
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotocatalytic C–C bond thio(seleno)esterification of 1,2-diketone-derived pro-aromatic intermediates†
Amit
Pal
,
Sudip
Sarkar‡
,
Aaron
Shibu‡
,
Prakash
Maity
and
Basudev
Sahoo
 *
*
School of Chemistry, Indian Institute of Science Education and Research Thiruvananthapuram, Thiruvananthapuram 695551, Kerala, India. E-mail: basudev@iisertvm.ac.in
First published on 28th February 2025
Abstract
We report an organophotocatalyst-enabled oxidant-free C–S/C–Se bond coupling of (un)symmetrical 1,2-diketones via pro-aromatic dihydroquinazolinones/benzothiazolines, employing readily accessible disulfides/diselenides. In this scalable and redox-neutral method, various dialkyl, di(hetero)aryl, and alkyl-aryl 1,2-diketones are expediently converted to S-aryl (S-alkyl) alkyl/(hetero)aryl thioesters and Se-alkyl aryl selenoesters with broad functional group compatibility in high efficiency.
1,2-Diketones constitute a unique class of carbonyl compounds that are prevalent in various natural products1 and synthetic materials2 as well as easily prepared in the laboratory (Scheme 1a).3 Due to their low-lying LUMO (π* + π*),4 1,2-dicarbonyl compounds serve as versatile synthetic intermediates in organic chemistry, including their use in the synthesis of heteroaromatic compounds and bisimine/bisoxime ligands among others.5 Compared to the traditional reactivity, the exploration of C(C
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)–C(CO) bond cleavage of 1,2-diketones is relatively underdeveloped in advanced organic synthesis.6–8 Notably, the cleavage of the C(CO)–C(α) bond of aliphatic ketones via different pro-aromatic intermediates is rapidly evolving, enabling researchers to devise various innovative methods for C–C and C–X bond formation.9 In this regard, Chen, Walsh, Shang and co-workers described a photoredox-catalyzed C–S/Se coupling of dihydroquinazolinones with thio(seleno)sulfonates, en route to thioethers and selenoethers (Scheme 1b).9d Later, Li et al. reported direct photo-excitation of a ketone-derived dihydropyrimidoquinolinone for C–S/Se coupling to deliver thioether/selenoether (Scheme 1b).9e Recently, 1,2-ketone-derived benzothiazolines7 and dihydroquinazolinones8 have emerged as efficient acyl radical precursors upon aromaticity-driven C(CO)–C(CO) bond cleavage, enabling access to a different set of molecular entities. In this respect, Zhu et al. and Akiyama et al. independently demonstrated the utilization of benzothiazolines in hydroacylation of alkenes or alkynylation of alkynyl hypervalent iodine reagents (Scheme 1b).7 Alternatively, Liao et al. and Martin et al. described dihydroquinazolinones in the C–C cross-coupling of aryl bromides toward ketone synthesis enabled by Ni and photoredox catalysis (Scheme 1b).8 Nonetheless, the full potential of these pro-aromatic intermediates from 1,2-diketones has yet to be realised, such as their application in carbon–heteroatom bond forming transformation.
O)–C(CO) bond cleavage of 1,2-diketones is relatively underdeveloped in advanced organic synthesis.6–8 Notably, the cleavage of the C(CO)–C(α) bond of aliphatic ketones via different pro-aromatic intermediates is rapidly evolving, enabling researchers to devise various innovative methods for C–C and C–X bond formation.9 In this regard, Chen, Walsh, Shang and co-workers described a photoredox-catalyzed C–S/Se coupling of dihydroquinazolinones with thio(seleno)sulfonates, en route to thioethers and selenoethers (Scheme 1b).9d Later, Li et al. reported direct photo-excitation of a ketone-derived dihydropyrimidoquinolinone for C–S/Se coupling to deliver thioether/selenoether (Scheme 1b).9e Recently, 1,2-ketone-derived benzothiazolines7 and dihydroquinazolinones8 have emerged as efficient acyl radical precursors upon aromaticity-driven C(CO)–C(CO) bond cleavage, enabling access to a different set of molecular entities. In this respect, Zhu et al. and Akiyama et al. independently demonstrated the utilization of benzothiazolines in hydroacylation of alkenes or alkynylation of alkynyl hypervalent iodine reagents (Scheme 1b).7 Alternatively, Liao et al. and Martin et al. described dihydroquinazolinones in the C–C cross-coupling of aryl bromides toward ketone synthesis enabled by Ni and photoredox catalysis (Scheme 1b).8 Nonetheless, the full potential of these pro-aromatic intermediates from 1,2-diketones has yet to be realised, such as their application in carbon–heteroatom bond forming transformation.
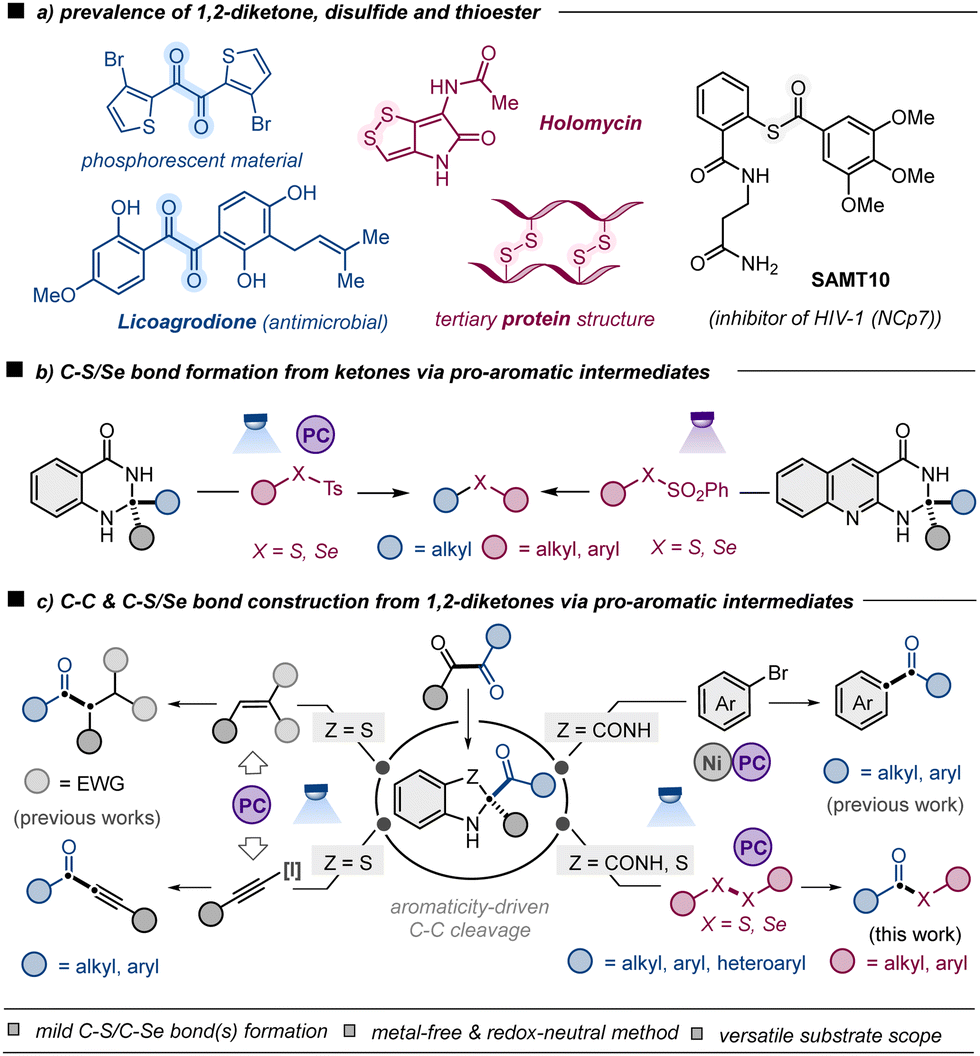 | ||
| Scheme 1 (a) Prevalence of 1,2-diketones, disulfides and thioesters, (b) C–S/Se bond formation from ketones via pro-aromatic intermediates, and (c) construction of C–C and C–S/Se bonds from 1,2-diketones via pro-aromatic intermediates. | ||
The thioester functionality is widely present in natural products, pharmaceutical drugs and polymers.10 Thio(seleno)esters have found extensive applications in native chemical ligation (NCL), biosynthesis of polyketides and non-ribosomal proteins as well as divergent organic synthesis.11 Recently, the exploitation of advanced strategies for synthesizing thio(seleno)esters has seen substantial momentum in organic synthesis.12 However, several of these methods involve: (a) the utilization of transition metal catalysts mediating acyl intermediate formation using toxic carbon monoxide;12a,f (b) CO alone at high pressure in the absence of metal catalyst;12g (c) the use of stoichiometric amount of oxidant;12h,j,k or (d) the utilization of ArSO2SR(f) reagents.12j,k The development of noble-metal-free photocatalytic protocols is in high demand to achieve sustainability and to avoid metal (e.g. iridium) contamination13 with compounds that are directly utilized in biological studies.
While C–C bond formation employing 1,2-diketone-derived pro-aromatic intermediates has recently been reported,7,8 C–X bond formation from the same has not been reported to the best of our knowledge. Intrigued by the emerging reports on thioetherification,9b,c in the current work we envisage developing a mild and efficient C–X (X = S, Se)-bond-forming transformation of 1,2-diketones with readily accessible disulfides and diselenides, en route to thioesters/selenoesters, enabled by organophotoredox catalysis (Scheme 1c). Nonpolar or low-polarity C(CO)–C(CO) and S–S/Se–Se bonds would be cleaved to form more polar C(CO)–S/Se bonds. This thio(seleno)esterification of envisaged method might be expected to preferentially fragment interior C–C skeleton rather than peripheral/terminal C–H, CHO, CO2H and related groups.
The envisaged plan was realized upon an extensive optimization study, employing benzil-derived dihydroquinazolinone (1a) and diethyl 3,3′-disulfanediyldipropionate (2h) as model substrates. Blue light irradiation of DHQ (1a, 1 equiv.) with disulfide (2h, 2.5 equiv.) in the presence of 4CzIPN (1 mol%) in DMF afforded the desired thioester product in 96% yield (Table 1, entry 1). Changing the photocatalyst to Rose Bengal or eosin Y resulted in decreased yields of the thioester formation (Table 1, entries 2 and 3). Several solvents were screened: DMF was found to be superior at facilitating the reaction, whereas DMSO, DME and CH2Cl2 were less effective (Table 1, entries 4–6). Using fewer equivalents of 2h led to diminished yields of 3ah (Table 1, entries 7 and 8). Purple LED was less effective than blue LED in promoting the reaction (Table 1, entry 9). Control experiments showed the indispensable role of photocatalyst and light (Table 1, entries 10 and 11).
|
|
|||
|---|---|---|---|
| Entry | Variation from standard conditions | Yieldb (%) of 3ah | |
| a Reaction conditions: 1a (0.1 mmol), 2h (0.25 mmol), 4CzIPN (1 mol%), DMF (1 mL), rt, 12 h, blue LEDs. b Isolated yield. 4CzIPN = 2,3,4,6-tetrakis(9H-carbazole-9-yl)-isophthalo-nitrile, RB = Rose Bengal, DMF = N,N-dimethylformamide, DMSO = dimethylsulfoxide, DME = 1,2-dimethoxyethane. | |||
| 1 | None | 96 |

|
| 2 | RB, instead of 4CzlPN | 57 | |
| 3 | Eosin Y, instead of 4CzlPN | 81 | |
| 4 | DMSO, instead of DMF | 71 | |
| 5 | 1,2-DME instead of DMF | 54 | |
| 6 | CH2Cl2, Instead of DMF | 59 | |
| 7 | 2h (2.0 equiv.), instead of 2h (2.5 equiv.) | 83 | |
| 8 | 2h (1.5 equiv.), instead of 2h (2.5 equiv.) | 67 | |
| 9 | Purple LEDs instead of Blue LEDs | 83 | |
| 10 | w/o 4CzlPN | 0 | |
| 11 | w/o light | 0 | |

Having optimized conditions established, we set out to assess the versatility of disulfides and diselenides in reacting with dihydroquinazolinone (1a) (Scheme 2). Initially, an array of disulfides, namely 2a–2f including n-octyl, n-butyl, isobutyl, tert-butyl, benzylic and homobenzylic thiols, were tested and provided thioester products 3aa–3af in good to excellent yields (Scheme 2A). Additionally, disulfide 2g and 2h containing a silyl ether and ester functional groups respectively successfully furnished thioester products 3ag and 3ah in a good yield under the optimized protocol. Delightfully, disulfides 2i and 2j featuring menthol ester and phthalimide, respectively, performed efficiently, affording the corresponding thioesters 3ai and 3aj in good yields (Scheme 2A). Furthermore, a spectrum of aryl disulfides (2k–2o), decorated with electron-rich and electron-poor functionalities at the para-position were employed, and furnished the desired thioester products 3ak–3ao in good to excellent yields (Scheme 2A). Moreover, disubstituted aryl and polyaryl disulfides 2p–2r were examined under the optimized conditions to deliver thioesters 3ap–3ar, respectively (Scheme 2A). Prompted by the emerging interest of selenoester in native chemical ligation, we became interested in expanding the scope of chalcogen esters; in our test, alkyl and aryl diselenides (2s–2u) reacted smoothly with dihydro-quinazolinone (1a and 1b) and satisfactorily delivered the corresponding selenoesters 3as–3au and 3bs (Scheme 2B).
 | ||
| Scheme 2 Reaction conditions: (A) 1a (0.2 mmol), 2a–2r (0.5 mmol), 4CzIPN (1 mol%), DMF (2 mL), rt, 12 h, blue LEDs; isolated yields. (B) 1a,1b (0.2 mmol), 2s–2u (0.5 mmol), 4CzIPN (1 mol%), DMF (2 mL), rt, 12 h, blue LEDs; isolated yields. | ||
Afterwards, we tested symmetrical and unsymmetrical 1,2-diketones in the form of dihydroquinazolinones or benzothiazolines (Scheme 3A). Initially, benzil was converted to dihydroquinazolinone 1a and benzothiazoline 1a’, which afforded parent thioester 3ah in good and comparable yields upon reaction with disulfide 2h under standard conditions. Likewise, another dihydroquinazolinone, namely 1b, reacted smoothly with disulfide 2h to provide thioester product 3bh in a good yield. While synthesis of dihydroquinazolinones from diaryl 1,2-diketones was attempted, these DHQs were not obtained in reasonable yield, presumably due to the competitive bisimine formation. We turned our attention to the synthesis of benzothiazolines from 1,2-diketones. Various benzothiazolines, namely 1c′–1h′, prepared from para- and meta-substituted electron-rich and electron-deficient diaryl-1,2-diketones effectively participated in this reaction, delivering desired thioesters 3c′h–3h′h in good to excellent yields and hence showcasing the functional group tolerance. Furthermore, benzothiazolines 1i′–1j′ from heteroaryl-1,2-diketones performed well under the optimized conditions to furnish 3i′h–3j′j in good yields. Dihydroquinazolinone 1k from aliphatic 1,2-diketone was also competent, with the desired thioester 3ko obtained in good yield. Gratifyingly, an unsymmetrical ketone was selectively converted to dihydroquinazolinone 1l in a decent yield, and the benzoyl motif of 1l transformed to furnish thioester 3ah in an excellent yield. To showcase the scalability and operational simplicity of the developed reaction, a one-pot telescopic synthesis of thioester 3ah was performed on a 1-mmol scale without purification of dihydroquinazolinone 1a (Scheme 3B).
 | ||
| Scheme 3 (A) Reaction conditions: 1a–1b, 1k, 1l, 1a′, 1c′–1k′ (0.2 mmol), 2h, 2i, 2o (0.5 mmol), 4CzIPN (1 mol%), DMF (2 mL), rt, 12 h, blue LEDs; isolated yields. AB = 2-aminobenzamide. (B) Scale up one-pot reaction. | ||
To elucidate the mechanistic intricacies here, initially a light OFF–ON experiment was conducted to indicate the need for continuous light irradiation for effective reaction.14 The photoluminescence quenching of 4CzIPN against the potential quenchers 1a, 1a′ and 2h and subsequent Stern–Volmer plot analysis were carried out, and revealed the substantial quenching ability of substrates 1a′ and 1a (Scheme 4A). Radical inhibition tests in the presence of TEMPO (4) radical scavenger or 1,1-diphenylethene (DPE, 6) provided indirect evidence of benzoyl rather than phenyl radical formation, with the corresponding adducts 5 and 7 having been detected (Scheme 4B). Furthermore, the standard reaction in the presence of Sanger's reagent (8) identified an alkyl aryl sulfide (9), providing evidence for the formation of thiolate in this reaction (Scheme 4C).
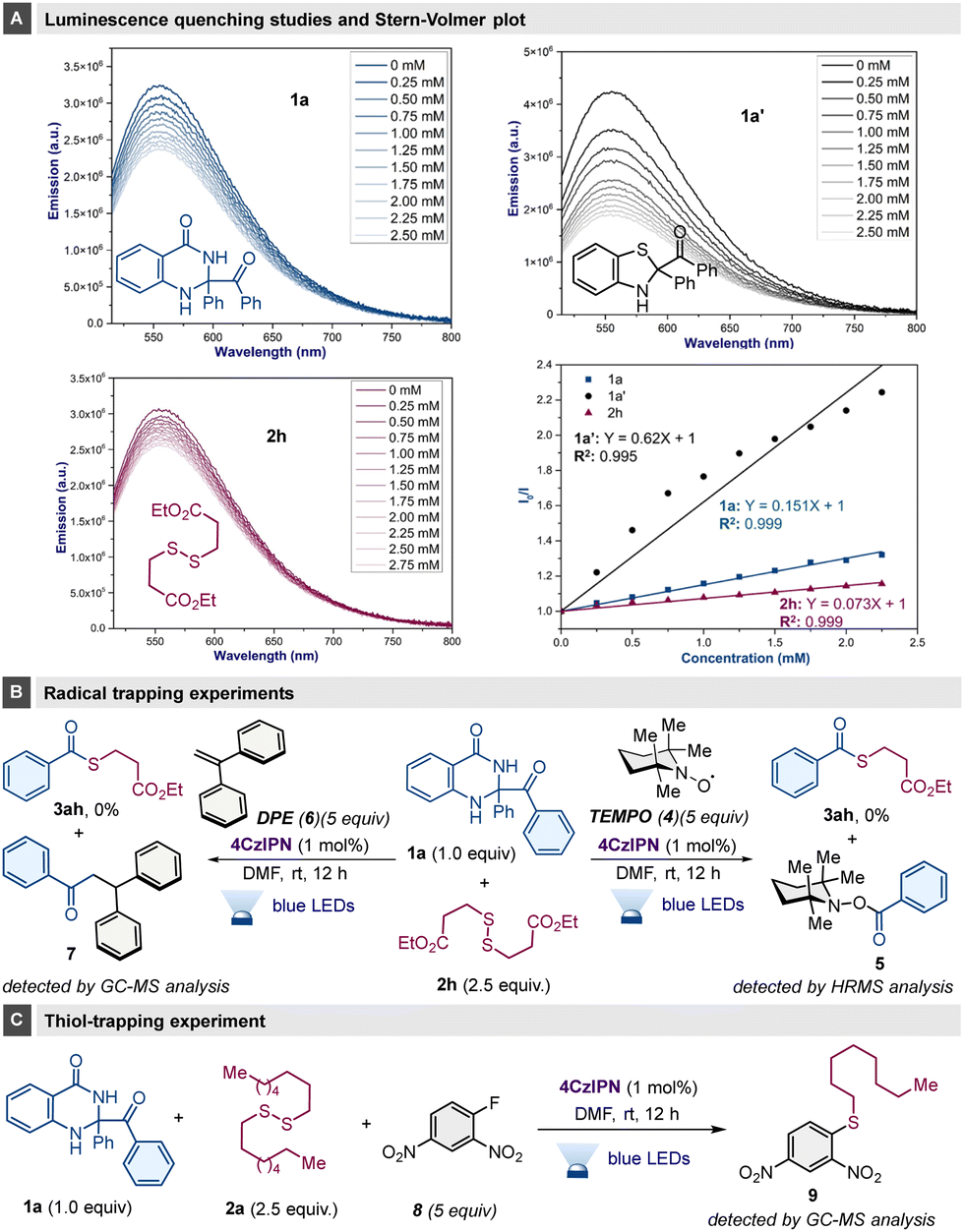 | ||
| Scheme 4 Mechanistic studies: (A) luminescence studies and Stern–Volmer plot; (B) radical trapping experiments; (C) thiol-trapping experiment. | ||
Based on the preliminary mechanistic information and literature reports, we have sketched a mechanistic scenario, shown in Scheme 5. Light irradiation of 4CzIPN leads to the formation of photo-excited 4CzIPN* (E4CzIPN*/4CzIPN˙− = +1.35 V vs. SCE),15 which is reductively quenched by dihydroquinazolinone 1a (E1a˙+/1a = +0.50 V vs. SCE)8a to generate radical cation 1a˙+ alongside the formation of 4CzIPN˙−. Fragmentation of the C–C bond of the radical cation releases benzoyl (Bz˙) radical, which is driven by aromatization.7,8 Next, addition of benzoyl (Bz˙) radical to the S–S bond of disulfide 2h results in the formation of the C–S bond of thioester 3ah and thiyl (RS˙) radical.12h Lastly, the reduction of thiyl (RS˙) (ERS˙/RS− = ∼ + 0.3 V vs. SCE)16 by 4CzIPN˙− (E4CzIPN/4CzIPN˙− = −1.21 V vs. SCE)15 liberates thiolate, regenerating 4CzIPN in the ground state. A similar mechanism for benzothiazoline is outlined in the ESI† (Fig. S12).14
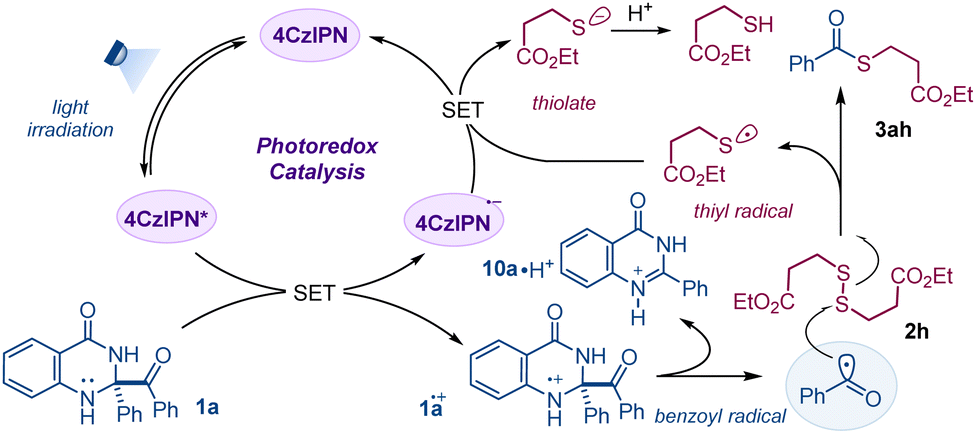 | ||
| Scheme 5 Mechanistic proposal. | ||
In summary, we report a mild and efficient thioester and selenoester synthesis from symmetrical and unsymmetrical 1,2-diketones and readily accessible disulfides/diselenides enabled by organic photoredox catalysis. While 1,2-diketone-derived dihydroquinazolinones and benzothiazolines were previously demonstrated for C–C bond formation, the currently developed reaction is essentially characterized by polar C–S/C–Se bond construction upon successive cleavage of the nonpolar or low-polarity C(CO)–C(CO) bond of 1,2-diketones and nonpolar S–S/Se–Se bond cleavage of disulfides/diselenides. This oxidant-free and scalable C–C activation protocol enables various dialkyl, diaryl, diheteroaryl and alkyl-aryl 1,2-diketones to be transformed to numerous S-aryl (S-alkyl) alkyl/aryl/heteroaryl thioesters as well as Se-aryl (Se-alkyl) aryl selenoesters with diverse functional group tolerance and in high efficiency.
B. S. gratefully acknowledges the SERB, India (File: SRG/2021/000572) for financial support and IISER Thiruvananthapuram for infrastructure support. A. P. thanks the Ministry of Education, India for PMRF and P. M. thanks the IISER Thiruvananthapuram for a junior research fellowship. The authors thank Ms Prabhubandana Patra (IISER Thiruvananthapuram) for assistance with the experiments.
Data availability
The data underlying this work are available in the published article and its ESI.†Conflicts of interest
The authors declare no competing financial interest.Notes and references
- (a) R. Worayuthakarn, S. Boonya-udtayan, E. Arom-oon, P. Ploypradith, S. Ruchirawat and N. Thasana, J. Org. Chem., 2008, 73, 7432 CrossRef CAS PubMed; (b) T. Manome, Y. Hara and M. A. Ishibashi, J. Nat. Med., 2023, 77, 370 CAS; (c) R. M. Wadkins, J. L. Hyatt, X. Wei, K. J. P. Yoon, M. Wierdl, C. C. Edwards, C. L. Morton, J. C. Obenauer, K. Damodaran, P. Beroza, M. K. Danks and P. M. Potter, J. Med. Chem., 2005, 48, 2906 CrossRef CAS PubMed.
- Y. Tani, K. Miyata, E. Ou, Y. Oshima, M. Komura, M. Terasaki, S. Kimura, T. Ehara, K. Kubo, K. Onda and T. Ogawa, Chem. Sci., 2024, 15, 10784 CAS.
- A. Kumar and V. Sridharan, Asian J. Org. Chem., 2021, 10, 1619 CrossRef CAS.
- J. Clayden, N. Greeves, S. Warren and P. Wothers, Organic Chemistry, Oxford University Press, 2001, p. 728 Search PubMed.
- (a) R. J. Wehrle, A. Rosen, T. V. Nguyen, K. Koons, E. Jump, M. Bullard, N. Wehrle, A. Stockfish, P. M. Hare, A. Atesin, T. A. Ateşin and L. Ma, ACS Omega, 2022, 7, 26650 CrossRef CAS PubMed; (b) S. Samanta, D. Roy, S. Khamarui and D. K. Maiti, Chem. Commun., 2014, 50, 2477 RSC.
- (a) K. N. Carter, J. Org. Chem., 1982, 47, 2208 CrossRef CAS; (b) T.-H. Hsieh, P.-Y. Liao, Y. T. Liu, C.-H. Wang, C.-C. Lin and T. C. Chien, J. Chin. Chem. Soc., 2018, 65, 325 CrossRef CAS.
- (a) L. Li, S. Guo, Q. Wang and J. Zhu, Org. Lett., 2019, 21, 5462 CrossRef CAS PubMed; (b) T. Uchikura, K. Moriyama, M. Toda, T. Mouri, I. Ibanez and T. Akiyama, Chem. Commun., 2019, 55, 11171 RSC; (c) T. Uchikura, M. Toda, T. Mouri, T. Fujii, K. Moriyama, I. Ibanez and T. Akiyama, J. Org. Chem., 2020, 85, 12715 Search PubMed.
- (a) S.-C. Lee, L.-Y. Li, Z.-N. Tsai, Y.-H. Lee, Y.-T. Tsao, P. G. Huang, C. K. Cheng, H.-B. Lin, T.-W. Chen, C.-H. Yang, C.-C. Chiu and H.-H. Liao, Org. Lett., 2022, 24, 85 Search PubMed; (b) X. Y. Lv, R. Abrams and R. Martin, Nat. Commun., 2022, 13, 2394 CrossRef CAS PubMed.
- Review: (a) S. Miñoza, I. L. Librando and H. H. Liao, Synlett, 2024, 1072 Search PubMed Selected reports; (b) X. Zhou, D. Pyle, Z. Zhang and G. Dong, Angew. Chem., Int. Ed., 2023, 62, e202213691 CrossRef CAS PubMed; (c) H.-J. Miao, J.-H. Zhang, W. Li, W. Yang, H. Xin, P. Gao, X.-H. Duan and L.-N. Guo, Chem. Sci., 2024, 15, 8993 RSC; (d) H. Wu, S. Chen, C. Liu, Q. Zhao, Z. Wang, Q. Jin, S. Sun, J. Guo, X. He, P. J. Walsh and Y. Shang, Angew. Chem., Int. Ed., 2024, 63, e202314790 CrossRef CAS PubMed; (e) J. Li, D. Zhang, L. Tan and C. J. Li, Angew. Chem., Int. Ed., 2024, 63, e202410363 CAS; (f) Q.-Z. Li, M.-H. He, R. Zeng, Y.-Y. Lei, Z.-Y. Yu, M. Jiang, X. Zhang and J.-L. Li, J. Am. Chem. Soc., 2024, 146, 22829 CrossRef CAS PubMed; (g) P. P. Mondal, S. Das, S. Venugopalan, M. Krishnan and B. Sahoo, Org. Lett., 2023, 25, 1441 CrossRef CAS PubMed; (h) X. Y. Lv, R. Abrams and R. Martin, Angew. Chem., Int. Ed., 2023, 62, e202217386 CrossRef CAS; (i) P. P. Mondal, A. Pal, A. K. Prakash and B. Sahoo, Chem. Commun., 2022, 58, 13202 RSC; (j) L. Li, L. Fang, W. Wu and J. Zhu, Org. Lett., 2020, 22, 5401 CrossRef CAS PubMed.
- (a) F. H. Al-Awadhi, L. A. Salvador-Reyes, L. A. Elsadek, R. Ratnayake, Q. Y. Chen and H. Luesch, ACS Chem. Neurosci., 2020, 11, 1937 CrossRef CAS PubMed; (b) Y. Yang, J. Zhu, M. Hassink, L. M. M. Jenkins, Y. Wan, D. H. Appella, J. Xu, E. Appella and X. Zhang, Emerging Microbes Infect., 2017, 6, e40 Search PubMed; (c) S. Aksakal, R. Aksakal and C. R. Becer, Polym. Chem., 2018, 9, 4507 RSC.
- (a) V. Agouridas, O. E. Mahdi, V. Diemer, M. Cargoët, J.-C. M. Monbaliu and O. Melnyk, Chem. Rev., 2019, 119, 7328–7443 CrossRef CAS PubMed; (b) J. Staunton and K. J. Weissman, Nat. Prod. Rep., 2001, 18, 380 RSC; (c) R. D. Süssmuth and A. Mainz, Angew. Chem., Int. Ed., 2017, 56, 3770 CrossRef PubMed; (d) V. Hirschbeck, P. H. Gehrtz and I. Fleischer, Chem. Eur. J., 2018, 24, 7092–7107 CAS.
- Reviews: (a) A. Pal, P. P. Mondal, F. Niloofar and B. Sahoo, Eur. J. Org. Chem., 2022, e202201159 CrossRef CAS; (b) X. Wang and Z.-B. Dong, Eur. J. Org. Chem., 2022, e202200452 CrossRef CAS . Selected reports: ; (c) S. Wu and P. Melchiorre, Angew. Chem., Int. Ed., 2024, 63, e202407520 CAS; (d) H. Wang, Z. Liu, A. Das, P. Bellotti, S. Megow, F. Temps, X. Qi and F. Glorius, Nat. Synth., 2023, 2, 1116–1126 CAS; (e) J. Su, A. Chen, G. Zhang, Z. Jiang and J. Zhao, Org. Lett., 2023, 25, 8033–8037 CrossRef CAS PubMed; (f) D. Y. de Albuquerque, W. K. O. Teixeira, M. do Sacramento, D. Alves, C. Santi and R. S. Schwab, J. Org. Chem., 2022, 87, 595–605 CrossRef PubMed; (g) B. Chen and X.-F. Wu, Org. Biomol. Chem., 2021, 19, 9654–9658 RSC; (h) J. Roy, P. P. Sen and S. R. Roy, J. Org. Chem., 2021, 86, 16965–16976 CrossRef PubMed; (i) S. Murakami, T. Nanjo and Y. Takemoto, Org. Lett., 2021, 23, 7650–7655 CrossRef CAS PubMed; (j) S.-H. Guo, X.-L. Zhang, G.-F. Pan, X.-Q. Zhu, Y.-R. Gao and Y.-Q. Wang, Angew. Chem., Int. Ed., 2018, 57, 1663–1667 CrossRef CAS PubMed; (k) B. Xu, D. Li, L. Lu, D. Wang, Y. Hu and Q. Shen, Org. Chem. Front., 2018, 5, 2163–2166 RSC.
- (a) L. Krasnov, S. Tatarin, D. Smirnov and S. Bezzubov, Sci. Data, 2024, 11, 870 CrossRef CAS PubMed; (b) S. F. He, W. C. Han, Y. Y. Shao, H. B. Zhang, W. X. Hong, Q.-H. Yang, Y.-Q. Zhang, R.-R. He and J. Sun, Bioorg. Chem., 2023, 141, 106867 CrossRef CAS PubMed.
- See ESI†.
- T.-Y. Shang, L.-H. Lu, Z. Cao, Y. Liu, W.-M. He and B. Yu, Chem. Commun., 2019, 55, 5408 RSC.
- E. L. Tyson, Z. L. Niemeyer and T. P. Yoon, J. Org. Chem., 2014, 79, 1427 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cc06735e |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |