
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePolyoxometalate photocatalysts: solar-driven activation of small molecules for energy conversion and greenhouse gas valorization†
Yinghao
Wang‡
 ,
Geqian
Fang‡
,
Vitaly V.
Ordomsky
* and
Andrei Y.
Khodakov
*
,
Geqian
Fang‡
,
Vitaly V.
Ordomsky
* and
Andrei Y.
Khodakov
*
University of Lille, CNRS, Centrale Lille, Université d’Artois, UMR 8181, UCCS – Unité de Catalyse et Chimie du Solide, F-59000, Lille, France
First published on 26th June 2025
Abstract
The efficient transformation of small molecules including CO2, H2O, CH4, and N2, critical to energy systems and environmental sustainability is fundamental to addressing global energy challenges and advancing a sustainable future. Key technologies driving this progress include photocatalytic CO2 reduction reaction (CO2RR), overall water splitting (OWS), direct selective methane conversion (DSMC), and nitrogen fixation reaction (NFR). The development of advanced photocatalysts is essential to accelerate sluggish reaction kinetics and improve the selectivity. Polyoxometalates (POMs), as a unique class of multi-electron transfer catalysts, have attracted considerable interest due to their tunable geometric and electronic structures, excellent redox properties, reversible electron storage capacity and stability. Their capacity to extend light absorption into the visible spectrum and offer specific active sites significantly enhances their potential for photocatalytic oxidation and reduction reactions. This review summarizes recent progress in utilizing POM-based materials for CO2RR, OWS, DSMC and NFR. Significant advancements in enhancing photocatalytic selectivity and activity have been achieved by managing charge generation and recombination, engineering band structures and active sites, and optimizing reaction parameters. The advantages, challenges, strategies and outlooks of POM-based materials for improving photocatalytic performance are discussed.
 Yinghao Wang | Yinghao Wang pursues his PhD and performs research about photocatalysis under the supervision of Dr Andrei Khodakov and Dr Vitaly Ordomsky at the UCCS-CNRS laboratory (France) from 2022 to 2025. His project at UCCS focuses on new materials and processes for photoconversion of methane and other small molecules at mild conditions. |
 Geqian Fang | Geqian Fang received his PhD degree from the Dalian Institute of Chemical Physics, Chinese Academy of Sciences, in 2023. He is now a post-doctoral researcher at the UCCS-CNRS laboratory (France). His research interests include the controlled tailoring of nano and atomic scale metal catalysts, and their applications in energy and environmental catalysis. |
 Vitaly V. Ordomsky | Dr Vitaly V. Ordomsky received his MSc (2006) and PhD (2009) degree from Moscow State University in zeolite catalysis. He was a postdoctoral researcher in the Department of Chemical Engineering at the Eindhoven University in the Netherlands, working on biomass conversion. He received a permanent position as a researcher in 2013 at CNRS (France) in the Energy group of UCCS laboratory in Lille University focusing on Fischer–Tropsch synthesis. From 2016 to 2019, he was a CNRS researcher in E2P2L laboratory in Shanghai. His research field is new nano-materials and processes for C1 chemistry. |
 Andrei Y. Khodakov | Andrei Y. Khodakov received his PhD in 1991 from the Zelinsky Institute of Organic Chemistry, USSR Academy of Sciences. He worked as a postdoc in France, the UK, and the USA (1992–1999), and has been a CNRS researcher in France since 1999. Since 2017, he is Senior Research Director at UCCS-CNRS laboratory, University of Lille and Centrale Lille Institute. His research focuses on catalytic conversion of fossil and renewable feedstocks into clean fuels and valuable chemicals, notably on Fischer–Tropsch synthesis, CO2 hydrogenation, and photocatalysis. |
1. Introduction
The growing demand for sustainable energy and the need to mitigate greenhouse gas emissions have driven research into the efficient conversion of simple molecules into value-added chemicals.1,2 The conversion of these molecules, such as CO2, CH4 and N2, is central to critical energy and environmental issues including carbon dioxide reduction reaction (CO2RR),3 overall water splitting (OWS),4 direct selective methane conversion (DSMC),5 and nitrogen fixation reaction (NFR).6 However, the conversion of these molecules is always affected by high thermodynamic barriers, slow reaction kinetics and insufficient selectivities, which need well-designed catalytic systems to drive the reaction under mild conditions. Among the various strategies explored, photocatalysis stands out as a promising approach that harnesses the inexhaustible power of solar energy.7–10 However, the semiconductors used in conventional photocatalysts, such as TiO2, ZnO, WO3 often suffer from limited solar absorption, often in the UV range, inefficient charge separation and poor selectivity to the target products.11–14 Therefore there remain significant challenges in the design of innovative photocatalysts that can further enhance solar energy conversion efficiency and broaden their applicability in photocatalytic processes.15–18Polyoxometalates (POMs) are generally classified as p-type semiconductors. They have well-defined structures and versatile functionalities. Since the first classical Keggin topologies were found in 1934, researchers have systematically identified and characterized other five classical POMs, including Dawson, Waugh, Silverton, Anderson, and Lindqvist. In the recent years, these POMs gradually become the most famous branches of the POM family. (Fig. 1)19–21 Unlike other conventional semiconductor materials, POMs with tunable electron storage capacity can facilitate efficient charge separation. Furthermore, their molecular properties allow precise control of electronic structures which can enable the rational design of photocatalysts to enhance light-harvesting.22–24 Last but not least, by ligand-to-metal charge transfer (LMCT) and combination with light-harvesting components, POMs could further broaden their applicability in solar energy and environmental applications.25 By incorporating cocatalysts, modifying ligand environments, or hybridizing with semiconductors, researchers have significantly improved the photocatalytic performance of POM-based materials for CO2RR, OWS, DSMC and NFR.
 | ||
| Fig. 1 Different structures of POMs. | ||
POMs have long been recognized as exceptional electrocatalysts due to their structurally flexible MOx frameworks, which can reversibly accommodate multiple electron transfers. The heteroatom substitution enables their highly tunable redox potentials and their remarkable structural stability under operating potentials. However, the intrinsically low electrical conductivity of POM clusters often requires their hybridization with conductive supports (e.g., carbon nanotubes, metal–organic frameworks) to achieve high current densities and Faradaic efficiencies. By contrast, in photocatalysis, POMs serve simultaneously as molecular photosensitizers and charge stores.26–28 In light of these dual roles, in this review, we focus specifically on the photocatalytic applications of POMs to highlight the unique photophysical and charge-storage functions that are not discussed in purely electrocatalytic surveys.
Recently published works on photocatalysis have shown remarkable progress in designing POM band structures and modifying surface chemical properties. For example, a combination of POMs, semiconductors and plasmonic nanoparticles has enhanced their ability of light absorption and charge transfer. The incorporation of transition metals or organic ligands helps POMs tailor their redox properties.29 The POM-based materials achieved high selective conversion of CO2 into C1–C2 products in CO2RR by producing and stabilizing the intermediates.30–33 Researchers designed more promising systems by rationally combining and adjusting POM catalysts, photosensitizers and sacrificial reagents for OWS.34–36 In DSMC and NFR, POMs improve the activation of C–H and N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N bonds by concerted proton-coupled electron transfer mechanisms and generating reactive oxygen species (ROS).37,38 This remarkable progress highlights the importance of rational catalyst design which could help the catalytic system to balance activity, selectivity and stability.
N bonds by concerted proton-coupled electron transfer mechanisms and generating reactive oxygen species (ROS).37,38 This remarkable progress highlights the importance of rational catalyst design which could help the catalytic system to balance activity, selectivity and stability.
This review provides a comprehensive analysis of recent progress in the design of POM-based materials for CO2RR, OWS, DSMC, and NFR. We examine their structure, reaction mechanisms, and performance, offering insights into the rational principles essential for developing photocatalysts with high potential in sustainable energy conversion and greenhouse gas mitigation. Additionally, we summarize recent advancements in POM-based materials and discuss future research directions, key challenges, and emerging opportunities in the field. Further information on POM photocatalysts for CO2RR, OWS, DSMC, and NFR discussed in the manuscript is available in Table S1 (ESI†).
2. Structure and properties of POMs
POMs are clusters of cationic and polyanionic entities composed of pretransition/transition metals and oxygen atoms. The basic fundamental units of POMs are polyhedra of metal–oxo (typically denoted as MOx with x = 5 or 6). The metal centers in these polyhedra are in high oxidation states and most of them are W6+, Mo6+, V5+, Ta5+ and Nb5+.39 In many POMs, MOx units adopt octahedral geometries. These octahedral units share oxygen atoms by edge or corner linkages to form large three-dimensional frameworks and different structural types. Polyhedral distortions are also common and lead to the variations in bonding and coordination. By selecting different heteroatoms (such as P, Si, or B) in the central positions and by replacing some of the metal centers, it is possible to tune the geometry, electronic properties and catalytic behavior of POMs.40The structural versatility of POMs has garnered significant research interest in the field of photocatalysis. The POM tunability allows researchers to precisely control the chemical properties of POMs, including redox potential, acidity, solubility and thermal stability.41–45 By designing heterojunctions with other semiconductors, creating Schottky barriers with metals, incorporating transition metal cations into their frameworks, the light absorption characteristics of POMs can be adjusted and make full use of the solar irradiation. POMs can reversibly accept and donate electrons and holes, enabling them to participate in multi-electron redox processes. The POM-modified clusters exhibit higher stability, better altered redox characteristics and wider light absorption. Many traditional POMs are active under ultraviolet (UV) light. This limitation curtails their photocatalytic efficiency because UV light represents only a minor fraction of the solar spectrum. Combining POMs with conductive polymers, carbon- and carbon-nitride nanomaterials capable of absorbing visible light can effectively overcome this limitation. In addition, embedding POM clusters into metal–organic frameworks (POMs@MOFs) or synthesizing POM-based MOFs (POMOFs) can also help to broaden the absorption spectrum, improve charge separation efficiency, and facilitate easier catalyst recovery.46–49 Notably, POMs can generate heteropolyblues (HPBs) with multiple valence species of reduced metal centers and oxide–ligand radicals. They can store multiple electrons and participate in subsequent redox reactions. HPBs can donate these stored electrons to substrates or co-reagents, then prevent the electron–hole recombination. Mechanistically, HPBs have been confirmed to facilitate challenging photocatalytic processes.50,51 Incorporating HPB into POMs-based systems can extend light absorption and improve charge separation. Consequently, POM-based materials have attracted significant interest52–56 for the photocatalytic conversion of small molecules, such as CO2, CH4, N2, and H2O, into valuable fuels and chemicals.
3. POM-based materials for photocatalytic CO2 reduction
Due to the significant environmental challenges posed by excessive CO2 emissions, photocatalytic CO2RR has garnered significant attention.57,58 The CO2 reduction is challenging due to thermodynamic and kinetic stability of this molecule. From a thermodynamic point of view, the energy of the conduction band minimum (CBM) of the semiconductor must be higher than the energy required for CO2 reduction and, most importantly, higher than the lowest electron-accepting bands or orbitals of the reduction catalyst. CO2 photocatalytic reduction is always accompanied by the oxidation of water or a sacrificial agent.59 Likewise, the energy of the valence band maximum (VBM) must be lower than that of the corresponding oxidative reactions, with H2O oxidation being a notable example. From a kinetic perspective, the high C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond dissociation energy (approximately 750 kJ mol−1) necessitates substantial energy for CO2 activation. Furthermore, the products of CO2RR could have a multitude of oxidation states. Intense competition from photocatalytic reactions leads to various products that can further diminish both the reaction rate and selectivity.8,60–62 Typically, the photocatalytic reduction of CO2 with H2O or sacrificial agents occurs at ambient temperature and pressure. In contrast, reduction with H2 requires harsher conditions and can often be viewed as a combination of photocatalysis and thermal catalysis.
O bond dissociation energy (approximately 750 kJ mol−1) necessitates substantial energy for CO2 activation. Furthermore, the products of CO2RR could have a multitude of oxidation states. Intense competition from photocatalytic reactions leads to various products that can further diminish both the reaction rate and selectivity.8,60–62 Typically, the photocatalytic reduction of CO2 with H2O or sacrificial agents occurs at ambient temperature and pressure. In contrast, reduction with H2 requires harsher conditions and can often be viewed as a combination of photocatalysis and thermal catalysis.
Photocatalysis involves photon absorption to promote an electron transfer from the valence band (VB) to the conduction band (CB) of a semiconducting material. This creates electrons and holes and the resulting charge separation and transport provides the necessary excitons for two half-reactions. The reactions may take place at the semiconductor itself or at a separate cocatalyst. It is crucial to prevent rapid electron–hole recombination, as this can significantly reduce the number of useful charge carriers reaching substrate-accessible interfaces, thereby lowering the quantum yield.63,64
The CO2 reduction requires multielectron transfer. The transfer of the first electron to CO2 is the most energy-demanding process, because it leads to partial bond breaking and a bend in the stable linear CO2 molecule (E0 = −1.90 V). The coupled 2H+/2e− reductions of CO2 to either HCOOH or to CO + H2O are more favorable:
| CO2 + 2H+ + 2e− → HCOOH − 0.61 V |
| CO2 + 2H+ + 2e− → CO + H2O −0.53 V |
Selectivity control in the CO2 reduction is difficult because several reactions have similar potentials.65
When designing photocatalysts, several crucial factors are need to be considered: (a) efficient separation of photoinduced electron–hole pairs and preventing recombination of charge carriers. (b) Broadening absorption spectrum to boost the efficiency of solar energy conversion. (c) Engineering of active sites to facilitate subsequent electron transfer and to produce target products (Fig. 2a).48,66–68 Many conventional semiconductors with large band gaps and short carrier lifetimes could be limited in their application in CO2RR photocatalytic processes because of the need for hard UV irradiation and fast charge carrier recombination. Incorporating POMs into catalyst materials is a tailored strategy to overcome these challenges.
 | ||
| Fig. 2 (a) Simple schematic diagram of the function of POMs in the CO2 photoreduction: efficient separation of photoinduced electron–hole pairs; a broader absorption spectrum; active surface sites effectively adsorb CO2 or directly playing the roles of catalysts. The blue cylinder designates POM-based materials. (b) Schematic energy-level diagram and electron transfer from [Ru(bpy)3]Cl2·6H2O to Ru2Ge2Mo20.72 Reproduced from ref. 72 with permission from Royal Society of Chemistry, copyright 2024. (c) Schematic representation of intensified Mo–O–Ni/Co bonds for promoting the migration of H* in Pt1-PMo12@NiCo-LDH and Pt1-PMo12/NiCo-LDH.74 Reproduced from ref. 74 with permission from John Wiley and Sons, copyright 2025. | ||
3.1. POMs as multifunctional electron “sponges”
Due to the unique capacity to accept photoinduced electrons and the ability to form p–n heterojunctions with other semiconductors, POMs can serve as “electron sponges” that can slow recombination of photoinduced electrons and holes and prolong their lifetimes.To better understand contribution of POMs for the separation of photoinduced electron and holes, we categorize POM-based catalysts into three main groups: pure POMs, metal/POM hybrids, and POM/MOF composites. Pure POMs can act as electron acceptors or form shallow electron traps, demonstrating excellent charge separation capabilities.
The interaction between CO2 and POM derivatives was first reported by Kozik69 with colleagues in 1988. This pioneering work demonstrated that integrating POMs with other materials could prevent the charge recombination and stabilize the crucial reaction intermediates. Lan70 and colleagues prepared photocatalysts by doping saturated and mono-/tri-lacunary Keggin-type SiWx POMs into Bi2WO6 and BiOBr by a two-step hydrothermal method. They exploited the redox active nature of POMs that can act as shallow electron traps due to the unoccupied W 5d orbitals in tungsten–oxygen clusters. These POMs can efficiently capture the photoinduced electrons from Bi2WO6 or BiOBr, and reduce electron–hole recombination. Further advancing the field, Xu et al.71 developed a facile electrosynthesis method to uniformly disperse Wells–Dawson- and Keggin-type POMs on TiO2 nanotube arrays. POMs prevented the charge recombination in TiO2 and enhanced the density of photoinduced holes needed for water oxidation by acting as electron sponges. These photoinduced holes enabled photoreduction of CO2 to CH3OH by using H2O as electron donor. The optimized P2Mo18O62@TiO2 catalyst achieved a 11 times higher rate of CH3OH production than pristine TiO2. In 2024, Li et al.72 synthesized a purely inorganic germanium–molybdenum–oxo cluster incorporating ruthenium, Na12Rb2[Ru2O2(GeMo10O36)2]·44H2O (Ru2Ge2Mo20) by hydrothermal methods. The unique sandwich structure was comprised by two [GeMo10O36]8− units and bridged by a Ru3+–oxo dimer. Under visible light, this catalyst achieved impressive yields of CH4 and CO. This special preparation method created spatially separated electron hole pathways that significantly prevented charge carrier recombination. Mechanistic studies revealed that the Ru centers mediated the electrons transfer from the [Ru(bpy)3]2+ to CO2. At the same time, the POM framework stabilized the reaction intermediates and avoided side reactions (Fig. 2b).
Incorporating metal centers into the POM structures is another method for modification electronic structures and improving redox properties to enhances charge separation. Feng et al.73 reported a one-pot solvothermal synthesis where two novel atomically precise silver clusters were stabilized by multidentate lacunary POM ligands. The Ag24(Si2W18O66)3 and Ag27(Si2W18O66)3 clusters with feature trefoil-propeller-shaped Ag cores, have 10 delocalized electrons, which are effectively stabilized by three C-shaped Si2W18O66 units. Femtosecond and nanosecond transient absorption spectroscopy reveals rapid charge transfer between the silver cores and the POM ligands. This efficient transfer helps clusters exhibit promising catalytic activity with high selectivity for the photocatalytic reduction of CO2 to formic acid. Besides photoinduced electrons, Cai74 and colleagues shown the crucial role of active hydrogen (H*) in photocatalytic CO2 methanation. The Pt1-PMo12@NiCo-LDH catalyst was developed by a Pt1-anchored polyoxometalate (Pt1-PMo12) integrated with NiCo layered double hydroxide (LDH) using in situ encapsulation-reassembly strategy. By leveraging the multiple redox-active sites in the Keggin-type PMo12 polyoxometalates play the role of electron–proton shuttle, the catalyst significantly enhances the proton-coupled electron transfer (PCET) during CO2 photoreduction. The Pt1-PMo12 component facilitated efficient H2O dissociation to generate active hydrogen species (H*), and H* species were rapidly transported to NiCo-LDH catalytic active sites through engineered Mo–O–Ni/Co interfacial bonds for the CO2 methanation (Fig. 2c).
Hybridization of MOFs with POM-based materials75,76 can leverage high surface areas, structural stability, and enhanced charge separation pathways. Du77 and colleagues developed a series of POM@CdMOF photocatalysts and integrated Keggin-type POMs (PW12, SiW12 and PMo12) into a Cd-MOF framework. They tried to exploit the unique electron sponge property of POMs to capture electrons. This mechanism accelerated charge separation by quickly transferring photoinduced electrons from the Cd-MOF to the POMs. Solé-Daura et al.29 combined microkinetic modeling with spectroscopic experiments and developed a Rh-functionalized UiO-67 MOF catalyst co-immobilized with Keggin-type POMs. POMs served as efficient electron reservoirs that capture electrons from the photoinduced [Ru(bpy)3]+, and then ensured the robust separation of electron–hole pairs. This effective charge separation prevented the recombination of electron–hole and maximizes utilization of photoinduced electrons.
Thus, by serving as “electron sponges” or electron reservoirs, POMs can obviously facilitate charge separation during photocatalytic process. POMs have been successfully integrated into a wide range of hybrid systems from inorganic clusters and MOFs to semiconductor arrays, and highly promoted the efficiency and selectivity of CO2 reduction or related processes. The rational design of POM-based structure and fine-tuning of interfacial interactions deserve further development in the future, which can optimize the electron transfer and catalytic performance.
3.2. Broadening the absorption spectrum
Most of POMs absorb irradiation in the UV range. A shift of absorption spectrum to visible range is crucial for driving highly efficient CO2 photocatalytic reduction under solar light. As early as 2017, Sun et al.78 first introduced noble-metal-free POMs (Co4) into graphitic carbon nitride (g-C3N4) to develop low-cost hybrid catalysts (Co4@g-C3N4). The optimized catalyst exhibited a CO2 conversion rate of 107 μmol g−1 h−1 and 94% selectivity to CO under visible-light. In this system, Co4 has broadened the visible light absorption range to generate photoinduced electrons, and to improve efficient electron transfer to g-C3N4. Building upon this early breakthrough, more strategies have been developed to broaden the light absorption spectrum for visible-light-induced CO2 reduction.Initial investigations of Anderson-type POM complexes revealed that the narrower bandgaps and the stronger visible-light absorption could significantly enhance the efficiency of charge transfer. Yang et al.79 in 2022 synthesized two one-dimensional (1D) Anderson-type POM-based metal–organic complexes, [Co(H2O)2DABT]2[CrMo6(OH)5O19] (Co–CrMo6) and [Zn(H2O)2DABT]2[CrMo6(OH)5O19] (Zn–CrMo6), by a hydrothermal method. Co–CrMo6 exhibited enhanced photocatalytic CO2 reduction due to its strong visible-light absorption (up to 750 nm) and narrow bandgap (2.00 eV), which improved light utilization efficiency and energy conversion (Fig. 3). As a result, they achieved a CO formation rate of 1935.3 μmol g−1 h−1 with a high selectivity.
 | ||
| Fig. 3 (a) Mechanism diagram for photocatalytic CO2 reduction by Co–CrMo6. (b) UV-vis diffuse reflection spectra and K–M function curve of Co–CrMo6. (c) UV-vis diffuse reflection spectra and K–M function curve of Zn–CrMo6.79 Reproduced from ref. 79 with permission from American Chemical Society, copyright 2022. | ||
Zhang et al.80 developed a Z-scheme heterojunction photocatalyst (SiW9Co3@UiO-67-NH2) for CO2 reduction to CO. The catalyst combined Keggin POM SiW9Co3 and UiO-67-NH2 MOF by in situ encapsulation. SiW9Co3 provided a strong visible-light absorption (absorption edge up to 782.6 nm) and a narrow bandgap (2.23 eV). The broad optical absorption range suggested that the heterojunctions could generate more charge carriers during photocatalysis. The strong visible-light absorption and interfacial interactions enhanced charge separation and catalytic performance. Yan et al. fabricated two Anderson type POM-based materials modified by cobalt, with the two-dimensional layered structures.81 By leveraging the tunable semiconductor structure, the incorporation of Co ions into the Anderson POM CrMo6 units enhanced visible light absorption due to the d–d transitions of Co2+. This successfully broadened the light absorption spectrum and enhanced the conversion efficiency from light to chemical energy.
3.3. Active sites engineering
The presence of co-catalysts in POMs enhances the CO2 reduction and shifts the selectivity to a specific reaction product. The ability to adsorb CO2 on the active surface sites plays a pivotal role in facilitating electron transfer or even directly catalyzing the CO2 reduction.40,52,82 In 2010, Neumann83 and coworkers first synthesized a Ru-substituted Keggin-type POM and demonstrated its application for CO2 reduction to CO with the triethylamine (Et3N) used as sacrificial agent. Experimental and computational research revealed that the Ru center played a critical role in activating CO2, while the [SiW11O39]8− fragment served as effective light-harvesting material and co-catalyst. Building on this early breakthrough, subsequent research focused on utilizing POM-based materials for CO2 activation by tailoring surface sites to enhance adsorption and electron transfer.In 2021, Du et al.84 synthesized a novel MOF and built a unique “cage-within-cage” structure by incorporating POM-derived MoO42− anions. This innovative structure created abundant active sites on surface and significantly enhances CO2 adsorption. By effectively capturing CO2, these sites facilitated the electron transfer processes and drove the selective photoreduction of CO2 to CO. In the same year, Yu et al.85 synthesized vanadoborate clusters modified by transition metals. Notably, incorporation of Co or Ni created distinct active surface sites that effectively adsorbed CO2 and promoted the photocatalytic process. By exploiting the efficient transfer properties of POMs, these catalysts increased conversion of CO2 into syngas and formates. Density functional theory (DFT) calculations further confirmed that the transition-metal centers served as the pivotal active sites for CO2 adsorption and activation. Mallick et al.86 replaced Na+ ions with Cu2+ ions via post-synthetic metalation to synthesize a copper-functionalized catalyst (Cux[POM]@TiO2). The enhanced CO2 adsorption capability of Cu2+ sites was due to the charge-balancing anionic surfaces of POMs ([PW11O39Ti]5− units). The POMs with the 6 nm TiO2 nanocore facilitated the η2-binding of CO2 to Cu2+, resulting in the formation of the critical [Cu-(η2-CO2)]− intermediate. This optimized method significantly helped the material to adsorb CO2 and dramatically enhanced the efficiency of electron transfer under UV light (Fig. 4a). Yu et al.87 reported design of new efficient core–shell nanocomposites for selective photocatalytic CO2 reduction. A combination of characterization techniques indicated that the CO2 to CO reduction rate reached 50 μmol g−1 h−1. A selectivity of 73% to CO occurred over zinc species highly dispersed on the heteropolyacid/titania core–shell nanocomposites acting as co-catalysts. In situ Fourier-Transform Infrared (In situ FTIR) investigation uncovered that the reaction involved surface zinc bicarbonates as key reaction intermediates. Li et al.88 synthesized a manganese modified POM photocatalyst, [Mn(en)2]6[V12B18O54(OH)6] (Mn6V12). This catalyst harnessed the oxygen-rich surface of POMs and enhanced the electron-accepting capability. The Mn modification introduced active Mn–N/O sites within the vanadium-borate POM framework. These sites optimized CO2 adsorption and directed electron transfer toward CO2 reduction with a CO production rate of 4625.0 μmol g−1 h−1. Li89 and collaborators designed a catalyst by morphological engineering, which integrated mono- (Co) or bimetallic (CoNi) species into dodecahedral POM frameworks (K3PW12O40). Through hydrothermal etching and annealing, the structure of POMs evolved from solid dodecahedra to yolk shell and hollow structures. The physical confinement enhanced CO2 adsorption capacity. The anchored Co/CoNi sites played a crucial role and facilitated chemical activation of CO2 (Fig. 4b). Alomar90 and colleagues developed a hybrid catalyst, which combined an γ-Anderson POM and manganese porphyrin within a covalent organic framework (COF). The materials were specifically tailored to enhance CO2 adsorption at the active sites on surface. The POMs ([PMo8O26]3−) provided structured anchoring points for CO2 on their oxygen-rich surfaces and confirmed the η2-binding. This adsorption facilitated multi-electron reduction by Mn(III)/Mn(II) and Mo(V)/Mo(IV) redox cycles. Finally, Benseghir et al. synthesized heterometallic molybdenum(V) phosphate catalysts using a hydrothermal method and integrated [Ru(bpy)3]2+ as a photosensitizer.91 They engineered sandwich-type structures, in which P4Mo6 units were connected by Mn2+ and Fe2+/Fe3+ centers. Crucially, the Mn(II) sites acted as active adsorption sites for CO2 and effectively anchored the CO2 molecules. This anchoring facilitated the electron transfer needed for CO2 reduction into CH4 under visible-light irradiation. In this system, POM clusters acted as electron shuttles and played a crucial role for intrinsic redox and charge transfer properties.
