
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Structure–activity relationships of DNA nanocarriers based on the amphipathic cell penetrating peptide transportan 10†
Lucas Rodrigues
de Mello
 a,
Tâmisa Seeko Bandeira
Honda
a,
Sang Won
Han
a,
Valeria
Castelletto
b,
Ian William
Hamley
b,
Ly
Porosk
c,
Ülo
Langel
cd and
Emerson Rodrigo
da Silva
*a
a,
Tâmisa Seeko Bandeira
Honda
a,
Sang Won
Han
a,
Valeria
Castelletto
b,
Ian William
Hamley
b,
Ly
Porosk
c,
Ülo
Langel
cd and
Emerson Rodrigo
da Silva
*a
aDepartamento de Biofísica, Universidade Federal de São Paulo, São Paulo 04062-000, Brazil. E-mail: er.silva@unifesp.br
bDepartment of Chemistry, University of Reading, Reading RG6 6AD, UK
cInstitute of Technology, University of Tartu, Tartu 50411, Estonia
dDepartment of Biochemistry and Biophysics, Stockholm University, Stockholm SE-10691, Sweden
First published on 6th August 2024
Abstract
Cell penetrating peptides (CPPs) have emerged as promising materials for the fabrication of synthetic nanovectors endowed with potential for improving the future landscape of gene therapy. A group of well-studied CPPs includes the transportan family, comprised of chimeric molecules combining segments derived from the antimicrobial wasp-venom mastoporan and the neuropeptide galanin. The success of these CPPs is supported by their effective use as the base for commercial peptide-based transfection reagents. Herein, we present a comprehensive study of the structure of peptiplexes formed between DNA fragments and transportan 10, a prototype example of amphipathic CPP. We conducted a thorough analysis of the self-aggregation of TP10, its secondary structure, and revealed details of its interaction with DNA. We employed atomic force microscopy-based nanospectroscopy to obtain single-particle data that revealed details of the conformations assumed by the peptide and DNA in the inner structure of nanoassemblies with different morphologies. Our structural results showed that TP10 exhibits self-aggregation capabilities and a strong propensity to assume α-helical conformations upon association with DNA strands. This behavior contrasts with that of prototype CPPs such as TAT-HIV and penetratin, potentially explaining why peptiplexes based on transportans demonstrate increased uptake compared to their cationic counterparts. Also, single-particle spectroscopy indicated that the secondary structure in peptiplexes is strongly dependent on the size and shape, reinforcing that controlled self-assembly is crucial for optimizing CPP-based nanotherapeutics. The peptiplexes were also evaluated for cell uptake efficiency and kinetics, revealing a logistic time–response increase in permeability, suggestive of cooperativeness. We anticipate that the findings presented here might contribute to refining structure–activity relationships of peptiplexes based on amphipathic CPPs, assisting the optimization of products based on this relevant class of CPPs with potential applications in therapeutic delivery systems.
Introduction
The fabrication of synthetic vectors based on nanoscopic carriers capable of vectorizing exogenous nucleic acids into cells is a cornerstone in the future development of gene therapy.1–3 These nanocarriers must not only fulfill the role of protecting the cargo against degradation by nucleases but also need to circumvent the innate defense and facilitate translocation through biological membranes.4 Materials commonly employed in the production of synthetic nanocarriers include liposomes,5,6 cubosomes,7 polymers,6,8 and more recently, cell-penetrating peptides (CPPs).9,10 Synthetic vehicles offer several advantages over viral vectors, including lower genotoxicity and cost, ease of synthesis, and scalability. In the case of CPPs, a notable benefit is their inherent capability to translocate biomembranes and to be tailored with specific target sequences, thereby enhancing the specificity of the resulting vectors. In addition, when the peptide sequences have amphiphilic features, they allow for controlling the self-assembly into supramolecular arrangements. In addition, these peptides assist in the formulation of peptiplexes, a term coined to designate complexes formed from the non-covalent association between nucleic acids and peptides.A group of well-studied CPPs includes the transportan family.11,12 This family comprises chimeric molecules combining segments of the amino terminus of the neuropeptide galanin covalently linked through a lysine residue to the C-terminal tail of mastoporan, an antimicrobial peptide found in wasp-venom (ESI, Fig. S1†).12 In recent years, transportans have been successfully used to improve the pharmacokinetics of antibiotics,13 modulate the elastic properties of lipid vesicles,14,15 perturb cancer cells,16,17 and even exhibit antimicrobial activity against parasites.18 The success of transportans demonstrated over the last decades has even led to the development of commercial reagents and several PepFect and NickFect CPPs.19–21 Transportan 10 (TP10), a processor of this family, corresponds to a shortened version of the original transportan and has become a prototype example of amphipathic CPP.11 The newer generations of PepFects and NickFects rely on the conjugation of fatty acids to amino acid sequences,22 which strongly affects both complexation and delivery efficacy; however, the peptide component is the leading part in the penetration process, thus justifying further investigations on the structure–activity relationship of this component. TP10 has the amino acid sequence AGYLLGKINLKALAALAKKIL-NH2 and thus simultaneously exhibits hydrophobic residues intercalated with lysine sites. This composition imparts amphipathic behavior to TP10, inspiring us to explore its potential self-assembly in the current study. Furthermore, the cationic charge conveyed by the presence of lysine residues makes TP10 a suitable candidate for association with nucleic acids via electrostatic attraction. The mechanisms of uptake used by transportans are a topic of ongoing research, and no consensus has emerged yet in the literature.11 Previous reports indicate that they enter cells through either endocytosis or membrane disturbance, with their prevalence depending on the cargo size. When transportans are conjugated to small nucleic acid sequences, the prevalent mechanism is membrane perturbation, likely leading to pore formation and internalization.23 In contrast, when long plasmid sequences are used, endocytosis has been found to play a major role in uptake.24
While TP10 has been extensively investigated in basic research studies and serves as the basis for commercial CPP-based products, structural analyses focusing on its self-assembly properties are still lacking in the literature. Furthermore, detailed knowledge on the structure of non-covalent complexes with nucleic acids is lacking. Herein, we aimed to provide a comprehensive characterization of both the structure of TP10/DNA peptiplexes and their interaction with cells. Notably, we analysed the self-aggregation of TP10, its secondary structure, and detailed how its interaction with DNA impacts the conformation of biomacromolecules in the inner structure of the resulting peptiplexes. We employed AFM-based infrared spectroscopy to obtain single-particle data that revealed details of the conformation assumed by the peptide and DNA in nanoassemblies with different morphologies. Furthermore, the peptiplexes were evaluated for their cytotoxicity and transfection efficiency. The internalization kinetics of peptiplexes was also evaluated, revealing a cooperative behavior underlying cell uptake. We anticipate that our findings will contribute to refining structure–activity relationships of peptiplexes based on amphipathic CPPs, facilitating the optimization of products based on this relevant class used in commercial formulations.
Materials and methods
Reagents and sample preparation
The transportan 10 peptide (AGYLLGKINLKALAALAKKIL-NH2) was custom synthesized by AminoTech (São Paulo, Brazil) using standard solid-phase techniques and the Fmoc strategy. It was delivered as a TFA salt, with a purity of 95.6% and a molecular weight (Mw) of 2180.4 Da (ESI, Fig. S2†). Linear calf thymus DNA was purchased from Sigma-Aldrich (product code D1501) and subjected to ultrasonication in a Diagenode bioruptor to break it into fragments of approximately 200 bp or Mw ≈ 132![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Da (ESI, Fig. S3B†). The resulting fragments were relatively stiff, as the length of a 200 bp DNA fragment corresponds to approximately 65 nm, which is close to the persistence length of DNA chains.25 The peptide and DNA stock solutions were prepared by weighing the powder or fibers in Eppendorf tubes and suspending them in ultrapure water to the desired concentration. The DNA stock solutions were left to rest in a refrigerator (4 °C) for 2–3 days with several vigorous vortexing cycles to homogenize the mixture. The complexes were formed by mixing predetermined volumes of stock solutions to attain the desired molar charge ratios. Under the conditions applied (near-neutral pH), TP10 was assigned a charge of +5 (corresponding to its lysine residues and the N-terminus), while DNA was assumed to bear one negative charge per phosphate group in the nucleotides (N+:P− ratio). The molecular mass of nucleotides used in the calculations was Mw = 660 Da per base pair.
000 Da (ESI, Fig. S3B†). The resulting fragments were relatively stiff, as the length of a 200 bp DNA fragment corresponds to approximately 65 nm, which is close to the persistence length of DNA chains.25 The peptide and DNA stock solutions were prepared by weighing the powder or fibers in Eppendorf tubes and suspending them in ultrapure water to the desired concentration. The DNA stock solutions were left to rest in a refrigerator (4 °C) for 2–3 days with several vigorous vortexing cycles to homogenize the mixture. The complexes were formed by mixing predetermined volumes of stock solutions to attain the desired molar charge ratios. Under the conditions applied (near-neutral pH), TP10 was assigned a charge of +5 (corresponding to its lysine residues and the N-terminus), while DNA was assumed to bear one negative charge per phosphate group in the nucleotides (N+:P− ratio). The molecular mass of nucleotides used in the calculations was Mw = 660 Da per base pair.
Steady-state fluorimetry
The aggregation of TP10 and peptiplexes formed between TP10 and DNA in ultrapure water was monitored using fluorescence assays. In this case, solutions containing different peptide concentrations were co-solubilized with 5 μM pyrene or 70 μM 1-anilino-8-naphthalene sulfonate (ANS). The fluorescence behavior of TP10 in the presence of these probes was tracked on a Hitachi F2500 fluorescence spectrophotometer, using an excitation wavelength (λexc.) of 338 nm for pyrene and 356 nm for ANS. Spectra were acquired at a rate of 300 nm min−1 from samples in quartz cuvettes with a 1 cm pathlength while under magnetic stirring. All experiments were performed at room temperature, using both excitation and emission slits of 5 nm.Circular dichroism (CD)
CD assays were carried out using a JASCO-810 spectropolarimeter, which was employed to obtain spectra in the far UV-range, spanning from 190 nm to 250 nm. Peptide solutions were prepared at appropriate concentrations and loaded into quartz cuvettes with a 1 mm pathlength, and the measurements were performed at room temperature. A total of 5 accumulations were recorded at 100 nm min−1, with steps of 0.5 nm. The absorbance channel was also monitored, and only data corresponding to A < 2 were selected for further analysis. To eliminate noise, the spectra were processed using FFT filters with a 7-point window. Background subtraction was performed by collecting spectra of the buffer under identical conditions to those used for the peptide solutions.Small-angle X-ray scattering
SAXS measurements were performed on the SAXS-1 beamline at LNLS (Campinas, Brazil). Samples containing about 300 μl of peptide solutions at various concentrations were placed between mica windows inside a 1 mm path length cell holder. For samples containing nucleic acids, the concentration of the DNA base pair was kept at 0.2 mM and the corresponding amount of TP10 was added to match the desired peptide-to-DNA molar ratios. The beam energy was set at 8.05 keV (1.54 Å), and the scattering was recorded using a Dectris Pilatus 300 K CCD detector. The setup was calibrated using silver behenate standards to obtain a q-range of 0.13 < q < 3.5 nm−1. Ten frames, each 30 seconds long, were collected and compared to each other. In the absence of radiation damage, they were averaged and background subtracted. The full range data fit was performed using the SASFit program library with the model equations detailed in the ESI.†Atomic force microscopy combined with infrared spectroscopy (AFM-IR)
Samples for AFM imaging were prepared by depositing small volumes of peptiplex solution (5:1 TP10/DNA) onto silicon substrates covered with a 100 nm Au layer. The substrates were left to rest for a few minutes inside a Petri dish, and then the excess of water was removed with filter paper. The samples were then left to dry overnight in a desiccator. The data were collected using an Anasys NanoIR2-s microscope. The instrument was used in contact mode, with samples illuminated by a tunable laser in the 950–1750 cm−1 range. This arrangement permitted the measurement of IR absorption profiles as a function of wavenumber at spatial resolutions determined by the tip radius (∼50 nm). All data were baseline subtracted (Au profile) and smoothed using a FFT filter (5 points of window). We also collected absorption maps from the samples. To this end, the infrared laser was tuned at specific wavenumbers (1260, 1670, or 1720 cm−1) during tip scanning across the surface. The spatial resolution was limited by the tip radius (50 nm), and the wavenumber resolution was set at 2 cm−1. Data treatment (smoothing of IR spectra and image enhancement) was performed using the AnalysisStudio software provided by Anasys while image enhancement was carried out using the Gwyddion software.
Cryo-electron transmission microscopy (cryo-TEM)
Cryo-TEM images were obtained using a JEOL 2100 FEG-TEM microscope equipped with a cryogenic module at LNNano in Campinas, Brazil. Samples were prepared by depositing about 10 μl of an aqueous solution containing peptiplexes at 5:1 TP10/DNA on copper grids. The peptide concentration was 0.4 mM. The grids were then vitrified with liquid ethane in a Vitrobot device. The microscope was operated at an acceleration voltage of 200 keV, and the data treatment was carried out using ImageJ software.
Cell culture and fluorescence imaging on confocal microscopy
For the cell assays, HeLa cells were cultured in Dulbecco's modified Eagle's medium (DMEM) with 10% fetal bovine serum and 2 mM glutamine (ThermoFisher) and maintained at 37 °C under an atmosphere of 5% CO2. The source of HeLa cells was the Rio de Janeiro Cell Bank (BCRJ code #0100, ATCC® CCL-2). For assays using paraformaldehyde (PFA)-preserved cells, we used 24-well plates with glass coverslips placed at the bottom, and 5 × 104 HeLa cells were seeded and incubated for 24 h to adhere to the coverslips. After incubation, the cells were washed three times with PBS to remove serum and cell debris. Peptide stock solutions were prepared below the critical aggregation concentration (CAC) in ultrapure water. Complexes were prepared by mixing 5 μg of fragmented DNA and the respective amount of TP10 to obtain the final molar ratios in 50 μl of ultrapure water, a methodology already applied in previous works. These peptiplexes were incubated at 37 °C for 30 minutes, and then transferred to wells containing 950 μl of DMEM without serum to evaluate the internalization of labeled DNA. We also prepared positive and negative controls consisting of DNA + Lipofectamine 2000™ and DNA alone, respectively. The controls were incubated in the same manner as the peptiplexes. After this, the samples were washed again three times with PBS, and fixation was performed using PBS with 4% PFA. Nuclear staining was performed using DAPI (Invitrogen, Carlsbad, CA, USA) in PBS for five minutes at room temperature. The coverslips were washed three times with PBS after each step. Labelling with Lysotracker deep red™ (Invitrogen, California) was achieved by adding 0.75 μM of the dye to the culture medium and incubating HeLa cells in this medium at 37 °C for 1.5 h prior to fixation with 4% PFA. For phalloidin-Texas red (Invitrogen, California) labeling, cells were permeabilized with Triton-X 0.1% for 15 min, washed twice with PBS, and the coverslips were incubated in phalloidin-Texas red at room temperature for 1 h, protected from light. After this period, the samples were washed again three times with PBS and analyzed using confocal microscopy (Leica TCS SP8, Mannheim, Germany).Flow cytometry
The percentage of internalized cells was determined using flow cytometry. The assays were carried out using the fixable viability dye eFluor780 (eBioscience, California, USA) to distinguish live and dead cells, and YOYO-1 was used for labeling DNA to formulate the peptiplexes. HeLa cells, at 5 × 104 cells per well, were cultivated in a 24-well plate and incubated at 37 °C with 5% CO2 overnight. On the day of the experiment, these cells were incubated for 4 h with the same concentrations and methodology used in microscopy assays: DNA without any carriers, DNA + Lipofectamine 2000, and the peptiplexes. The mass of DNA was kept at 5 μg throughout the experiments. After incubation, the cells were washed with PBS, detached with trypsin 0.25% + EDTA (Gibco, Grand Island, NY, USA), inactivated with culture media containing serum, centrifuged, and resuspended in 100 μL of PBS. The resulting cell suspension was incubated with the fixable viability dye eFluor780 according to the recommendations of the manufacturer. Flow cytometry was performed using a BD LSRFortessa™ (model 4.2.4.2, BD Biosciences, New Jersey, USA). To evaluate the percentage of YOYO-1 internalization, the cells were analyzed based on forward and side scatter parameters, a method used to determine cell population and debris exclusion. From the initial population, the live cells were identified and used to evaluate the percentage of YOYO-1 positive events. To ensure cut-off reliability and statistical accuracy, we excluded samples that did not provide at least 2000 events in the last gate. The use of peptiplexes above the 5:1 molar charge ratio resulted in significant clustering and cell size alteration. This hindered data collection and further analysis. All experiments were conducted in duplicate on different days.
Uptake kinetics of TP10/DNA peptiplexes
To monitor cellular uptake, live HeLa cells were imaged. Initially, 3 × 105 HeLa cells were added onto a 25 mm coverslip in a 6-well plate with DMEM and 10% serum and incubated for 24 h. Post-incubation, the cells were washed three times with PBS, stained with Hoechst 33342 for 10 min, washed again three times with PBS, and placed in an Attofluor™ coverslip holder (Invitrogen, Carlsbad, CA, USA) for live cell imaging. Hank’ balanced salt solution, simulating the extracellular fluid, was used to maintain the cells during imaging. The coverslip holder was placed into a chamber with controlled temperature (37 °C) and CO2 (5%), loaded with 5:1 molar ratio TP10/DNA (YOYO-1 labelled) peptiplexes, and data collection was immediately started. Images were registered every 60 s with a Zeiss LCM 780 microscope.
Results and discussion
Self-assembly and the secondary structure
We first performed a structural characterization of TP10 and its peptiplexes with DNA. Since TP10 has amphiphilic features, alternating domains enriched with hydrophobic residues of lysine along the amino acid strand (see ESI, Fig. S1†), we examined the self-aggregation behaviour by determining the critical aggregation concentration (CAC) above which self-assemblies are formed in solution. These assays were performed by monitoring the fluorescence of extrinsic probes in preparations containing various TP10 concentrations. Pyrene is a highly hydrophobic probe, whose fluorescence is modulated by changes in the polarity of its surrounding microenvironment.26 From the ratio between vibronic bands appearing at 372 and 384 nm, I1/I3, it is possible to assess the formation of aggregates. Fig. 1A shows a series of fluorescence spectra from TP10 samples co-solubilized with 5 μM pyrene. The analysis of the I1/I3 ratio as a function of peptide concentration revealed two emission regimes: at lower TP10 concentrations, the ratio remained around 0.9 or above, while at higher concentrations, an inflexion point was observed, and the ratio decreased to about 0.8. The outline of the data was fitted according to a sigmoidal function and linear regressions made across the near-constant fluorescence plateau, and the decline phase indicated a crossover point at ∼0.9 mM (∼2 mg mL−1), which is interpreted as the onset of the transition between fluorescence regimes. To complement the measurements above, we also conducted fluorescence assays using ANS as an extrinsic probe. Like pyrene, ANS fluorescence is highly sensitive to polarity; however, in this case, emission is affected not only in terms of intensity but also in terms of wavelength.27,28 Therefore, by examining the emission maxima in the ANS spectra, an independent mechanism for evaluating peptide aggregation can be obtained, thus improving the robustness of CAC determination. In Fig. 1B, photographs of ANS solutions containing different amounts of TP10 clearly exhibit different colours under UV light. At low peptide concentrations, the emission maximum remains at around 505 nm and the solution exhibits a yellowish glow. In contrast, when higher amounts of TP10 are present in the mixture, a hypsochromic effect occurs and the fluorescence maximum shifts to about 475 nm, resulting in a vivid turquoise glow. This colour change is indicative of hydrophobic clusters in the sample. The plot of shifted wavelengths as a function of peptide concentration revealed a smoother changeover occurring approximately at 0.4 mM, which is within the same order of magnitude of the CAC value identified in pyrene assays. Therefore, the two methods described above provide independent measurements for critical concentrations, thus strengthening the robustness and reliability. By averaging the critical values obtained from these independent assays, it is reasonable to estimate a CAC of 0.65 ± 0.25 mM for TP10.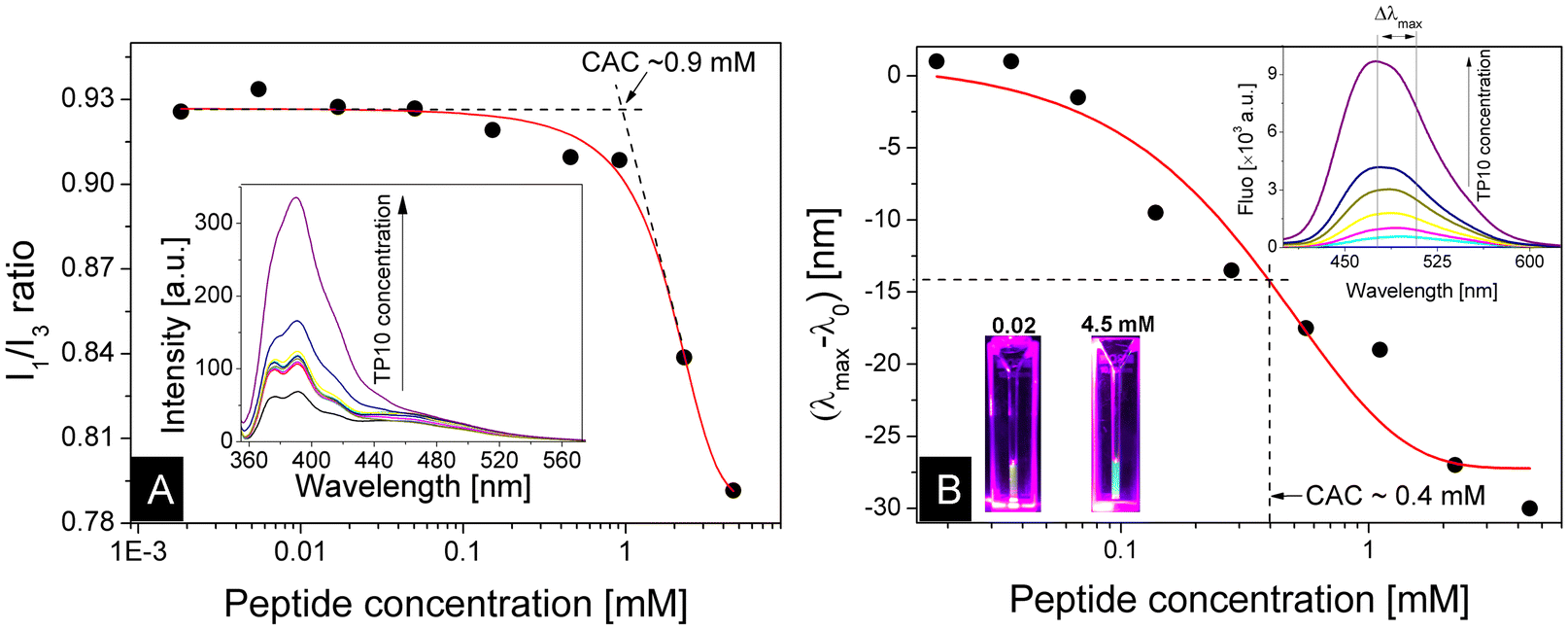 | ||
| Fig. 1 Steady state fluorimetry measurements for assessing critical aggregation concentrations. (A) Ratio between pyrene vibronic bands (I1 and I3) analysed as a function of TP10 concentration. The crossover of linear regressions performed across distinct emission regimes indicates a CAC. (B) ANS assays displaying shifts in emission maxima as a function of peptide concentration. The solid line is a sigmoidal fit whose inflexion point is interpreted as a CAC estimation. Insets show fluorescence spectra and photographs from solutions with different TP10 concentrations under UV light. | ||
In the subsequent stage of our analysis, we performed circular dichroism (CD) experiments to examine the secondary structure assumed by TP10 either below or above the CAC. As depicted in Fig. 2A, CD data indicate that the conformation of the peptide chains in aqueous solution is dependent on the concentration. At concentrations below the CAC, i.e., 0.2 mM, the spectrum exhibits a negative signal at 203 nm, indicative of random coiled conformations.29 At concentrations near the CAC, i.e., 0.4 mM, a redshift is observed, and the rotation minimum is displaced to ∼207 nm, but the overall shape of the spectrum retains its disordered signature. In contrast, at concentrations well-above the CAC, i.e., 2.2 mM, the CD spectrum exhibits a distinctive pattern, characterized by a positive peak at 193 nm, a prominent minimum at 210 nm, and a secondary minimum at 221 nm. This signature is indicative of α-helices,29,30 suggesting that aggregation has a strong impact on the secondary structure of TP10 and that the hydrophobic environment created within the aggregates stimulates folding of the peptide into ordered structures. The conformational transition of TP10 into α-helices has been observed upon interaction with the microenvironment of biomimetic membranes,24,31 thereby underscoring the significance of hydrophobicity in guiding and stabilizing helical structures. In addition, the substantial increase of ellipticity amplitude demonstrates the growth of chirality, consistent with the confinement of peptide chains in ordered environments. To further experimentally validate the propensity of TP10 to form α-helices, we also examined its secondary structure in the presence of trifluoroethanol (TFE), an alcohol known to induce conformational changes in peptides by prompting structuration into helices.32,33 In Fig. 2B, a series of CD spectra from 0.2 mM TP10 solutions prepared with TFE fractions varying from 0 to 50% by volume are shown. It is clearly observed that the CD signature transitions from random coiled conformations at low TFE contents to α-helices at high TFE contents, with spectra showing the growth of a positive band at 195 nm and minima at 210 and 220 nm. Moreover, the increase in the amplitude of CD signals indicates a strengthening of chirality, further supporting structuration. The α-helix signature becomes even more evident when analysing the induced CD, obtained by subtracting the reference spectrum of the sample prepared in water from the data of samples containing TFE (ESI, Fig. S5A†). The analysis of amplitude differences between the distinctive α-helix signals at 195 nm and 220 nm as a function of %TFE reveals a characteristic pattern of cooperative ligand–protein interactions (inset in Fig. 2B).34 This result reinforces the notion that TFE actively participates in the peptide environment, favouring H-bond formation and transformation into α-helices.32 Therefore, we have further experimental evidence that the formation of α-helices is favoured when TP10 strands are in a hydrophobic environment. The above experimental findings are consistent with the supplementary assays conducted using bioinformatics tools dedicated to predicting the three-dimensional structure of proteins from the primary amino acid sequence.35,36 Results presented in Fig. S4 (ESI†) demonstrate that the C-terminal portion comprising TP10 from the wasp-venom mastoporan consistently conforms to α-helices, while the N-terminal region derived from the neuropeptide galanin also shows a probability of adopting random conformations (see configurations II and IV in Fig. S4A†). These findings also align with previous structural information derived from NMR data.31 The helix wheel projection, Fig. S4B,† indicates the formation of a hydrophobic face opposing a lysine cluster, thereby imparting amphipathicity to the α-helical conformation and further assisting stability. As discussed below, this amphipathic distribution of the helical structure potentially enhances interactions between lysine clusters and phosphate groups (anionic) in the DNA backbone. Simultaneously, it might facilitate association between the adjacent peptide helices through the contact of hydrophobic faces.
 | ||
| Fig. 2 Circular dichroism from peptides and peptide/DNA solutions. (A) CD spectra from TP10 samples at the concentrations indicated. (B) CD data from 0.2 mM TP10 solutions prepared with different TFE fractions. Inset: CD amplitude, [θ]195–[θ]220, as a function of %TFE. (C) CD from a 0.1 mM TP10 solution titrated with DNA aliquots, where [DNA] represents the base pair concentration. (D) Spectra from TP10/DNA complexes (2:1 charge ratio) during an up–down temperature cycle. | ||
We assessed the formation of TP10–DNA complexes by preparing peptide and nucleic acid mixtures at different charge ratios to determine the ratio required for the formation of stable complexes. Electrophoretic runs, conducted with both fragmented DNA (∼200 bp) and non-sonicated DNA ranging from a few hundred base pairs to over 20 kbp, showed that full complexation occurs near a 1:1 molar charge ratio, regardless of DNA size (ESI, Fig. S3B†). This suggests that the charge ratio is a more critical parameter for complexation than peptide concentrations.
CD assays were also conducted to analyse the binding of nucleotides to TP10. Fig. 2C shows a series of spectra from a 0.1 mM TP10 solution titrated with DNA fragments. The data reveal that the CD signature is strongly affected by the addition of DNA to the peptide solution, with a negative band indicative of random coiled conformations near 207 nm diminishing with increasing DNA concentration. Concomitantly, the spectra acquire α-helix characteristics, with the emergence of a pronounced negative peak at 222 nm and the increase in a positive band near to 200 nm. Additionally, the plot shown in Fig. 2C remarkably features isosbestic points at 214 and 265 nm, suggesting two-state populations (α-helix and random coiled) of biomolecular assemblies. The isosbestic point around 265 nm might indicate charge interactions between the phosphate backbone of DNA and cationic motifs in TP10,37 thereby indicating that electrostatic attraction also plays a significant role in the formation of TP10/DNA nanoparticles. The inset in Fig. 2C shows the monitoring of the peak at 210 nm as a function of [DNA], revealing a sigmoidal behaviour that was fitted to the logistic equation:
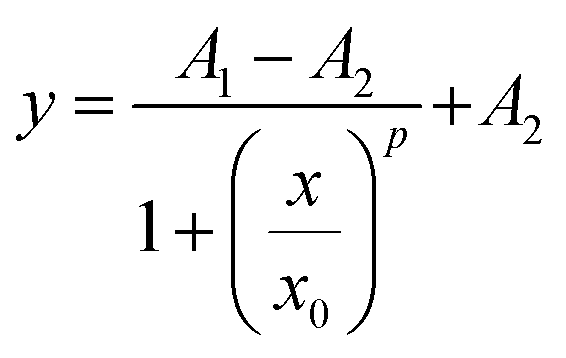 | (1) |
In the following, we examined the endpoint of our titration series to investigate stability upon temperature change. Fig. 2D illustrates the CD spectra resulting from an up–down temperature cycle performed in the 2:1 TP10/DNA sample. It is evident that the α-helix shape found at room temperature disappears upon heating. Specifically, the negative bands at 210 and 222 nm diminish, while a negative peak at 205 nm emerges, indicating transitions to disordered conformations. Interestingly, this process is found to be irreversible as the disordered signature persists (or even improves) upon cooling back to room temperature (dashed lines in Fig. 2D). We hypothesize that such irreversibility might be associated with transformations in the DNA structure. The examination of the induced CD, shown in Fig. S5B (ESI†), reveals similarities to spectra in the literature reporting transitions from B-DNA to Z-DNA forms upon heating.38 In this case, we tentatively propose that the more compact scaffolding and left-handedness of Z-DNA hinder its association with TP10.
Nanoscale structure
After evaluating the secondary structure of TP10 self-assemblies and their peptiplexes, we shifted our focus to the nanoscale characterization of aggregates using small-angle X-ray scattering (SAXS). Scattering profiles from these experiments are presented in Fig. 3A and B, and fitting models, along with the best fitting parameters, are detailed in the ESI (eqn (S1)–(S5) and Table S1†). | ||
| Fig. 3 (A) SAXS data of solutions containing DNA (bottom curve) or TP10 at the indicated concentrations. (B) SAXS curves of TP10/DNA peptiplexes prepared at various peptide-to-DNA ratios, with the DNA concentration maintained at 0.2 mM. The inset provides detailed information on the high-q range of the 125:1 formulation. The curves were shifted for clarity, and red lines represent data fitting according to models described in the ESI.† (C) Cryo-EM micrograph of 5:1 charge ratio peptiplexes. The sizes of aggregates are measured and indicated by red arrows. (D) Molecular docking between DNA and TP10 modelled with the assistance of PatchDock. | ||
In Fig. 3A, the bottom curve shows SAXS data collected from a sample containing DNA fragments at a concentration of 2.3 mM (base pair concentration). The curve exhibits an almost linear profile in a log–log representation, scaling with q−1, indicative of cylindrical structures.39 This behavior is fully consistent with the expected shape for stiff DNA fragments, as described in the Materials and methods section. To quantify the size of the particles, we fitted the data using the Porod cylinder form factor (eqn (S1)†). Data fitting confirmed the existence of rod-like structures with a radius of 0.91 ± 0.28 nm, consistent with the dimensions expected for the DNA duplex (diameter ∼2 nm).25,40 For TP10 solutions at concentrations closer to the CAC, 0.5 mM peptide, the scattering displays a linear descent in the high-q region, a near flat plateau in the intermediate range, and an upward turn in the low-q region. This indicates that the sample is mostly populated by free peptide strands in coexistence with a few larger aggregates. The fitting was performed straightforwardly using the summation of a power law plus the generalized Gaussian coil form factor (eqn (S2)–(S4)†), accounting for larger aggregates with scattering in the low-q range and free peptide chains with scattering in the mid-to-high q-range. The power law component revealed an exponent of −1.95, suggesting the presence of mass fractal aggregates,39,41,42 while the radius of gyration of the Gaussian coils was found at 0.7 nm and the Flory parameter was found to be ν = 0.39, compatible with collapsed TP10 strands.39,43 Upon increasing the peptide concentration, the shape of the curve changes significantly and the near flat plateau in the mid-range region disappears, hinting that the profile becomes dominated by scattering from aggregates. For solutions at both 1.4 and 2.3 mM TP10 concentrations, the fitting revealed consistent power law exponents of approximately −1.9, still supporting the presence of mass fractals (see Table S1†). Additionally, the fitting indicated collapsed chains with radii of gyration of around 1.5 nm, highlighting a consistent structural characteristic across both concentrations.
Fig. 3B reveals that SAXS curves from TP10/DNA peptiplexes are markedly different from those of the individual components displayed in Fig. 3A. Initially, one observes that scattering profiles from TP10/DNA mixtures could not be properly described in terms of linear combinations of their constituents, highlighting the emergence of new levels of organization upon complexation. The low-q Guinier region is not attained, indicating that characteristic sizes in the aggregates surpass the measurement window.44 The lowest q-value measured in our experiments was q = 0.13 nm−1, corresponding to a direct-space size of approximately 45 nm (Dmax = 2π/qmin). Therefore, the sizes of peptiplexes produced here are beyond this value. A striking feature of these curves occurs in the low-angle region, where the decay is characterized by a Porod behavior scaling with ∼q−4, indicating surface fractals.39,45,46 Interestingly, the high-q range presents different features depending on the peptide-to-DNA ratio, with interference peaks appearing when the fraction of the peptide in the mixture is sufficiently high. Data from samples prepared at the 5:1 charge ratio do not exhibit interference peaks, thus suggesting the absence of internal order in the aggregates. Employing a fitting procedure similar to that used for peptide solutions, these data were described with the summation of a power law plus the generalized Gaussian chain form factor. The results indicated a scaling exponent of −3.96, suggestive of smooth interfaces (dimensionality Ds = 2.04), along with unbound chains with a radius of gyration of 1.2 nm. Cryo-EM imaging from peptiplexes prepared at the 5:1 charge ratio, depicted in Fig. 3C, unveiled samples populated by flake-like aggregates with lateral dimensions varying from several tens to hundreds of nanometers. This observation is consistent with nanoscale information derived from SAXS data. DLS measurements (ESI, Fig. S6†) confirm that TP10/DNA peptiplexes form heterogeneous mixtures. Most of the population comprises particles with sizes ranging from several tens to a few hundred nanometers, coexisting with a few large aggregates reaching the micrometer range.
Upon increasing the TP10:DNA proportion to 25:1 and 125:1 charge ratios, interference peaks in the high-q region underscored the appearance of order in the local structure of peptiplexes. Unfortunately, it is not easy to devise an analytical model that captures the entire range of these data to offer an accurate description of both larger scale and local structures. In this case, we have thus employed an empirical formula, detailed in eqn (S5) in the ESI.† This functional equation incorporates power laws to describe Porod exponents in the low-to-intermediate q-range, while broad peak functions allow for fitting interference peaks in the high-q region. The formula offers valuable information on the dimensionality of fractal aggregates, as well as on the repeat distances and correlation lengths of ordered domains within these aggregates. It has been effectively used in previous studies to describe scattering from soft matter systems.46,47 The fitting of data from samples containing a 25:1 charge ratio mixture indicated that the low-q range is characterized by a scaling exponent equal to −3.8, suggesting an increase of roughness in surface fractals (dimensionality Ds = 2.2). Additionally, the need to use a power law with an exponent of 0.39 hinted that mid-range domains at length scales of the order of 5–10 nm exhibit globular shapes (see Table S1†).47 The fit of the broad peak component revealed that the peak is centered at q0 = 1.62 nm, which corresponds to a repeat distance of 3.9 nm, while the correlation length associated with this repeat is short, ξ = 4.7 nm. We interpret that the repeat distance is likely associated with the mean separation between DNA strands within the inner structure of the peptiplexes, in agreement with observations found in other peptiplex systems.47 In the case of mixtures prepared at a 125:1 charge ratio, the scaling exponent in the low-q range was −3.48, confirming the trend of an increase in surface roughness (dimensionality Ds = 2.52) upon growth of the peptide fraction. Fitting of the high-q range revealed two peaks centered at q0 = 1.63 and q1 = 2.11 nm−1, corresponding to repeats of 3.9 and 3.0 nm, respectively. The correlation lengths associated with these interference peaks are ξ = 6.7 and 1.9 nm, respectively. Therefore, it seems that two ordered domains emerge at higher peptide fractions, one of them maintaining the interstrand spacing between DNA duplexes, while in the other, the separation decreases. It is also interesting to note that the distance between DNA chains observed in peptiplexes based on TP10 is significantly larger than observations made in peptiplexes involving the cationic CPP penetratin.46,47 In fact, in penetratin/DNA peptiplexes, separations of 2.9 nm were found in formulations prepared at a 5:1 charge ratio. Here, not only the separations are much larger, but the peptide ratio required for the emergence of internal order was much higher (25:1 charge ratio). In light of these findings, we propose that this effect may arise from the distinct peptide length, which, in the case of TP10, is greater than in the case of penetratin. Moreover, charge density likely plays a role, with TP10 demonstrating a lower charge density (and thus weaker condensing power) than penetratin.
To gain further insights into the role of electrostatic interactions in complexation, we conducted a docking assay using computational tools. The interaction between TP10 strands and a 10 bp DNA duplex was evaluated. The model was initially docked using PatchDock and further refined using the Firedock web server.48,49 In Fig. 3D, the top-scoring model resulting from these tests is depicted. It can be observed that TP10 chains preferably intercalate in the minor groove region of the DNA strand, which is the site with the highest electronegative density of the double helix.
Fiber diffraction experiments conducted on dried stalks of TP10/DNA peptiplexes unequivocally validated the presence of ordered arrangements in the internal structure (ESI, Fig. S7†). Pronounced reflections were identified at positions corresponding to repeat distances of 0.46 and 1 nm, respectively, linked to the separation between adjacent strands and the spacing between β-sheets in peptide aggregates.50 Hence, TP10/DNA peptiplexes demonstrate a propensity to form well-ordered arrays at the local structural level. We propose that the amphipathic features exhibited by the TP10 helix (refer to Fig. S4A†), with lysine residues situated on one face of the helix and hydrophobic residues on the opposite side, favour the association with DNA and the formation of nanoparticles. This association is likely facilitated through electrostatic attraction between lysine residues and phosphates, and subsequent hydrophobic interactions between the nonpolar faces of adjacent helix strands contribute to the development of complex nanoparticles. The role of amphipathicity in the formation of complexes with DNA is further reinforced by the enhanced efficacy in forming stable nanoparticles demonstrated by NickFects and PepFects, which are stearylated analogs of TP10.22
The next step of our nanoscale analyses involved atomic force microscopy imaging combined with infrared spectroscopy (AFM-IR). This technique allows for the simultaneous collection of topography data and infrared spectra from single particles, thereby providing spatially resolved vibrational information.51,52 In addition, it enables the construction of absorbance maps at specific wavelengths, offering insights into the distribution of domains with distinct compositions or structural organization across the surface.
In Fig. 4A, a topography image shows TP10/DNA nanoparticles formed on a gold substrate. The image unveils the formation of a few clustered assemblies, approximately 50 nm in height, exhibiting lateral sizes that reach several hundreds of nanometers (ESI, Fig. S8†). Alongside these bigger assemblies, it is also possible to discern tiny aggregates with sizes from tens to a hundred nanometers and heights of only a few nanometers. The dimensions of these smaller species are in good agreement with cryo-EM data, whereas the larger clustered structures likely arise from drying effects on the gold surface. In Fig. 4B–D, absorbance maps at specific wavelengths reveal diverse distributions along the substrate. These observations indicate heterogeneity, suggesting that assemblies with different sizes and shapes exhibit differences in the structure and composition. In Fig. 4E and F, we present infrared spectra from tiny particles and bigger assemblies, respectively. The collection of infrared spectra from single particles spread across the substrate disclosed variations in the vibrational profiles, confirming a diversity of composition and ordering. The data exhibit strong peaks in the 950–1300 cm−1 range, commonly assigned to resonances in phosphate and deoxyribose groups, earning these features the designation of “DNA fingerprint”.53,54 A second group of relevant vibrations is found in the range between 1600 and 1750 cm−1. This encompasses the amide I band region, situated between 1600 and 1700 cm−1,55,56 where resonances related to peptide bonds are concentrated. In addition, the 1700–1750 cm−1 range contains resonances attributed to carbonyl groups situated in the nucleobases.53,57
 | ||
| Fig. 4 AFM-IR assays on TP10/DNA samples (1:1 ratio). Top row: topography data (A) are shown along with absorption maps at 1720 (B), 1670 (C), and 1260 cm−1 (D). Middle row: absorbance spectra from the regions indicated in the topography image. The regions corresponding to vibrations characteristic of specific functional groups are shaded in different colors: the DNA phosphate/sugar fingerprint in green, the amide I band in yellow, and the carbonyl in nucleobases in lilac. (E) Infrared spectra from small (oligomeric) aggregates. (F) Infrared data from larger nanoflakes. In the bottom row, the mean second derivative spectra of oligomeric structures and nanoflake assemblies are shown (G). The positions of the resonances are shaded in gray, and the corresponding wavenumbers are indicated. | ||
To enhance resolution and facilitate peak discrimination, we computed the mean second derivative of the spectral regions highlighted in Fig. 4E and F. The results, shown in Fig. 4G, revealed the presence of 6 resonances in the DNA fingerprint region (identified by minima in the 2nd derivative spectra). Resonances at 1006, 1036, 1078, and 1260 cm−1 are particularly intense. The assignments of these peaks are summarized in Table 1, and they predominantly correlate with stretching vibrations in the PO2 groups of the DNA backbone.53 These peaks consistently appeared at the same positions for both small particles and larger aggregates; however, varying intensities suggest that DNA might assume distinct conformational modes in the different types of particles. Additionally, minor vibrations are observed at 970 cm−1 and 1186 cm−1. The resonance at 970 cm−1, assigned to PO2 bending,53,54 is slightly blue-shifted in spectra from small aggregates, also supporting variation in DNA conformation. The analysis of the amide I band reveals that this range is dominated by an intense vibration at 1660 cm−1, which can be assigned to α-helices.56,58 This observation agrees with circular dichroism experiments performed in solution, which indicated a strong propensity of TP10 for structuration into helices. The appearance of weaker peaks at 1640 and 1688 cm−1 indicates that β-sheets and β-turns also appear in dry aggregates, consistent with information derived from fiber X-ray diffraction (ESI, Fig. S7†). Unlike the behaviour observed in the DNA fingerprint region, the size of aggregates has only minor effects on peak intensities in the amide I band, suggesting a consistent conformation of peptide chains in both types of assemblies. Finally, the carbonyl range (also associated with DNA groups) presents noticeable peaks at 1709 and 1740 cm−1, attributed to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching in nucleobases.54 The variations between smaller and larger aggregates are pronounced in this region, supporting the change in DNA conformation within these assemblies.
O stretching in nucleobases.54 The variations between smaller and larger aggregates are pronounced in this region, supporting the change in DNA conformation within these assemblies.
| Region | Wavenumber [cm−1] | Assignment | Ref. |
|---|---|---|---|
| DNA fingerprint | 970 | PO2 bending | Mello and Vidal53 |
| 1006 | Phosphate/ribose linkage | Mello and Vidal53 | |
| 1036 | Symmetric stretching phosphodiester | Mello and Vidal53 | |
| 1078 | Symmetric PO2 stretching | Mello and Vidal53 | |
| 1186 | Symmetric PO2 stretching | Tomić et al.54 | |
| 1260 | Asymmetric PO2 stretching | Tomić et al.54 | |
| Amide I | 1640 | β-Sheet | Kong and Yu56 |
| 1660 | α-Helix | Kong and Yu56 | |
| 1688 | β-Turns | Kong and Yu56 | |
| Carbonyl | 1709 | Guanine CO stretching |
Mello and Vidal53 |
| 1740 | CO stretching in nucleobases |
Mello and Vidal53 | |
The maps shown in Fig. 4B–D show the absorbance distribution of wavelengths in the three main spectral regions detected in the infrared profile of our samples. Interestingly, absorbances related to the DNA fingerprint and carbonyl resonances are comparatively stronger in the smaller aggregates than in the bigger assemblies formed on the surface (refer also to the maps shown in ESI, Fig. S8†). In contrast, absorbances related to the amide I band are relatively higher in the larger clusters, suggesting a higher concentration of TP10 within them. We hypothesize that these larger aggregates result from the coalescence of peptide self-assemblies that originated around nucleation centers formed through the condensation of TP10 strands onto DNA chains. As a result, the outer envelope is formed by a thicker peptide shell, while the nucleic acid charge remains inside the structure.
From the structural data presented above, we are now able to discuss the complexation pathway leading to the formation of TP10/DNA peptiplexes. The association between TP10 and DNA can be explained in terms of traditional mechanisms of complexation between oppositely charged polyelectrolytes.59,60
A schematic representation illustrating the proposed mechanism is shown in Fig. 5. According to this proposition, complexation occurs through a multistep process triggered by electrostatic attraction between positive charges on peptides and anionic groups on the DNA backbone.61 Initially, both DNA and TP10 are free in solution, surrounded by water solvation layers and counterion clouds that neutralize the overall charge. Upon their association, these counterions and water molecules from the solvation shells are released into the bulk solution, leading to a high gain in translational entropy that compensates for the conformational entropy loss of the biomolecules (yellow-orange arrows).61,62 This association further proceeds with the condensation of peptide chains surrounding the DNA strands and through concomitant peptide–peptide interactions (red-pink arrows). As a result, small clusters are produced inside which the DNA fragments are protected by condensed peptide layers, consistent with the DLS data (Fig. S6†), which indicated the presence of extensive fractions of nanoscopic aggregates in our samples. The stability of the DNA payload depends on its protection within the complexes. In the case of the DNA fragments analyzed here, the formation of peptide envelopes around the payload is presumably less costly.63 This process is much more challenging when long DNA chains are involved due to high energy costs in constructing a peptide wrapping around their typically folded structures.64 This “packing issue” is likely one of the major reasons behind the greater success of CPP-based peptiplexes in internalizing small RNA sequences compared to kilobase-long plasmids.11
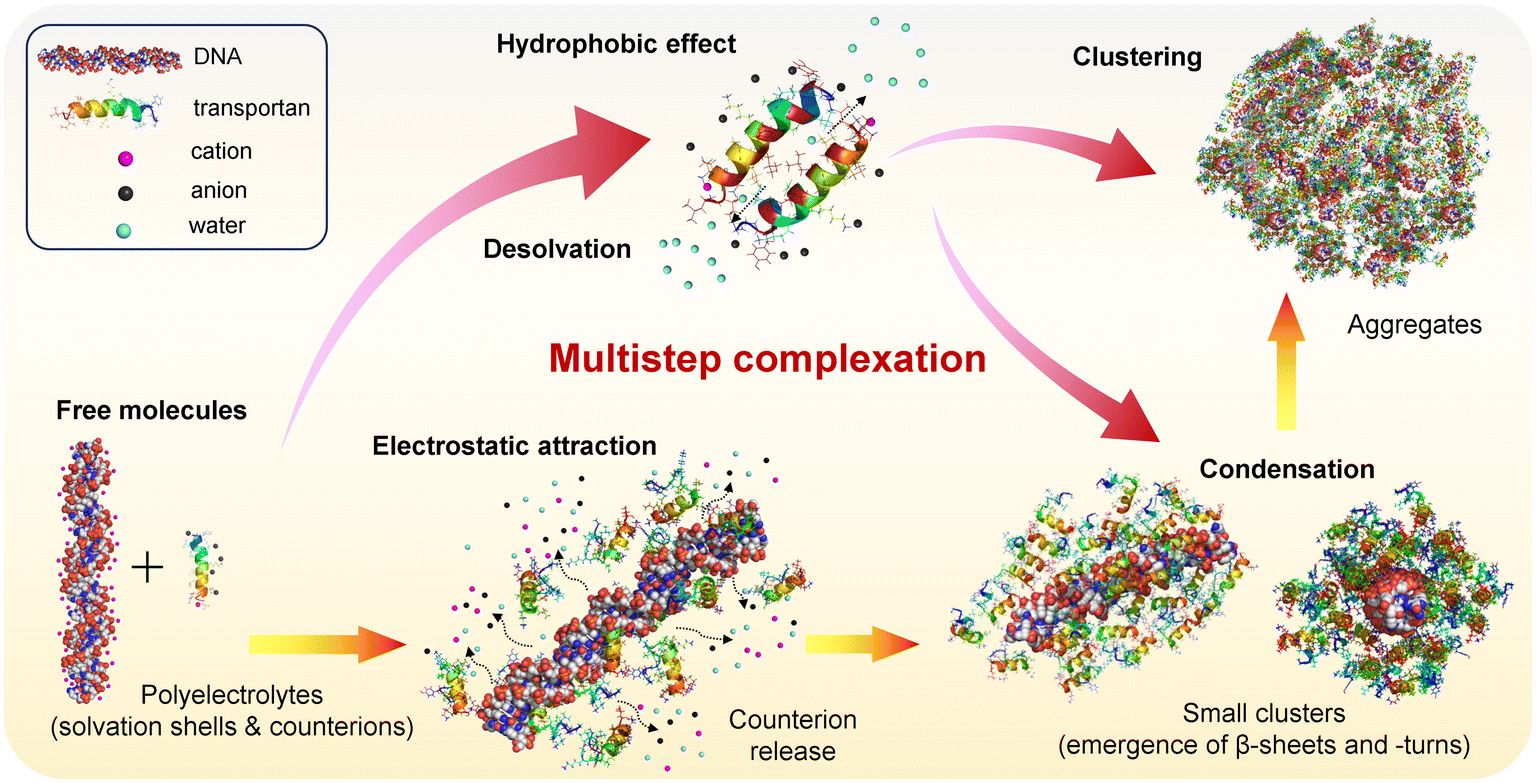 | ||
| Fig. 5 Schematic illustration (not to scale) of the proposed pathway for complexation between TP10 and DNA. The process is triggered by electrostatic attraction between cationic TP10 chains and anionic DNA strands. The release of counterions and water molecules from the solvation shells drives entropy gain and thermodynamic stability. The condensation of peptide chains around DNA results in small clusters that protect the load. The coalescence of these clusters forms larger aggregates, as revealed by AFM-IR, SAXS, and DLS assays (yellow-orange arrows). The association between peptide strands, occurring concomitantly to peptide–DNA complexation (red-pink arrows), is driven mainly by hydrophobic effects between non-polar residues (highlighted in brown) on the hydrophobic face of α-helices. The desolvation of TP10 strands is the primary factor leading to thermodynamic stability. | ||
As shown by our fluorometry data (Fig. 1), TP10 exhibits self-aggregation properties due to its amphipathic nature, with peptide–peptide interactions driven primarily by hydrophobic effects between residues on the hydrophobic faces of the peptide helices (ESI, Fig. S4†). This process also results in entropy gain due to the desolvation of peptide chains, as demonstrated experimentally in a recent report.65 It should be noted that while electrostatic attraction, hydrophobic effects, and entropic gain are driving forces behind peptide–peptide and peptide–DNA associations, other noncovalent interactions such as H-bonding and van der Waals forces are also crucial for stabilizing the complexes.62 Finally, the clustering of small complexes leads to the formation of larger aggregates, consistent with the data observed in AFM images.
Biological assays and delivery of peptiplexes to HeLa cells
After determining the molecular interaction between TP10 and DNA, and the nanoscale structure of the resulting peptiplexes, we analysed the cell-penetrating capabilities of TP10 and the delivery of DNA by peptiplexes into HeLa cells, a common model for gene delivery.66 For this purpose, DNA fragments used in the formulation of peptiplexes were conjugated to YOYO-1, a fluorescent probe impermeable to cell membranes, serving as a reporter in this case. The cells were labelled with DAPI for nucleus staining, phalloidin-Texas red for cytoskeleton staining, and Lysotracker deep red for discriminating the endosomal system. We began our analysis by performing fluorescence microscopy experiments with cells incubated with TP10/DNA complexes. Exploratory assays revealed that peptiplexes at a 5:1 charge molar ratio displayed the highest internalization rates, a phenomenon we suspect may be linked to the more relaxed inner structure of these peptiplexes compared to formulations with higher TP10 fractions (see the Nanoscale structure section for SAXS analyses). Indeed, a high degree of order in the internal structure of peptiplexes also implies greater cohesion of the DNA payload within the vector, leading to increased difficulty in the release of DNA from the peptiplex. In addition, we also observed that higher TP10 fractions led to cell clustering in flow cytometry assays, hindering proper quantitative analysis. HeLa cells were also incubated with Lipofectamine and uncomplexed DNA, serving as positive and negative controls, respectively.
The resulting fluorescence micrographs are presented in Fig. 6, illustrating the capacity of TP10 to deliver the labelled DNA to HeLa cells, as observed in the YOYO-1 channel. Samples incubated only with DNA, without the peptide, exhibited no signal in the green channel, indicating that nucleotides cannot penetrate the cells without a transfection reagent. In contrast, cells incubated with Lipofectamine complexes exhibited green fluorescence, indicating DNA penetration into the cells. The same behavior was observed in cells incubated with TP10 peptiplexes, confirming the uptake capabilities of the peptide.
 | ||
| Fig. 6 Confocal microscopy images illustrating HeLa cells treated with 5:1 charge ratio TP10/DNA peptiplexes alongside controls of uncomplexed DNA and Lipofectamine (incubation time = 4 h). Channel representations include DAPI in blue, YOYO-1 in green, Lysotracker in magenta, and phalloidin-Texas red marking the cytoskeleton. The bottom row displays the merged channels. | ||
The examination of a cell at a higher magnification, depicted in Fig. 7, reveals interesting aspects of the distribution of the DNA load across the intracellular space. Firstly, we observe that the peptiplexes were not only homogeneously distributed throughout both the cytoplasm and nucleus but also exhibited colocalization with the phalloidin and DAPI channels, as indicated by the zones pointed out by white arrows in Fig. 7.67 It should be noted that the reporter DNA is covalently linked to the YOYO-1 fluorophore; therefore, dye leaching is not an issue and the greenish glow observed across the cells can be associated with the spread of DNA fragments in the cytoplasm and membranes. Therefore, TP10/DNA peptiplexes were able to reach the entirety of the cell. Another interesting feature was that, although some degree of colocalization with the Lysotracker channel could be observed, most peptiplexes were not entrapped in endosomal compartments, as observed in other CPP-based systems reported in the literature.68,69 In fact, the green signal of the labelled DNA was found to be well spread across the cell, and in several regions, it lacked colocalization with the magenta channel that reports the endosome marker Lysotracker. The zones of exclusion between green and magenta channels are indicated by yellow arrows in Fig. 7. Hence, TP10/DNA peptiplexes exhibited the ability to undergo endosomal escape.69 Previous literature has demonstrated that transportans conjugated to short nucleic acid sequences induce membrane perturbation as a key step for uptake.23 Given that our complexes are made of relatively small DNA fragments, we hypothesize that membrane perturbation is the preferred route of internalization. Moreover, the capacity of transportans to disrupt membranes has been conjectured as an explanation for the endosomal escape exhibited by peptiplexes based on this CPP group.24
 | ||
| Fig. 7 Magnified confocal microscopy image showing a HeLa cell stained with DAPI, YOYO-1, phalloidin-Texas red, and Lysotracker. White arrows indicate regions of colocalization between DAPI and phalloidin with YOYO-1, while yellow arrows point to regions illustrating the absence of colocalization between YOYO-1 and Lysotracker. Scale bar: 10 μm. | ||
After qualitatively assessing cell uptake by TP10/DNA peptiplexes, we sought to quantify the internalization rate and evaluate cytotoxicity through flow cytometry experiments. Cell cytometry assays were carried out using eFluor780, a commercial reagent used for live and dead assays in flow cytometry, and the fluorescence of YOYO-1 conjugated to the DNA load in peptiplexes was evaluated. All tests were performed in duplicate on different days, and representative dot plots for samples incubated with DNA, Lipofectamine + DNA, and 5:1 charge ratio TP10/DNA peptiplexes are shown in Fig. 8, whereas the corresponding replicates are found in ESI, Fig. S9.† Live/dead analysis (top row) indicated cytocompatibility with viability rates above 95%. Dead cells, killed by heat shock, were used as a positive control to determine the eFluor780 fluorescence and gate establishment. The high biocompatibility of peptiplexes formed between TP10 and the model DNA fragments investigated here is consistent with previous studies showing low cytotoxicity and low immunogenic levels of TP10 or complexes formed between nucleic acids and PepFects, which have the same amino acid composition as TP10 but with a stearoyl chain attached to the N-terminus or a lysine side chain.70 These studies reported negligible in vitro cytotoxicity of transportan-based formulations against THP-1 cells (human acute monocytic leukemia cells) and minimal serum levels of the cytokines IL-1β, IL-6, and TNF-α 24 or 48 hours after in vivo administration of 5 mg kg−1 doses.70,71
 | ||
| Fig. 8 Representative flow cytometry data showing live/dead assays performed with the fluorophore eFluor780 (top row), and uptake quantification testing carried out for cells positive for YOYO-1 (bottom row). (A)–(C) show cytometry data from live/dead assays, whereas (E)–(G) display plots from DNA uptake. Figures (D) and (H) show averaged percentages from independent duplicates (mean ± SD). | ||
The resulting internalization ratios, defined by the percentage of cells positive for YOYO-1 fluorescence, are shown in the bottom row of Fig. 7. Not surprisingly, naked DNA exhibited negligible uptake with only 0.9 ± 0.4% of cells positive for YOYO-1. In contrast, cells incubated with TP10/DNA peptiplexes demonstrated an average internalization rate of 44 ± 8%, roughly representing two-thirds of the rate observed with the gold standard reagent Lipofectamine (78.2 ± 9%).
The last assay investigating the internalization of the peptiplexes involved real-time monitoring of the accumulation of peptiplexes into living cells by confocal microscopy. The results showing a time-series of photographs are depicted in Fig. 9A (see also the ESI,† supplementary film). In the first minute of monitoring, it was already possible to observe a few accumulated clusters of DNA (labelled with YOYO-1, green channel) in the HeLa cells. This process was initially slow but continuous over time and, after 15 to 30 min, the cells became more populated by DNA-enriched domains. At 60 min, there was a relevant increase in intensity with most cells exhibiting at least a small DNA cluster, demonstrating internalization of the load. The apex was reached at 120 min, maintaining a near-constant behaviour up to the end of the experiment at 180 min. The analysis of the integrated intensity of the green channel as a function of time, depicted in Fig. 9B, reveals a logistic profile, and the data could be adequately fitted with the time–response form of eqn (1). The midpoint of the curve, indicating the time lapse required for the half-maximal effect, was found at 63.0 ± 0.6 min, while the saturation region is reached at around 120 minutes after the administration of peptiplexes. Additionally, the Hill coefficient was found to be p = 2.10 ± 0.04, also suggesting positive cooperativeness for cell uptake.72 Interestingly, the characteristic time identified for the internalization of peptiplexes is notably larger than the timeframes observed for free TP10 chains (approximately 3 minutes).12 This discrepancy could be potentially related to the greater size of the TP10/DNA complexes compared to peptide strands. Putting the findings together, we tentatively propose that the internalization kinetics of peptiplexes features a lag phase, possibly involving the activation of membrane sites mediating the process. Once activation occurs, uptake accelerates during a phase of increased cell permeability. Beyond 120 minutes, an equilibrium concentration is reached, or these membrane sites might saturate, hindering an increase in DNA concentration within the cells.
 | ||
| Fig. 9 Time lapse of a sample incubated with TP10/DNA peptiplexes. (A). Representative images taken at the indicated time points (minutes). The blue channel reports Hoechst 33342 fluorescence and shows the nuclei of living cells, while the green channel reports the DNA loaded in peptiplexes labelled with YOYO-1. (B) Integrated intensity of the green channel as a function of time. Data have been fitted according to a logistic function. | ||
Potential pharmaceutical attributes of TP10/DNA peptiplexes
The production of DNA vectors based on CPPs may result in formulations with interesting pharmaceutical attributes such as enhanced targetability, ease of preparation, cost-effectiveness, and stability.11 Peptides offer several advantages in terms of chemical stability compared to formulations based on macromolecules, especially those of biological origin like proteins and antibodies.73 Peptides are typically shipped in lyophilized powder form, can be transported at room temperature, and have extended shelf life under standard refrigeration conditions. In the case of TP10, except for tyrosine and asparagine residues, which have a moderate tendency to undergo oxidation or deamination, the other amino acids are quite stable. The absence of residues like cysteine, methionine, or tryptophan thus represents an advantage in terms of stability compared to other traditional CPPs such as penetratin. Utilizing TP10, a comprehensive array of peptiplexes could be synthesized, subsequently augmented with glucose, and thereafter subjected to lyophilization to significantly prolong shelf life, remarkably, even at room temperature.In terms of material balance, the amount of peptide required in these formulations can be relatively high due to their lower molecular weight and charge, which may be considered a disadvantage. To transport 1 mg of DNA using complexes at a 5:1 charge ratio, approximately 8.5 mg of TP10 is needed, resulting in a current cost of around one hundred and fifty US dollars. While this cost remains relatively high compared to cationic polymers commonly used in delivery systems such as branched polyethyleneimine (PEI) or poly(amidoamine) (PAMAM) dendrimers, it is more affordable than Lipofectamine for instance. Additionally, certain cell lines, especially primary cells, exhibit high cytotoxicity when transfected with Lipofectamine or polymer-based complexes.74,75 Therefore, the mild transportation and storage requirements, potential for enhanced targetability, and low levels of toxicity and immunogenicity so far reported in the literature mitigate this disadvantage, potentially making transportan-based peptiplexes competitive for translation into pharmaceutical formulations.76
Conclusions
We presented a comprehensive study of the structure of peptiplexes formed between the cell-penetrating peptide (CPP) transportan 10 and DNA fragments. The organization of these non-covalent systems was analyzed in detail, covering from the molecular level to the mesoscopic scale. Through fluorescence studies, we identified that the amphipathic nature of TP10 shows self-aggregation capabilities at critical aggregation concentrations of approximately 0.65 ± 0.25 mM. Additionally, we confirmed the high propensity of TP10 to assume α-helical conformations in hydrophobic environments. This finding strongly correlates with bioinformatic analyses, which consistently indicated that the mastoporan-derived C-terminal region of TP10 transforms into amphipathic helices opposing a lysine cluster on one side to a hydrophobic face on the opposite side. This characteristic of transportan 10 contrasts with other classes of CPPs, such as cationic and hydrophobic ones, which have previously shown a strong inclination towards β-sheets at the secondary structure level. Notably, this propensity was observed both for the peptide alone and in complexation with DNA, potentially linked to the higher penetration efficacy displayed by amphipathic CPPs compared to their cationic counterparts. Due to their high stability, β-sheet arrangements are commonly observed in CPP complexes, representing a structurally more entangled and challenging configuration to destabilize compared to α-helices. Hence, the efficacy manifested by transportans may be related to their remarkable structural versatility and lower energetic cost during cargo release stages. Such flexibility is further supported by the absence of high levels of ordering found in peptiplexes exhibiting relatively low peptide:DNA ratios.
A contribution from our study was using AFM-based nanospectroscopy to reveal nuanced conformational changes of nucleic acids in different polymorphs. This finding suggests that polymorphic variability in peptiplexes potentially affects the conformation of the nucleic acid payload and should be considered in the formulation of these systems. Finally, our study presented data on the internalization kinetics of TP10-based peptiplexes, indicating a logistic time–response profile. This behavior suggests the presence of a short activation phase which is followed by a rapid uptake phase presumably linked to a significant increase in permeability.
In summary, the results presented here shed light on the structure–activity relationships of transportan peptiplexes. Considering the archetypal nature of TP10 as a representative of amphiphilic CPPs and its potential for translation in pharmaceutical formulations, we anticipate that our findings will contribute to the refinement of vectors based on the transportan family.
Data availability
Raw data from structural analyses, PDB files for schematic illustrations, and confocal image panels from cell assays have been deposited in the Mendeley repository under https://doi.org/10.17632/ymmg7sjygh.1.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This study was mostly supported by the Sao Paulo Research Foundation, FAPESP, under grants #2022/03056-6 to ERS and a PhD fellowship to LRM (#2019/19719-1). The Brazilian Council for Scientific Research, CNPq, is acknowledged for a research productivity fellowship (307443/2022-1 to ERS). The Brazilian Nanotechnology National Laboratory (LNNano) is acknowledged for granting access to TEM and AFM-IR instruments, respectively, under proposals: #20210417 and #20240148, and we are grateful to Dr Carlos Costa (AFM) and Dr Antonio Borges for assistance (TEM). We thank the Brazilian Synchrotron Laboratory for awarding us with beamtime at the SAXS-1 beamline (proposal 20190106). IWH and VC were supported by EPSRC (UK) Fellowship grant EP/V053396/1. We thank Dr Sandrine Marriot (Solid State Physics Laboratory, Orsay) for her assistance with DLS measurements and the Ph.D. student Thiago C. Lourenço (Biophysics Dept, UNIFESP) for performing electrophoretic runs.References
- C. E. Dunbar, K. A. High, J. K. Joung, D. B. Kohn, K. Ozawa and M. Sadelain, Gene Therapy Comes of Age, Science, 2018, 359(6372), eaan4672, DOI:10.1126/science.aan4672.
- M. Ramamoorth and A. Narvekar, Non Viral Vectors in Gene Therapy- An Overview, J. Clin. Diagn. Res., 2015, 9(1), GE01, DOI:10.7860/JCDR/2015/10443.5394.
- T. Wirth, N. Parker and S. Ylä-Herttuala, History of Gene Therapy, Gene, 2013, 525(2), 162–169, DOI:10.1016/j.gene.2013.03.137.
- C. Wang, C. Pan, H. Yong, F. Wang, T. Bo, Y. Zhao, B. Ma, W. He and M. Li, Emerging Non-Viral Vectors for Gene Delivery, J. Nanobiotechnol., 2023, 21(1), 272, DOI:10.1186/s12951-023-02044-5.
- C. Zylberberg, K. Gaskill, S. Pasley and S. Matosevic, Engineering Liposomal Nanoparticles for Targeted Gene Therapy, Gene Ther., 2017, 24(8), 441–452, DOI:10.1038/gt.2017.41.
- B. B. M. Garcia, O. Mertins, E. R. da Silva, P. D. Mathews and S. W. Han, Arginine-Modified Chitosan Complexed with Liposome Systems for Plasmid DNA Delivery, Colloids Surf., B, 2020, 193, 111131, DOI:10.1016/j.colsurfb.2020.111131.
- O. Mertins, P. D. Mathews and A. Angelova, Advances in the Design of PH-Sensitive Cubosome Liquid Crystalline Nanocarriers for Drug Delivery Applications, Nanomaterials, 2020, 10(5), 963, DOI:10.3390/nano10050963.
- J. W. Salameh, L. Zhou, S. M. Ward, C. F. Santa Chalarca, T. Emrick and M. L. Figueiredo, Polymer-Mediated Gene Therapy: Recent Advances and Merging of Delivery Techniques, WIREs Nanomed. Nanobiotechnol., 2020, 12(2), e1598, DOI:10.1002/wnan.1598.
- J. Geng, X. Xia, L. Teng, L. Wang, L. Chen, X. Guo, B. Belingon, J. Li, X. Feng, X. Li, W. Shang, Y. Wan and H. Wang, Emerging Landscape of Cell-Penetrating Peptide-Mediated Nucleic Acid Delivery and Their Utility in Imaging, Gene-Editing, and RNA-Sequencing, J. Controlled Release, 2022, 341, 166–183, DOI:10.1016/j.jconrel.2021.11.032.
- L. Porosk, I. Gaidutšik and Ü. Langel, Approaches for the Discovery of New Cell-Penetrating Peptides, Expert Opin. Drug Discovery, 2020, 1–13, DOI:10.1080/17460441.2021.1851187.
- Ü. Langel, Cell-Penetrating Peptides and Transportan, Molecules, 2021, 13(987), 1–31 Search PubMed.
- M. Pooga, M. Hällbrink, M. Zorko and Ü. Langel, Cell Penetration by Transportan, FASEB J., 1998, 12(1), 67–77, DOI:10.1096/fj.1530-6860.
- J. Ruczyński, I. Rusiecka, K. Turecka, A. Kozłowska, M. Alenowicz, I. Gągało, A. Kawiak, P. Rekowski, K. Waleron and I. Kocić, Transportan 10 Improves the Pharmacokinetics and Pharmacodynamics of Vancomycin, Sci. Rep., 2019, 9(1), 3247, DOI:10.1038/s41598-019-40103-w.
- M. M. R. Moghal, M. Z. Islam, S. Sharmin, V. Levadnyy, M. Moniruzzaman and M. Yamazaki, Continuous Detection of Entry of Cell-Penetrating Peptide Transportan 10 into Single Vesicles, Chem. Phys. Lipids, 2018, 212, 120–129, DOI:10.1016/j.chemphyslip.2018.02.001.
- J. Ruczynski, I. Rusiecka, K. Turecka, A. Kozlowska, M. Alenowicz, I. Gagalo, A. Kawiak, P. Rekowski, K. Waleron, I. Kocic, N. Ptaszynska, K. Gucwa, K. Olkiewicz, M. Heldt, M. Serocki, A. Stupak, D. Martynow, D. Debowski, A. Gitlin-Domagalska, J. Lica, A. Legowska, S. Milewski, K. Rolka, M. M. R. Moghal, M. Z. Islam, F. Hossain, S. K. Saha and M. Yamazaki, Role of Membrane Potential on Entry of Cell-Penetrating Peptide Transportan 10 into Single Vesicles, Sci. Rep., 2020, 21(13), 57–69, DOI:10.1038/s41598-019-40103-w.
- P. J. Cosme, J. Ye, S. Sears, E. P. Wojcikiewicz and A. C. Terentis, Label-Free Confocal Raman Mapping of Transportan in Melanoma Cells, Mol. Pharm., 2018, 15(3, SI), 851–860, DOI:10.1021/acs.molpharmaceut.7b00601.
- S. Anselmo, G. Sancataldo, H. M. Nielsen, V. Fodera and V. Vetri, Peptide-Membrane Interactions Monitored by Fluorescence Lifetime Imaging: A Study Case of Transportan 10, Langmuir, 2021, 37(44), 13148–13159, DOI:10.1021/acs.langmuir.1c02392.
- L. Aguiar, M. Machado, M. Sanches-Vaz, M. Prudencio, N. Vale and P. Gomes, Coupling the Cell-Penetrating Peptides Transportan and Transportan 10 to Primaquine Enhances Its Activity against Liver-Stage Malaria Parasites, MedChemComm, 2019, 10(2), 221–226, 10.1039/c8md00447a.
- G. Carreras-Badosa, J. Maslovskaja, K. Periyasamy, E. Urgard, K. Padari, H. Vaher, L. Tserel, M. Gestin, K. Kisand, P. Arukuusk, C. Lou, U. Langel, J. Wengel, M. Pooga and A. Rebane, NickFect Type of Cell-Penetrating Peptides Present Enhanced Efficiency for MicroRNA-146a Delivery into Dendritic Cells and during Skin Inflammation, Biomaterials, 2020, 262, DOI:10.1016/j.biomaterials.2020.120316.
- H. Margus, P. Arukuusk, U. Langel and M. Pooga, Characteristics of Cell-Penetrating Peptide/Nucleic Acid Nanoparticles, Mol. Pharm., 2016, 13(1), 172–179, DOI:10.1021/acs.molpharmaceut.5b00598.
- L. Porosk, H. H. Härk, R. N. Bicev, I. Gaidutšik, J. Nebogatova, E.-J. Armolik, P. Arukuusk, E. R. da Silva and Ü. Langel, Aggregation Limiting Cell-Penetrating Peptides Derived from Protein Signal Sequences, Int. J. Mol. Sci., 2023, 24(5), 4277, DOI:10.3390/ijms24054277.
- L. Pärnaste, P. Arukuusk, K. Langel, T. Tenson and Ü. Langel, The Formation of Nanoparticles between Small Interfering RNA and Amphipathic Cell-Penetrating Peptides, Mol. Ther. – Nucleic Acids, 2017, 7, 1–10, DOI:10.1016/j.omtn.2017.02.003.
- E. Bárány-Wallje, J. Gaur, P. Lundberg, Ü. Langel and A. Gräslund, Differential Membrane Perturbation Caused by the Cell Penetrating Peptide Tp10 Depending on Attached Cargo, FEBS Lett., 2007, 581(13), 2389–2393, DOI:10.1016/j.febslet.2007.04.046.
- L. Vasconcelos, F. Madani, P. Arukuusk, L. Paernaste, A. Graslund and U. Langel, Effects of Cargo Molecules on Membrane Perturbation Caused by Transportan10 Based Cell-Penetrating Peptides, Biochim. Biophys. Acta, Bioenerg., 2014, 1838(12), 3118–3129, DOI:10.1016/j.bbamem.2014.08.011.
- G. S. Manning, The Persistence Length of DNA Is Reached from the Persistence Length of Its Null Isomer through an Internal Electrostatic Stretching Force, Biophys. J., 2006, 91(10), 3607–3616, DOI:10.1529/biophysj.106.089029.
- J. Aguiar, P. Carpena, J. A. Molina-Bolívar and C. Carnero Ruiz, On the Determination of the Critical Micelle Concentration by the Pyrene 1:3 Ratio Method, J. Colloid Interface Sci., 2003, 258(1), 116–122, DOI:10.1016/S0021-9797(02)00082-6.
- L. de Mello, L. Porosk, T. Lourenço, B. Garcia, C. Costa, S. Han, J. Souza, U. Langel and E. da Silva, Amyloid-like Self-Assembly of a Hydrophobic Cell-Penetrating Peptide and Its Use as a Carrier for Nucleic Acids, ACS Appl. Bio Mater., 2021, 4(8), 6404–6416, DOI:10.1021/acsabm.1c00601.
- A. Hawe, M. Sutter and W. Jiskoot, Extrinsic Fluorescent Dyes as Tools for Protein Characterization, Pharm. Res., 2008, 25(7), 1487–1499, DOI:10.1007/s11095-007-9516-9.
- B. Nordén, A. Rodger and T. Dafforn, Linear Dichroism and Circular Dichroism: A Textbook on Polarized-Light Spectroscopy, Royal Society of Chemistry, Cambridge, Royal Society of Chemistry (Great Britain), 2010 Search PubMed.
- N. J. Greenfield, Using Circular Dichroism Spectra to Estimate Protein Secondary Structure, Nat. Protoc., 2006, 1(6), 2876–2890, DOI:10.1038/nprot.2006.202.
- E. Bárány-Wallje, A. Andersson, A. Gräslund and L. Mäler, NMR Solution Structure and Position of Transportan in Neutral Phospholipid Bicelles, FEBS Lett., 2004, 567(2–3), 265–269, DOI:10.1016/j.febslet.2004.04.079.
- K. Shiraki, K. Nishikawa and Y. Goto, Trifluoroethanol-Induced Stabilization of the Alpha-Helical Structure of Beta-Lactoglobulin: Implication for Non-Hierarchical Protein Folding, J. Mol. Biol., 1995, 245(2), 180–194, DOI:10.1006/jmbi.1994.0015.
- M. M. L. Ferreira, S. E. G. de Souza, C. C. da Silva, L. E. A. Souza, R. N. Bicev, E. R. da Silva and C. R. Nakaie, Pyroglutamination-Induced Changes in the Physicochemical Features of a CXCR4 Chemokine Peptide: Kinetic and Structural Analysis, Biochemistry, 2023, 62(17), 2530–2540, DOI:10.1021/acs.biochem.3c00124.
- C. A. Hunter and H. L. Anderson, What Is Cooperativity?, Angew. Chem., Int. Ed., 2009, 48(41), 7488–7499, DOI:10.1002/anie.200902490.
- X. Zhou, W. Zheng, Y. Li, R. Pearce, C. Zhang, E. W. Bell, G. Zhang and Y. Zhang, I-TASSER-MTD: A Deep-Learning-Based Platform for Multi-Domain Protein Structure and Function Prediction, Nat. Protoc., 2022, 17(10), 2326–2353, DOI:10.1038/s41596-022-00728-0.
- R. Gautier, D. Douguet, B. Antonny and G. Drin, HELIQUEST: A Web Server to Screen Sequences with Specific Alpha-Helical Properties, Bioinformatics, 2008, 24(18), 2101–2102, DOI:10.1093/bioinformatics/btn392.
- C. M. Goodman, N. S. Chari, G. Han, R. Hong, P. Ghosh and V. M. Rotello, DNA-Binding by Functionalized Gold Nanoparticles: Mechanism and Structural Requirements, Chem. Biol. Drug Des., 2006, 67(4), 297–304, DOI:10.1111/j.1747-0285.2006.00372.x.
- A. Parkinson, M. Hawken, M. Hall, K. J. Sanders and A. Rodger, Amine Induced Z-DNA in Poly(DG-DC)·poly(DG-DC): Circular Dichroism and Gel Electrophoresis Study, Phys. Chem. Chem. Phys., 2000, 2(23), 5469–5478, 10.1039/B005801G.
- J. S. Pedersen, Analysis of Small-Angle Scattering Data from Colloids and Polymer Solutions: Modeling and Least-Squares Fitting, Adv. Colloid Interface Sci., 1997, 70, 171–210, DOI:10.1016/S0001-8686(97)00312-6.
- I. Koltover, K. Wagner and C. R. Safinya, DNA Condensation in Two Dimensions, Proc. Natl. Acad. Sci. U. S. A., 2000, 97(26), 14046–14051, DOI:10.1073/pnas.97.26.14046.
- J. Teixeira, Small-Angle Scattering by Fractal Systems, J. Appl. Crystallogr., 1988, 21, 781–785 CrossRef.
- J.-M. Lin, T.-L. Lin, U.-S. Jeng, Z.-H. Huang and Y.-S. Huang, Aggregation Structure of Alzheimer Amyloid-β(1–40) Peptide with Sodium Dodecyl Sulfate as Revealed by Small-Angle X-Ray and Neutron Scattering, Soft Matter, 2009, 5(20), 3913–3919, 10.1039/B908203D.
- G. Beaucage, Small-Angle Scattering from Polymeric Mass Fractals of Arbitrary Mass-Fractal Dimension, J. Appl. Crystallogr., 1996, 29, 134–146, DOI:10.1107/s0021889895011605.
- B. Hammouda, SANS from Pluronic P85 in D-Water, Eur. Polym. J., 2010, 46(12), 2275–2281, DOI:10.1016/j.eurpolymj.2010.10.012.
- R. Besselink, T. M. Stawski, A. E. S. Van Driessche and L. G. Benning, Not Just Fractal Surfaces, but Surface Fractal Aggregates: Derivation of the Expression for the Structure Factor and Its Applications, J. Chem. Phys., 2016, 145(21), 211908, DOI:10.1063/1.4960953.
- L. R. De Mello, I. W. Hamley, V. Castelletto, B. B. M. Garcia, S. W. Han, C. L. P. De Oliveira and E. R. Da Silva, Nanoscopic Structure of Complexes Formed between DNA and the Cell-Penetrating Peptide Penetratin, J. Phys. Chem. B, 2019, 123(42), 8861–8871, DOI:10.1021/acs.jpcb.9b05512.
- B. B. S. Souza, T. C. Lourenço, B. B. Gerbelli, P. L. Oseliero Filho, C. L. P. Oliveira, A. Miranda and E. R. da Silva, A Biophysical Study of DNA Condensation Mediated by Histones and Protamines, J. Mol. Liq., 2022, 368, 120745, DOI:10.1016/j.molliq.2022.120745.
- D. Schneidman-Duhovny, Y. Inbar, R. Nussinov and H. J. Wolfson, PatchDock and SymmDock: Servers for Rigid and Symmetric Docking, Nucleic Acids Res., 2005, 33, W363–W367, DOI:10.1093/nar/gki481 , (web server issue).
- E. Mashiach, D. Schneidman-Duhovny, N. Andrusier, R. Nussinov and H. J. Wolfson, FireDock: A Web Server for Fast Interaction Refinement in Molecular Docking, Nucleic Acids Res., 2008, 36, W229–W232, DOI:10.1093/nar/gkn186 , (web server issue).
- L. Serpell, Amyloid Structure, Essays Biochem., 2014, 56, 1–10, DOI:10.1042/bse0560001.
- J. J. Schwartz, D. S. Jakob and A. Centrone, A Guide to Nanoscale IR Spectroscopy: Resonance Enhanced Transduction in Contact and Tapping Mode AFM-IR, Chem. Soc. Rev., 2022, 51(13), 5248–5267, 10.1039/D2CS00095D.
- F. S. Ruggeri, J. Habchi, S. Chia, R. I. Horne, M. Vendruscolo and T. P. J. Knowles, Infrared Nanospectroscopy Reveals the Molecular Interaction Fingerprint of an Aggregation Inhibitor with Single Aβ42 Oligomers, Nat. Commun., 2021, 12(1), 688, DOI:10.1038/s41467-020-20782-0.
- M. L. S. Mello and B. C. Vidal, Changes in the Infrared Microspectroscopic Characteristics of DNA Caused by Cationic Elements, Different Base Richness and Single-Stranded Form, PLoS One, 2012, 7(8), e43169–e43169, DOI:10.1371/journal.pone.0043169.
- K. Serec, S. D. Babić, R. Podgornik and S. Tomić, Effect of Magnesium Ions on the Structure of DNA Thin Films: An Infrared Spectroscopy Study, Nucleic Acids Res., 2016, 44(17), 8456–8464, DOI:10.1093/nar/gkw696.
- A. Barth, Infrared Spectroscopy of Proteins, Biochim. Biophys. Acta, Bioenerg., 2007, 1767(9), 1073–1101, DOI:10.1016/j.bbabio.2007.06.004.
- J. Kong and S. Yu, Fourier Transform Infrared Spectroscopic Analysis of Protein Secondary Structures, Acta Biochim. Biophys. Sin., 2007, 39(8), 549–559, DOI:10.1111/j.1745-7270.2007.00320.x.
- K. Serec, S. D. Babić and S. Tomić, Magnesium Ions Reversibly Bind to DNA Double Stranded Helix in Thin Films, Spectrochim. Acta, Part A, 2022, 268, 120663, DOI:10.1016/j.saa.2021.120663.
- A. Barth, The Infrared Absorption of Amino Acid Side Chains, Prog. Biophys. Mol. Biol., 2000, 74(3–5), 141–173 CrossRef CAS PubMed . DOI: S0079-6107(00)00021-3[pii].
- J. R. Vieregg, M. Lueckheide, A. B. Marciel, L. Leon, A. J. Bologna, J. R. Rivera and M. V. Tirrell, Oligonucleotide–Peptide Complexes: Phase Control by Hybridization, J. Am. Chem. Soc., 2018, 140(5), 1632–1638, DOI:10.1021/jacs.7b03567.
- L. R. Mello, I. W. Hamley, V. Castelletto, B. B. M. Garcia, T. C. Lourenço, S. V. Vassiliades, W. A. Alves, S. W. Han and E. R. Silva, Self-Assembly and Intracellular Delivery of DNA by a Truncated Fragment Derived from the Trojan Peptide Penetratin, Soft Matter, 2020, 16(20), 4746–4755, 10.1039/D0SM00347F.
- T. C. Lourenço, L. R. de Mello, M. Y. Icimoto, R. N. Bicev, I. W. Hamley, V. Castelletto, C. R. Nakaie and E. R. da Silva, DNA-Templated Self-Assembly of Bradykinin into Bioactive Nanofibrils, Soft Matter, 2023, 19(26), 4869–4879, 10.1039/D3SM00431G.
- B. B. M. Garcia, O. Mertins, E. R. da Silva and S. W. Han, Influence of the Degree of Arginine Substitution on Chitosan-N-Arginine-Based Chitosomes: Insights for Improved Gene Delivery Systems, J. Drug Delivery Sci. Technol., 2024, 92, 105368, DOI:10.1016/j.jddst.2024.105368.
- E. R. Silva, G. Cooney, I. W. Hamley, W. A. Alves, S. Lee, B. F. O'Connor, M. Reza, J. Ruokolainen and D. Walls, Structural Behaviour and Gene Delivery in Complexes Formed between DNA and Arginine-Containing Peptide Amphiphiles, Soft Matter, 2016, 12(45), 9158–9169, 10.1039/c6sm01618a.
- A. E. Neitzel, G. X. De Hoe and M. V. Tirrell, Expanding the Structural Diversity of Polyelectrolyte Complexes and Polyzwitterions, Curr. Opin. Solid State Mater. Sci., 2021, 25(2), 100897, DOI:10.1016/j.cossms.2020.100897.
- C. Yuan, R. Xing, J. Cui, W. Fan, J. Li and X. Yan, Multistep Desolvation as a Fundamental Principle Governing Peptide Self-Assembly Through Liquid–Liquid Phase Separation, CCS Chem., 2024, 6(1), 255–265, DOI:10.31635/ccschem.023.202302990.
- J. Mueller, I. Kretzschmar, R. Volkmer and P. Boisguerin, Comparison of Cellular Uptake Using 22 CPPs in 4 Different Cell Lines, Bioconjugate Chem., 2008, 19(12), 2363–2374, DOI:10.1021/bc800194e.
- H. Mazloom-Farsibaf, F. Farzam, M. Fazel, M. J. Wester, M. B. M. Meddens and K. A. Lidke, Comparing Lifeact and Phalloidin for Superresolution Imaging of Actin in Fixed Cells, PLoS One, 2021, 16, 1–13, DOI:10.1371/journal.pone.0246138.
- A. Sahni, Z. Qian and D. Pei, Cell-Penetrating Peptides Escape the Endosome by Inducing Vesicle Budding and Collapse, ACS Chem. Biol., 2020, 15(9), 2485–2492, DOI:10.1021/acschembio.0c00478.
- A. Erazo-Oliveras, N. Muthukrishnan, R. Baker, T.-Y. Wang and J.-P. Pellois, Improving the Endosomal Escape of Cell-Penetrating Peptides and Their Cargos: Strategies and Challenges, Pharmaceuticals, 2012, 5(11), 1177–1209, DOI:10.3390/ph5111177.
- J. Suhorutsenko, N. Oskolkov, P. Arukuusk, K. Kurrikoff, E. Eriste, D.-M. Copolovici and Ü. Langel, Cell-Penetrating Peptides, PepFects, Show No Evidence of Toxicity and Immunogenicity In Vitro and In Vivo, Bioconjugate Chem., 2011, 22(11), 2255–2262, DOI:10.1021/bc200293d.
- T. Lehto, O. E. Simonson, I. Mager, K. Ezzat, H. Sork, D. M. Copolovici, J. R. Viola, E. M. Zaghloul, P. Lundin, P. M. D. Moreno, M. Mae, N. Oskolkov, J. Suhorutsenko, C. I. E. Smith and S. E. L. Andaloussi, A Peptide-Based Vector for Efficient Gene Transfer In Vitro and In Vivo, Mol. Ther., 2011, 19(8), 1457–1467, DOI:10.1038/mt.2011.10.
- W. W. Focke, I. van der Westhuizen, N. Musee and M. T. Loots, Kinetic Interpretation of Log-Logistic Dose-Time Response Curves, Sci. Rep., 2017, 7(1), 2234, DOI:10.1038/s41598-017-02474-w.
- O. Al Musaimi, L. Lombardi, D. R. Williams and F. Albericio, Strategies for Improving Peptide Stability and Delivery, Pharmaceuticals, 2022, 15(10), 1283, DOI:10.3390/ph15101283.
- T. Wang, L. M. Larcher, L. Ma and R. N. Veedu, Systematic Screening of Commonly Used Commercial Transfection Reagents towards Efficient Transfection of Single-Stranded Oligonucleotides, Molecules, 2018, 23(10), 2564, DOI:10.3390/molecules23102564.
- V. Kafil and Y. Omidi, Cytotoxic Impacts of Linear and Branched Polyethylenimine Nanostructures in A431 Cells, Bioimpacts, 2011, 1(1), 23–30, DOI:10.5681/bi.2011.004.
- L. Porosk, J. Nebogatova, H. H. Härk, B. Vunk, P. Arukuusk, U. Toots, M. Ustav, Ü. Langel and K. Kurrikoff, Predicting Transiently Expressed Protein Yields: Comparison of Transfection Methods in CHO and HEK293, Pharmaceutics, 2022, 14(9), 1949, DOI:10.3390/pharmaceutics14091949.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4pm00065j |
| This journal is © The Royal Society of Chemistry 2024 |