
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEosin, blue LEDs and DIPEA are employed in a simple synthesis of (poly)cyclic O,O- and N,O-acetals†
Ioannis
Papadopoulos‡
,
Artemis
Bosveli‡
 ,
Tamsyn
Montagnon
,
Ioannis
Zachilas
,
Dimitris
Kalaitzakis
and
Georgios
Vassilikogiannakis
*
,
Tamsyn
Montagnon
,
Ioannis
Zachilas
,
Dimitris
Kalaitzakis
and
Georgios
Vassilikogiannakis
*
Department of Chemistry, University of Crete, Vasilika Vouton, 71003, Iraklion, Crete, Greece. E-mail: vasil@uoc.gr
First published on 30th April 2024
Abstract
A simple procedure for the synthesis of (poly)cyclic O,O- and N,O-acetals from various enol ethers, N-acyl enamines or Boc-protected enamines has been developed. The key step is a photocatalytic Stork–Ueno-type cylization using the very simple metal-free conditions of catalytic eosin, diisopropylamine in the green solvent ethanol with blue LED irradition.
Rigid (poly)cyclic structures, rich in sp3 carbons and bearing appropriately positioned heteroatoms, constitute highly privileged compounds for a variety of reasons. In the pressing search for new drugs, increasing the proportion of sp3-carbons and chiral centres within candidate compounds has been shown to increase rates of success.1 Strategically placed heteroatoms can improve a compound's solubility in biological fluids, its binding to a target protein2 or its ability to act as a ligand for transportation of metal cations across membranes.3 Moreover, when all these characteristics are included as part of a rigid polycyclic/spirocyclic structure there are additional benefits, such as, greater specificity (lower receptor promiscuity) and a reduced entropy penalty upon binding.1c,4 It was within this context that we hoped to develop a new and sustainable approach to (poly)cyclic O,O- and N,O-acetals; pharmacophores which occur in some very potent natural products and drugs.5 We wanted a general and mild method for their synthesis that wouldn’t place restrictions on the substrates that could be employed, and, by sustainable, we mean a highly efficient (cascade reaction sequence/one pot),6 metal-free process using a green solvent with visible spectrum light as the energy source.7
We saw the potential for a very simple solution to these challenges if some interesting reactivity that had been observed during our investigations into a biomimetic synthesis of pyridones, could be adapted and applied to this new task.8 During the pyridone work, we had seen how an adjacent oxygen functionality could facilitate the formation of a carbon-centred radical through homolysis of a C–I bond under very mild photocatalytic conditions (eosin,9 diisopropylethylamine DIPEA, blue LEDs). We wanted to see whether we could apply a similar strategy to a Stork–Ueno-type cyclization. Success, however, might require both an adaptation of the propagation mechanism and an alternative co-oxidant (for returning EY˙− to EY). While the Stork–Ueno disconnection has been used to make cyclic acetals,10 existing methodologies are associated with a number of negative issues that have become increasingly important in modern synthetic chemistry, such as; complex substrate preparation;10a use of metals (stoichiometric10d,e and/or toxic; for example, Bu3SnH10a,b); a need for high energy ultraviolet light;10c or a sensitive catalyst.10f What is more, we wanted to be the first to unite the substrate synthesis and cyclization steps into a one pot protocol (Scheme 1).
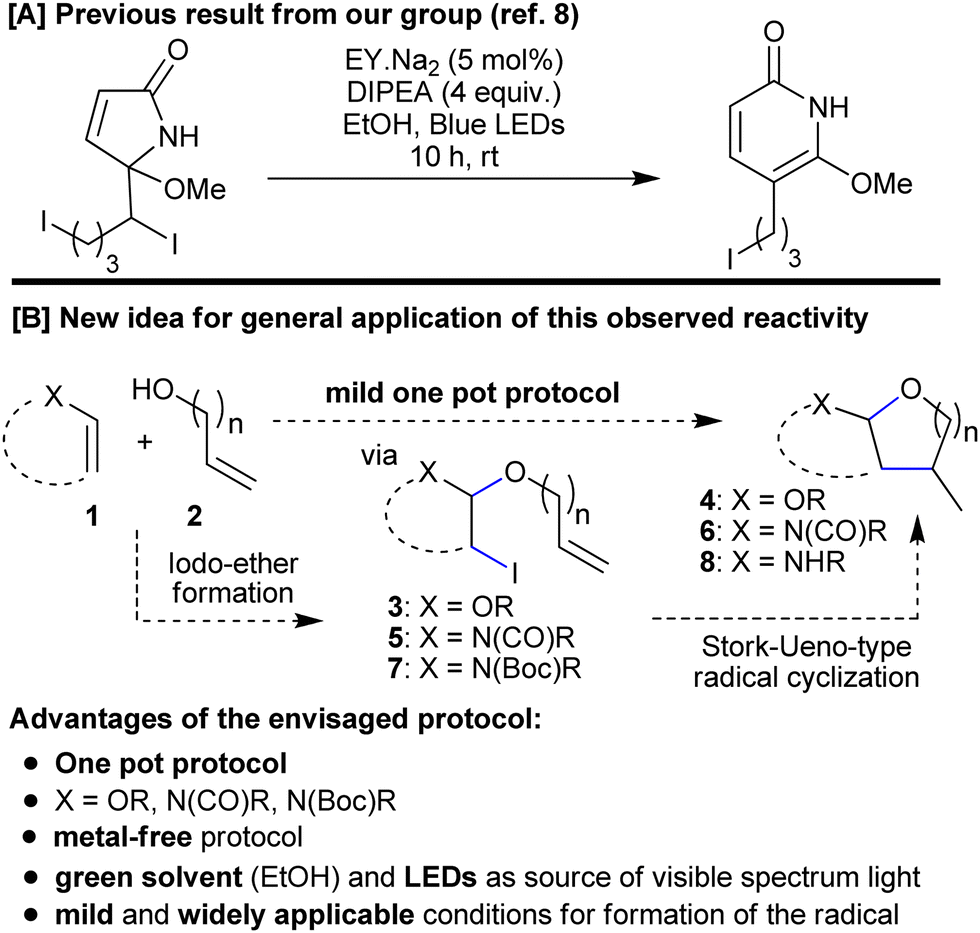 | ||
| Scheme 1 [A] Observed reactivity that formed the basis for the new idea. [B] Generalized concept for new one pot methodology targeting (poly)cyclic O,O- and N,O-acetals. | ||
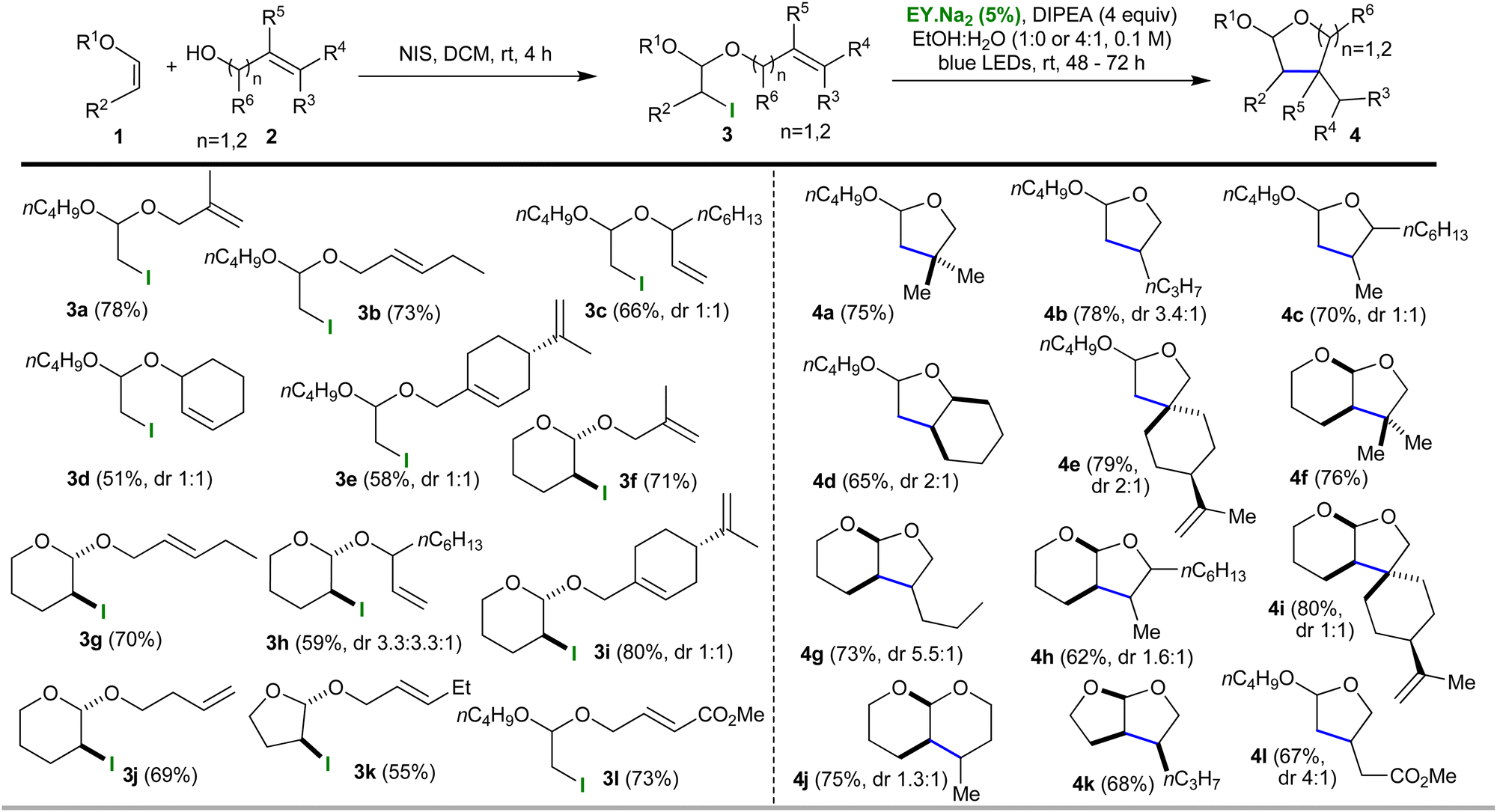
Our group is experienced in using photocatalytic one pot protocols to increase the efficiency and sustainability of synthetic methods.8,11 The first task is always to validate individual steps before attempting to combine them; in this case, we also had to investigate the mechanistic adjustments previously mentioned (the nature of the propagation steps and the potential need for a co-oxidant). To this end, we synthesised iodo-acetal 3a from vinyl ether 1a using N-iodosuccinimide and the allylic alcohol 2a (DCM, dark, rt, 4h, yield 78%, Table 1). Next, we sought to obtain proof of principle by using photocatalysis to transform iodo-acetal 3a into cyclic acetal 4a. Somewhat to our surprise, we found that the simplest envisaged conditions worked with no need for any further additives. The conditions employed were eosin (EY.Na2, 5 mol%) and DIPEA (4 equiv.) in EtOH at rt with 72 h irradiation from blue LEDs. 1H-NMR analysis of the crude mixture showed complete conversion of 3a to 4a had occurred and 4a was obtained in a yield of 75% (see; ESI†).
|
With the proof of principle in hand we sought to generate further examples. To this end, iodo-acetals 3b–3k were synthesised using the established conditions. Yields ranged from 51–80%, and, as expected, the cyclic enol ethers all afforded iodo-acetals that had a trans relationship between the iodo group and alcohol-derived side chain. With respect to other stereocentres, inseparable mixtures of diastereoisomers were observed in the products in the ratios shown in Table 1. Each iodo-acetal (3a–3k) was then subjected to the photocatalytic conditions (5 mol% EY.Na2, 4 equiv. DIPEA, in either EtOH or EtOH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O (4:1) at rt with blue LED irradiation) and monitored using either tlc or 1H NMR. With respect to the solvent, for the majority of the substrates EtOH:H2O was employed because this appeared to reduce the reaction times required (to 48 h); however, for a limited number of substrates (such as, 3a) the use of dry EtOH gave a cleaner result (albeit after a longer 72 h reaction time). Cyclised O,O-acetal products 4a–4k were formed in yields ranging from 65 to 80% and with the diastereoisomeric ratios shown in Table 1. The stereochemically simple iodo-acetals, 3f and 3k, each gave a single isomer, and, in the case of the former, NOE studies confirmed the expected relative stereochemistry of this product (4f, cis relationship for the bridgehead hydrogens). The simplicity of the reaction meant it could be scaled-up easily (1 mmol) with no effect on the yield.
:H2O (4:1) at rt with blue LED irradiation) and monitored using either tlc or 1H NMR. With respect to the solvent, for the majority of the substrates EtOH:H2O was employed because this appeared to reduce the reaction times required (to 48 h); however, for a limited number of substrates (such as, 3a) the use of dry EtOH gave a cleaner result (albeit after a longer 72 h reaction time). Cyclised O,O-acetal products 4a–4k were formed in yields ranging from 65 to 80% and with the diastereoisomeric ratios shown in Table 1. The stereochemically simple iodo-acetals, 3f and 3k, each gave a single isomer, and, in the case of the former, NOE studies confirmed the expected relative stereochemistry of this product (4f, cis relationship for the bridgehead hydrogens). The simplicity of the reaction meant it could be scaled-up easily (1 mmol) with no effect on the yield.
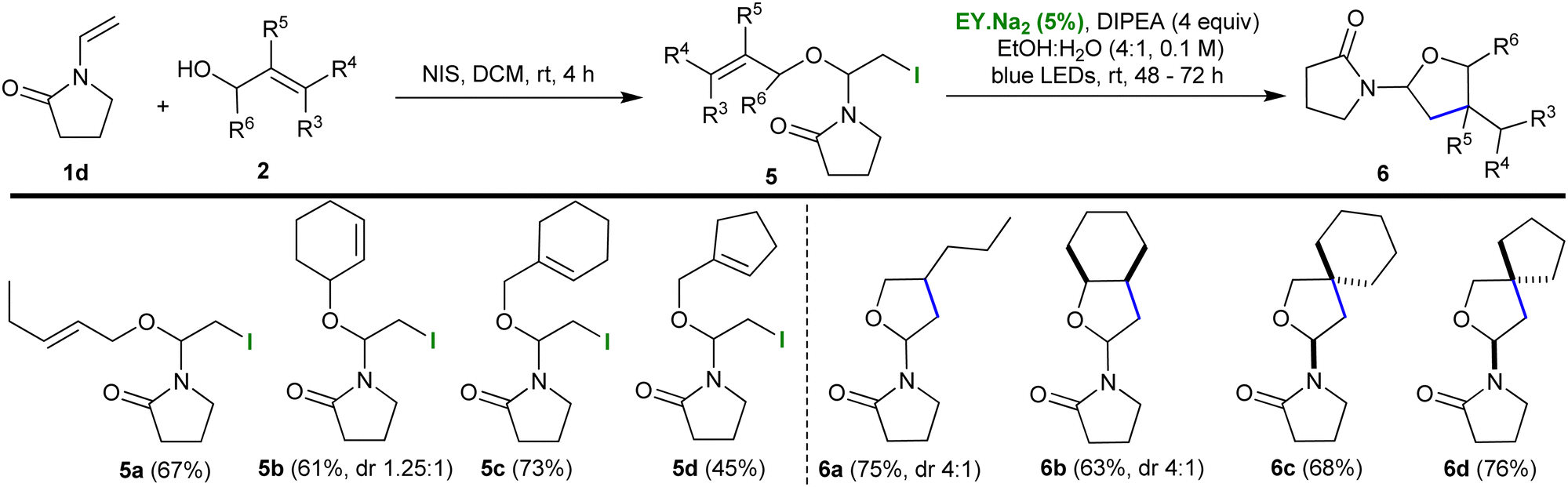
Next, the scope of the protocol was expanded by exchanging enol ether 1 with vinyl pyrrolidin-2-one 1d (Table 2). The previously developed conditions were employed to synthesize the iodo-ether motif; namely, 1d was reacted with the corresponding alcohol 2 and NIS in DCM for 4 h (yields 45–73%). The reaction could be scaled-up (to 2 mmol) with no effect on the yield. The resulting iodoalkoxy pyrrolidinones 5 were subjected to the optimised photocatalytic protocol which resulted in their smooth cyclization to the polycyclic pyrrolidinone-tetrahydrofurans 6 (yields 63–76%). Thus, the presence of an electron deficient nitrogen in the substrate was shown not to impede the reaction. Both 6a and 6b were formed as an inseparable mix of diastereoisomers with a dr of 4:1.
|
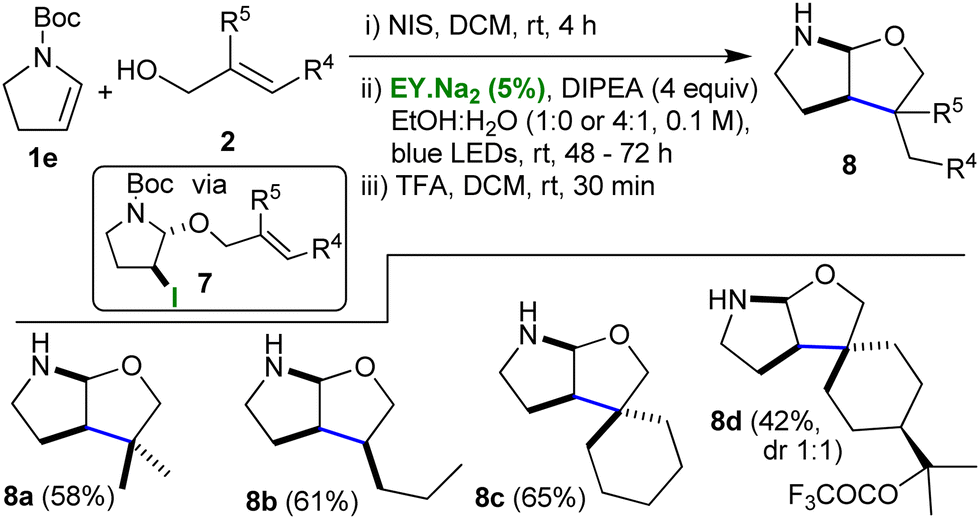
A third iteration expanded the scope further when the Boc- protected enamine 1e was used as the initial substrate (Table 3). Once again, the previously developed conditions were applied and first the iodoalkoxy pyrrolidines 7 were obtained (yields 61–73%), and, then, following application of the photocatalytic protocol with in situ deprotection, the cyclised N,O-acetals 8 were isolated (yields 42–65%).
|
The impact of the new multistep protocol we had developed would be significantly enhanced if we could convert it into a simple one pot procedure. We were assisted in this task by the simplicity of the individual steps and it was with minimal effort that we were able to combine the steps to give an easy and versatile one pot procedure for the synthesis of polycyclic O,O- and N,O-acetals (Scheme 2 and ESI†).
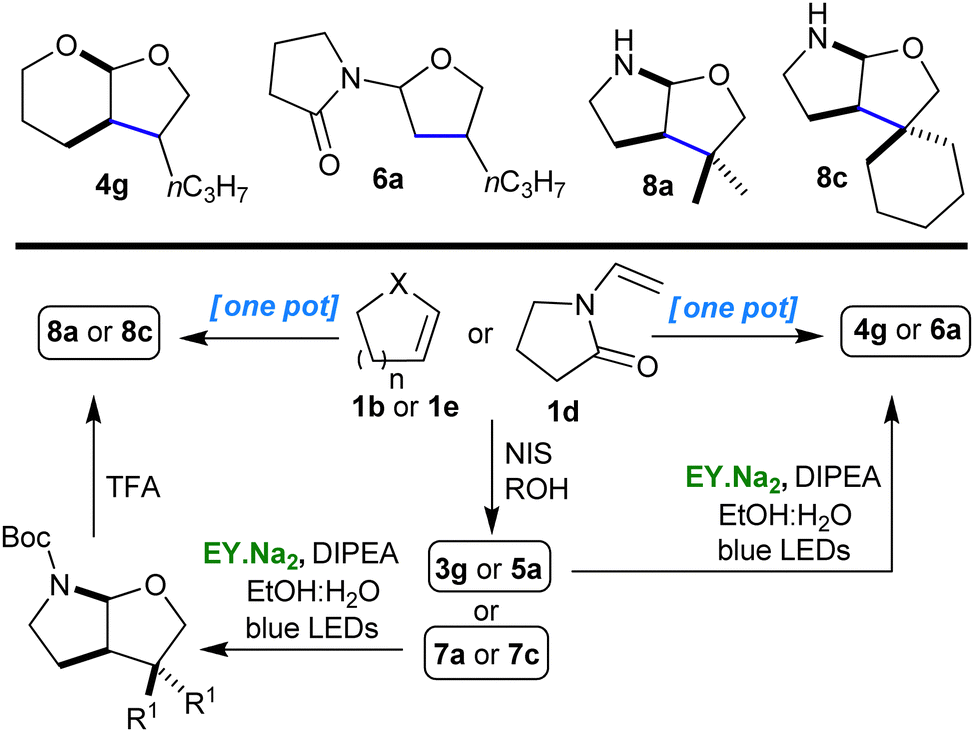 | ||
| Scheme 2 One pot procedure for the synthesis of O,O- and N,O-acetals (of types 4, 6 & 8) from 1b, 1d & 1e. | ||
On the basis of these results and our experience from other projects,8,11a as well as the seminal work of Leonori,12 we derived a logical mechanism in which we are proposing that DIPEA is fulfilling up to five different roles (roles 1–5, Scheme 3). Beginning with roles 1–3 which are supported by precedent8,11a,12 and by the following experiments: Emission quenching experiments, which were undertaken in order to derive Stern–Volmer plots, clearly show that DIPEA quenches the photocatalyst's excited state (role 1) at a substantially higher rate than representative iodo-acetal substrates (see, ESI†). Eosin's reduction potential (EEY/EY˙− = −1.06 V)13 suggests that it is not capable of direct reduction of a C–I bond and further proof that direct reduction by the photocatalyst (PC˙−) is not likely, came from testing a range of other photocatalysts. Specifically, both riboflavin and 9,10-dicyanoanthracene (DCA) that are capable of oxidising DIPEA (Eox = +0.68 V vs. SCE14), but which have considerably lower reduction potentials (−0.7915 and −0.91 V,13 repectively) still catalysed the reaction albeit with lower conversions accompanied by larger amounts of the by-product 4a′ (entries 7 & 8, Scheme 3). Moreover, switching the base to ones still capable of quenching the excited triplet state of EY confirmed that only those bearing an α–CH bond, needed for the formation of a radical at this α-position (B, e.g. DIPEA and 1,2,2,6,6-pentamethylpiperidine), could promote the reaction (role 3, entries 3 and 10–14). Roles 4 and 5 are in line with what is known in the literature where reduction of imminium cations by photocatalysts has been studied16 and amines as potential HAT donors has been explored.17 By- product 4a′ likely arises via a secondary cycle in which radical II reacts with starting material 3 (halogen atom transfer XAT) also producing radical I. Finally, 4a′ slowly converts to product (as seen by 1H NMR monitoring of the reactions) via analogous steps to those seen in the primary cycle (4a′ + B → II + A,12 followed by II + DIPEA → 4a). Further mechanistic details, including radical trapping experiments, are presented in the ESI.†
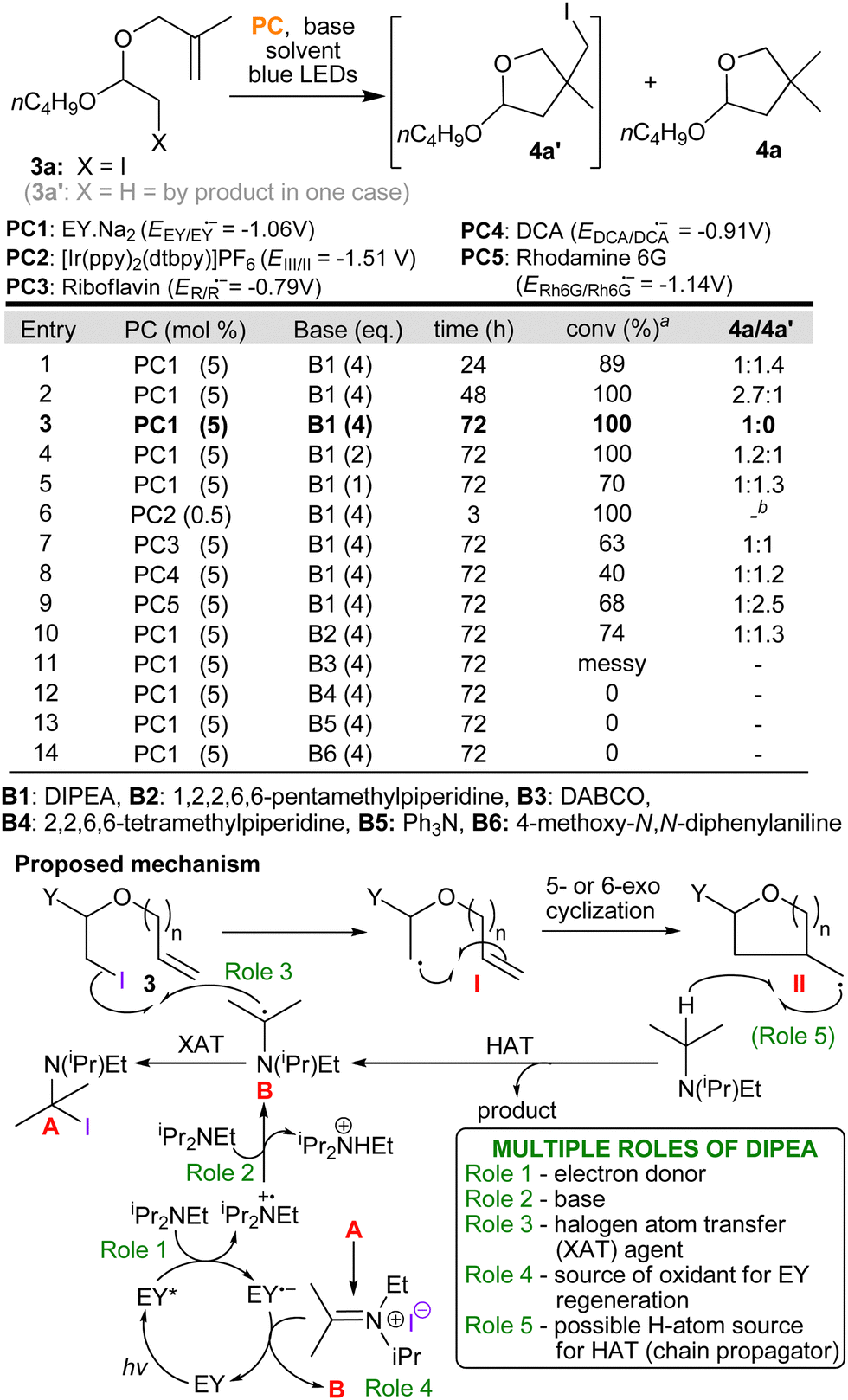 | ||
| Scheme 3 Experiments undertaken to obtain proof of principle, optimise the protocol and confirm mechanistic detail. aDetermined by 1H-NMR. bIn this reaction 4a was produced alongside significant quantities of reduced starting material 3a′ with 4a:3a′ = 4.8:1. | ||
Herein we have presented a very user-friendly methodology for synthesizing (poly)cyclic O,O- and N,O-acetals from enol ethers, N-acyl enamines or Boc-protected enamines, using photocatalysis. The developed protocol uses very simple conditions (eosin, DIPEA, EtOH:H2O, blue LEDs), is metal-free and uses a green solvent system.
This research has been co-financed by the European Regional Development Fund of the European Union and Greek national funds through the Operational Program Competitiveness, Entrepreneurship and Innovation, under the call RESEARCH – CREATE – INNOVATE (project code: T2EDK-02364).
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) F. Lovering, J. Bikker and C. Humblet, J. Med. Chem., 2009, 52, 6752 CrossRef CAS PubMed; (b) P. A. Clemens, N. A. Bodycombe, H. A. Carrinski, J. A. Wilson, A. F. Shamji, B. K. Wagner, A. N. Koehler and S. L. Schreiber, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 18787 CrossRef PubMed; (c) F. Lovering, Med. Chem. Commun., 2013, 4, 515 RSC.
- For O,O-acetal protein binding examples, see; (a) D. L. Surleraux, A. Tahri, W. G. Verschueren, G. M. Pille, H. A. De Kock, T. H. Jonckers, A. Peeters, S. De Meyer, H. Azijn, R. Pauwels, M.-P. de Bethune, N. M. King, M. Prabu-Jeyabalan, C. A. Schiffer and P. B. T. P. Wigerinck, J. Med. Chem., 2005, 48, 1813 CrossRef CAS PubMed; (b) A. K. Ghosh, S. Kulkarni, D. D. Anderson, L. Hong, A. Baldridge, Y. F. Wang, A. A. Chumanevich, A. Y. Kovalevsky, Y. Tojo, M. Amano, Y. Koh, J. Tang, I. T. Weber and H. Mitsuya, J. Med. Chem., 2009, 52, 7689 CrossRef CAS PubMed.
- D. Łowicki and A. Huczyński, BioMed Res. Int., 2013, 742149 Search PubMed.
- (a) E. M. Carreira and T. C. Fessard, Chem. Rev., 2014, 114, 8257 CrossRef CAS PubMed; (b) Y. Zheng, C. M. Tice and S. B. Singh, Bioorg. Med. Chem. Lett., 2014, 24, 3673 CrossRef CAS PubMed; (c) K. Hiesinger, D. Dar’in, E. Proschak and M. Krasavin, J. Med. Chem., 2021, 64, 150 CrossRef CAS PubMed.
- (a) For example: a Drugbank search 20/9/2023 yields the following examples amongst others; monensin (natural product, veterinary drug), Darunavir (HIV protease inhibitor, approved human drug), ribostamycin (natural product, WHO critical antimicrobial agent) and cedazuridine (approved human drug); (b) Paeonilide is a bioactive natural product belonging to the ginkgolide family, see; K. Harrar and O. Reiser, Chem. Commun., 2012, 48, 3457 RSC.
- (a) S. E. Denmark and A. Thorarensen, Chem. Rev., 1996, 96, 137 CrossRef CAS PubMed; (b) K. C. Nicolaou, T. Montagnon and S. A. Snyder, Chem. Commun., 2003, 551 RSC; (c) O. S. Magne, Curr. Green Chem., 2014, 1(3), 216–226 CrossRef; (d) Y. Hayashi, Chem. Sci., 2016, 7, 866 RSC.
- H. C. Erythropel, J. B. Zimmerman, T. M. de Winter, L. Petitjean, F. Melnikov, C. H. lam, A. W. Lounsbury, K. E. Mellor, N. Z. Janković, Q. Tu, L. N. Pincus, M. M. Falinski, W. Shi, P. Coish, D. L. Plata and P. T. Anastas, Green Chem., 2018, 20, 1929 RSC.
- D. Kalaitzakis, A. Bosveli, T. Montagnon and G. Vassilikogiannakis, Chem. – Eur. J., 2022, 28, e202200322 CrossRef CAS PubMed.
- A. Bosveli, T. Montagnon, D. Kalaitzakis and G. Vassilikogiannakis, Org. Biomol. Chem., 2021, 19, 3303 RSC.
- (a) Y. Ueno, K. Chino, M. Watanabe, O. Moriya and M. Okawara, J. Am. Chem. Soc., 1982, 104, 5564 CrossRef CAS; (b) G. Stork, R. Mook, S. A. Biller and S. D. Rychnovsky, J. Am. Chem. Soc., 1983, 105, 3741 CrossRef CAS; (c) J. Cossy, J. L. Ranaivosata and V. Bellosta, Tetrahedron Lett., 1994, 35, 8161 CrossRef CAS; (d) R. Yanada, Y. Koh, N. Nishimori, A. Matsumura, S. Obika, H. Mitsuya, N. Fujii and Y. Takemoto, J. Org. Chem., 2004, 69, 2417 CrossRef CAS PubMed; (e) J. Salom-Roig, F. Dénès and P. Renaud, Synthesis, 2004, 1903 Search PubMed; (f) F. Wu, L. Wang, D. A. Nicewicz, J. Chen and Y. Huang, iScience, 2020, 23, 101395 CrossRef CAS PubMed.
- For recent representative examples, see; (a) A. Bosveli, N. Griboura, I. Kampouropoulos, D. Kalaitzakis, T. Montagnon and G. Vassilikogiannakis, Chem. – Eur. J., 2023, 29, e202301713 CrossRef CAS PubMed; (b) L.-P. Apostolina, A. Bosveli, A. Profyllidou, T. Montagnon, V. Tsopanakis, M. Kaloumenou, D. Kalaitzakis and G. Vassilikogiannakis, Org. Lett., 2022, 24, 8786 CrossRef CAS PubMed; (c) D. Kalaitzakis, A. Bosveli, K. Sfakianaki, T. Montagnon and G. Vassilikogiannakis, Angew. Chem., Int. Ed., 2021, 60, 4335 CrossRef CAS PubMed.
- T. Constantin, M. Zanini, A. Regni, N. S. Sheikh, F. Juliá and D. Leonori, Science, 2020, 367, 1021 CrossRef CAS PubMed.
- N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075 CrossRef CAS PubMed.
- U. Pischel, K. Zhang, B. Hellrung, E. Haselbach, P.-A. Muller and W. M. Nau, J. Am. Chem. Soc., 2000, 122, 2027 CrossRef CAS.
- S. L. J. Tan and R. D. Webster, J. Am. Chem. Soc., 2012, 134, 5954 CrossRef CAS PubMed.
- For a lead reference, see; J. A. Leitch, T. Rossolini, T. Rogova, J. A. P. Maitland and D. J. Dixon, ACS Catal., 2020, 10, 2009 CrossRef CAS.
- For leading references, see; (a) J. Lalevée, X. Allonas and J.-P. Fouassier, J. Am. Chem. Soc., 2002, 124, 9613 CrossRef PubMed; (b) L. Capaldo, D. Ravelli and M. Fagnoni, Chem. Rev., 2022, 122, 1875 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cc01175a |
| ‡ These authors have contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |