styrene (6) and p-F-styrene (3) as substrates under ALTADENA conditions.§ The 19F signal enhancement factors are summarised in Table S1 (ESI†), and the corresponding spectra are shown in Fig. S46–S50 (ESI†). As illustrated in Fig. 3b, both catalysts 1 and 2 show PHIP transfer to 19F, with enhancement factors of up to 190 obtained for o-F-styrene with catalyst 2 in THF-d8. In C6D6, polarization transfer to 19F with catalyst 1 is even more efficient, leading to an enhancement factor of 775 obtained for p-F-styrene. This again demonstrates the influence of the solvent on the signal enhancement. Comparison of enhancement factors for o- and p-fluoro substituents (30 vs. 95 for catalyst 1 and 190 vs. 200 for catalyst 2) does not allow us to establish any clear trend with respect to the efficiency of polarization transfer as a function of styrene substitution. This is consistent with the results reported by Kuhn et al. who investigated 19F PHIP polarization transfer for o- and p-F-styrenes with a Rh(I) catalyst.14c To the best of our knowledge, this is the first report of iron-mediated PHIP transfer to 19F. It is interesting to note that PHIP can be observed with 1 despite the presence of the low-lying triplet states, the redox non-innocent nature of the chelating ligand and the absence of an Fe–H functional group (for the proposed mechanism, see the ESI†).
To gain further mechanistic insight into the catalytic hydrogenation, we investigated the reactivity of complex 2 toward styrenes and H2 over a broader temperature range using NMR spectroscopy. Under H2 pressure (7 bar, −40 °C to RT), 2 remained unchanged, although it readily formed the iron deuteride 2-d1 with D2, consistent with previous reports (see the ESI†). This selective reactivity, along with the observed PHIP in hydrogenation products, points towards the following mechanism (Fig. 4): (i) rapid formation of an η2-olefin complex (Int. 1), especially in the presence of excess substrate; (ii) formation of a σ-H2 complex (Int. 3) following the coordination of para-H2 and (iii) selective transfer of the coordinated dihydrogen to the coordinated olefin to give rise to a hyperpolarised substrate. As a competing pathway, reversible olefin insertion into the Fe–H bond to give Int. 2 is conceivable, but would probably not give rise to a hyperpolarised product. This pathway is also likely responsible for the observed styrene polymerisation reaction at higher catalyst loadings. Further mechanistic studies are required to determine to which extent the insertion pathway to give Int. 2 is also involved in the productive catalytic hydrogenation pathway. Catalytic hydrogenation with D2 gives predominantly the 1,2-deuterated product, with minor amounts of terminal isotopomers (see the ESI†), suggesting reversible β-hydride elimination.
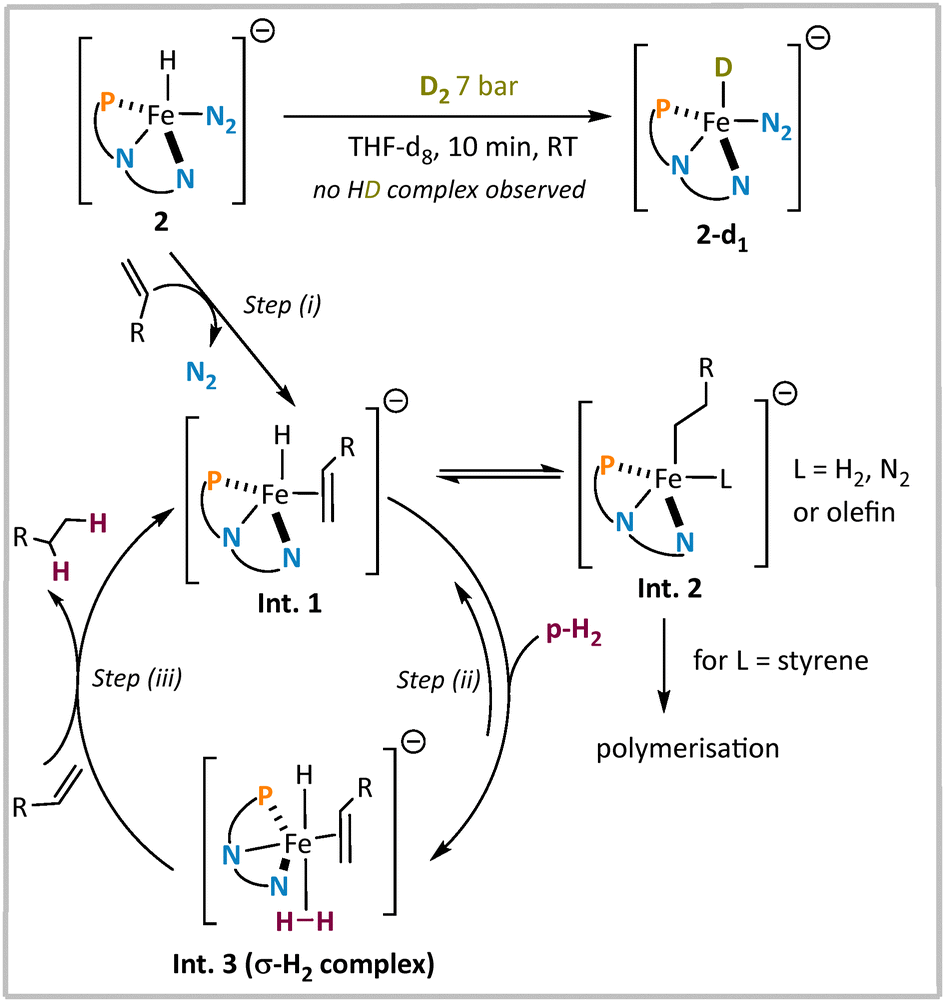 | ||
| Fig. 4 Proposed mechanism for the hydrogenation of olefins in the presence of 2. | ||
In conclusion, we have introduced two highly active iron catalysts, capable of hydrogenating aromatic and aliphatic olefins at loadings down to 0.1 mol%. Conducting the hydrogenation experiments on propene and p-F-styrene in the presence of p-H2 allowed achieving enhancement factors of up to 270 and 2250, respectively, in THF-d8 at 1.4 T (60 MHz benchtop spectrometer). We have also demonstrated for the first time that iron-based systems are capable of PHIP transfer to 19F, with enhancement factors of up to 775 in C6D6 and up to 200 in THF-d8 at 1.4 T. Interestingly, this work shows that in principle, both open-shell singlet iron complexes with low lying triplet states (1) and closed-shell iron complexes (2) can act as PHIP catalysts, demonstrating that ligand field strength is not the only parameter that governs the design of iron based PHIP catalysts. The current work lays the groundwork for a broader investigation of polarisation transfer to other heteronuclei using iron catalysis based on redox-active ligands, with the aim of replacing and complementing the established approaches based on noble metals (e.g. Rh and Ir).
This work was supported by the Deutsche Forschungsgemeinschaft (DFG grant RO5979/2-1 to D.-A. R.). J. K., B. Z., C. M. T., M. E., G. B. and T. G. thank the CRC 1487 “Iron Upgraded” funded by the DFG for financial support (project no. 443703006). Computational resources were provided by the state of Baden-Württemberg through bwHPC and the DFG (INST 40/575-1 FUGG, JUSTUS 2 cluster). The authors acknowledge Dr Eckhard Bill for measuring the Mössbauer spectrum of 2. D.-A. R. is grateful to Prof. Lutz H. Gade for generous support over the years.
Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article, including experimental details and copies of the NMR spectra, have been included in the ESI.†Notes and references
- (a) G. Buntkowsky, F. Theiss, J. Lins, Y. A. Miloslavina, L. Wienands, A. Kiryutin and A. Yurkovskaya, RSC Adv., 2022, 12, 12477–12506 RSC; (b) A. I. Silva Terra, D. A. Taylor and M. E. Halse, Prog. Nucl. Magn. Reson. Spectrosc, 2024, 144–145, 153–178 CrossRef CAS; (c) A. D. Robinson, F. Hill-Casey, S. B. Duckett and M. E. Halse, Phys. Chem. Chem. Phys., 2024, 26, 14317–14328 RSC; (d) T. Castaing-Cordier, D. Bouillaud, J. Farjon and P. Giraudeau, in Annual Reports on NMR Spectroscopy, ed. G. A. Webb, Academic Press, 2021, vol. 103, pp. 191–258 Search PubMed; (e) F. Theiss, J. Lins, J. Kergassner, L. Wienands, S. Doller and G. Buntkowsky, J. Magn. Reson., 2024, 362, 107673 CrossRef CAS PubMed.
- (a) F. Reineri, T. Boi and S. Aime, Nat. Commun., 2015, 6, 5858 CrossRef CAS; (b) O. G. Salnikov, N. V. Chukanov, A. N. Pravdivtsev, D. B. Burueva, S. V. Sviyazov, K. Them, J. B. Hovener and I. V. Koptyug, ChemPhysChem, 2025, e202401119 Search PubMed.
- (a) B. J. Tickner and V. V. Zhivonitko, Chem. Sci., 2022, 13, 4670–4696 RSC; (b) W. Wang, Q. Wang, J. Xu and F. Deng, ACS Catal., 2023, 13, 3501–3519 CrossRef CAS; (c) S. B. Duckett and N. J. Wood, Coord. Chem. Rev., 2008, 252, 2278–2291 CrossRef CAS; (d) A. N. Pravdivtsev, B. J. Tickner, S. Glöggler, J.-B. Hövener, G. Buntkowsky, S. B. Duckett, C. R. Bowers and V. V. Zhivonitko, ACS Catal., 2025, 15, 6386–6409 CrossRef CAS; (e) T. C. Eisenschmid, R. U. Kirss, P. P. Deutsch, S. I. Hommeltoft, R. Eisenberg, J. Bargon, R. G. Lawler and A. L. Balch, J. Am. Chem. Soc., 1987, 109, 8089 CrossRef CAS.
- (a) F. Theiss, L. Wienands, J. Lins, M. Alcaraz-Janssen, C. M. Thiele and G. Buntkowsky, Sci. Rep., 2023, 13, 10117 CrossRef CAS; (b) J. Lins, Y. A. Miloslavina, O. Avrutina, F. Theiss, S. Hofmann, H. Kolmar and G. Buntkowsky, J. Am. Chem. Soc., 2024, 146, 35175–35184 CrossRef CAS PubMed.
- (a) M. Leutzsch, L. M. Wolf, P. Gupta, M. Fuchs, W. Thiel, C. Fares and A. Fürstner, Angew. Chem., Int. Ed., 2015, 54, 12431–12436 CrossRef CAS PubMed; (b) C. Godard, S. B. Duckett, S. Polas, R. Tooze and A. C. Whitwood, J. Am. Chem. Soc., 2005, 127, 4994–4995 CrossRef CAS; (c) S. B. Duckett, C. L. Newell and R. Eisenberg, J. Am. Chem. Soc., 1994, 116, 10548–10556 CrossRef CAS; (d) A. Manoharan, P. J. Rayner, W. Iali, M. J. Burns, V. H. Perry and S. B. Duckett, ChemMedChem, 2018, 13, 352–359 CrossRef CAS.
- A. Manoharan, P. J. Rayner, W. Iali, M. J. Burns, V. H. Perry and S. B. Duckett, ChemMedChem, 2018, 13, 352–359 CrossRef CAS PubMed.
- (a) M. V. Joannou, J. M. Hoyt and P. J. Chirik, J. Am. Chem. Soc., 2020, 142, 5314–5330 CrossRef CAS PubMed; (b) L. E. Hertwig, T. Bender, F. J. Becker, P. Jäger, S. Demeshko, S. J. Gross, J. Ballmann and D.-A. Roşca, ACS Catal., 2023, 13, 6416–6429 CrossRef CAS.
- (a) S. C. Bart, E. Lobkovsky and P. J. Chirik, J. Am. Chem. Soc., 2004, 126, 13794–13807 CrossRef CAS PubMed; (b) A. Archer, M. W. Bouwkamp, M.-P. Cortez, E. Lobkovsky and P. J. Chirik, Organometallics, 2006, 25, 4269–4278 CrossRef CAS; (c) R. J. Trovitch, E. Lobkovsky, E. Bill and P. J. Chirik, Organometallics, 2008, 1470–1478 CrossRef CAS; (d) R. P. Yu, J. M. Darmon, J. M. Hoyt, G. W. Margulieux, Z. R. Turner and P. J. Chirik, ACS Catal., 2012, 2, 1760–1764 CrossRef CAS; (e) J. C. Ott, C. K. Blasius, H. Wadepohl and L. H. Gade, Inorg. Chem., 2018, 57, 3183–3191 CrossRef CAS; (f) Y. Sunada, H. Ogushi, T. Yamamoto, S. Uto, M. Sawano, A. Tahara, H. Tanaka, Y. Shiota, K. Yoshizawa and H. Nagashima, J. Am. Chem. Soc., 2018, 140, 4119–4134 CrossRef CAS.
- (a) L. González-Sebastián, A. Reyes-Sanchez and D. Morales-Morales, Organometallics, 2023, 42, 2426–2446 CrossRef; (b) D. Wei and C. Darcel, Chem. Rev., 2019, 119, 2550–2610 CrossRef CAS PubMed; (c) L. Alig, M. Fritz and S. Schneider, Chem. Rev., 2019, 119, 2681–2751 CrossRef CAS PubMed.
- H. U. Blaser, F. Spindler and M. Thommen, in The Handbook of Homogeneous Hydrogenation, ed. J. G. de Vries, C. J. Elsevier, Wiley-VCH: Weinheim, 2008, Ch. 37, pp. 1279–1324 Search PubMed.
- (a) L. Yang, X. Li, Y. Chen, X. Zheng and K. Sun, Sci. Rep., 2024, 14, 20925 CrossRef CAS PubMed; (b) T. Das, S.-C. Kweon, I. W. Nah, S. W. Karng, J.-G. Choi and I.-H. Oh, Cryogenics, 2015, 69, 36–43 CrossRef CAS.
- (a) A. M. Balu, S. B. Duckett and R. Luque, Dalton Trans., 2009, 5074 RSC; (b) L. Kaltschnee, A. N. Pravdivtsev, M. Gehl, G. Huang, G. L. Stoychev, C. Riplinger, M. Keitel, F. Neese, J.-B. Hövener, A. A. Auer, C. Griesinger, S. Shima and S. Glöggler, Nat. Catal., 2024, 7, 1417–1429 CrossRef CAS; (c) D. Schott, P. Callaghan, J. Dunne, S. B. Duckett, C. Godard, J. M. Goicoechea, J. N. Harvey, J. P. Lowe, R. J. Mawby, G. Müller, R. N. Perutz, R. Poli and M. K. Whittlesey, Dalton Trans., 2004, 3218 RSC.
- D. C. Najera and A. R. Fout, J. Am. Chem. Soc., 2023, 145, 21086 CrossRef CAS PubMed.
- (a) U. Bommerich, T. Trantzschel, S. Mulla-Osman, G. Buntkowsky, J. Bargon and J. Bernarding, Phys. Chem. Chem. Phys., 2010, 12, 10309–10312 RSC; (b) M. Plaumann, U. Bommerich, T. Trantzschel, D. Lego, S. Dillenberger, G. Sauer, J. Bargon, G. Buntkowsky and J. Bernarding, Chem. – Eur. J., 2013, 19, 6334–6339 CrossRef CAS; (c) L. T. Kuhn, U. Bommerich and J. Bargon, J. Phys. Chem. A, 2006, 110, 3521–3526 CrossRef CAS; (d) A. S. Kiryutin, K. L. Ivanov, A. V. Yurkovskaya, R. Kaptein and H.-M. Vieth, Z. Phys. Chem., 2012, 226, 1343–1362 CrossRef CAS.
- (a) N. I. Regenauer, H. Wadepohl and D.-A. Roşca, Inorg. Chem., 2022, 61, 7426–7435 CrossRef CAS PubMed; (b) N. I. Regenauer, H. Wadepohl and D. A. Roşca, Chem. – Eur. J., 2022, e202202172 CrossRef CAS PubMed.
- S. B. Duckett and R. E. Mewis, Acc. Chem. Res., 2012, 45, 1247–1257 CrossRef CAS.
- J. Eills, D. Budker, S. Cavagnero, E. Y. Chekmenev, S. J. Elliott, S. Jannin, A. Lesage, J. Matysik, T. Meersmann, T. Prisner, J. A. Reimer, H. Yang and I. V. Koptyug, Chem. Rev., 2023, 123, 1417–1551 CrossRef CAS PubMed.
- (a) O. Bondar, E. Cavallari, C. Carrera, S. Aime and F. Reineri, Catal. Today, 2022, 397–399, 94–102 CrossRef CAS; (b) T. Gutmann, T. Ratajczyk, S. Dillenberger, Y. Xu, A. Grünberg, H. Breitzke, U. Bommerich, T. Trantzschel, J. Bernarding and G. Buntkowsky, Solid State Nucl. Magn. Reson., 2011, 40, 88–90 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5cc02409a |
| ‡ These authors contributed equally to this work. |
| § m-F-Styrene undergoes polymerisation under these reaction conditions and is not a suitable substrate for PHIP. See the ESI.† |
| This journal is © The Royal Society of Chemistry 2025 |