
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Relaxation and diffusion of an ionic plasticizer in amorphous poly(vinylpyrrolidone)†
Lara
Röwekamp
a,
Kevin
Moch
 *a,
Merve
Seren
a,
Philipp
Münzner
a,
Roland
Böhmer
a and
Catalin
Gainaru
ab
*a,
Merve
Seren
a,
Philipp
Münzner
a,
Roland
Böhmer
a and
Catalin
Gainaru
ab
aFakultät Physik, Technische Universität Dortmund, D-44221 Dortmund, Germany. E-mail: kevin.moch@tu-dortmund.de
bChemical Sciences Division, Oak Ridge National Laboratory, Oak Ridge, Tennessee 37831, USA
First published on 18th April 2024
Abstract
The present work focuses on the dynamics of the ionic constituents of 1-propyl-3-methyl-imidazolium-bis-(trifluormethylsulfonyl)-imide (PT), a paradigmatic ionic liquid, as an additive in poly(vinylpyrrolidone) (PVP). Hence, the resulting product can be regarded as a polymer electrolyte as well as an amorphous dispersion. Leveraging dielectric spectroscopy and oscillatory shear rheology, complemented by differential scanning calorimetry, the spectral shapes and the relaxation maps of the supercooled PVP-PT mixtures are accessed in their full compositional range. The study also presents dielectric and shear responses of neat PVP with a molecular weight of 2500 g mol−1. We discuss the plasticizing role of the PT additive and the decoupling between ionic dynamics and segmental relaxation in these mixtures. The extracted relaxation times, steady-state viscosities, and conductivities are employed to estimate the translational diffusivities of the ionic penetrants by means of the Stokes–Einstein, Nernst–Einstein, and Almond–West relations. While some of the estimated diffusivities agree with each other, some do not, pointing to the importance of the chosen hydrodynamic approximations and the type of response considered for the analysis. The present extensive dielectric, rheological, and calorimetric study enables a deeper understanding of relaxation and transport of ionic ingredients in polymers, particularly in the slow-dynamics regime which is difficult to access experimentally by direct-diffusivity probes.
I. Introduction
Many biotechnological applications rely on the retarded release of active ingredients that are embedded in amorphous solid dispersions or on the liberation of drugs and pesticides from polymeric carriers.1–3 These phenomena require a detailed characterization of the underlying diffusion processes that take place in the amorphous matrices. Numerous theoretical approaches have been developed in the context of plasticizer transport, yet their predictive power leaves room for improvements. This is understandable, for instance in view of the long-known and still debated mutual decoupling of various relaxation processes.4–14 Decoupling phenomena are not at all confined to the field of polymers, but observed in a diverse array of condensed matter systems.15–19 In fact, for instance in the field of polymer electrolytes and other ion conducting systems decoupling of charge and structure dynamics is highly desired.20–23Therefore, further experimental studies of small-molecule diffusion in polymers are warranted. Consequently, a wide variety of techniques have been devised for this purpose. Many of them employ light as a probe such as in studies using forced Rayleigh scattering,24,25 fluorescence recovery after photobleaching,26–29 fluorescence nonradiative energy transfer,30 and fluorescence correlation microscopy.31 Among various other approaches, nuclear magnetic resonance was also exploited.32 Furthermore, dielectric and viscosimetric techniques were explored to assess diffusion in neat supercooled liquids.33,34
In the present work, these latter approaches are exploited for amorphous solid dispersions based on polyvinylpyrrolidone (PVP). This biocompatible polymer plays a prominent role. e.g., in the field of pharmaceutical formulations. PVP was studied in pure and hydrated forms for various molecular weights.35–46 PVP-based amorphous solid dispersions featuring pharmaceuticals such as nifedipine,47,48 indomethacin,49–51 and others were also investigated.52
Another area of research, which is particularly relevant for energy storage technologies, is the transport of penetrants that act as electric charge carriers in polymer matrices.23,53–55 The workhorse material here is polyethylene oxide,56 a semicrystalline polar polymer doped with various salts in a rather reduced concentration range.57 While the resulting polymer electrolytes mitigate several drawbacks of inorganic conductors by providing mechanical flexibility and good contact with the electrodes, they often suffer from reduced electrochemical stability.58 For example, the degradation of polyethylene oxide above 4 V precludes its use in combination with high-voltage electrodes for high performance batteries.59 This situation warrants the current search for alternative polar polymers with a reduced degree of crystallization propensity, such as PVP, as ion matrices for energy storage applications.
In the present work, we first study neat PVP with a molecular weight near 2500 g mol−1 (on average featuring 23 monomer units per chain) and then mixtures of an ionic liquid with PVP. While many applications have emerged for mixtures of polymers and ionic liquids,60,61 here we employ them to track the ion diffusion via dielectric spectroscopy. Thus, we exploit that mass and charge transport are intimately coupled62,63 in ionic liquids such as 1-propyl-3-methyl-imidazolium-bis(trifluormethylsulfonyl)-imide (for brevity here denoted PT, previously also abbreviated as PMIM-TFSI).
Many active ingredients are however not intrinsically charged, but often display a residual conductivity only, mostly originating from minute ionic contaminants left over from their production. This calls for tests to find out whether charge transport and the diffusion-related mass transport are coupled or not. Interestingly, for several viscous liquids it has been demonstrated how rheological timescales can be used as a suitable predictor of diffusion phenomena over a temperature range down to the calorimetric glass transition at T = Tg.34
Therefore, in the present article, we generalize the approach from that work and test it for a polymer electrolyte. Thus, we examine the extent to which rheology in conjunction with conductometry can be exploited to track the diffusion of PT in the PVP matrix. More specifically, based on dielectrically detected DC conductivities, we use the Nernst–Einstein relation to estimate the plasticizer's diffusion coefficients and compare the results with those from a rheology based determination using the Stokes–Einstein equation. Apart from these classical approaches relying on steady-state responses we also employed the original64 and the recently introduced modified Almond–West concepts. Here, the elementary diffusivity length of the plasticizer is assessed based on the material's frequency-dependent conductivity65 as well as on its fluidity.34
While the dielectric and rheological properties of PT66 have been examined already, it appears that neat PVP with a molecular weight of 2500 g mol−1 was not studied previously in these respects. Therefore, after providing the experimental details, we present and discuss our results regarding the dynamics of neat PVP. Then, based on measurements and analyses of the dielectric and shear mechanical spectra for the PVP-PT mixtures, we compare their spectral widths, glass transition temperatures, viscosities, and electrical conductivities. Finally, based on the relaxation times for all systems, we compute the related diffusivities and discuss the relative merits of the various approaches used to this end.
II. Experimental details
For the present experiments we use PT (nominal purity >98%, Sigma Aldrich) and PVP (also termed PVP K-12 = Kollidon 12PF from BASF). The latter is specified by the supplier with a weight average molecular weight of 2000–3000 g mol−1.Prior to the measurements, pure PVP was dried overnight at 80 °C and melted at 180 °C directly on the lower plate of the dielectric or the rheology cell. Then, for the dielectric measurements the (room-temperature) upper electrode was put onto the sample, while for the rheological measurements the upper plate was priorly heated to 180 °C.
To prepare the ionic liquid mixtures, the appropriately weighed components PVP powder and liquid PT were mixed, stirred, and dried at 100 °C until a clear (slightly yellowish) liquid was formed. The premixed PVP powders were melted at 160 °C because at higher temperatures, the samples will quickly develop a brownish color. As soon as the mixtures have become liquid they are kept under vacuum at a temperature of 80 °C. For the ionic-liquid mixtures we studied molar PT fractions (with respect to the PVP monomers) of x = 0.10, 0.16, 0.33, and 0.66, corresponding to weight fractions of w = 28.6, 41.2, 64.0, and 87.8 wt%, respectively.
The dielectric response was probed using an Alpha-A impedance analyzer from Novocontrol and thermostatted using a Quatro unit. The samples were investigated in a parallel-plate cell made of invar and sapphire.67 For the shear mechanical oscillation or rotation experiments, an MCR 502 rheometer from Anton-Paar was employed. The gap between the two shear plates was 1 mm.
Differential scanning calorimetry (DSC) was conducted with a Q2000 from TA Instruments. The onset glass transitions were determined for a heating rate of 10 K min−1. For the mixtures, the glass transition temperatures range from that of pure PVP (Tg = 375 K;68 this work: Tg = 373 K) to that of neat PT (Tg = 180 K).66 Further experimental details as well as the DSC thermograms are given as ESI.†
III. Results and discussion
A. Dynamics of neat PVP
The segmental relaxation of PVP was probed using dielectric spectroscopy and Fig. 1 shows the results obtained for the dielectric function, ε* = ε′ − iε′′. In panel (a) the typical relaxation step is observed in ε′. As the temperature is lowered, it moves through the frequency window, signaling the slowdown of the segmental motion. Towards lower frequencies an upturn in ε′ is seen. This is the hallmark of blocking electrode effects which in this polymer arise as a consequence of a residual ionic conduction. The conductivity itself is seen more directly as a low-frequency feature in ε′′ which, in particular at low temperatures, see Fig. 1(b), masks the dielectric loss peak completely. For sufficiently broad spectra, one can nevertheless render the underlying process visible by using the logarithmic derivative69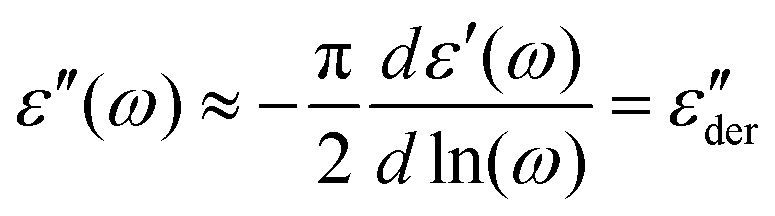 | (1) |
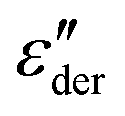 peaks is demonstrated in Fig. 1(c).
peaks is demonstrated in Fig. 1(c).
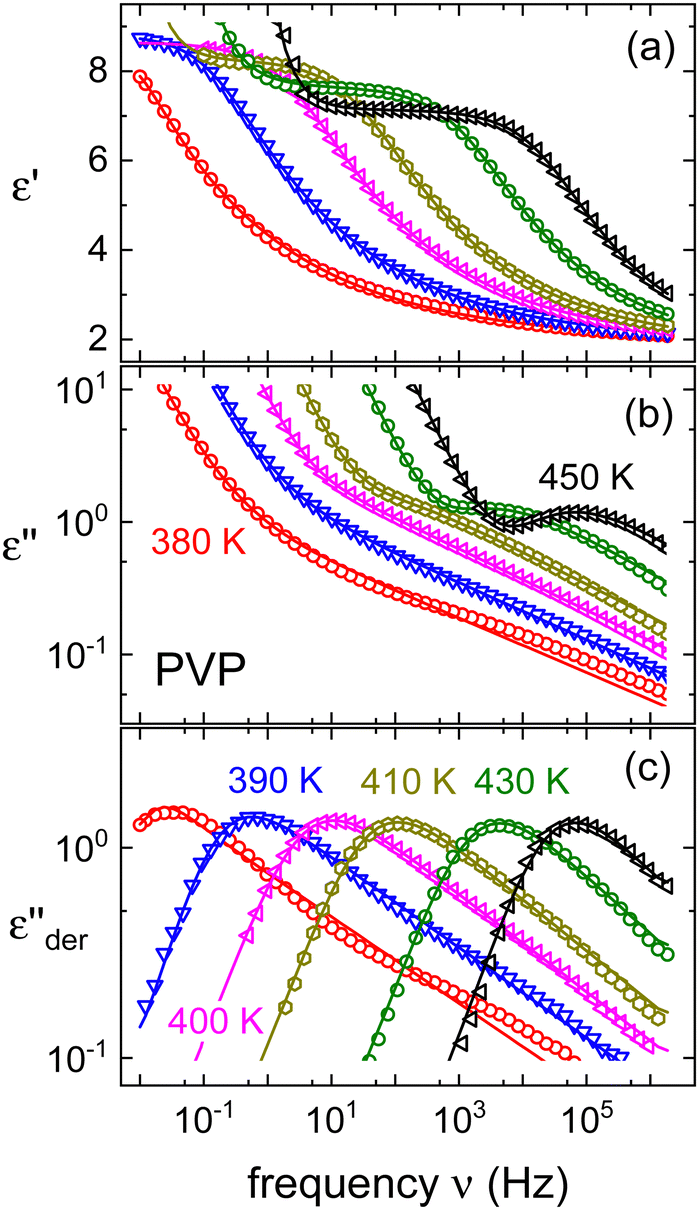 | ||
Fig. 1 (a) Real and (b) imaginary part of the dielectric function of PVP K-12. The lines in panel (a) and (b) are fits using eqn (2), those in (a) are augmented by the electrode polarization term described in the text. Panel (c) presents 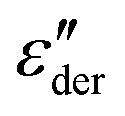 , cf.eqn (1), and the lines are fits using eqn (2). , cf.eqn (1), and the lines are fits using eqn (2). | ||
For a more quantitative analysis we employ the empirical Havriliak–Negami (HN) function72
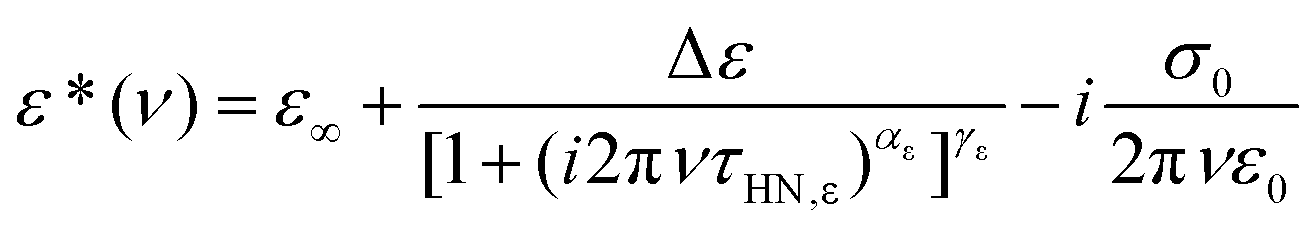 | (2) |
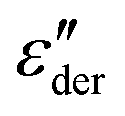 , in order to gain a better impression of the fitted spectral peak shape.
, in order to gain a better impression of the fitted spectral peak shape.
The dielectric relaxation time τε corresponding to the position of maximum loss can be assessed from75
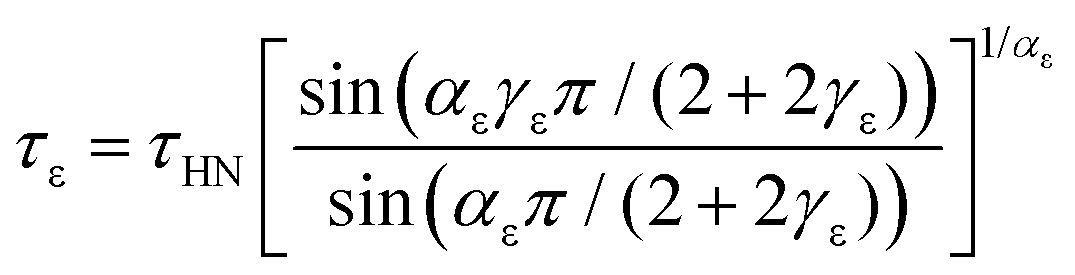 | (3) |
The relaxation times resulting for neat PVP will be summarized together with those from the mixtures, in Section III.C. This comparison will also involve results from DSC (see the ESI†) and the rheological experiments which we present in Fig. 2. There, we show the real and the imaginary part of the complex shear mechanical modulus G* = G′ + iG′′ of neat PVP. The present data cover dynamics in the range of the segmental relaxation. In Fig. 2(a) one recognizes that toward high frequencies G′ approaches a plateau modulus, G∞. Furthermore, in Fig. 2(b) well resolved shear loss peaks are detected over a wide range of temperatures.
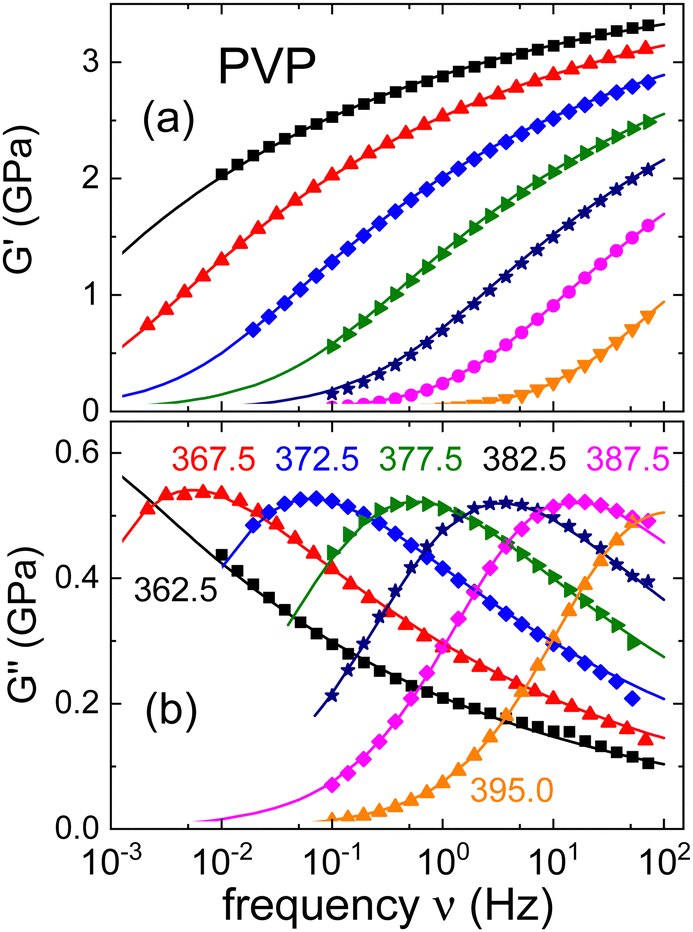 | ||
| Fig. 2 (a) Real and (b) imaginary part of the shear modulus of PVP K-12. The lines are fits using eqn (4) and the numbers indicate temperatures in Kelvin. | ||
To describe the rheological data we employed a (suitably adapted) HN function
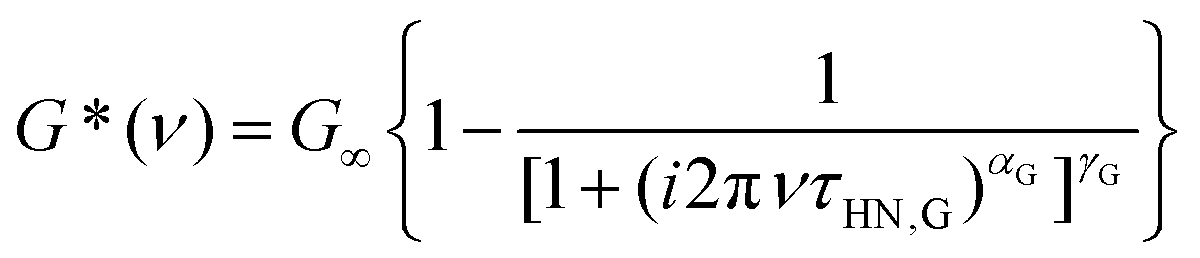 | (4) |
Analogous to eqn (2), the exponents appearing in eqn (4) describe the shape of the rheological spectra. From the fits shown as solid lines in Fig. 2 we find αG = 0.68 ± 0.05 and γG = 0.25 ± 0.03. Furthermore, τHN,G denotes the characteristic shear relaxation time, and a relation analogous to eqn (3) holds for the rheological time constants τG.
B. Dielectric and shear mechanical spectra for PVP mixed with PT
As an example for a mixture of PVP with PT, in Fig. 3 we present dielectric data for PVP0.9PT0.1. The permittivity spectra, shown in panel (a), resemble those of the neat polymer. However, due to the plasticizing effect of PT, in the binary melt the dynamics is faster so that their spectra were measured at lower temperatures. Furthermore, since the admixture of PT introduces numerous charge carriers into the system, for PVP0.9PT0.1 the relative contribution of the electrical conductivity is larger than that of neat PVP. Thus, even at high temperatures the dielectric loss peaks of PVP0.9PT0.1 are fully swamped. Therefore, in Fig. 3(b), we present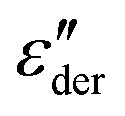 instead of ε′′. But even then, the
instead of ε′′. But even then, the 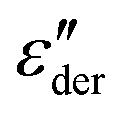 spectra display a significant broadening in the peak region, which may indicate a dynamical disparity between the two kinds of ions or between their translational and reorientational (since the present ions carry permanent dipole moments)76 degrees of freedom.
spectra display a significant broadening in the peak region, which may indicate a dynamical disparity between the two kinds of ions or between their translational and reorientational (since the present ions carry permanent dipole moments)76 degrees of freedom.
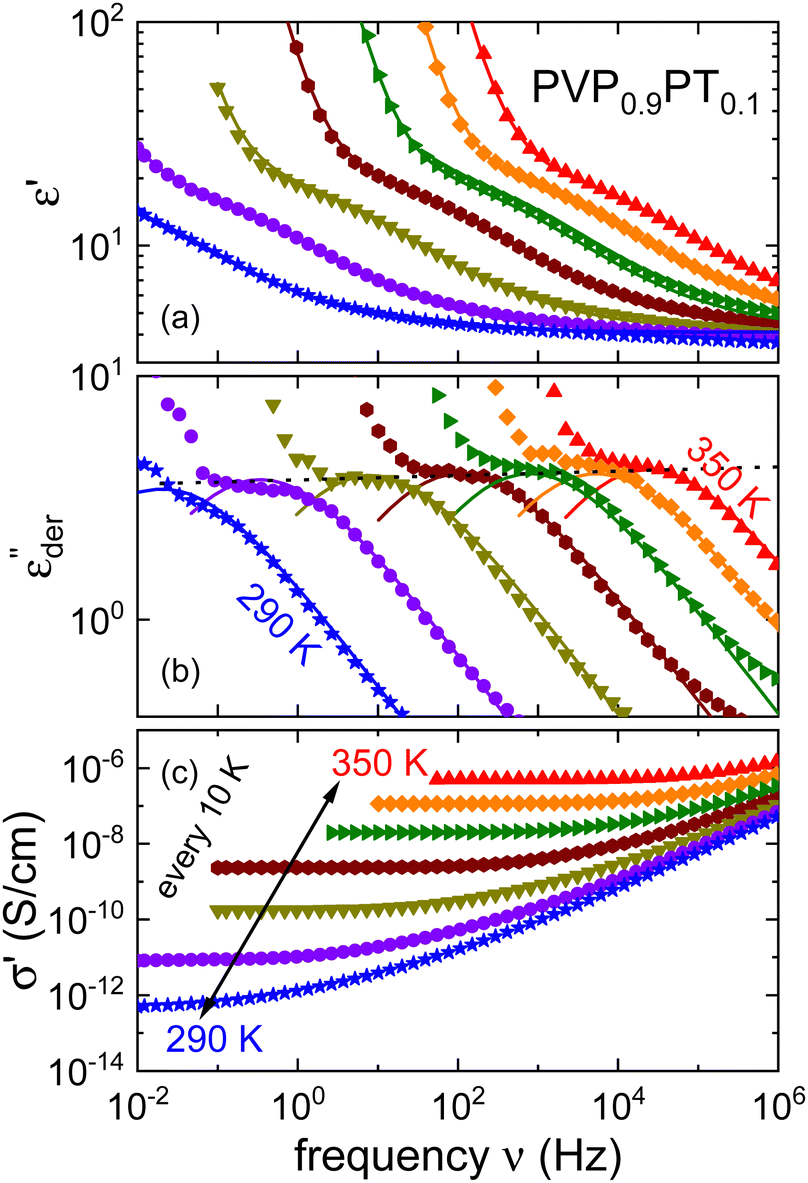 | ||
Fig. 3 Dielectric data of PVP0.9PT0.1 presented in terms of (a) ε′, (b) 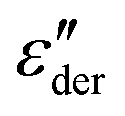 , and (c) σ′(ν). The lines are fits as described in Fig. 1. For this mixture the fit parameters, cf.eqn (2), are γε,eff = αεγε = 0.42 ± 0.05 and s = 1.5. , and (c) σ′(ν). The lines are fits as described in Fig. 1. For this mixture the fit parameters, cf.eqn (2), are γε,eff = αεγε = 0.42 ± 0.05 and s = 1.5. | ||
By contrast to the 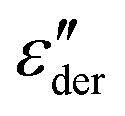 spectra of neat PVP, for PVP0.9PT0.1 a low-frequency flank cannot be discerned. Hence, for a quantitative description of the data in terms of eqn (2), the exponent αε was fixed. Here, we have chosen a value of 0.5 which provides a good description in the peak region of the
spectra of neat PVP, for PVP0.9PT0.1 a low-frequency flank cannot be discerned. Hence, for a quantitative description of the data in terms of eqn (2), the exponent αε was fixed. Here, we have chosen a value of 0.5 which provides a good description in the peak region of the 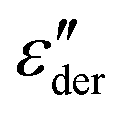 spectra. Fig. 3(a) and (b) show that this approach provides a satisfactory fit to the spectra for frequencies near the peak and higher. Similarly broadened
spectra. Fig. 3(a) and (b) show that this approach provides a satisfactory fit to the spectra for frequencies near the peak and higher. Similarly broadened 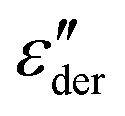 peaks, typically devoid of a clearly discernible maximum, are observed for all PVP1−xPTx samples. The corresponding ε′, ε′′, and
peaks, typically devoid of a clearly discernible maximum, are observed for all PVP1−xPTx samples. The corresponding ε′, ε′′, and 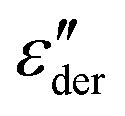 spectra are summarized as ESI.† As will be discussed in detail in Section III.C, for all mixtures the spectral widths (more precisely the high-frequency flanks of their dielectric spectra) are similarly broad.
spectra are summarized as ESI.† As will be discussed in detail in Section III.C, for all mixtures the spectral widths (more precisely the high-frequency flanks of their dielectric spectra) are similarly broad.
Returning to PVP0.9PT0.1, a close inspection of Fig. 3(a) reveals that upon cooling the relaxation strength slightly decreases and consequently also the amplitude of the 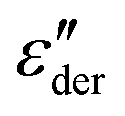 spectra, see Fig. 3(b). This anti-Curie-type temperature evolution of the dielectric strength is much more pronounced for the samples with x = 0.33 and 0.66, see the ESI.† This anti-Curie effect is known from the highly viscous regime of glassforming electrolytes,66,77–79 where the dielectric absorption is largely governed by the conductivity relaxation of the ionic species. This observation also rationalizes why this effect is particularly pronounced in more highly PT doped PVP samples.
spectra, see Fig. 3(b). This anti-Curie-type temperature evolution of the dielectric strength is much more pronounced for the samples with x = 0.33 and 0.66, see the ESI.† This anti-Curie effect is known from the highly viscous regime of glassforming electrolytes,66,77–79 where the dielectric absorption is largely governed by the conductivity relaxation of the ionic species. This observation also rationalizes why this effect is particularly pronounced in more highly PT doped PVP samples.
The frequency dependent conductivity is directly related to the dielectric permittivity via σ*(ν) = 2πνε0ε*(ν). Fig. 3(c) displays the real part of the complex conductivity. From the low-frequency (ν → 0) plateau of these σ′(ν) curves one can extract the DC conductivity σ0 in a straightforward manner. The temperature evolution of σ0, together with that of τε, extracted from the peak of the 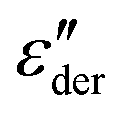 spectra is discussed in Sections III.C and III.D, respectively.
spectra is discussed in Sections III.C and III.D, respectively.
Shear loss spectra, G′′(ν), of PVP1−xPTx with PT fractions of x = 0.10, 0.33, and 0.66 are presented in Fig. 4. The corresponding G′ spectra and the full set of shear data for x = 0.16 are provided as ESI.†Fig. 4 shows that the G′′(ν) responses for x = 0.1 and x = 0.66 are rather broad. Nevertheless, their shear loss spectra can still satisfactorily be described using eqn (4).
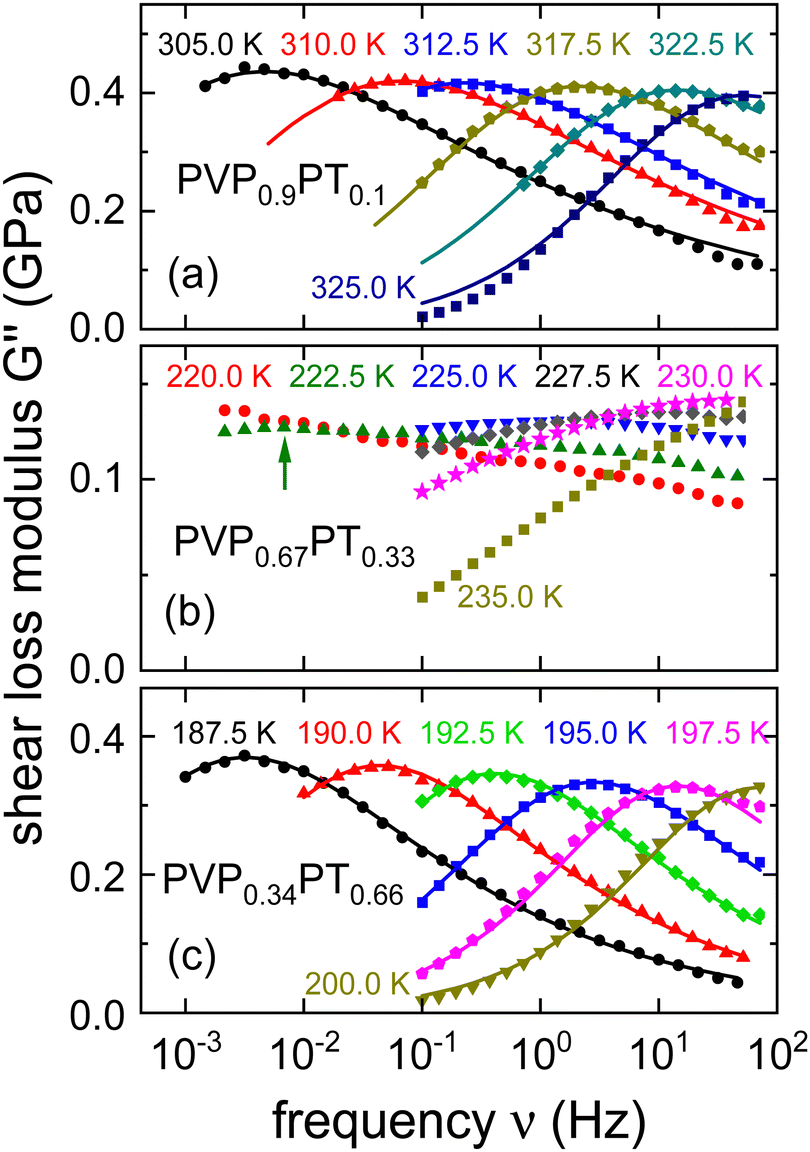 | ||
| Fig. 4 Shear loss modulus spectra measured for PVP1−xPTx with x given by (a) 0.1, (b) 0.33, and (c) 0.66. The lines are fits using eqn (4) with αG = 0.54 ± 0.04 and γG = 0.38 ± 0.04 for x = 0.1 and with αG = 0.55 ± 0.04 and γG = 0.56 ± 0.03 for x = 0.66. The arrow in panel (b) highlights the approximate position of the broad low-temperature peak that can be identified for 222.5 K. | ||
The spectra for x = 0.33 display, however, a much larger width and obviously they develop a two-peak structure toward low temperatures which also in other contexts signaled the existence of a microphase separation.80 This circumstance renders a reliable determination of time constants cumbersome. Corroborating the conjecture of a microphase separation, we mention that for x = 0.33 a bimodal calorimetric feature is observed (see the ESI†). Near 220 K, the separation of the processes that are comprised in the rheological two-peak structure is on the order of 3 decades. In view of the complexity of the rheological spectra for x = 0.33, we refrained from quantitatively describing their shape and restrict ourselves to determining time constants τG from the G′′ maxima, whenever possible.
Apart from frequency dependent measurements which mostly cover the low-frequency or high-viscosity range of these glassforming binary melts, we also gathered rotational viscosities, extending the probed range down to values of about 102 Pa s.
C. Comparison of spectral widths, glass transitions, viscosities, and electrical conductivities
After introducing the data base for the different PVP mixtures and in order to prepare for the analyses regarding the charge and molecular diffusion within them, we first compare the width of their rheological and dielectric spectra, their glass transition temperatures, as well as their DC conductivities and zero-shear viscosities.To be able to compare the shapes of the different spectra, it is advantageous to rescale them appropriately. Following ref. 81, G′′ masterplots were constructed with respect to a reference temperature Tref. For the masterplots presented in Fig. 5(a), Tref has been chosen close to Tg, so that the spectra can be considered isochronal with respect to a structural relaxation time of roughly 100 s. The resulting masterplots, further normalized with respect to their peak amplitude  and peak frequency, νmax, are shown in Fig. 5(a). Except for PVP0.67PT0.33, cf.Fig. 4(b), for which the construction of a master curve is far from unambiguous, the spectral width of all mixtures are similar to each other and with respect to PVP. Only the G′′ spectrum of neat PT66 is significantly narrower than for the samples containing PVP. Hence, disregarding PVP0.67PT0.33 and slightly simplifying, one may say that the spectral width of the shear response is essentially governed by the segmental relaxation of the polymeric component.
and peak frequency, νmax, are shown in Fig. 5(a). Except for PVP0.67PT0.33, cf.Fig. 4(b), for which the construction of a master curve is far from unambiguous, the spectral width of all mixtures are similar to each other and with respect to PVP. Only the G′′ spectrum of neat PT66 is significantly narrower than for the samples containing PVP. Hence, disregarding PVP0.67PT0.33 and slightly simplifying, one may say that the spectral width of the shear response is essentially governed by the segmental relaxation of the polymeric component.
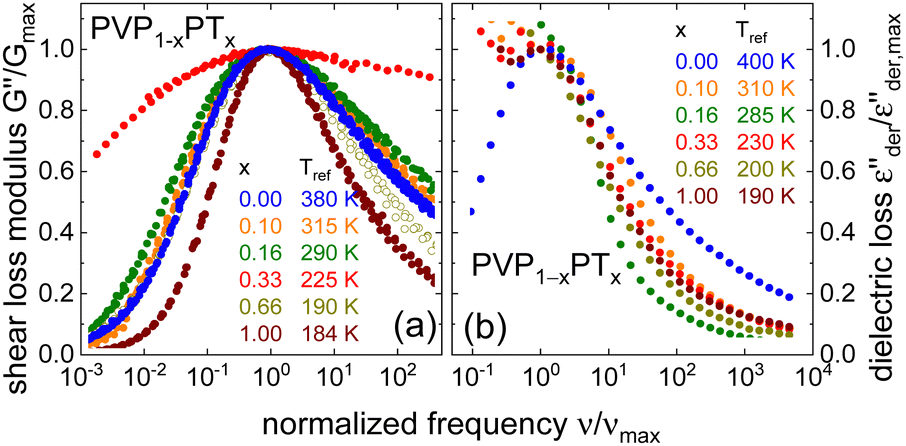 | ||
| Fig. 5 Amplitude- and frequency-scaled (a) rheological and (b) dielectric spectra of PVP1−xPTx. The plots essentially correspond to isochronal representations with rheological time constants of ∼102 s and dielectric time constants of ∼10−2 s. | ||
For the dielectric (derivative) spectra, where in Fig. 5(b) the displayed frequency window is shifted to higher values, it is more appropriate to choose a shorter reference time scale. The spectra displayed in Fig. 5(b) are isochronal with respect to about 10−2 s. These dielectric loss data again emphasize that only the high-frequency flank of the spectra is well defined. Interestingly, these flanks are broadest for PVP. If one considers that for PVP the dielectric loss peaks reflect structural rearrangements, while for PVP-PT they reflect the conductivity relaxation, this difference is highly plausible.
Furthermore, Fig. 6 shows that PT acts as plasticizer in PVP, thereby significantly reducing Tg of the mixtures with increasing x. Based on the weight fraction w of the components and on the specific heat steps Δcp of PVP (0.37 J g−1 K−1)40 and PT (1.3 J g−1 K−1),82 the observed composition dependence of Tg is well approximated by the Gordon–Taylor equation83
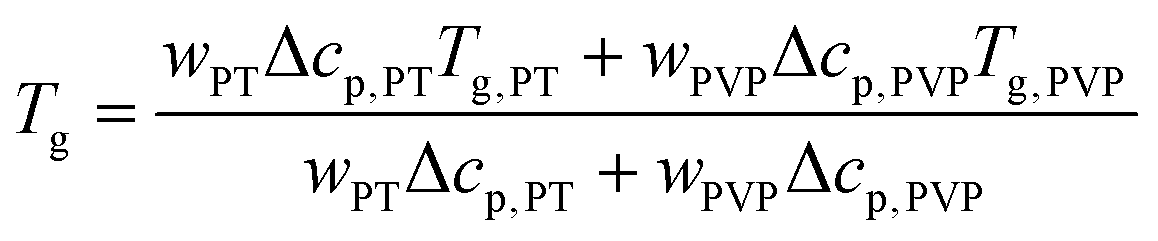 | (5) |
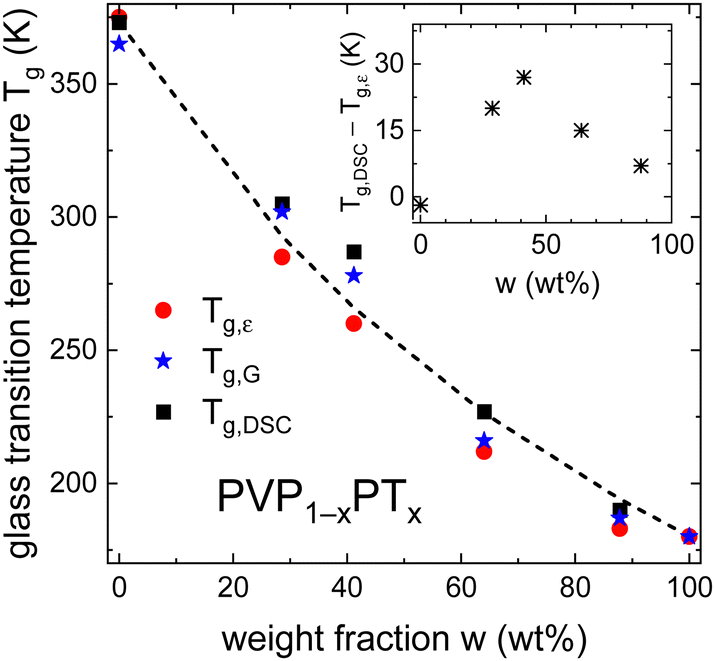 | ||
| Fig. 6 Composition dependent glass transition temperatures Tg of PVP1−xPTx as determined from dielectric spectroscopy, rheology, and DSC experiments. The lines are calculated using eqn (5) and the parameters given above that equation. The inset shows that for intermediate weight fractions w, Tg,DSC and Tg,ε differ significantly. | ||
In the PT-containing electrolytes, the concentration of the (intrinsic) charge carriers should be directly proportional to x. Therefore, it is interesting to find out whether this expectation is reflected in the conductivity of these mixtures and whether, like in other ionic liquids, factors like the glass transition temperature84 or the fragility85 need to be considered in addition. To examine these parameters, we present a Tg,ε-scaled plot, see Fig. 7(a), where we compile the temperature dependent DC conductivities for all PT-containing samples. This Angell plot reveals that the curvature and even the scaled values of the DC conductivities σ0 are the same for all PT mixtures. In this context it is important to recall that Tg,ε reflects the freezing of the dielectric degrees of freedom, which for decoupled systems corresponds to ionic rearrangements.
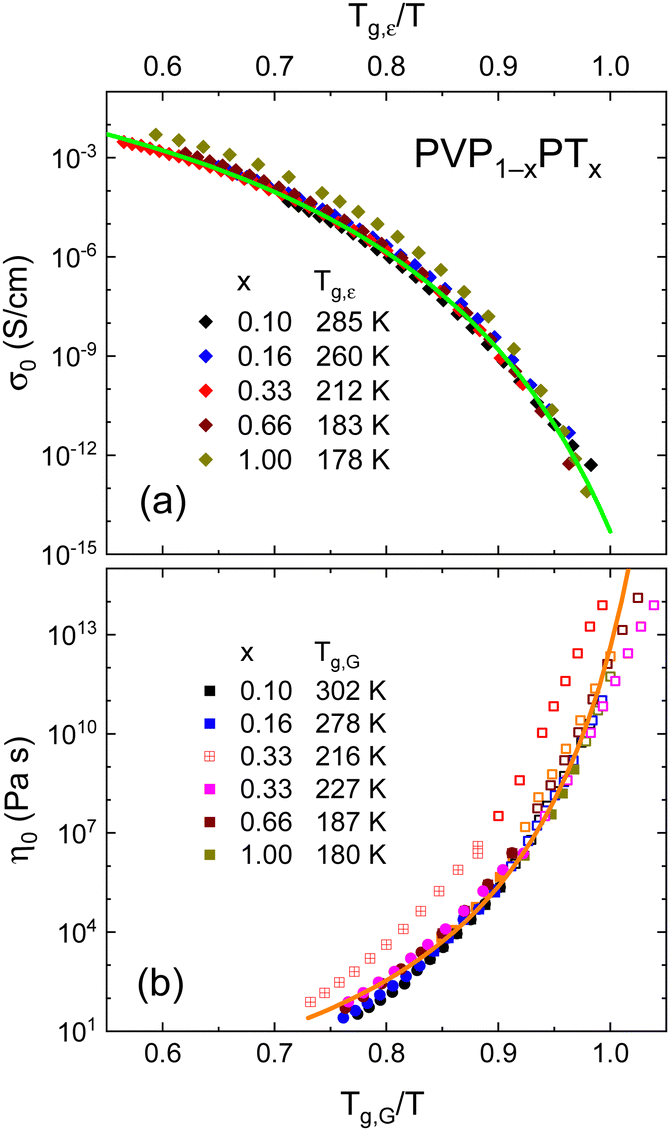 | ||
| Fig. 7 (a) DC conductivities σ0 and (b) shear viscosities η0 for all currently studied samples are shown as symbols on reduced inverse temperature scales. The lines are fits (a) using eqn (6) and (b) using eqn (7). In panel (b), the circles refer to viscosities from rotation experiments, all others are from oscillation experiments. The open symbols result from master-curve constructions. The PVP0.67PT0.33 data are scaled once with Tg,G = 216 K (crossed symbols) and once with Tg = 227 K as taken from DSC. Data for neat PT are from ref. 66. | ||
The solid line in Fig. 7(a) demonstrates that the DC conductivities σ0 of the binary mixtures can be described using the Vogel–Fulcher law
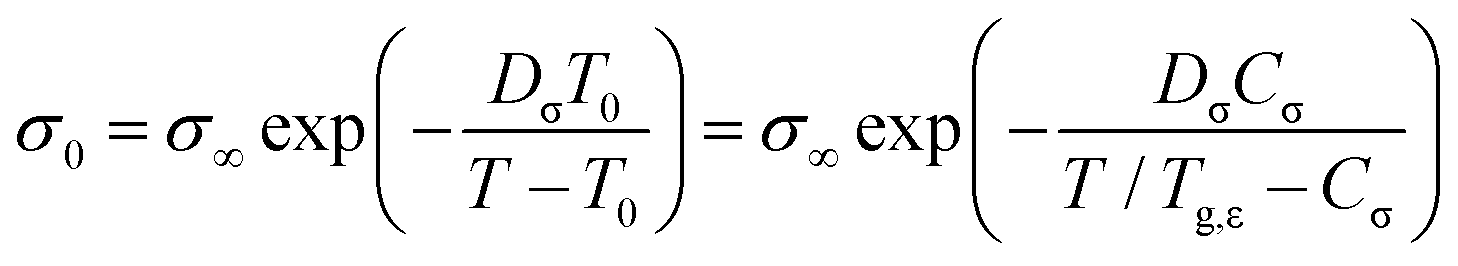 | (6) |
Fig. 7(b) provides a similar compilation for the shear viscosities of PVP1−xPTx as determined using rotational and oscillatory measurements. Here, Tg,G was used for rescaling, and in this representation all viscosity datasets appear to be in good agreement with each other. Like for the DC conductivities σ0, cf.eqn (6), after appropriate rescaling, also the zero-shear viscosities η0 essentially obey a suitably adapted Vogel–Fulcher law
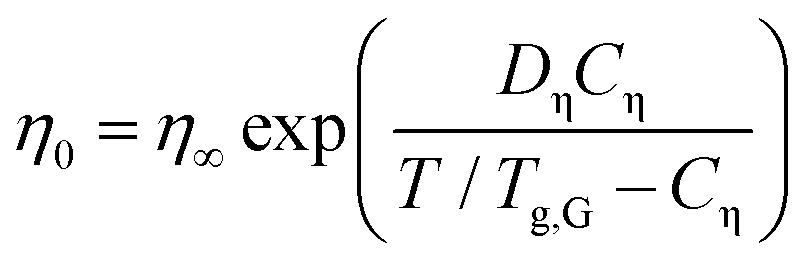 | (7) |
D. Relaxation and diffusion
With the steady-state transport-related quantities η0 and σ0 summarized in Fig. 7 at hand, several well-known hydrodynamics-based approximations have been employed to estimate the corresponding diffusivities, at least in the regime where the melt is sufficiently fluid. With the viscosity given, one may start from the Stokes–Einstein (SE) relation,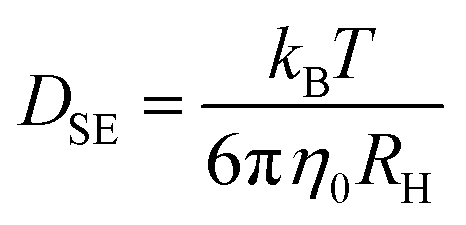 | (8) |
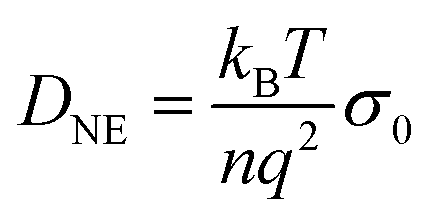 | (9) |
A different approach of extracting translational diffusion coefficients is based on insights developed by Almond and West (AW) in the framework of describing the electrical conductivity in inorganic solid electrolytes.64 Basically, AW suggested to identify the crossover frequency, at which σ′(ν) changes from DC to AC conductivity with an ionic ‘hopping rate’ Γ. If the effective hopping distance l is known, then one can exploit Einstein's relation to obtain
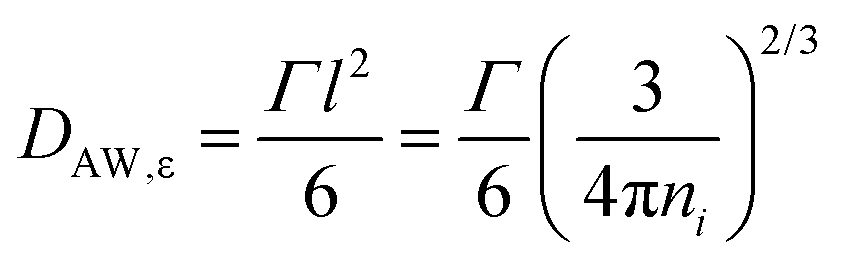 | (10) |
This formalism assumes that l can be estimated in a model-independent way from the ionic number density ni according to l = [3/(4πni)]1/3,63 see Table 1, where l is taken to represent the effective radius of the Coulombic cage which extends around each ion. Accordingly, Γ−1 can be identified with τε, except for neat PVP. Here, τε reflects solely dipolar reorientations and is not related to ions (translationally) escaping from a Coulombic cage.
| x | n i (1027 m−3) | R H (Å) | l (Å) |
|---|---|---|---|
| 0.10 | 0.986 | 0.3 | 6.2 |
| 0.16 | 1.593 | 0.1 | 5.3 |
| 0.33 | 2.543 | 0.1 | 4.5 |
| 0.66 | 3.187 | 0.3 | 4.2 |
| 1.00 | 4.312 | 0.3 | 3.8 |
To tackle, in a generalized manner, also situations in which dielectric spectroscopy is not or not clearly providing the relevant information, recently a “rheological” AW formalism has been introduced to characterize the molecular flow in liquids. Here, the crossover frequency is not determined from the conductivity, but from the shear fluidity response.34 In this approach, ni is given by the molecular number density, Γ−1 can be identified with τG,34 and the resulting diffusivity is called DAW,G. Since in the present work the matrix is not a simple liquid but a polymer, in the following the number density relevant for the microscopic flow events will be identified with that of the monomeric units. Note that nm = 6.502 × 1027 m−3 for neat PVP.
To discuss the applicability of these approaches for the PVP1−xPTx mixtures, Fig. 8(a) collects the dielectric time scales τε, the rheological time scales τG, and the ones estimated from DSC, τDSC. As described in the ESI,† the calculation of τDSC follows the procedure reviewed by Hodge.86Fig. 8(a) shows that the calorimetric and rheological time scales agree relatively well, but they differ from the dielectric ones. For x = 0.1 we find that τε becomes shorter than τG, signaling an additive-matrix decoupling as pointed out in relation to the inset of Fig. 6.
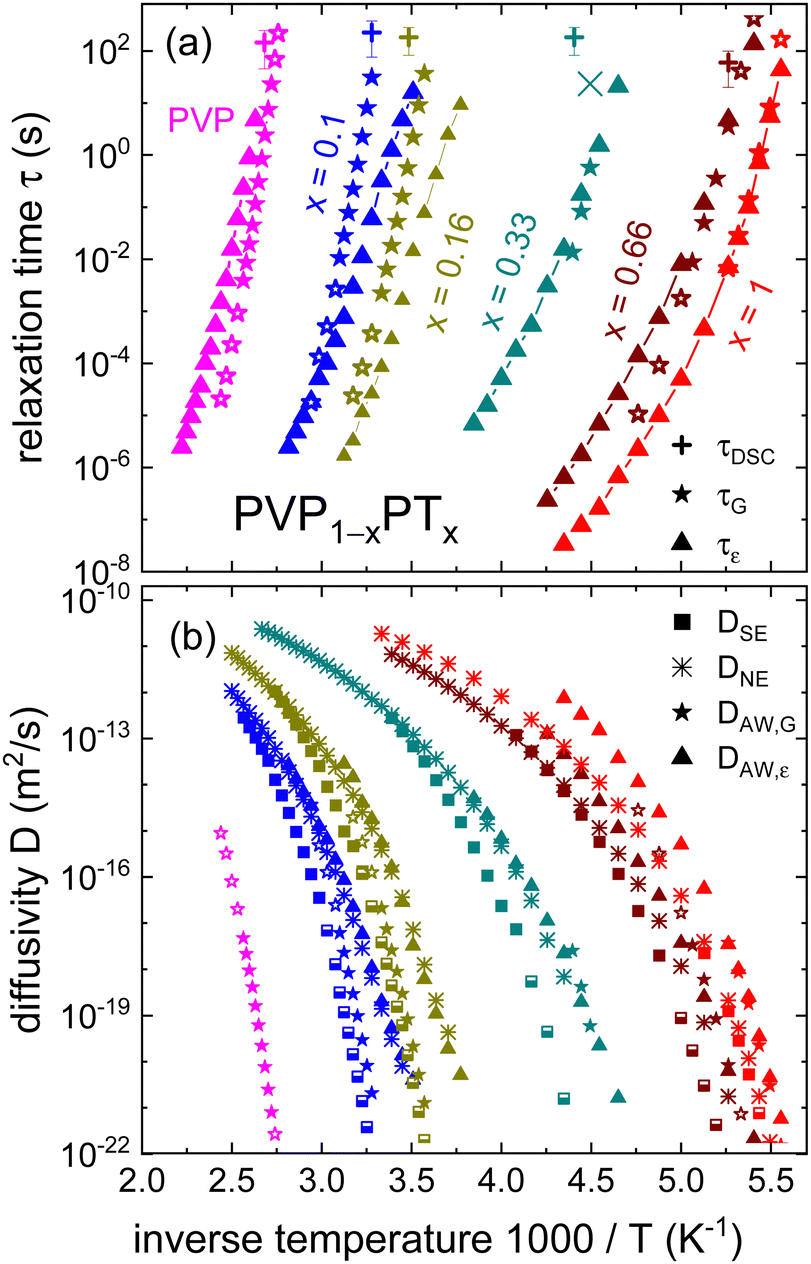 | ||
| Fig. 8 (a) Arrhenius plot of the time scales τε, τG, and τDSC determined for PVP1−xPTx in the present work, except for neat PT (x = 1) where τε was taken from ref. 66. Full symbols reflect time scales directly from the loss peaks, open symbols are from shear modulus master curves. The × symbol reflects the relaxation time corresponding to the low-frequency shear loss peak marked in Fig. 4(b) for x = 0.33. (b) The diffusion coefficient DSE (solid squares) was calculated using eqn (8) from RH in Table 1 and from η0. For the half-filled squares, η0 was extracted from the shift factors used for the shear modulus master curves. DNE (asterisks) was computed using eqn (9) from σ0 as shown in Fig. 7(a) and ni in Table 1. The calculation of DAW is based on eqn (10) and the data presented in panel (a): For DAW,G (stars) we used Γ = 1/τG and for DAW,ε we used Γ = 1/τε. The open stars refer to time scales from the shear modulus master curves. For the determination of DAW,ε for neat PVP see the text. | ||
In Fig. 8(a) another hallmark of this decoupling phenomenon63 becomes obvious: the temperature variation of the dielectric time scales changes from a relatively strong one above, to a weaker one below the temperature Tg,G at which the rheological dynamics freeze in. As observed in Fig. 8(a), the degree of decoupling increases for x = 0.16 (in agreement with the inset of Fig. 6), but decreases for x = 0.33 and 0.66. In the regime where PT is highly concentrated, the ions themselves constitute the matrix or at least the dominant fraction of it. The decoupling is also reflected by the Walden-type relation, η0 ∝ σ0−ξ, which, except for x = 1, reveals fractional exponents ξ (see Fig. S7, ESI†). Such a fractional relationship or an enhanced translational dynamics are observed also in many other glassforming materials (see ref. 4–19).
The diffusivities DSE, DNE, and DAW,ε assessed according to eqn (8), (9), and (10), respectively, as well as DAW,G estimated according to the rheological counterpart of eqn (10) are shown in Fig. 8(b). These reflect the rheological and conductivity (ionic translational) responses of PVP1−xPTx. For the compositions containing the ionic liquid, RH was obtained by imposing an agreement between DSE and DNE at the highest investigated temperatures. The corresponding RH values are included in Table 1, together with the calculations for the number density ni of the ion pairs.
For the current DNE estimates the correlated charge-carrier dynamics, which is known to reduce the conductivity in concentrated electrolytes,63,87 have been neglected. Consequently, the extracted RH values may be underestimated, at least for the systems with large concentrations of ionic liquid. Similar rheological cooperativity effects have been recently advocated to explain the large differences between DSE and DAW,G determined for several van der Waals liquids near their glass transition.34 Cooperativity effects may also rationalize the relatively large contrast between DSE and DAW,G seen in Fig. 8(b) for x = 0.66 and 1. These samples correspond to “liquid”-matrix materials. Since in ionic liquids mass and charge flows are coupled with each other,66 one may expect similarly large differences between the conductivity-related diffusivities DNE and DAW,ε, as also revealed by Fig. 8(b). However, this observation contrasts with the knowledge gained from investigations of weakly and moderately supercooled regimes of ionic liquids that charge correlation effects are not that strong in these materials.62,63 Further investigations, preferably involving direct diffusivity measurements of deeply supercooled ionic liquids would be beneficial in clarifying these seemingly contrasting observations.
Despite the complexity implied by the, in general, differently temperature-dependent time scales that are summarized in Fig. 8(b), several common trends can be recognized:
(i) For all mixtures, the lowest diffusivity estimates are those based on the SE relation, eqn (8). This is barely surprising, considering that the macroscopic viscosity η0 corresponds to the terminal flow of the polymer chains. Although these are not very long (note the relatively low molecular weight of PVP), the probed η0 should be larger than the microscopic viscosity governing the local friction of the monomers (x = 0) or of ionic plasticizers (0 < x < 1). Additionally, in the high-viscosity regime the SE approach is to be considered with caution since it neglects the decoupling phenomenon. The effects of a dynamic decoupling that were mentioned in the introduction section were shown early on to play a significant role for the diffusion of nonionic additives near the glass transition temperature of polymeric materials.5,24
(ii) For the low-concentration electrolytes (x = 0.1 and 0.16), the NE predictions based on the macroscopic conductivity agree excellently with the AW calculations which are based on the microscopic ionic relaxation time τε. This observation confirms that the elementary step for single-ion diffusion corresponds to the radius l of the Coulombic cage. For the low-charge concentrations l is much larger than the effective steric radius of the monomeric units.
(iii) The agreement between the collective (NE based) and the single-particle (AW based) diffusivities breaks down at high charge concentrations (x = 0.66 and 1). The relationship DNE < DAW,ε prevailing in this regime indicates that cooperative effects emerge which suppress the conductivity, an effect usually quantified in terms of the Haven ratio.
(iv) At variance with the situation for x = 0.1 and 0.16, for the electrolytes with x = 0.66 and 1 the diffusivities DAW,ε and DAW,G are similar. This finding indicates again that local-charge and matrix dynamics are decoupled at low x and more coupled in the ion-rich materials.
(v) The time scales from DSC are typically close to those extrapolated from the rheological measurements with the most significant deviation observed for x = 0.33. However, for this sample the relaxation time from the low-frequency shear peak, see the crosses (×) in Fig. 8(a), is not too far from τDSC.
IV. Concluding remarks
To summarize, the present work explores the frequency-dependent dielectric and shear rheological responses of PVP with a molecular weight of 2500 g mol−1, and of mixtures of this polymer with the ionic liquid PT. Differential scanning calorimetry has additionally been employed. The dielectric and mechanical responses of PVP are dominated by the structural relaxation process of this material. As is typical for neat glassforming systems, the characteristic time scales extracted from the analysis of the shear modulus function of PVP are somewhat smaller than those from its dielectric compliance. However, with the addition of an ionic liquid the dielectric response of PVP-PT becomes faster than its mechanical counterpart. Corroborated by the significant differences found between the glass transition temperatures accessed via calorimetry and those estimated from the dielectric dynamics, and by the sigmoidal-like variation of the logarithm of dielectric time scales as a function of inverse temperature, this observation indicates that a decoupling of charge dynamics from segmental rearrangements emerges in PVP-PT at low and intermediate ionic concentrations.Although signs of crystallization have not been detected for any of the investigated systems, an anomalously broad shear response and a bimodality of the calorimetric response indicate the emergence of a microscopic phase separation for a molar PT concentration near 33%. Disregarding this concentration, the analysis of the glass transition temperatures for the other mixtures reveals that the ionic-liquid additive is a good plasticizer for PVP. This is clearly seen from the concentration dependent Tg which can be described well by the Gordon–Taylor equation. Free parameters are not necessary for this approach, since the corresponding specific heat steps are directly available from calorimetric investigations.
The parameterization of the dielectric and rheological spectra allowed us to extract dynamic and transport coefficients such as the characteristic times, steady-state viscosities, and steady-state conductivities. Using these parameters, we estimated the translational diffusivities of the ionic additive PVP, based on the Stokes–Einstein and Nernst–Einstein relations as well as on the conductivity and mechanical versions of the Almond–West formalism. Although the present case – with the dynamics of the additive being faster than the matrix rearrangements – constitutes a clear deviation from the hydrodynamic limit, these approaches provide several interesting observations: first, for all materials, the smallest diffusivities are those estimated using the SE relation. Second, the NE predictions that are based on the macroscopic conductivity and the AW calculations based on the microscopic ionic relaxation time agree well at low ionic concentration, while this agreement breaks down for large ion densities.
Our study reveals that mixing ionic liquids with polar polymers enables the investigation of charge dynamics in the full concentration range by overcoming the dissociation and solubility issues of salt-doped polymer electrolytes. The results obtained in this work thus are relevant for understanding and controlling the microscopic transport in amorphous dispersions and polymer electrolytes near their glass transition.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The Deutsche Forschungsgemeinschaft supported this project under Grant No. 461147152. We thank Ali Mansuri (Department of Biochemical and Chemical Engineering, TU Dortmund University) for assistance while taking the DSC data. C. G. acknowledges partial support from “Fast and Cooperative Ion Transport in Polymer-Based Materials (FaCT)”, an Energy Frontier Research Center funded by the U.S. Department of Energy, Office of Science, Basic Energy Sciences for his contribution to data analysis and interpretation, preparation of figures and writing of the manuscript.References
- Disordered Pharmaceutical Materials, ed. Descamps, M., Wiley-VCH Verlag, Weinheim, Germany, 2016 Search PubMed.
- M. Rams-Baron, R. Jachowicz, E. Boldyreva, D. Zhou, W. Jamroz and M. Paluch, Amorphous Drugs Benefits and Challenges, Springer, Switzerland, 2018 Search PubMed.
- S. N. M. Yusoff, A. Kamari and N. F. A. Aljafree, A review of materials used as carrier agents in pesticide formulations, Int. J. Environ. Sci. Technol., 2016, 13, 2977 CrossRef.
- E. Rössler, Indications for a change of diffusion mechanism in supercooled liquids, Phys. Rev. Lett., 1990, 65, 1595 CrossRef PubMed.
- M. T. Cicerone, F. R. Blackburn and M. D. Ediger, Anomalous diffusion of probe molecules in polystyrene: evidence for spatially heterogeneous segmental dynamics, Macromolecules, 1995, 28, 8224–8232 CrossRef CAS.
- M. T. Cicerone, P. A. Wagner and M. D. Ediger, Translational Diffusion on Heterogeneous Lattices: A Model for Dynamics in Glass Forming Materials, J. Phys. Chem. B, 1997, 101, 8727–8734 CrossRef CAS.
- G. Diezemann, H. Sillescu, G. Hinze and R. Böhmer, Rotational correlation functions and apparently enhanced translational diffusion in a free-energy landscape model for the α-relaxation in glass-forming liquids, Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top., 1998, 57, 4398–4410 CrossRef CAS.
- K. L. Ngai, Alternative Explanation of the Difference between Translational Diffusion and Rotational Diffusion in Supercooled Liquids, J. Phys. Chem. B, 1999, 103, 10684–10694 CrossRef CAS.
- F. H. Stillinger and J. A. Hodgdon, Translation-rotation paradox for diffusion in fragile glass-forming liquids, Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top., 1994, 50, 2064–2068 CrossRef CAS PubMed.
- N. Ramesh, P. K. Davis, J. M. Zielinski, R. P. Danner and J. L. Duda, Application of Free-Volume Theory to Self Diffusion of Solvents in Polymers below the Glass Transition Temperature: A Review, J. Polym. Sci. B Polym. Phys., 2011, 49, 1629–1644 CrossRef CAS.
- G. Tarjus and D. Kivelson, Breakdown of the Stokes–Einstein relation in supercooled liquids, J. Chem. Phys., 1995, 103, 3071–3073 CrossRef CAS.
- F. Puosi, A. Pasturel, N. Jakse and D. Leporini, Communication: fast dynamics perspective on the breakdown of the Stokes-Einstein law in fragile glassformers, J. Chem. Phys., 2018, 148, 131102 CrossRef CAS PubMed.
- S. P. Bhardwaj and R. Suryanarayanan, Molecular Mobility as an Effective Predictor of the Physical Stability of Amorphous Trehalose, Mol. Pharmaceutics, 2012, 9, 3209–3217 CrossRef CAS PubMed.
- A. Mansuri, M. Völkel, T. Feuerbach, J. Winck, A. W. P. Vermeer, W. Hoheisel and M. Thommes, Modified Free Volume Theory for Self-Diffusion of Small Molecules in Amorphous Polymers, Macromolecules, 2023, 56, 3224–3237 CrossRef CAS.
- K. V. Edmond, M. T. Elsesser, G. L. Hunter, D. J. Pine and E. R. Weeks, Decoupling of rotational and translational diffusion in supercooled colloidal fluids, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 17891–17896 CrossRef CAS PubMed.
- K. L. Ngai, On Enhanced Translational Diffusion or the Fractional Stokes−Einstein Relation Observed in a Supercooled Ionic Liquid, J. Phys. Chem. B, 2006, 110, 26211–26214 CrossRef CAS PubMed.
- K. L. Ngai and S. Capaccioli, An explanation of the differences in diffusivity of the components of the metallic glass Pd43Cu27Ni10P20, J. Chem. Phys., 2013, 138, 094504 CrossRef CAS PubMed.
- V. Dubey, S. Dueby and S. Daschakraborty, Breakdown of the Stokes-Einstein relation in supercooled water: the jump-diffusion perspective, Phys. Chem. Chem. Phys., 2021, 23, 19964–19986 RSC.
- M. Roos, M. Ott, M. Hofmann, S. Link, E. Rössler, J. Balbach, A. Krushelnitsky and K. Saalwächter, Coupling and Decoupling of Rotational and Translational Diffusion of Proteins under Crowding Conditions, J. Am. Chem. Soc., 2016, 138, 10365–10372 CrossRef CAS PubMed.
- C. A. Angell, Dynamic processes in ionic glasses, Chem. Rev., 1990, 90, 523–542 CrossRef CAS.
- J. C. Dyre, P. Maass, B. Roling and D. L. Sidebottom, Fundamental questions relating to ion conduction in disordered solids, Rep. Prog. Phys., 2009, 72, 046501 CrossRef.
- J. Habasaki, C. León and K. L. Ngai, Dynamics of Glassy, Crystalline and Liquid Ionic Conductors: Experiments, Theories, Simulations, Springer, Switzerland, 2017 Search PubMed.
- C. Gainaru, R. Kumar, I. Popov, A. Rahman Md, M. Lehmann, E. Stacy, V. Bocharova, B. G. Sumpter, T. Saito, K. S. Schweizer and A. P. Sokolov, Mechanisms Controlling the Energy Barrier for Ion Hopping in Polymer Electrolytes, Macromolecules, 2023, 56, 6051–6059 CrossRef CAS.
- D. Ehlich and H. Sillescu, Tracer Diffusion at the Glass Transition, Macromolecules, 1990, 23, 1600–1610 CrossRef CAS.
- A. V. Veniaminov and H. Sillescu, Polymer and dye probe diffusion in poly(methyl methacrylate) below the glass transition studied by forced Rayleigh scattering, Macromolecules, 1999, 32, 1828–1837 CrossRef CAS.
- F. R. Blackburn, C. Y. Wang and M. D. Ediger, Translational and rotational motion of probes in supercooled 1,3,5-tris(naphthyl)benzene, J. Phys. Chem., 1996, 100, 18249–18257 Search PubMed.
- M. T. Cicerone and M. D. Ediger, Enhanced Translation of Probe Molecules in Supercooled O-terphenyl: Signature of Spatially Heterogeneous Dynamics?, J. Chem. Phys., 1996, 104, 7210–7218 CrossRef CAS.
- J. Rajesh Rajian, W. Huang, R. Richert and E. L. Quitevis, Enhanced Translational Diffusion of Rubrene in Sucrose Benzoate, J. Chem. Phys., 2006, 124, 14510 CrossRef PubMed.
- Y. Hwang and M. D. Ediger, Enhanced translational diffusion of rubrene and tetracene in polysulfone, J. Polym. Sci., Part B: Polym. Phys., 2003, 34, 2853–2861 CrossRef.
- D. B. Hall, D. D. Deppe, K. E. Hamilton, A. Dhinojwala and J. M. Torkelson, Probe translational and rotational diffusion in polymers near Tg: roles of probe size, shape, and secondary bonding in deviations from Debye–Stokes–Einstein scaling, J. Non-Cryst. Solids, 1998, 235–237, 48–56 CrossRef CAS.
- N. L. Mandel, S. Lee, K. Kim, K. Paeng and L. J. Kaufman, Single molecule demonstration of Debye–Stokes–Einstein breakdown in polystyrene near the glass transition temperature, Nat. Commun., 2022, 13, 3580 CrossRef CAS PubMed.
- P. Medick, M. Vogel and E. A. Rössler, Large angle jumps of small molecules in amorphous matrices analyzed by 2D exchange NMR, J. Magn. Reson., 2002, 159, 126–136 CrossRef CAS.
- I. Chang and H. Sillescu, Heterogeneity at the Glass Transition: Translational and Rotational Self-Diffusion, J. Phys. Chem. B, 1997, 101, 8794–8801 CrossRef CAS.
- C. Gainaru, S. Ahlmann, L. S. Röwekamp, K. Moch, S. P. Bierwirth and R. Böhmer, Rheology Based Estimates of Self- and Collective Diffusivities in Viscous Liquids, J. Chem. Phys., 2021, 155, 011101 CrossRef CAS PubMed.
- S. Cerveny, Á. Alegría and J. Colmenero, Broadband dielectric investigation on poly(vinylpyrrolidone) and its water mixtures, J. Chem. Phys., 2008, 128, 044901 CrossRef PubMed.
- A. Elhoussiny, A. Ward, S. Mansour and S. Abd-El-Messieh, Biodegradable blends based on polyvinylpyrrolidone for insulation purposes, J. Appl. Polym. Sci., 2012, 124, 3879–3891 CrossRef.
- S. K. Jain and G. P. Johari, Dielectric Studies of Molecular Motions in the Glassy States of Pure and Aqueous Poly(vinylpyrrolidone), J. Phys. Chem., 1988, 92, 5851–5854 CrossRef CAS.
- T. Sun and H. E. King, Aggregation Behavior in the Semidilute Poly(N-vinyl-2-pyrrolidone)/Water System, Macromolecules, 1996, 29, 3175–3181 CrossRef CAS.
- D. Sen, S. Mazumder, P. Sengupta, A. K. Ghosh and V. Ramachandhran, Small-Angle X-Ray Scattering Study of Porous Polysulfone and Poly(VinylPyrrolidone)/Polysulfone Blend Membranes, J. Macromol. Sci., Part B: Phys., 2000, 39, 235–243 CrossRef.
- M. M. Knopp, N. E. Olesen, P. Holm, P. Langguth, R. Holm and T. Rades, Influence of Polymer Molecular Weight on Drug–polymer Solubility: A Comparison between Experimentally Determined Solubility in PVP and Prediction Derived from Solubility in Monomer, J. Pharmaceutical Sci., 2015, 104, 2905–2912 CrossRef CAS PubMed.
- R. Busselez, A. Arbe, S. Cerveny, S. Capponi, J. Colmenero and B. Frick, Component dynamics in polyvinylpyrrolidone concentrated aqueous solutions, J. Chem. Phys., 2012, 137, 084902 CrossRef PubMed.
- K. Sasaki, M. Takatsuka, N. Shinyashiki and K. L. Ngai, Relating the dynamics of hydrated poly(vinyl pyrrolidone) to the dynamics of highly asymmetric mixtures and polymer blends, J. Mol. Liq., 2021, 333, 115907 CrossRef CAS.
- S. A. Ingole and A. Kumbharkhane, Temperature dependent Broadband dielectric relaxation study of Aqueous Polyvinylpyrrolidone (PVP K-15, K-30 & K-90) using a TDR, Phys. Chem. Liq., 2021, 59, 806–816 CrossRef CAS.
- N. Shinyashiki, M. Miyara, S. Nakano, W. Yamamoto, M. Ueshima, D. Imoto, K. Sasaki, R. Kita and S. Yagihara, Dielectric relaxation strength and magnitude of dipole moment of poly(vinyl pyrrolidone) in polar solutions, J. Mol. Liq., 2013, 181, 110–114 CrossRef CAS.
- M. Nakada, H. Ishida and Y. Fusuhima, Structural and dynamical characterisation of intermediate water interacting polyvinyl pyrrolidone, Materialia, 2020, 12, 100743 CrossRef CAS.
- M. Nakada, T. Yamada, K. Ikeda and T. Otomo, Static and Dynamic Structure Analysis of Intermediate Water on Polyvinyl Pyrrolidone using Neutron Scattering, JPS Conf. Proc., 2021, 33, 011080, DOI:10.7566/JPSCP.33.011080.
- K. Kothari, V. Ragoonanan and R. Suryanarayanan, The role of polymer concentration on the molecular mobility and physical stability of nifedipine solid dispersions, Mol. Pharmaceutics, 2015, 12, 1477–1484 CrossRef CAS PubMed.
- X. Yuan, D. Sperger and E. J. Munson, Investigating Miscibility and Molecular Mobility of Nifedipine-PVP Amorphous Solid Dispersions Using Solid-State NMR Spectroscopy, Mol. Pharmaceutics, 2014, 11, 329–337 CrossRef CAS PubMed.
- X. Yuan, T. X. Xiang, B. D. Anderson and E. J. Munson, Hydrogen bonding interactions in amorphous indomethacin and its amorphous solid dispersions with poly(vinylpyrrolidone) and poly-(vinylpyrrolidone-co-vinyl acetate) studied using 13C solid-state NMR, Mol. Pharmaceutics, 2015, 12, 4518–4528 CrossRef CAS PubMed.
- S. Mohapatra, S. Samanta, K. Kothari, P. Mistry and R. Suryanarayanan, Effect of Polymer Molecular Weight on the Crystallization Behavior of Indomethacin Amorphous Solid Dispersions, Cryst. Growth Des., 2017, 17, 3142–3150 CrossRef CAS.
- A. Mathers, F. Hassouna, M. Klajmon and M. Fulem, Comparative Study of DSC-Based Protocols for API–Polymer Solubility Determination, Mol. Pharmaceutics, 2021, 18, 1742–1757 CrossRef CAS PubMed.
- A. Mansuri, P. Münzner, A. Heermant, F. Patzina, T. Feuerbach, J. Winck, A. W. P. Vermeer, W. Hoheisel, R. Böhmer, C. Gainaru and M. Thommes, Molecular Dynamics and Diffusion in Amorphous Solid Dispersions Containing Imidacloprid, Mol. Pharmaceutics, 2023, 20, 2067–2079 CrossRef CAS PubMed.
- I. Osada, H. de Vries, B. Scrosati and S. Passerini, Ionic-Liquid-Based Polymer Electrolytes for Battery Applications, Angew. Chem., Int. Ed., 2016, 55, 500–513 CrossRef CAS PubMed.
- J. Muldoon, C. B. Bucur, N. Boaretto, T. Gregory and V. di Noto, Polymers: Opening Doors to Future Batteries, Polym. Rev., 2015, 55, 208–246 CrossRef CAS.
- J. Yi, S. Guo, P. He and H. Zhou, Status and Prospects of Polymer Electrolytes for Solid-State Li−O2 (Air) Batteries, Energy Environ. Sci., 2017, 10, 860–884 RSC.
- V. Bocharova and A. P. Sokolov, Perspectives for Polymer Electrolytes: A View from Fundamentals of Ionic Conductivity, Macromolecules, 2020, 53, 4141–4157 CrossRef CAS.
- D. Devaux, R. Bouchet, D. Glé and R. Denoyel, Mechanism of ion transport in PEO/LiTFSI complexes: Effect of temperature, molecular weight and end groups, Solid State Ionics, 2012, 227, 119–127 CrossRef CAS.
- L. Edman, M. M. Doeff, A. Ferry, J. Kerr and L. C. De Jonghe, Transport Properties of the Solid Polymer Electrolyte System P(EO)nLiTFSI, J. Phys. Chem. B, 2000, 104, 3476–3480 CrossRef CAS.
- Y. Xia, T. Fujieda, K. Tatsumi, P. P. Prosini and T. Sakai, Thermal and electrochemical stability of cathode materials in solid polymer electrolyte, J. Power Sources, 2001, 92, 234–243 CrossRef CAS.
- D. M. Correia, L. C. Fernandes, P. M. Martins, C. García-Astrain, C. M. Costa, J. Reguera and S. Lanceros-Méndez, Ionic Liquid–Polymer Composites: A New Platform for Multifunctional Applications, Adv. Funct. Mater., 2020, 30, 1909736, DOI:10.1002/adfm.201909736.
- S. N. Pedro, C. S. R. Freire, A. J. D. Silvestre and M. G. Freire, The Role of Ionic Liquids in the Pharmaceutical Field: An Overview of Relevant Applications, Int. J. Mol. Sci., 2020, 21, 8298 CrossRef CAS PubMed.
- J. R. Sangoro, A. Serghei, S. Naumov, P. Galvosas, J. Kärger, C. Wespe, F. Bordusa and F. Kremer, Charge Transport and Mass Transport in Imidazolium-Based Ionic Liquids, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2008, 77, 051202 CrossRef CAS PubMed.
- C. Gainaru, E. W. Stacy, V. Bocharova, M. Gobet, A. P. Holt, T. Saito, S. Greenbaum and A. P. Sokolov, Mechanism of conductivity relaxation in liquid and polymeric electrolytes: Direct link between conductivity and diffusivity, J. Phys. Chem. B, 2016, 120, 11074–11083 CrossRef CAS PubMed.
- D. P. Almond and A. R. West, Mobile ion concentrations in solid electrolytes from an analysis of a.c. conductivity, Solid State Ionics, 1983, 9/10, 277–282 CrossRef.
- B. Roling, C. Martiny and S. Bruckner, Ion transport in glass: Influence of glassy structure on spatial extent of nonrandom ion hopping, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 63, 214203 CrossRef.
- C. A. Thomann, P. Münzner, K. Moch, J. Jacquemin, P. Goodrich, A. Sokolov, R. Böhmer and C. Gainaru, Tuning the dynamics of imidazolium-based ionic liquids via hydrogen bonding. I. The viscous regime, J. Chem. Phys., 2020, 153, 194501 CrossRef CAS PubMed.
- H. Wagner and R. Richert, Equilibrium and non-equilibrium type β-relaxations: D-sorbitol versus o-terphenyl, J. Phys. Chem. B, 1999, 103, 4071–4077 CrossRef CAS.
- Y. Sun, J. Tao, G. G. Z. Zhang and L. Yu, Solubilities of Crystalline Drugs in Polymers: An Improved Analytical Method and Comparison of Solubilities of Indomethacin and Nifedipine in PVP, PVP/VA, and PVAc, J. Pharm. Sci., 2010, 99, 4023–4031 CrossRef CAS PubMed.
- M. Wübbenhorst and J. van Turnhout, Analysis of Complex Dielectric Spectra. I. One-Dimensional Derivative Techniques and Three-Dimensional Modelling, J. Non-Cryst. Solids, 2002, 305, 40–49 CrossRef.
- A. J. Staverman and F. Schwarzl, in Physik der Hochpolymeren; Band IV, ed. Stuart, H. A., Springer, Berlin, 1955, pp. 1–121 Search PubMed.
- H. C. Booij and G. P. J. M. Thoone, Generalization of Kramers-Kronig transforms and some approximations of relations between viscoelastic quantities, Rheol. Acta, 1982, 21, 15–24 CrossRef.
- C. J. F. Böttcher and P. Bordewijk, Theory of electric polarization II: Dielectrics in time-dependent fields, Elsevier, Amsterdam, 1978 Search PubMed.
- S. Emmert, M. Wolf, R. Gulich, S. Krohns, S. Kastner, P. Lunkenheimera and A. Loidl, Electrode polarization effects in broadband dielectric spectroscopy, Eur. Phys. J. B, 2011, 83, 157–165 CrossRef CAS.
- P. Ben Ishai, M. S. Talary, A. Caduff, E. Levy and Y. Feldman, Electrode polarization in dielectric measurements: a review, Meas. Sci. Technol., 2013, 24, 102001 CrossRef CAS.
- F. Kremer and A. Schönhals, Broadband Dielectric Spectroscopy, Springer, Berlin, Heidelberg, 2003 Search PubMed.
- K. Wendler, F. Dommert, Y. Y. Zhao, R. Berger, C. Holm and L. Delle Site, Ionic liquids studied across different scales: A computational perspective, Faraday Discuss., 2012, 154, 1359–6640 RSC.
- F. Wieland, V. Bocharova, P. Münzner, W. Hiller, R. Sakrowski, C. Sternemann, R. Böhmer, A. P. Sokolov and C. Gainaru, Structure and dynamics of short-chain polymerized ionic liquids, J. Chem. Phys., 2019, 151, 034903 CrossRef CAS PubMed.
- P. Münzner, L. Hoffmann, R. Böhmer and C. Gainaru, Deeply supercooled aqueous LiCl solution studied by frequency-resolved shear rheology, J. Chem. Phys., 2019, 150, 234505 CrossRef PubMed.
- S. Ahlmann, P. Münzner, K. Moch, A. P. Sokolov, R. Böhmer and C. Gainaru, The relationship between charge and molecular dynamics in viscous acid hydrates, J. Chem. Phys., 2021, 155, 014505 CrossRef CAS PubMed.
- S. P. Bierwirth, C. Gainaru and R. Böhmer, Coexistence of two structural relaxation processes in monohydroxy alcohol–alkyl halogen mixtures: Dielectric and rheological studies, J. Chem. Phys., 2018, 149, 044509 CrossRef PubMed.
- S. P. Bierwirth, C. Gainaru and R. Böhmer, Communication: Correlation of terminal relaxation rate and viscosity enhancement in supramolecular small-molecule liquids, J. Chem. Phys., 2018, 148, 221102 CrossRef CAS PubMed.
- M. A. Rocha, M. Bastos, J. A. Coutinho and L. M. N. B. F. Santos, Heat capacities at 298.15 K of the extended [CnC1im][Ntf2] ionic liquid series, J. Chem. Thermodyn., 2012, 53, 140–143 CrossRef CAS.
- M. Gordon and J. S. Taylor, Ideal Copolymers and the Second-Order Transitions of Synthetic Rubbers. I. Noncrystalline Copolymers, Rubber Chem. Technol., 1953, 26, 323–335 CrossRef CAS.
- P. Sippel, P. Lunkenheimer, S. Krohns, E. Thoms and A. Loidl, Importance of liquid fragility for energy applications of ionic liquids, Sci. Rep., 2015, 5, 13922 CrossRef CAS PubMed.
- R. Böhmer, K. L. Ngai, C. A. Angell and D. J. Plazek, Nonexponential Relaxations in Strong and Fragile Glass Formers, J. Chem. Phys., 1993, 99, 4201–4209 CrossRef.
- I. M. Hodge, Enthalpy relaxation and recovery in amorphous materials, J. Non-Cryst. Solids, 1994, 169, 211–266 CrossRef CAS.
- J. O. Isard, The Haven ratio in glasses, J. Non-Cryst. Solids, 1999, 246, 16–26 CrossRef CAS.
Footnote |
† Electronic supplementary information (ESI) available: As supporting information we present loss modulus spectra for PVP0.84PT0.16, G′, ε′, and 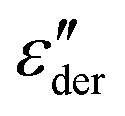 spectra for PVP1−xPTx with x = 0.16. 0.33, and 0.66, as well as details regarding the DSC measurements and how they were analyzed. Furthermore, the data in Fig. 7 are presented in a Walden-type plot. See DOI: https://doi.org/10.1039/d4cp01001a spectra for PVP1−xPTx with x = 0.16. 0.33, and 0.66, as well as details regarding the DSC measurements and how they were analyzed. Furthermore, the data in Fig. 7 are presented in a Walden-type plot. See DOI: https://doi.org/10.1039/d4cp01001a |
| This journal is © the Owner Societies 2024 |