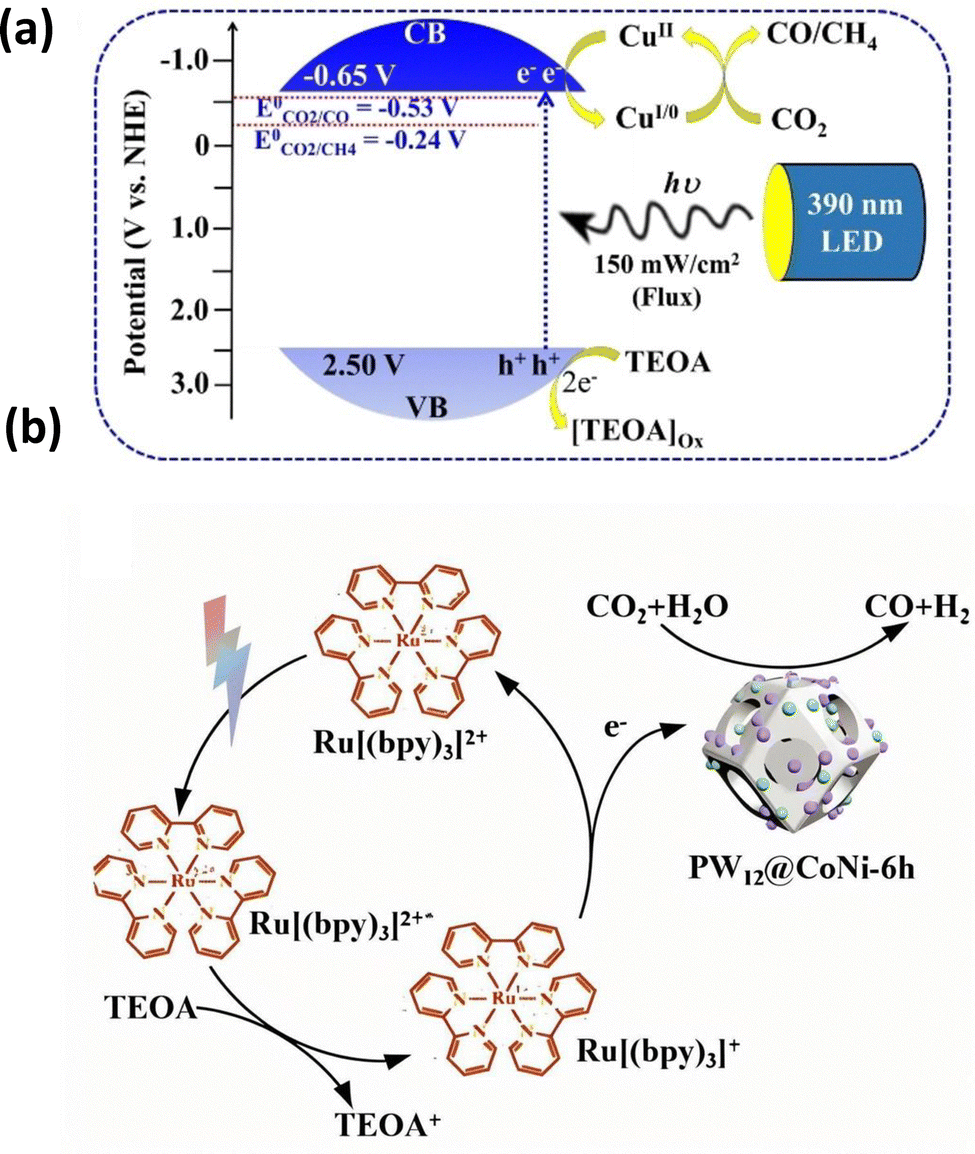 | ||
| Fig. 4 (a) Schematic presentation of the band positions of Cux[POM]@TiO2 along with photochemical CO2 reduction scheme in the presence of 390 nm LED lamp.86 Reproduced from ref. 86 with permission from John Wiley and Sons, copyright 2024. (b) PW12@CoNi in photosensitizer system CO2 reduction mechanism diagram.89 Reproduced from ref. 89 with permission from Elsevier, copyright 2024. | ||
Besides optimizing the CO2 adsorption, POMs can also directly activate CO2. Talbi92 and collaborators identified the tetrabutylammonium salt of the tetranuclear [Ni4(H2O)2(PW9O34)2]10− cluster in nickel-substituted POMs. The Ni4 POM could accumulate two electrons and four protons under light irradiation, enabling direct interaction with CO2 by Ni2+ centers. The protonation of the POM decreased its reduction potential and facilitated electron transfer from the reduced photosensitizer ([Ru(bpy)3]+) for CO2 activation. Notably, CO2 bubbling caused phase separation. This process segregated POMs from bulky TBA+ counter-ions. As a result, the Ni2+ active sites became more accessible and significantly improved the catalytic performance. Zhu et al.93 developed a PWx/g-C3N4 catalyst (x = 12, 11, 9) by integrating lacunary POMs with g-C3N4. The tri-lacunary PW9/g-C3N4 achieved 80% CH4 selectivity and 40.8 μmol g−1 h−1 by leveraging POMs oxygen-rich active sites and high charge density. At the same time, CO intermediates were also stabilized. These defects enhanced CO adsorption by π-bond interactions with W atoms and directing sequential hydrogenation of CH4. Continuing this trend, Wang et al.94 developed a composite catalyst (PTi2W10@EB-TFP). They achieved this by encapsulating a titanium-substituted polyoxometalate (PTi2W10) into a EB-TFP COF by an ion-exchange method. PTi2W10 provided Lewis acidic Ti4+ sites to activate epoxides. At the same time, EB-TFP's Br− ions and nitrogen-based basic sites adsorbed and polarized CO2, driving its cycloaddition. Experimental results have shown a major effect of the dual-site synergy. As a result, the catalyst achieves a 98.62% yield of cyclic carbonates without external cocatalysts.
Thus, POMs possess well-defined active centers, tunable energy band structures, and strong light absorption capabilities, enabling reversible and stepwise multiple-electron transfer without structural alterations. These properties make POMs highly promising for CO2 photocatalysis and offer potential solutions for environmental challenges and sustainable energy needs. However, significant hurdles remain, including low efficiency, stability concerns, and scalability limitations, which must be overcome for practical applications.
While POM catalysts have attracted much attention for their application in photocatalytic CO2 reduction, their utilization for conversion of other simple molecules deserves further development. In the following section, we introduce a selection of pioneering studies that demonstrate the potential of POM-based photocatalysts for converting water, methane and nitrogen.
4. Photocatalytic reactions involving other small molecules
4.1. Water splitting
There are a few reports about applications of POM materials for photocatalytic water splitting. The group of Lv34 used a comprehensive strategy to utilize the properties of POMs for driving photocatalytic hydrogen evolution reaction (HER). Their studies systematically exploited the geometries, electron storage capabilities and electron transfer characteristics of POMs. They synthesized a cage-like and POM-based organic structure Ni16L6(SiW9)4. This structure was constructed from Ni4-substituted Keggin clusters and flexible tartaric acid ligands, which significantly enhanced the accessibility of active sites and increased turnover number (TONs) up to 6834 within 5 h and 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 over 96 h. The progress in molecular orbital engineering produced twin-Dawson-type POMs. The modulation of LUMO levels by Mo incorporation resulted in TON of 2277 and 4722.5, respectively.35 Beyond these systems, the group expanded their approach by designing composite catalysts that integrated POMs with other functional materials. Tri-nickel-containing POM@MOF composites (Ni3P2W16@NU-1000 and Ni3PW10@NU-1000) with strong host–guest interactions were prepared. These composites were paired with ascorbic acid as a sacrificial electron donor and exhibited HER rates as high as 13051 μmol g−1 h−1.36
500 over 96 h. The progress in molecular orbital engineering produced twin-Dawson-type POMs. The modulation of LUMO levels by Mo incorporation resulted in TON of 2277 and 4722.5, respectively.35 Beyond these systems, the group expanded their approach by designing composite catalysts that integrated POMs with other functional materials. Tri-nickel-containing POM@MOF composites (Ni3P2W16@NU-1000 and Ni3PW10@NU-1000) with strong host–guest interactions were prepared. These composites were paired with ascorbic acid as a sacrificial electron donor and exhibited HER rates as high as 13051 μmol g−1 h−1.36
Additionally, by coupling water-soluble CdSe quantum dots with a series of Ni-substituted POMs, it was demonstrated that the catalytic activity was closely related to the Ni centers—the Ni4P2-based system achieving a TON of 9000 after 12 h and a hydrogen production rate of 138 mmol g−1 h−1 (Fig. 5).95 Finally, an innovative dual-functional system combining Ni4P2 with CdS nanorods was developed. This system could drive efficient HER and facilitated the selective photooxidative dehydrogenative coupling of 4-methoxythiophenol into 4-methoxydiphenyl disulfide at the same time.96
 | ||
| Fig. 5 (a) Proposed mechanism of Ni4P2-based system visible-light-driven H2 evolution. (b) Long-term photocatalytic hydrogen production experiment over the system including 2μM CdSe-MPA QDs(540), 2μM Ni4P2, 0.2M ascorbic acid (AA) in 6.0mLH2O at pH 4.5 upon irradiation with 520nm LED.95 Reproduced from ref. 95 with permission from Elsevier, copyright 2022. | ||
In addition to HER, POMs have been increasingly explored for the oxygen evolution reaction (OER).26 In 2008, Hill and Bonchio groups independently reported the first homogeneous POM-based molecular photocatalyst for water oxidation, a tetra-ruthenium(IV)-substituted cluster [Ru4O4(OH)2(H2O)4(γ-SiW10O36)2]10− (Ru4SiPOM).97 Under neutral conditions with [Ru(bpy)3]3+ as oxidant, the catalyst achieved a TON of ∼18 and a Turnover Frequency (TOF) of 0.45–0.6 s−1, marking a key breakthrough in POM oxygen evolution reaction development. Recently, Li and co-workers introduced a crystalline β-octamolybdate cluster, MV2[β-Mo8O26],98 in which methyl viologen cations (MV) serve both as counter-ions and as strong π-polarization agents. By selecting a well-defined β-Mo8O264− cluster and pairing it with a π-conjugated organic cation (MV2+), the authors harness electron-transfer photochromism to create and lock in active “oxygen-hole” sites on the POMo scaffold. This stable charge-separated state (with a lowered Fermi level) directly promotes O–O bond formation, thereby significantly boosting OER performance in neutral electrolytes.
Generally, these works have shown a strategic exploitation of redox properties and structural tunability of POMs. To achieve highly efficient, robust, and multifunctional solar-driven HER, the electron-storage capabilities and tunable redox potentials of POMs must be fully leveraged in the design of POM-based materials. These properties make POMs promising for solar-driven water splitting.
4.2. Methane conversion
In addition to CO2 reduction and water splitting, recent advances in POMs-based materials have expanded the scope of sustainable reactions of methane. Methane conversion reactions are extensively studied, yet the potential of POMs in this field remains underexplored, warranting further investigation.99–101 Direct and selective methane photocatalytic oxidation into carbon monoxide was observed at ambient conditions.102 The composite catalysts on the basis of zinc, tungstophosphoric acid and titania exhibited exceptional performance in this reaction, a high carbon monoxide selectivity and a quantum efficiency of 7.1% at 362nm. The reaction proceeded via intermediate formation of surface methyl carbonates. Furthermore, a photochemical looping strategy was developed for highly selective stoichiometric conversion of methane to ethane at ambient temperature over silver–heteropolyacid–titania catalysts.103 A stoichiometric reaction of methane with highly dispersed cationic silver occurred under illumination. Recombination of the generated methyl radicals led to selective, and almost quantitative formation of ethane. Additionally, we developed Ag- and Pd-phosphotungstic acid (HPW)/TiO2 nanocomposites for photocatalytic methane coupling to ethane.37
A combination of ex/in situ techniques such as XAS, EPR and DFT modeling demonstrated that AgPW/TiO2 was relied on Ag+ species for methane activation and Pd/NPW/TiO2 utilized metallic Pd clusters. Addition of water boosted the activity by promoting ˙OH generation from H2O which is crucial for methane activation. The Keggin structure and redox versatility of POMs were used to stabilize the cationic Ag+ and disperse the Pd clusters on the catalyst surface. At the same time, p–n heterojunctions of POM with TiO2 were formed to optimize charge separation and prevent electron–hole recombination. Tunable acidity of POMs and strong interaction with TiO2 further facilitated the adsorption and activation of methane (Fig. 6a–c).
 | ||
| Fig. 6 (a) STEM-HAADF images of (Pd/NPW)/TiO2. Color code: O – white, P – orange, W – green. (b) Methane photocatalytic coupling on (Pd/NPW)/TiO2 nanocomposites in dry and aqueous environments. (c) EPR spectra of DMPO spin trapping of (Pd/NPW)/TiO2 and TiO2 under methane, *Signals of DMPO/˙CH3, °Signals of DMPO/˙OH.37 Reproduced from ref. 37 with permission from Elsevier, copyright 2024. (d) Atomic-resolved HAADF-STEM image of Pt/NPW/TiO2 (3:10). The inset image represents the monomer of Keggin-structured NPW with isolated Pt atoms anchored at the 4-fold hollow site of W–O–W, color code: red (oxygen), gray (tungsten), blue (phosphorus), purple (platinum). (e) 13C NMR spectra of liquid products of (Pt/NPW)/TiO2 (3:5) with 12CH4 + 12CO, 13CH4 + 12CO, and 12CH4 + 13CO. (f) Scheme of the generation of photoexcited charge carriers by Pt/NPW.104 Reproduced from ref. 104 with permission from American Chemical Society, copyright 2023. | ||
Our group recently reported104 a POM/TiO2-supported photocatalyst for selective synthesis of acetic acid from methane and CO in aqueous medium at ambient temperature:
| CH4 + CO + H2O → CH3COOH + H2 |
In this POM/TiO2 catalyst, Pt single atoms were anchored on ammonium phosphotungstate sub-nanoclusters. The catalytic performance was enabled by the unique properties of POM-derived clusters, where the Keggin structure facilitated electron storage and transfer. The catalyst achieved 90% liquid-phase selectivity for acetic acid, with a concentration of 5.7 mmol L−1 and a TON of 99 (Fig. 6d–f).
Thus, POMs can provide redox-active centers, tunable acid–base properties, and create heterojunctions with other semiconductors, which efficiently enhance charge separation. They enable the selective activation of methane under mild conditions and facilitate the valorization of methane into value-added chemicals. Numerous promising avenues remain to be explored for further development in this field.
4.3. Nitrogen fixation
POMs have also started to show promise as catalysts in photocatalytic nitrogen conversion. Li et al.38 developed an dual active site photocatalyst by anchoring Mo72Fe30 POM clusters onto a UiO-66. In this work, the POMs clusters substituted traditional bidentate linkers within the MOF structure and exposed additional unsaturated metal sites that possessed unoccupied d-orbitals with strong Lewis acidity. These newly available active sites effectively interacted with nitrogen molecules by accepting electrons from the 3σg bonding orbital of N2 and exhibited considerably enhanced N2 adsorption. Moreover, the “electron sponge” property of the POMs improved charge carrier separation. This directional flows of electrons and holes suppressed their recombination and also facilitated the efficient oxidation of N2 to HNO3. Consequently, unsaturated MOF sites captured more effectively N2 (Fig. 7). | ||
| Fig. 7 Schematic illustration of the efficient storage and conversion of N2 to HNO3 by POM@MOF (taking UiO-66 and Mo72Fe30 as an example).38 Reproduced from ref. 38 with permission from John Wiley and Sons, copyright 2023. | ||
Similarly, Xiao et al.105 and colleagues developed an SiW12/K-C3N4 innovative photocatalyst. They covalently linked the POM cluster (SiW12) with KOH-modified graphitic carbon nitride (K-C3N4) and then created a heterojunction with unique redox-active properties. In this design, the phosphate bridge served as an efficient electron transport channel and could rapidly transfer the photoinduced electrons from K-C3N4 to SiW12. The electron storage and transfer capability of the POM enhanced water oxidation, facilitated the nitrogen adsorption and activation and boosted the efficiency of photocatalytic nitrogen fixation. These works converged on the critical role of POMs in enhancing electron dynamics and active-site exposure for N2 activation. Further expanding this strategy, Li et al.106 developed a series of Keggin type POM based ZIF-67 composite materials. Their catalysts were prepared by a simple room-temperature mixing cobalt nitrate, POM clusters and 2-methylimidazole to form a robust ZIF-67 framework. The materials had high porosity and abundant active sites for effective N2 adsorption by integrating electron-rich POM clusters into ZIF-67. Similar with other reaction, under visible-light irradiation, the POMs were photo-reduced to generate a plentiful supply of electrons that were swiftly transferred to the adsorbed N2 and activated it.
In summary, POMs enhance nitrogen fixation by offering Lewis acidic sites, with electron-sponge behavior and structural tunability for in situ heterojunction formation. Similar to DSMC, POMs have not yet been extensively explored in the field of nitrogen fixation. We recently discovered that POMs could generate a significant number of oxygen vacancies upon irradiation. These vacancies significantly weaken the NN triple bond, thereby promoting NFR. Consequently, POMs hold considerable promise for future research in this field.
5. Conclusions and perspectives
POMs-based materials as multifunctional photocatalysts have been extensively explored for energy and greenhouse gas reactions of simple molecules. In CO2RR, POMs play several roles: acting as 'electron sponges' to facilitate efficient charge separation, serving as photosensitizers to broaden light absorption, and functioning as cocatalysts to greatly increase CO2 conversion and control selectivity towards CO and C1–C2 products. In OWS, multi-electron storage and transfer capabilities of POM can simultaneously enhance the hydrogen and oxygen evolution reactions. In DSMC, POMs help to activate the strong C–H bonds and to enable methane highly selective conversion into other value-added chemicals such as CO, ethane, or acetic acid. Similarly, in NFR, POMs-based materials improve N2 adsorption and activation by providing abundant active sites and photoinduced charge carriers to lower the reaction barriers.However, current photocatalytic systems still face numerous challenges, including rapid recombination of charge carriers, low quantum yields, insufficient selectivity, poor stability and productivity, and the need for sacrificial agents. Besides, the application of POM-based materials for other photocatalytic reactions has not been widely studied. In particular, there is a lack of research on redox coupling reactions of C1 molecules and reactions of multi-carbon compounds. Except for Keggin, Anderson, and Lindqvist types of POMs, the photocatalytic applications of other types of POMs have not been deeply developed. The unique redox properties, tunable electronic structures, and extended light absorption capabilities of POMs offer tremendous opportunities and intriguing directions for innovative research. Three research directions may be pivotal for further development. First, POM-based materials need the rational design of structures to utilize the properties of captured electrons. To meet the various requirements of a photocatalyst for different reduction reactions, POM-based materials can be purposefully designed. Many strategies can be considered, such as heteroatom substitution, transition-metal incorporation, and the creation of hybrid systems with semiconductors and organic complexes to construct heterojunctions or metal–organic frameworks. In this context, previously published works have identified the electron sponge behavior of POMs, which can be helpful for the efficient separation of photoinduced electron–hole pairs.
Second, POM-based materials can enhance the adsorption of reactants and act as co-catalysts. In addition, appropriately introducing structural defects such as oxygen vacancies, can create additional active sites in POMs. A significant promoting effect is expected on the adsorption and activation of certain gas molecules, especially N2. Modifying the acid–base properties of the materials can make them more effective for adsorbing gases.
Third, in situ characterization, isotope labelling experiments, DFT and artificial Intelligence (AI)-driven data analysis should be further used for comprehensive understanding of the relationships between structure and activity in POM-based materials and the reaction mechanism. Time-resolved spectroscopy, synchrotron-based X-ray absorption/diffraction, and electron microscopy enable researchers to observe dynamic changes in active sites and reaction intermediates under operating conditions. In situ spectroscopy will greatly accelerate the optimization of catalysts by combining these experimental results with AI-driven data analysis and computational modeling. Machine learning algorithms and high-throughput screening can also identify the correlations of POMs structure and function and predict their catalyst performance. We hope that the published works and integrated strategies reviewed here will provide valuable ideas for photocatalytic conversion of small molecules in energy and greenhouse gas reactions. Future research will increasingly unlock the potential of POMs-based materials in photocatalysis beyond reactions involving small molecules.
Author contributions
Yinghao Wang: conceptualization, writing. Geqian Fang: conceptualization, writing. Vitaly V. Ordomsky: conceptualization, writing, review and editing. Andrei Y. Khodakov: conceptualization, writing, review and editing.Conflicts of interest
There are no conflicts to declare.Data availability
No primary research results generated as part of this review.Acknowledgements
The authors acknowledge financial support from French National Agency for Research (Projects SolarMethaCell ANR-22-CE05-0019, PulseCoMeth ANR-22-CE50-0018, PEPR LUMA Synflux-Lumicals). Y. W. is grateful to China Scholarship Council for providing him a PhD stipend.Notes and references
- M. Aresta, A. Dibenedetto and A. Angelini, Chem. Rev., 2014, 114, 1709–1742 CrossRef CAS PubMed.
- V. L. Sushkevich, D. Palagin, M. Ranocchiari and J. A. van Bokhoven, Science, 2017, 356, 523–527 CrossRef CAS PubMed.
- H. Rao, L. C. Schmidt, J. Bonin and M. Robert, Nature, 2017, 548, 74–77 CrossRef CAS PubMed.
- T. Takata, J. Jiang, Y. Sakata, M. Nakabayashi, N. Shibata, V. Nandal, K. Seki, T. Hisatomi and K. Domen, Nature, 2020, 581, 411–414 CrossRef CAS PubMed.
- X. Meng, X. Cui, N. P. Rajan, L. Yu, D. Deng and X. Bao, Chem, 2019, 5, 2296–2325 CAS.
- Y. Feng, L. Jiao, X. Zhuang, Y. Wang and J. Yao, Adv. Mater., 2025, 37, e2410909 CrossRef PubMed.
- W. Tu, Y. Zhou and Z. Zou, Adv. Mater., 2014, 26, 4607–4626 CrossRef CAS PubMed.
- X. Xiang, F. Pan and Y. Li, Adv. Compos. Hybrid Mater., 2017, 1, 6–31 CrossRef.
- Y. Guo and C. Hu, J. Mol. Catal. A: Chem., 2007, 262, 136–148 CrossRef CAS.
- C. Onneken, T. Morack, J. Soika, O. Sokolova, N. Niemeyer, C. Muck-Lichtenfeld, C. G. Daniliuc, J. Neugebauer and R. Gilmour, Nature, 2023, 621, 753–759 CrossRef CAS PubMed.
- C. B. Ong, L. Y. Ng and A. W. Mohammad, Renewable Sustainable Energy Rev., 2018, 81, 536–551 CrossRef CAS.
- Y. Wang, Q. Wang, X. Zhan, F. Wang, M. Safdar and J. He, Nanoscale, 2013, 5, 8326–8339 RSC.
- Y. Huang, Z. Guo, H. Liu, S. Zhang, P. Wang, J. Lu and Y. Tong, Adv. Funct. Mater., 2019, 29, 1903490 CrossRef CAS.
- G. Fang, W. Yu, X. Wang and J. Lin, Chem. Commun., 2024, 60, 11034–11051 RSC.
- M. Gryszel, R. Rybakiewicz and E. D. Głowacki, Adv. Sustainable Syst., 2019, 3, 1900027 CrossRef.
- Y. Cheng, Y. Zhang, Z. Wang, R. Guo, J. You and H. Zhang, Nanoscale, 2023, 15, 18571–18580 RSC.
- X. Chen, H. Wu, X. Shi and L. Wu, Nanoscale, 2023, 15, 9242–9255 RSC.
- B. Xie, D. Hu, P. Kumar, V. V. Ordomsky, A. Y. Khodakov and R. Amal, Joule, 2024, 8, 312–333 CrossRef CAS.
- Y. Chen, S. Tian, Z. Qin, J. Zhang, Y. Cao, S. Chu, L. Lu and G. Li, Nanoscale, 2019, 11, 22270–22276 RSC.
- R. Wan, P. Ma, M. Han, D. Zhang, C. Zhang, J. Niu and J. Wang, Dalton Trans., 2017, 46, 5398–5405 RSC.
- J. Zhang, Y. Huang, G. Li and Y. Wei, Coord. Chem. Rev., 2019, 378, 395–414 CrossRef CAS.
- R. Li, W. Zhang and K. Zhou, Adv. Mater., 2018, 30, e1705512 CrossRef PubMed.
- T. Hisatomi, J. Kubota and K. Domen, Chem. Soc. Rev., 2014, 43, 7520–7535 RSC.
- J. L. White, M. F. Baruch, J. E. Pander Iii, Y. Hu, I. C. Fortmeyer, J. E. Park, T. Zhang, K. Liao, J. Gu, Y. Yan, T. W. Shaw, E. Abelev and A. B. Bocarsly, Chem. Rev., 2015, 115, 12888–12935 CrossRef CAS PubMed.
- H. Park, H.-i Kim, G.-h Moon and W. Choi, Energy Environ. Sci., 2016, 9, 411–433 RSC.
- N. Li, J. Liu, B. X. Dong and Y. Q. Lan, Angew. Chem., Int. Ed., 2020, 59, 20779–20793 CrossRef CAS PubMed.
- A. Ebrahimi, L. Krivosudský, A. Cherevan and D. Eder, Coord. Chem. Rev., 2024, 508, 215764 CrossRef CAS.
- K. Dashtian, S. Shahsavarifar, M. Usman, Y. Joseph, M. R. Ganjali, Z. Yin and M. Rahimi-Nasrabadi, Coord. Chem. Rev., 2024, 504, 215644 CrossRef CAS.
- A. Solé-Daura, Y. Benseghir, M.-H. Ha-Thi, M. Fontecave, P. Mialane, A. Dolbecq and C. Mellot-Draznieks, ACS Catal., 2022, 12, 9244–9255 CrossRef.
- Y. Bo, C. Gao and Y. Xiong, Nanoscale, 2020, 12, 12196–12209 RSC.
- X. Chang, T. Wang and J. Gong, Energy Environ. Sci., 2016, 9, 2177–2196 RSC.
- S. Bai, J. Jiang, Q. Zhang and Y. Xiong, Chem. Soc. Rev., 2015, 44, 2893–2939 RSC.
- L. Liu, S. Wang, H. Huang, Y. Zhang and T. Ma, Nano Energy, 2020, 75, 104959 CrossRef CAS.
- J. Li, Y. Feng, F. Fu, X. Xin, G. Yang and H. Lv, Chin. Chem. Lett., 2024, 35, 108736 CrossRef CAS.
- M. Chi, Y. Zeng, Z.-L. Lang, H. Li, X. Xin, Y. Dong, F. Fu, G.-Y. Yang and H. Lv, ACS Catal., 2024, 14, 5006–5015 CrossRef CAS.
- L. Jiao, Y. Dong, X. Xin, L. Qin and H. Lv, Appl. Catal., B, 2021, 291, 120091 CrossRef CAS.
- Y. Wang, C. Dong, D. Hu, R. Tokarz-Sobieraj, D. Rutkowska-Zbik, K. Ben Tayeb, A. Addad, V. Butenko, E. de la Torre Miranda, M. N. Tran, P. Simon, O. Safonova, V. Briois, V. V. Ordomsky and A. Y. Khodakov, Appl. Catal., B, 2024, 358, 124400 CrossRef CAS.
- X. Li, L. Yang, Q. Liu, W. Bai, H. Li, M. Wang, Q. Qian, Q. Yang, C. Xiao and Y. Xie, Adv. Mater., 2023, 35, e2304532 CrossRef PubMed.
- S. Omwoma, W. Chen, R. Tsunashima and Y.-F. Song, Coord. Chem. Rev., 2014, 258–259, 58–71 CrossRef CAS.
- A. Dolbecq, E. Dumas, C. R. Mayer and P. Mialane, Chem. Rev., 2010, 110, 6009–6048 CrossRef CAS PubMed.
- K. Kamata, Bull. Chem. Soc. Jpn., 2015, 88, 1017–1028 CrossRef CAS.
- N. Narkhede, S. Singh and A. Patel, Green Chem., 2015, 17, 89–107 RSC.
- L. M. Sanchez, H. J. Thomas, M. J. Climent, G. P. Romanelli and S. Iborra, Catal. Rev.: Sci. Eng., 2016, 58, 497–586 CrossRef CAS.
- S. S. Wang and G. Y. Yang, Chem. Rev., 2015, 115, 4893–4962 CrossRef CAS PubMed.
- K. Kamata and K. Sugahara, Catalysts, 2017, 7, 345 CrossRef.
- R. Dehghani, S. Aber and F. Mahdizadeh, Clean: Soil, Air, Water, 2018, 46, 1800413 Search PubMed.
- G. Paille, M. Gomez-Mingot, C. Roch-Marchal, B. Lassalle-Kaiser, P. Mialane, M. Fontecave, C. Mellot-Draznieks and A. Dolbecq, J. Am. Chem. Soc., 2018, 140, 3613–3618 CrossRef CAS PubMed.
- N. I. Gumerova and A. Rompel, Nat. Rev. Chem., 2018, 2, 0112–0131 CrossRef CAS.
- Z. X. Yang, F. Gong, D. M. Lin and Y. Huo, Coord. Chem. Rev., 2023, 492, 215205 CrossRef CAS.
- L. Gan, G. He, Y. Liu, W. Li and J. Li, Appl. Catal., B, 2024, 349, 123895 CrossRef CAS.
- S. Berbeć, S. Żołądek, P. J. Kulesza and B. Pałys, J. Electroanal. Chem., 2019, 854, 113537 CrossRef.
- Y. Cao, Q. Chen, C. Shen and L. He, Molecules, 2019, 24, 2069 CrossRef CAS PubMed.
- T. Ishizuka, S. Ohkawa, H. Ochiai, M. Hashimoto, K. Ohkubo, H. Kotani, M. Sadakane, S. Fukuzumi and T. Kojima, Green Chem., 2018, 20, 1975–1980 RSC.
- L. X. Cai, S. C. Li, D. N. Yan, L. P. Zhou, F. Guo and Q. F. Sun, J. Am. Chem. Soc., 2018, 140, 4869–4876 CrossRef CAS PubMed.
- A. Patel, N. Narkhede, S. Singh and S. Pathan, Catal. Rev.: Sci. Eng., 2016, 58, 337–370 CrossRef CAS.
- A. Troupis, A. Hiskia and E. Papaconstantinou, Angew. Chem., Int. Ed., 2002, 41, 1911 CrossRef CAS PubMed.
- T. Inoue, A. Fujishima, S. Konishi and K. Honda, Nature, 1979, 277, 637–638 CrossRef CAS.
- M. Corda, S. A. Chernyak, M. Marinova, J.-C. Morin, M. Trentesaux, V. A. Kondratenko, E. V. Kondratenko, V. V. Ordomsky and A. Y. Khodakov, ACS Catal., 2024, 14, 17244–17252 CrossRef CAS.
- Y. Zhou, A. Sadia Traore, D. V. Peron, A. J. Barrios, S. A. Chernyak, M. Corda, O. V. Safonova, A. Iulian Dugulan, O. Ersen, M. Virginie, V. V. Ordomsky and A. Y. Khodakov, J. Energy Chem., 2023, 85, 291–300 CrossRef CAS.
- Y. Y. Lee, H. S. Jung and Y. T. Kang, J. CO2 Util., 2017, 20, 163–177 CrossRef CAS.
- B. J. Soden, W. D. Collins and D. R. Feldman, Science, 2018, 361, 326–327 CrossRef CAS PubMed.
- A. K. Fellenberg, A. Addad, S. A. Chernyak, Y. Zhou, M. Corda, D. O. De Souza, O. V. Safonova, V. Martin-Diaconescu, V. V. Ordomsky, G. Ji and A. Y. Khodakov, Cell Rep. Phys. Sci., 2024, 5, 101926 CrossRef CAS.
- X. Yu, V. V. Ordomsky and A. Y. Khodakov, ChemCatChem, 2020, 12, 740–749 CrossRef CAS.
- C. Dong, Y. Wang, Z. Deng, W. Wang, M. Marinova, K. Ben Tayeb, J. C. Morin, M. Dubois, M. Trentesaux, Y. G. Kolyagin, M. N. Tran, V. Martin-Diaconescu, O. Safonova, J. Zaffran, A. Y. Khodakov and V. V. Ordomsky, Nat. Commun., 2024, 15, 8210 CrossRef CAS PubMed.
- S. Navarro-Jaen, M. Virginie, J. Bonin, M. Robert, R. Wojcieszak and A. Y. Khodakov, Nat. Rev. Chem., 2021, 5, 564–579 CrossRef CAS PubMed.
- S. Bhatt and S. Saha, Prog. Solid State Chem., 2023, 72, 100430 CrossRef CAS.
- C. M. Wolff, P. D. Frischmann, M. Schulze, B. J. Bohn, R. Wein, P. Livadas, M. T. Carlson, F. Jäckel, J. Feldmann, F. Würthner and J. K. Stolarczyk, Nat. Energy, 2018, 3, 862–869 CrossRef CAS.
- J. J. Walsh, A. M. Bond, R. J. Förster and T. E. Keyes, Coord. Chem. Rev., 2016, 306, 217–234 CrossRef CAS.
- S. H. Szczepankiewicz, C. M. Ippolito, B. P. Santora, T. J. Van De Ven, G. A. Ippolito, L. Fronckowiak, F. Wiatrowski, T. Power and M. Kozik, Inorg. Chem., 1998, 37, 4344–4352 CrossRef CAS PubMed.
- J. Lan, Q. Zhu, X. Gu, M. Ma, D. Li, K. Huang, X. Fu, Y. Zhu and Y. Zhang, Sep. Purif. Technol., 2023, 321, 124228 CrossRef CAS.
- S. Y. Xu, W. Shi, J. R. Huang, S. Yao, C. Wang, T. B. Lu and Z. M. Zhang, Angew. Chem., Int. Ed., 2024, 63, e202406223 CrossRef CAS PubMed.
- K. Li, Y. Hong, X. Ma, Y. Zhao, S. Zhang, P. Ma, J. Niu and J. Wang, Inorg. Chem. Front., 2024, 11, 6044–6051 RSC.
- Y. Feng, F. Fu, L. Zeng, M. Zhao, X. Xin, J. Liang, M. Zhou, X. Fang, H. Lv and G. Y. Yang, Angew. Chem., Int. Ed., 2024, 63, e202317341 CrossRef CAS PubMed.
- A. Cai, G. Hu, W. Chen, S. An, B. Qi and Y. F. Song, Small, 2025, 21, e2410343 CrossRef PubMed.
- G. Fang, J. N. Hu, L. C. Tian, J. X. Liang, J. Lin, L. Li, C. Zhu and X. Wang, Angew. Chem., Int. Ed., 2022, 61, e202205077 CrossRef CAS PubMed.
- G. Fang, F. Wei, J. Lin, Y. Zhou, L. Sun, X. Shang, S. Lin and X. Wang, J. Am. Chem. Soc., 2023, 145, 13169–13180 CrossRef CAS PubMed.
- J. Du, Y.-Y. Ma, W.-J. Cui, S.-M. Zhang, Z.-G. Han, R.-H. Li, X.-Q. Han, W. Guan, Y.-H. Wang, Y.-Q. Li, Y. Liu, F.-Y. Yu, K.-Q. Wei, H.-Q. Tan, Z.-H. Kang and Y.-G. Li, Appl. Catal., B, 2022, 318, 121812 CrossRef CAS.
- J. Zhou, W. Chen, C. Sun, L. Han, C. Qin, M. Chen, X. Wang, E. Wang and Z. Su, ACS Appl. Mater. Interfaces, 2017, 9, 11689–11695 CrossRef CAS PubMed.
- J. B. Yang, J. H. Pan, Y. H. Zhu, J. L. Wang, H. Mei and Y. Xu, Inorg. Chem., 2022, 61, 11775–11786 CrossRef CAS PubMed.
- P. Zhang, T. Wang, H. Ma, R. Ma, Z. Xia, Q. Yang, X. Yang, G. Xie and S. Chen, Inorg. Chem., 2023, 62, 20401–20411 CrossRef CAS PubMed.
- P. Yan, X. Li, J. Wang, J. Pan, H. Xu, Y. Zhu, Q. Chen, H. Mei and Y. Xu, Mol. Catal., 2024, 561, 114162 CrossRef CAS.
- C. Streb, Dalton Trans., 2012, 41, 1651–1659 RSC.
- A. M. Khenkin, I. Efremenko, L. Weiner, J. M. Martin and R. Neumann, Chemistry, 2010, 16, 1356–1364 CrossRef CAS PubMed.
- Z.-Y. Du, Y.-Z. Yu, N.-F. Li, Y.-S. Xue, L.-X. Xu, H. Mei and Y. Xu, Sustainable Energy Fuels, 2021, 5, 3876–3883 RSC.
- X. Yu, C. C. Zhao, J. X. Gu, C. Y. Sun, H. Y. Zheng, L. K. Yan, M. Sun, X. L. Wang and Z. M. Su, Inorg. Chem., 2021, 60, 7364–7371 CrossRef CAS PubMed.
- L. Mallick, K. Samanta and B. Chakraborty, Chemistry, 2024, 30, e202400428 CrossRef CAS PubMed.
- X. Yu, S. Moldovan, V. V. Ordomsky and A. Y. Khodakov, Nanoscale Adv., 2019, 1, 4321–4330 RSC.
- G. Li, Y. Gu, R. Ren, S. Li, H. Zhu, D. Xue, X. Kong, Z. Zheng, N. Liu, B. Li and J. Zhang, Nanoscale, 2024, 16, 12550–12558 RSC.
- B. Li, M. Chen, Q. Hu, J. Zhu, X. Yang, Z. Li, C. Hu, Y. Li, P. Ni and Y. Ding, Appl. Catal., B, 2024, 346, 123733 CrossRef CAS.
- T. S. Alomar, B. A. Tayyab, M. Nadeem, N. AlMasoud, A. A. Al-wallan, H. M. Asif and Z. M. El-Bahy, J. Photochem. Photobiol., A, 2025, 461, 116171 CrossRef CAS.
- Y. Benseghir, A. Solé-Daura, P. Mialane, J. Marrot, L. Dalecky, S. Béchu, M. Frégnaux, M. Gomez-Mingot, M. Fontecave, C. Mellot-Draznieks and A. Dolbecq, ACS Catal., 2021, 12, 453–464 CrossRef.
- K. Talbi, F. Penas-Hidalgo, A. L. Robinson, P. Gotico, W. Leibl, P. Mialane, M. Gomez-Mingot, M. Fontecave, A. Solé-Daura, C. Mellot-Draznieks and A. Dolbecq, Appl. Catal., B, 2024, 345, 123681 CrossRef CAS.
- Q. Zhu, Z. Li, T. Zheng, X. Zheng, S. Liu, S. Gao, X. Fu, X. Su, Y. Zhu, Y. Zhang and Y. Wei, Angew. Chem., Int. Ed., 2025, 64, e202413594 CrossRef CAS PubMed.
- Z. Wang, T. Wang, Y. Zhao, Q. Ye and P. He, J. Catal., 2025, 442, 115908 CrossRef CAS.
- M. Zhang, X. Xin, Y. Feng, J. Zhang, H. Lv and G.-Y. Yang, Appl. Catal., B, 2022, 303, 120893 CrossRef CAS.
- M. Ren, T. Liu, Y. Dong, Z. Li, J. Yang, Z. Diao, H. Lv and G.-Y. Yang, Chin. J. Catal., 2024, 61, 312–321 CrossRef CAS.
- Y. V. Geletii, B. Botar, P. Kögerler, D. A. Hillesheim, D. G. Musaev and C. L. Hill, Angew. Chem., Int. Ed., 2008, 47, 3896–3899 CrossRef CAS PubMed.
- D.-H. Li, X.-Y. Zhang, J.-Q. Lv, P.-W. Cai, Y.-Q. Sun, C. Sun and S.-T. Zheng, Angew. Chem., Int. Ed., 2023, 62, e202312706 CrossRef CAS PubMed.
- D. Hu, V. V. Ordomsky and A. Y. Khodakov, Appl. Catal., B, 2021, 286, 119913 CrossRef CAS.
- C. Dong, D. Hu, K. Ben Tayeb, P. Simon, A. Addad, M. Trentesaux, D. O. de Souza, S. Chernyak, D. V. Peron, A. Rebai, J.-F. Guillemoles, X. Wallart, B. Grandidier, A. Y. Khodakov, N. Naghavi and V. V. Ordomsky, Appl. Catal., B, 2023, 325, 122340 CrossRef CAS.
- D. Hu, C. Dong, S. Belhout, S. Shetty, H. Ng, P. Brasseur, L. S. Bezerra, K. Ben Tayeb, P. Simon, A. Addad, M. Virginie, R. Wojcieszak, V. V. Ordomsky, P. H. C. Camargo and A. Y. Khodakov, Mater. Today Energy, 2023, 36, 101358 CrossRef CAS.
- X. Yu, V. De Waele, A. Lofberg, V. Ordomsky and A. Y. Khodakov, Nat. Commun., 2019, 10, 700 CrossRef CAS PubMed.
- X. Yu, V. L. Zholobenko, S. Moldovan, D. Hu, D. Wu, V. V. Ordomsky and A. Y. Khodakov, Nat. Energy, 2020, 5, 511–519 CrossRef CAS.
- C. Dong, M. Marinova, K. B. Tayeb, O. V. Safonova, Y. Zhou, D. Hu, S. Chernyak, M. Corda, J. Zaffran, A. Y. Khodakov and V. V. Ordomsky, J. Am. Chem. Soc., 2023, 145, 1185–1193 CrossRef CAS PubMed.
- C. Xiao, L. Zhang, K. Wang, H. Wang, Y. Zhou and W. Wang, Appl. Catal., B, 2018, 239, 260–267 CrossRef CAS.
- X. H. Li, P. He, T. Wang, X. W. Zhang, W. L. Chen and Y. G. Li, ChemSusChem, 2020, 13, 2769–2778 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5cc01494h |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |