
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
An artificial viral capsid decorated with a DNA aptamer internalizing into lymphoma cells†
Kentarou
Sakamoto
a,
Kohsuke
Uchiyama
a,
Takashi
Iwasaki
b,
Hiroshi
Inaba
 ac and
Kazunori
Matsuura
*ac
ac and
Kazunori
Matsuura
*ac
aDepartment of Chemistry and Biotechnology, Graduate School of Engineering, Tottori University, Tottori 680-8552, Japan. E-mail: ma2ra-k@tottori-u.ac.jp
bDepartment of Bioresources Science, Graduate School of Agricultural Sciences, Tottori University, Tottori 680-8553, Japan
cCentre for Research on Green Sustainable Chemistry, Tottori University, Tottori 680-8552, Japan
First published on 9th June 2023
Abstract
Tumor-specific drug-delivering nanocarriers could be a promising modality for next-generation tumor therapy. Here we developed a Burkitt lymphoma-specific DNA aptamer-labeled nanocarrier using the β-Annulus peptide, which forms a spherical nanoassembly called artificial viral capsid. Dynamic light scattering and transmission electron microscopy of the DNA aptamer-decorated artificial viral capsid showed the formation of spherical assemblies with a diameter of approximately 50–150 nm. The artificial viral capsid was selectively internalized into the Burkitt lymphoma cell line, Daudi, and doxorubicin complexed with the capsid selectively killed Daudi cells.
1. Introduction
Aptamers have received attention as artificial biomolecular elements that specifically recognize the special structure of a target molecule specifically.1 DNA aptamers are short sequences of artificial DNA that bind to a specific target and are mainly obtained via an in vitro selection method, which is also known as SELEX (Systematic Evolution of Ligands by EXponential enrichment).2 Using this method, more specific DNA sequences are obtained by repeating (1) selection of sequences bound to target molecules from the randomized DNA library, (2) polymerase chain reaction amplification, and (3) condensing sequences with higher affinity. DNA aptamers recognize target molecules by forming their own conformation determined by their sequence; for example, thrombin-binding DNA aptamers form a G-quadruplex structure that recognizes their target, thrombin.3,4Burkitt lymphoma is one of the most aggressive human cancers, and a drug delivery system (DDS) for the treatment of Burkitt lymphoma is highly desired. Tan et al. developed a DNA aptamer that can selectively bind to the Burkitt lymphoma cell line, Ramos cells.5 Among their selected DNA aptamers, TE02 (43 base) showed the highest binding affinity toward Ramos cells with a dissociation constant of 0.76 ± 0.13 nM. Using TE02 they developed a circular bispecific aptamer which can bind to both Ramos cells and T cells to induce T cell-mediated tumor-specific killing,6 and an aptamer-nanoparticle strip biosensor to detect Ramos cells for rapid and low-cost detection.7 Its further application as a probe for early diagnosis of Burkitt lymphoma is anticipated.
The conjugation of aptamers to various nanomaterials, such as liposomes, inorganic nanoparticles, and viral capsids enables highly selective cell-targeted drug delivery.8,9 For example, Kang et al. reported that liposomes decorated with the sgc8 aptamer, which specifically binds to leukemia cells, did not bind to nontarget cells but did bind to CCRF-CEM cells, the target leukemia cancer cells.10 In addition, Francis et al. developed aptamer-decorated MS2 viral capsids that target the tyrosine kinase receptor on Jurkat T cells.11
Natural spherical viral capsids with icosahedral symmetry self-assembled from protein subunits have gained attention as nanomaterials with specific sizes, aggregation numbers, and inner cavities.12 These properties enhance the possibility of using viral capsids as DDS carriers.13,14 Recently, the artificial design of symmetrical assembled protein/peptide subunits has enabled the construction of virus-like nanocapsules.15–18 To date, there have been several reports in that the protein/peptide nanocapsules are applied to in-cell experiments. Plasmid DNA was intracellularly delivered using viral capsid-like nanospheres self-assembled from de novo designed peptides.19–21 Hilvert et al. recently demonstrated that Aquifex aeolicus lumazine synthase spontaneously forms self-assembled capsid-like protein shells in E. coli that efficiently encapsulate its own mRNA by laboratory evolution.22–24 We also succeeded in creating an “artificial viral capsid” with a size of around 30–50 nm in water from self-assembly of a 24 a.a. β-Annulus peptide, which participates in the formation of the inner skeleton of the tomato bushy stunt virus.15,25 Small angle x-ray scattering measurements revealed that the interior of the capsid is hollow.25 ζ potential measurements revealed that the N- and C-termini of the peptide direct to the inner and outer surfaces of the capsid, respectively.26 By using these features, we succeeded in encapsulating DNA,26 mRNA,27 and His-tagged protein28,29 into the artificial viral capsid via N-terminal modification. We also created capsids decorated with gold nanoparticles,30 coiled-coil peptides,31 single strand DNA,32 human serum albumin,33 and RNase S protein.34,35
In this study, we developed a DNA aptamer-decorated artificial viral capsid and utilized it as a Burkitt lymphoma cell line-specific DDS carrier. A Burkitt lymphoma cell-recognizing DNA aptamer TE02 was conjugated to the C-terminus of the β-Annulus peptide via a maleimide-activated carboxylic acid linker (Fig. 1). This conjugate (β-Annulus-TE02) self-assembled into the TE02-decorated artificial viral capsid and this capsid specifically internalized into the Burkitt lymphoma cell line, Daudi.
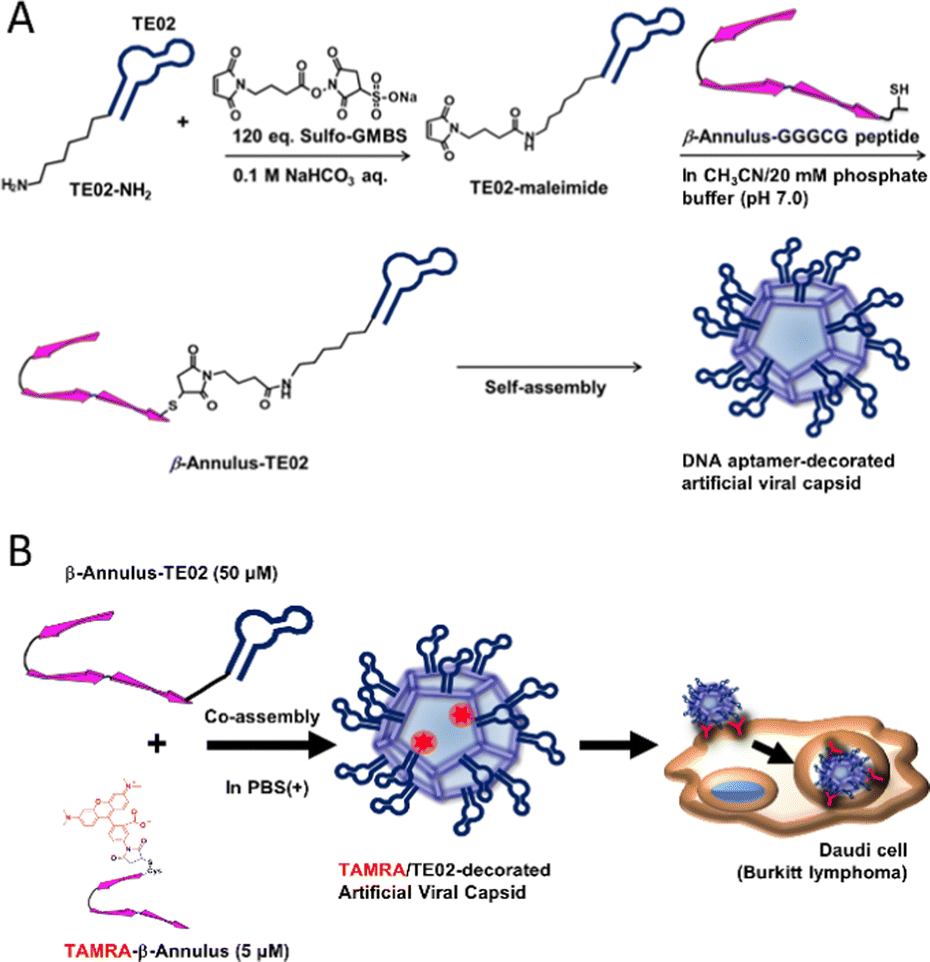 | ||
| Fig. 1 A schematic view of (A) formation of a DNA aptamer-decorated artificial viral capsid and (B) Burkitt lymphoma-specific internalization of the DNA aptamer-decorated artificial viral capsid. | ||
2. Materials and methods
General methods
All reagents, including chemicals, materials, and cell culture media were obtained from commercial sources and used without further purification. 5′-H2N-(CH2)6-DNA was synthesized and purchased from BEX Co. Ltd. Ultrapure water with a high resistivity (>18 MΩ cm) was purified using a Millipore Purification System (Milli-Q water) and was used as a solvent for the peptides and conjugates. Reversed-phase high performance liquid chromatography (RP-HPLC) was performed at ambient temperature using a Shimadzu LC-6AD liquid chromatography system equipped with a UV/vis detector (220 nm, Shimadzu SPD-10AVvp) and Inertsil WP300 C18 (GL Science) columns (250 × 4.6 mm or 250 × 20 mm). Matrix-assisted laser desorption ionization-time-of-flight mass spectra were obtained using an Autoflex T2 instrument (Bruker Daltonics) in linear/positive mode with α-cyano-4-hydroxy cinnamic acid (α-CHCA) or 3-hydroxypicolinic acid/diammonium hydrogen citrate = 8/1 as the matrix.Solid-phase peptide synthesis
The peptide Ile-Asn(Trt)-His(Trt)-Val-Gly-Gly-Thr(tBu)-Gly-Gly-Ala-Ile-Met-Ala-Pro-Val-Ala-Val-Thr(tBu)-Arg(Pbf)-Gln(Trt)-Leu-Val-Gly-Ser-Gly-Gly-Gly-Cys(Trt)-Gly-Alko-PEG resin was synthesized on Fmoc-Gly-Alko-PEG resin (500 mg, 0.125 mmol, Watanabe Chemical Ind. Ltd.) using Fmoc-based coupling reactions (4 equiv. of Fmoc amino acids). An N-methylpyrrolidone (NMP) solution containing (1-cyano-2-ethoxy-2-oxoethylidenaminooxy)dimethylamino-morpholino-carbenium hexafluorophosphate (COMU) and diisopropylamine was used as the coupling reagent. Fmoc deprotection was performed using 20% piperidine in N,N-dimethylformamide (DMF). The coupling reaction and Fmoc deprotection were monitored using TNBS and a chloranil test kit (Tokyo Chemical Industry Co, Ltd). Peptidyl-resin was washed with NMP and dried in vacuo. Peptides were deprotected and cleaved from the resin using a cocktail of trifluoroacetic acid (TFA)/1,2-ethanedithiol/triisopropylamine/thioanisole/water = 8.25/0.25/0.1/0.5/0.5 (mL) at room temperature for 3 h. The reaction mixtures were filtered from the resins, and the peptide was precipitated by adding methyl tert-butyl ether (MTBE). The supernatant was decanted, and the precipitated peptide was dried in vacuo after three times of repetitive washing with MTBE. The crude peptide was purified using RP-HPLC on a C18 column eluting with a linear gradient of CH3CN/water containing 0.1% TFA (5/95 to 100/0 over 95 min). The mass of the purified peptide, β-Annulus-GGGCG, was confirmed using matrix-assisted laser desorption ionization-time-of-flight mass spectrometry (MALDI-TOF MS, matrix; α-CHCA, m/z = 2637 ([M+H]+calc. = 2638)).Synthesis of DNA-maleimide
DNA with an amino group via a hexamethylene chain at the 5′ end (TE02-NH2-(5′-H2N-(CH2)6-TAGGCAGTGGTTTGACGTCCGCATGTTGGGAATAGCCACGCCT-3′) or random DNA-NH2 (5′-H2N-(CH2)6-AAAAAAAACCCCCCCCCCGGGGGGGGGGGGGGTTTTTTTTTTT-3′), 0.9 mM in 20 μL deionized water) was coupled with N-(4-maleimidobutyryloxy) sulfosuccinimide (Sulfo-GMBS, 120 equiv.) in the presence of 100 mM NaHCO3 at 25 °C for 4 h. The solution was then dialyzed using a dialysis tube (Spectra/por7, molecular weight cut off (MWCO) 1 kDa, SPECTRUM) to remove excess Sulfo-GMBS, and the concentrations of TE02-maleimide and random DNA-maleimide were quantified using UV spectroscopy (JASCO FP-8200).Synthesis of the β-annulus-DNA conjugate
The Michael reaction between DNA-maleimide and Cys residue in β-Annulus-GGGCG was performed as follows: DNA-maleimide and β-Annulus-GGGCG (16 equiv.) were mixed in 50% acetonitrile/20 mM phosphate buffer, vortexed, and sonicated until complete dissolution. The mixture was then agitated at 40 °C for 70 h and the sample was purified by RP-HPLC using a C18 column eluting with a linear gradient of CH3CN/0.1 M ammonium formate aq. (5/95 to 100/0 over 95 min). The masses of the purified products were identified using MALDI-TOF MS (matrix; 3-hydroxypicolinic acid/diammonium hydrogen citrate = 8/1, β-Annulus-TE02; m/z = 16359 (([M + H]+calc. = 16273), β-Annulus-random DNA; m/z = 16376 (([M+H]+calc. = 16273)).Synthesis of TAMRA-labeled β-Annulus
TAMRA-β-Annulus was synthesized based on a previous study.29 In brief, Cys-β-Annulus (Cys-Ile-Asn-His-Val-Gly-Gly-Thr-Gly-Gly-Ala-Ile-Met-Ala-Pro-Val-Ala-Val-Thr-Arg-Gln-Leu-Val-Gly-Ser) was synthesized using the same procedure mentioned above and purified using RP-HPLC. The peptide was then coupled with TAMRA-5-maleimide (tetramethylrhodamine-5-maleimide, 4 equiv.) in sodium phosphate buffer (pH 7.0) in the presence of 1 mM Tris(2-carboxyethyl)phosphine hydrochloride) in the dark at 25 °C for 6 h. After dialysis (Spectra/por7, MWCO 1 kDa), peptide was purified by RP-HPLC using a C18 column eluting with a linear gradient of CH3CN/water containing 0.1% TFA (5/95 to 100/0 over 100 min) and identified by MALDI-TOF MS (matrix; α-CHCA, m/z = 2893 ([M+H]+calc. = 2892)).Synthesis of TAMRA-labeled TE02
TAMRA-TE02 was synthesized by coupling TE02-NH2 with 5-TAMRA, SE (5-carboxytetramethylrhodamine, succinimidyl ester, 10 equiv.) in 10 mM sodium phosphate buffer (pH 7.0) to yield TAMRA-TE02. The sample was purified by RP-HPLC using a C18 column eluted with a linear gradient of CH3CN/0.1 M ammonium formate aq. (5/95 to 100/0 over 95 min) and dialyzed (Spectra/por7, MWCO 1 kDa). TAMRA labeling was confirmed, and the concentration was calculated using UV-vis spectroscopy.Dynamic light scattering (DLS)
The powder of β-Annulus-TE02 was dissolved in 10 mM sodium phosphate buffer (pH 7.0) to produce 50, 25, and 5 μM solutions. The solutions were then sonicated for 5 min and incubated at 25 °C for 30 min. DLS measurements were conducted using a Zetasizer Nano ZS (Malvern) instrument with an incident He-Ne laser (633 nm) at 25 °C. The correlation times of the scattered light intensities were measured several times, and the means of the diffusion coefficient were calculated. Hydrodynamic diameters of the particles were calculated using Stokes–Einstein equations.Transmission electron microscopy (TEM)
Aliquots (5 μL) of the DLS samples were applied to hydrophilized carbon-coated Cu-grids (C-SMART Hydrophilic TEM grids, ALLIANCE Biosystems) for 1 min. The sample was then removed using filter paper, the grid was stained for 1 min using a staining solution (2% phosphotungstic acid (Na3(PW12O40)(H2O)n). After removing the staining solution using filter paper, the grids were dried in vacuo. TEM (JEOL JEM 1400 Plus) was used to analyze the grids at an accelerating voltage of 80 kV.Circular dichroism (CD)
The circular dichroism spectra of the peptide conjugates (20 μM) in 10 mM sodium phosphate buffer (pH 7.0) were recorded at a JASCO J-820 spectrometer using a quartz optical cell with a 1 mm path length at 25 °C. Temperature-dependent changes in CD spectra were also recorded using the same instrument by heating from −10 °C to 90 °C (5 °C min−1).Cell culture
Daudi cells (B lymphoblast cell line from a Burkitt lymphoma patient), purchased from the RIKEN BioResource Research Center (Ibaraki, Japan), were cultured in Dulbecco's modified Eagle's medium (D-MEM) supplemented with 10% (v/v) heat-inactivated fetal bovine serum (FBS) (D-MEM(+)) containing 1x penicillin–streptomycin–neomycin (PSN) antibiotic mixture (Gibco). HeLa cells (human epithelial carcinoma cell line), purchased from the RIKEN BioResource Research Center, were cultured in D-MEM(+) containing 1x PSN antibiotic mixture, 1 mM sodium pyruvate, and 1% (v/v) MEM nonessential amino acids (Sigma). These cells were maintained at 37 °C, in a humidified 5% CO2 incubator and subculture was conducted every 3–4 days.Confocal laser scanning microscopy (CLSM) observation
Daudi cells were collected by centrifugation (800 g, 5 min) and washed twice with phosphate-buffered saline (PBS(−)) by repeated resuspension/centrifugation (800 g, 5 min). The cell pellet was then resuspended in 25 μM β-Annulus-TE02 and 5 μM TAMRA-β-Annulus in phosphate-buffered saline containing 1 mM CaCl2 and MgCl2 (PBS(+)) and incubated at 37 °C for 1 h. Centrifugation (800 g, 5 min) was used to collect the cells, which were then washed twice with PBS(−). After resuspension in D-MEM(+), the cells were seeded onto single well glass-based dishes (IWAKI), and CLSM observation (Olympus FV10i) was conducted. TAMRA was excited at 559 nm and observed through a 600 nm emission band-pass filter (red).Flow cytometry
Centrifugation (800 g, 5 min) was used to collect aliquots of cell suspension prepared for CLSM examination. The pellet was then resuspended in PBS(−) containing 2% FBS. The suspension was then transferred to a plastic tube and subjected to flow cytometry analysis using a Gallios Cell Analyzer (BECKMAN COULTER). About 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 gated events were analyzed in each sample.
000 gated events were analyzed in each sample.
Cell viability assay
HeLa cells and Daudi cells were seeded into 96 well cell culture plate (IWAKI) at 10,000 cells/50 μL per well. Peptide powder was dissolved into PBS(+) containing doxorubicin hydrochloride (DOX, Tokyo Chemical Industry) and incubated at 37 °C for 30 min to produce solution of TE02-decorated or unmodified artificial viral capsid complexed with DOX (150 μM β-Annulus or β-Annulus-TE02, 1 μM DOX in 10 μL PBS(+)). After incubation, the solution was added to cell suspension and incubated at 37 °C for 24 h (final concentration; peptide, 25 μM; DOX, 0.167 μM). After incubation, 10 μL WST-8 solution (Cell Counting Kit-8, DOJINDO) was added to each well and incubated at 37 °C for 1 h and absorbance at 450 nm was measured using microplate reader (Infinite 200 F Plex, TECAN).3. Results and discussion
Synthesis of β-annulus-DNA conjugates
β-Annulus-TE02 was synthesized by conjugating β-Annulus-GGGCG (INHVGGTGGAIMAPVAVTRQLVGSGGGCG-OH) and TE02 bearing an amino group via a hexamethylene chain at the 5′ end (TE02-NH2) using Sulfo-GMBS, maleimide-activated carboxylic acid linker (Fig. 1A). In brief, the amino group of TE02-NH2 was first coupled with Suflo-GMBS (120 eq.) in the presence of sodium bicarbonate to produce TE02-maleimide. The Michael reaction proceeded between TE02-maleimide and the thiol group of Cys in β-Annulus-GGGCG. As a control, β-Annulus-random conjugate was prepared as well as β-Annulus-TE02 using random DNA-NH2. To visualize cellular localization of the conjugate, TAMRA-β-Annulus was synthesized by labeling Cys-β-Annulus with TAMRA-5-maleimide. Labelling of TE02 with TAMRA was conducted using TE02-NH2 and 5-TAMRA, SE to produce TAMRA-TE02. The products were purified using RP-HPLC and the masses were identified using MALDI-TOF MS (Sup. Fig. 1).Self-assembly of β-Annulus-TE02 into the TE02-decorated artificial viral capsid
The formation of nanoparticles by self-assembly of β-Annulus-TE02 was confirmed using DLS (Fig. 2A) and TEM (Fig. 2B). β-Annulus-TE02 was dissolved in 10 mM phosphate buffer (pH 7.0) to produce 50, 25 and 5 μM β-Annulus-TE02 solutions. The solutions were sonicated for 5 min and incubated for 1 h at 25 °C. Assemblies were observed around 50 nm (25 μM; 55 ± 16 nm, 5 μM; 53 ± 12 nm) in the 25 and 5 μM solutions, which are similar to artificial viral capsid reported in the previous studies.25–35 However, in the 50 μM solution, nanostructures with the size of 222 ± 46 nm were observed, indicating aggregation of multiple capsids probably due to the interaction among DNAs on the capsids. These results suggest that at 25 μM and 5 μM β-Annulus-TE02 forms discrete artificial viral capsids. In the TEM images, individual spherical assemblies with sizes of around 50–150 nm were observed in 25 μM (see also Fig. S2A, ESI†) and 5 μM solutions, whereas spherical assemblies with sizes of around 200 nm aggregates were observed in 50 μM solution. These results are consistent with DLS measurements, indicating the formation of a spherical artificial viral capsid structure similar to that of β-Annulus (Fig. S2B, ESI†). TE02 only did not form any structure that could be observed by TEM (Fig. S2C, ESI†).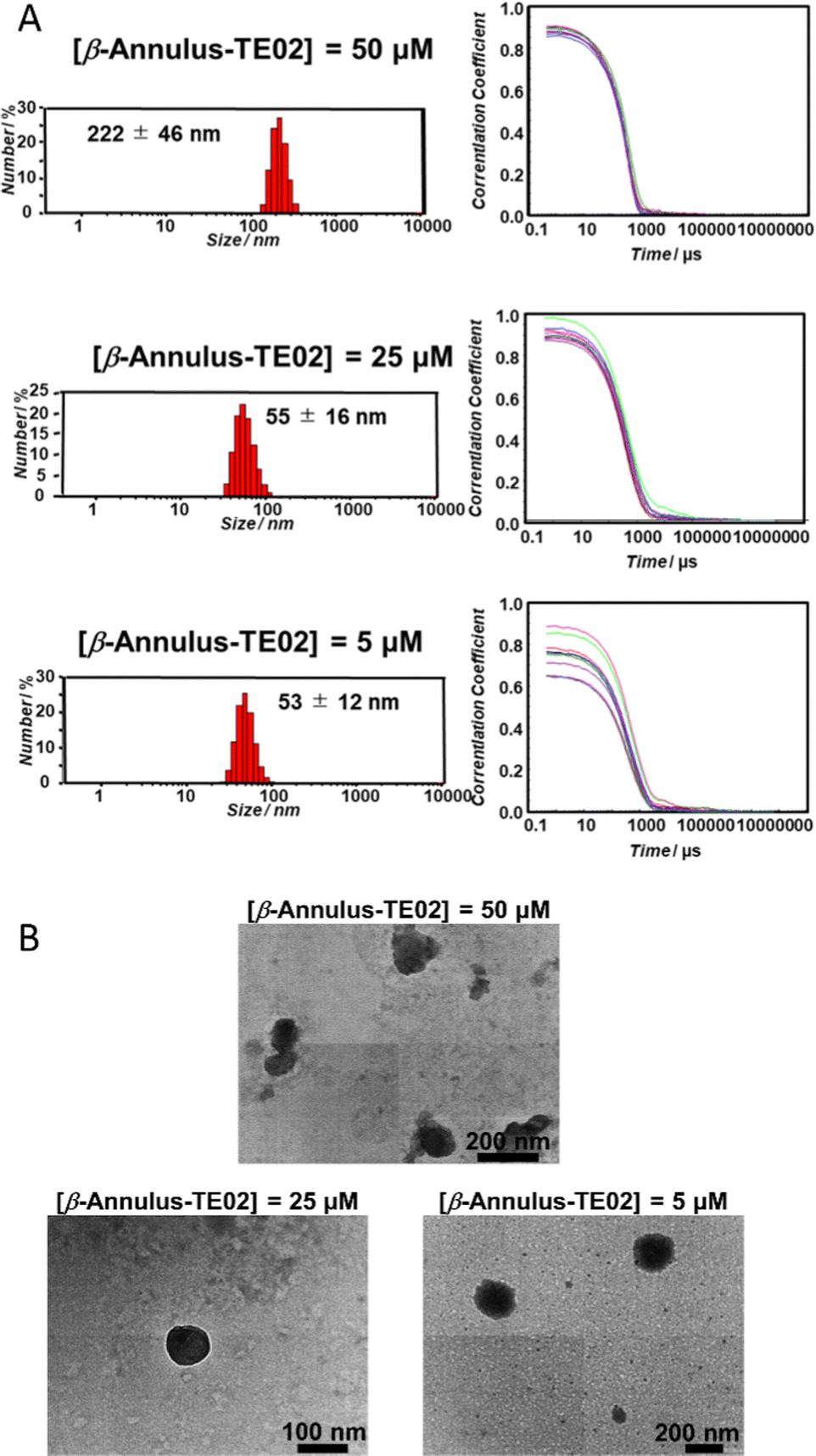 | ||
| Fig. 2 Nanoparticle formation of β-Annulus-TE02. (A) Size distribution (left) and autocorrelation curve (right) obtained from DLS and (B) TEM images of β-Annulus-TE02 in 10 mM phosphate buffer. | ||
Furthermore, the secondary structure of TE02 decorating the artificial viral capsid was analyzed using CD spectra (Fig. 3). The CD spectrum of β-Annulus-TE02 showed typical B-type DNA double-helical structure similar to TE02 alone, implying that TE02 on the artificial viral capsid retains its original B-type DNA double-helical hairpin structure (Fig. 3A). We measured CD spectra at various temperatures to investigate the thermostability of the helical hairpin structure of TE02 and β-Annulus-TE02 (Fig. 3B). The increase in temperature decreased the molecular ellipticity at 270 nm in both samples, indicating denaturation of the double-helical hairpin. The difference spectrum between β-Annulus-TE02 and TE02 suggests that the peptide part of β-Annulus-TE02 partly retains its original secondary structure similar to β-Annulus-GGGCG (Fig. S3, ESI†): the difference between them may be due to the effect of the maleimido linker or steric hindrance between β-Annulus and TE02. The Tm values of TE02 and β-Annulus-TE02 were determined to be 40 °C and 45 °C, respectively, and these results suggested that TE02 on the artificial viral capsid was more stable than TE02 alone because of the dense display of TE02 on the capsid.
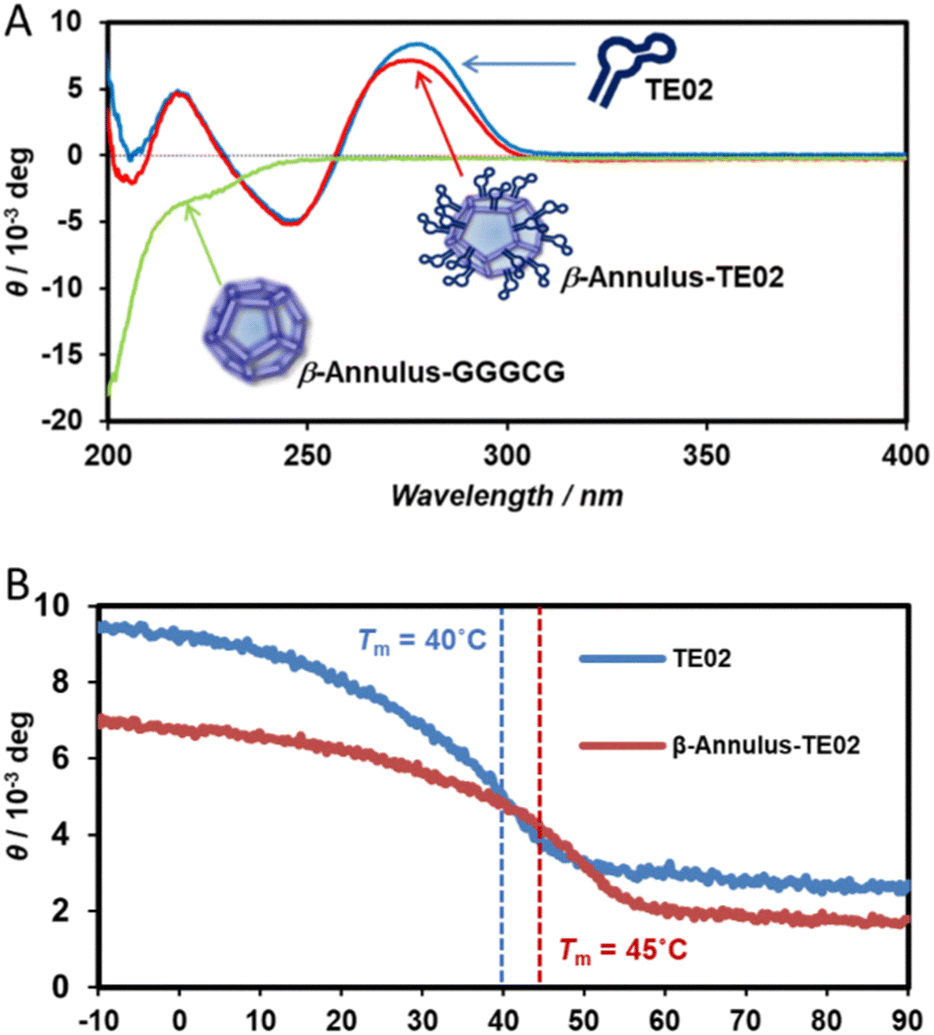 | ||
| Fig. 3 (A) CD spectra of 20 μM β-Annulus-TE02 (red), β-Annulus-GGGCG (green), and TE02 (blue) in 10 mM phosphate buffer at 25 °C. (B) Temperature-dependence in ellipticity at 270 nm of TE02 (blue) and β-Annulus-TE02 (red). | ||
Internalization of TE02-decorated artificial viral capsid into Burkitt lymphoma cells
The internalization of the TE02-decorated capsid was evaluated using coassembly of TAMRA-conjugated β-Annulus (TAMRA-β-Annulus) and β-Annulus-TE02 (1:10 (mol:mol)). The formation of the assembled nanoparticles was confirmed using DLS (Fig. S4, ESI†). Daudi, a Burkitt lymphoma cell line, was treated by the TE02-decorated capsid co-assembled from 2.5 μM TAMRA-β-Annulus and 25 μM β-Annulus-TE02 (TAMRA/β-Annulus-TE02) in PBS(+) for 1 h. Cells were then washed twice with PBS(−) in a centrifugation step, and resuspended cells in PBS(+) were seeded on glass bottom dishes. TAMRA fluorescence in cells was observed using a CLSM (Fig. 4). As controls, cells were treated with the capsid co-assembled from 2.5 μM TAMRA-β-Annulus and 25 μM β-Annulus (TAMRA/β-Annulus) or 2.5 μM TAMRA-β-Annulus and 25 μM β-Annulus-random DNA (TAMRA/β-Annulus-random DNA) or 2.5 μM TAMRA-TE02 and 25 μM TE02 (TAMRA/TE02). In cells treated with the TE02-decorated capsid (TAMRA/β-Annulus-TE02), the fluorescence of TAMRA was located in the cell interior. The fluorescence of cells treated with control samples was also detected, but the intensity was weaker than that of cells with TAMRA/β-Annulus-TE02. The selectivity of TE02-decorated capsid toward Burkitt lymphoma cells was also investigated in comparison to Daudi and HeLa cells using CLSM (Fig. 4E). HeLa cells treated with TAMRA/β-Annulus-TE02 showed a subtle punctate signal, whereas Daudi cells treated with TAMRA/β-Annulus-TE02 showed strong fluorescence signals in the cytosol, indicating that TAMRA/β-Annulus-TE02 has the selectivity toward Burkitt lymphoma cells because of the binding selectivity of the TE02 aptamer. Since the surface of cells is negatively charged and the TE02-decorated capsid is also negatively charged, it can be thought that TE02-decorated capsid cannot interact with nontarget cells. Therefore, the off-target effect e.g., cytotoxicity and cellular uptake toward normal cells in terms of the DDS carrier might be negligible.
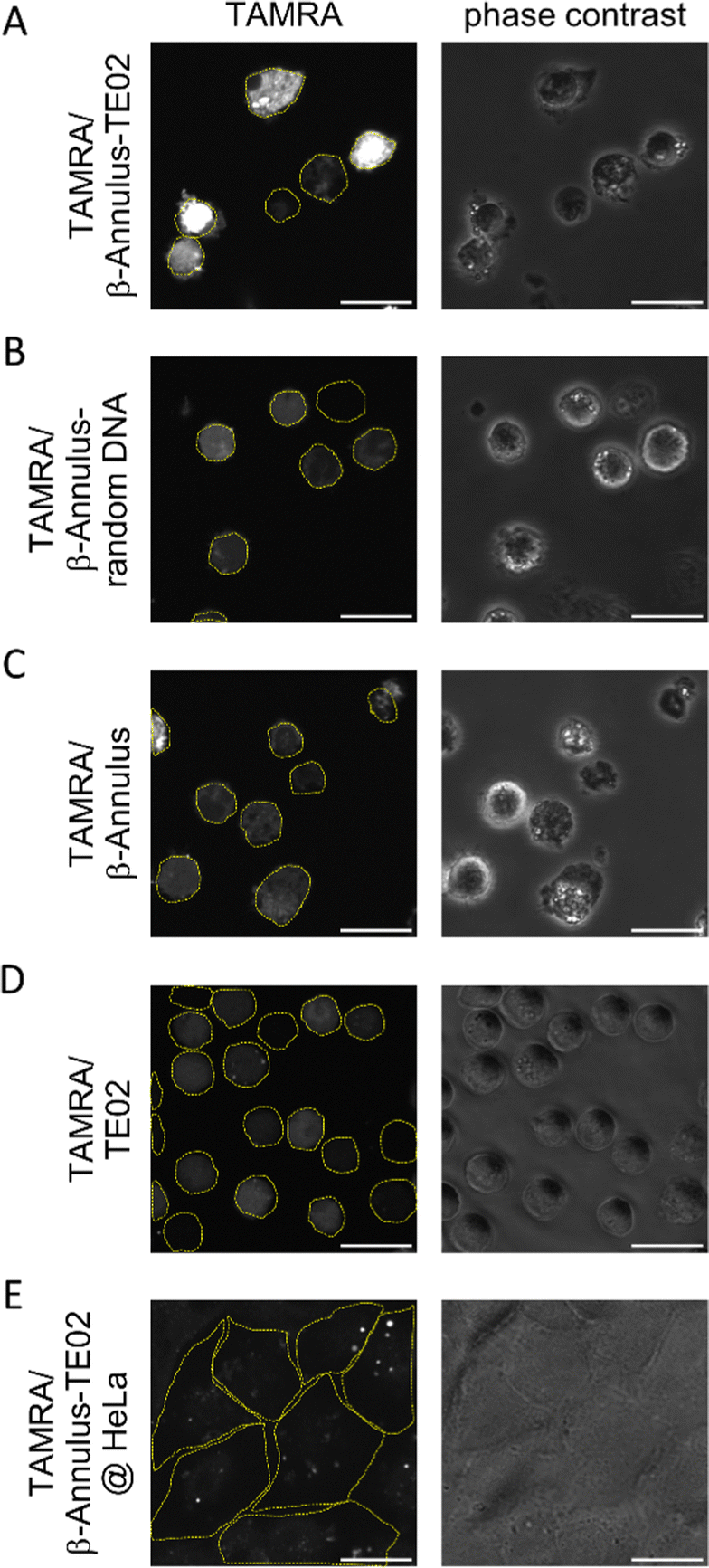 | ||
| Fig. 4 (A)–(D) CLSM images of Daudi cells treated using (A) TE02-decorated capsid (TAMRA/β-Annulus-TE02), (B) random DNA-decorated capsid (TAMRA/β-Annulus-random DNA), (C) unmodified capsid (TAMRA/β-Annulus) and (D) TAMRA/TE02. (E) CLSM image of HeLa cells treated using TE02-decorated capsid (TAMRA/β-Annulus-TE02). TAMRA fluorescence was shown as white. Yellow dashed lines; cellular boundaries. Scale bars; 20 μm. | ||
We used flow cytometry analysis to quantify internalization efficiency (Fig. 5). The Daudi cells were treated in the same method for microscopic examination, and the pellet of the cells was resuspended in PBS(−) containing 2% FBS before the flow cytometry analysis was conducted. When treated with TAMRA/β-Annulus-TE02, many cell groups showed a higher fluorescence intensity than that of TAMRA/β-Annulus-random DNA, indicating that the secondary structure of TE02 is important for binding to the Burkitt lymphoma cells. In contrast, TAMRA-TE02-treated cells did not have such a cell group with higher fluorescence intensity, implying the advantage of artificial viral capsids in terms of multivalent binding to lymphoma cells.
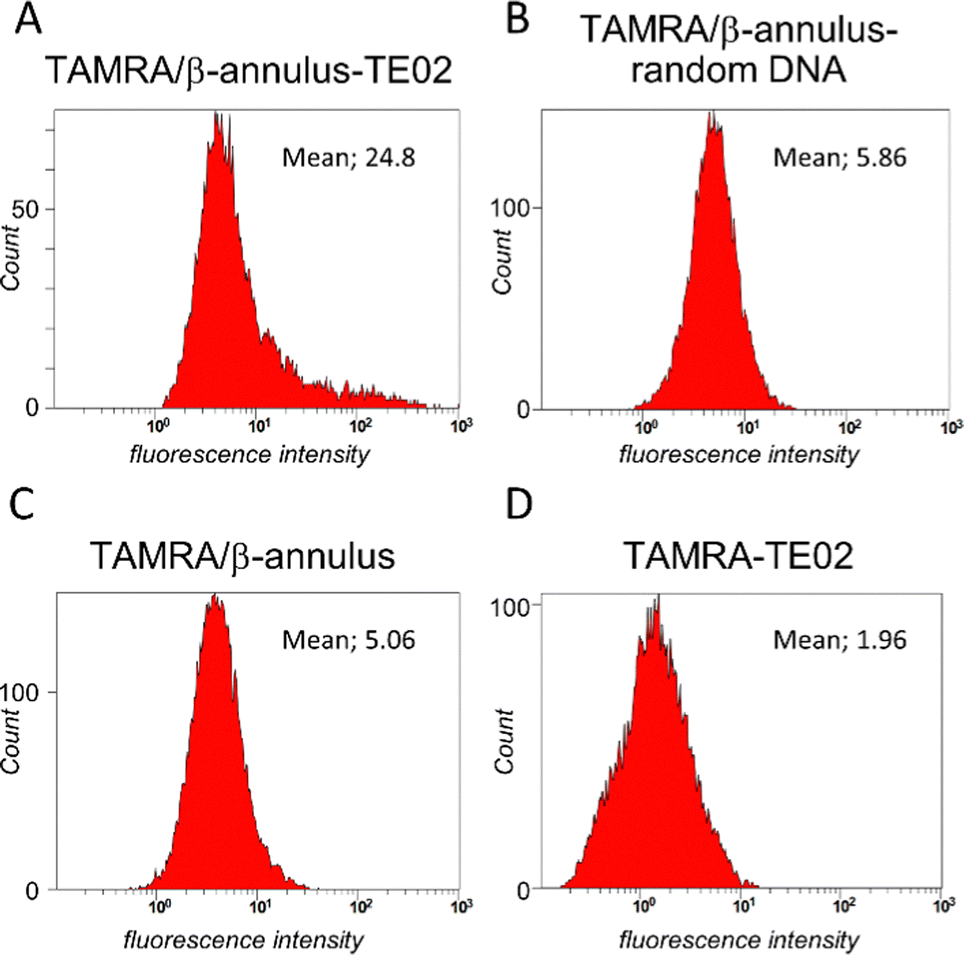 | ||
| Fig. 5 Flow cytometry analysis of Daudi cells treated with (A) TE02-decorated capsid (TAMRA/β-Annulus-TE02), (B) random DNA-decorated capsid (TAMRA/β-Annulus-random DNA), (C) unmodified capsid (TAMRA/β-Annulus) and (D) TAMRA-TE02. | ||
To investigate the applicability of the TE02-decorated artificial viral capsid as a lymphoma cell-specific drug carrier, we treated HeLa or Daudi cells with doxorubicin (DOX), one of the most used anticancer drugs, complexed with the TE02-decorated capsid and performed cell viability assay (Fig. 6). While at HeLa cells DOX showed no cytotoxicity, at Daudi cells DOX showed moderate cytotoxicity (∼25% cell death). In addition, half of Daudi cells was killed by DOX complexed with the TE02-decorated capsid, whereas DOX complexed with the unmodified capsid was not so effective. This result indicates the in vitro applicability of the TE02-decorated capsid for Burkitt lymphoma-specific cell killing.
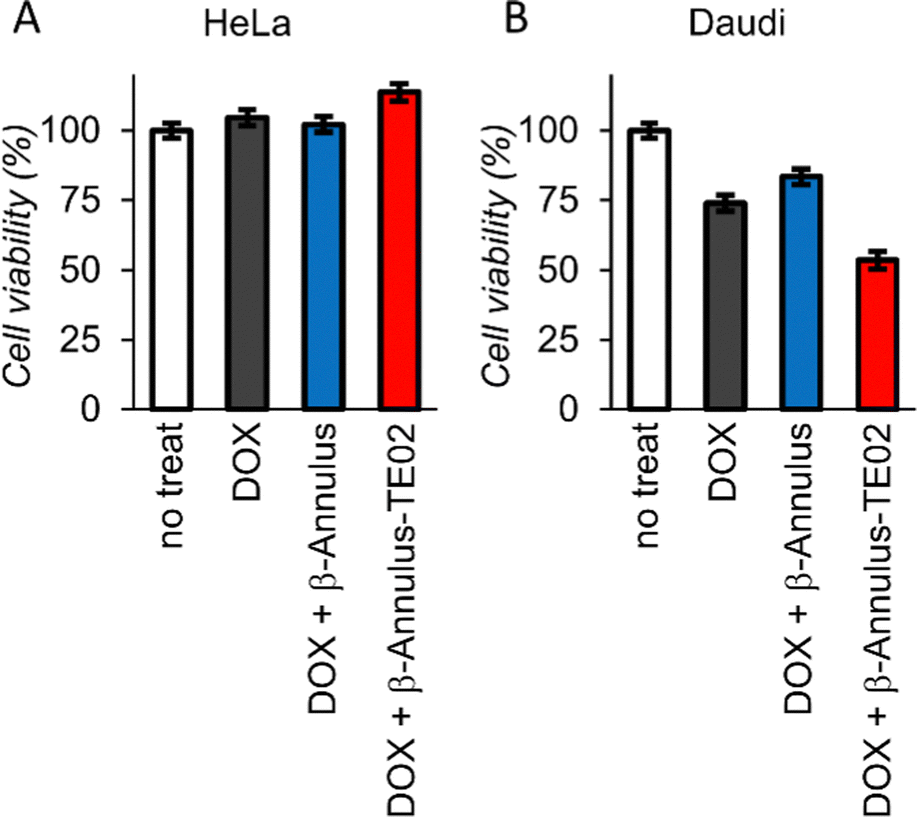 | ||
| Fig. 6 Cell viability assay of (A) HeLa cells and (B) Daudi cells treated with DOX (0.167 μM) complexed with β-Annulus or β-Annulus-TE02 (25 μM). | ||
4. Conclusions
In this study, we created a Burkitt lymphoma cell-specific TE02 aptamer-decorated artificial viral capsid. The capsid was formed in phosphate buffer, and the TE02 aptamer retained its original B-type DNA double-helical hairpin structure on the capsid. The TAMRA-labeled TE02-decorated capsid was internalized into its target cell, Daudi, and the fluorescence of TAMRA was located in the cell interior. The specific cell killing toward Burkitt lymphoma cells was also demonstrated compared with HeLa cells.Since Burkitt lymphoma is a highly aggressive cancer, a new type of treatment methodology is urgently needed. Targeting Burkitt lymphoma using a DDS carrier would be a promising therapeutic method with a high cure rate and low toxicity,36 and the Burkitt lymphoma-targeting TE02 aptamer-decorated nanoparticles that we developed could be one answer for the DDS carrier toward Burkitt lymphoma. Compared to other polymer- and lipid-based DDS carriers, this peptide/DNA-based nanocarrier has higher biocompatibility compared to polymer-based DDS carriers and reduces accumulation in the liver compared to lipid-based DDS carriers because of complexation of lipid with Apolipoprotein E in the blood.37 This report indicates the applicability of a cell-specific aptamer-coated peptide-based nanocarrier toward the cell-specific internalization. This strategy could also be applied to other peptide/protein-based nanocarriers mentioned above.19–21 We are now considering encapsulating other drugs or nucleic acid therapeutics in a tumor-selective aptamer-decorated capsid to achieve tumor-specific cell killing in vitro in the future.
Author contributions
K. S. performed cellular experiment and wrote the paper with K. M. K. U. synthesized the peptide and performed in vitro experiments. T. I. distributed the Daudi cells to us and supervised the flow cytometry experiment. H. I. supervised whole experiment with K. M. K. M. directed the project, designed the experiments and wrote the manuscript with K. S.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by a Grant-in-Aid for Scientific Research (B) (JSPS KAKENHI Grant No. JP 22H02199), a Grant-in-Aid for Scientific Research (S) (JSPS KAKENHI Grant No. JP22H04975), a Grant-in-Aid for JSPS Fellows (JSPS KAKENHI Grant No. JP21J01604), and AGC Research Collaboration System.Notes and references
- X. Fang and W. Tan, Acc. Chem. Res., 2010, 43, 48–57 CrossRef CAS PubMed.
- M. Blind and M. Blank, Mol. Ther. – Nucleic Acids, 2015, 4, e223 CrossRef PubMed.
- L. C. Bock, L. C. Griffin, J. A. Latham, E. H. Vermaas and J. J. Toole, Nature, 1992, 355, 564–566 CrossRef CAS PubMed.
- R. F. Macaya, P. Schultze, F. W. Smith, J. A. Roe and J. Feigon, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 3745–3749 CrossRef CAS PubMed.
- Z. Tang, D. Shangguan, K. Wang, H. Shi, K. Sefah, P. Mallikratchy, H. W. Chen, Y. Li and W. Tan, Anal. Chem., 2007, 79, 4900–4907 CrossRef CAS PubMed.
- G. J. Tong, S. C. Hsiao, Z. M. Carrico and M. B. Francis, J. Am. Chem. Soc., 2009, 131, 11174–11178 CrossRef CAS PubMed.
- Y. Yang, X. Sun, J. Xu, C. Cui, H. Safari Yazd, X. Pan, Y. Zhu, X. Chen, X. Li, J. Li and W. Tan, ACS Nano, 2020, 14, 9562–9571 CrossRef CAS PubMed.
- Y. Zhang, H. Hong and W. Cai, Curr. Med. Chem., 2011, 18, 4185–4194 CrossRef CAS PubMed.
- X. Li, Q. Zhao and L. Qiu, J. Controlled Release, 2013, 171, 152–162 CrossRef CAS PubMed.
- H. Kang, M. B. O’Donoghue, H. Liu and W. Tan, Chem. Commun., 2010, 46, 249–251 RSC.
- G. Liu, X. Mao, J. A. Phillips, H. Xu, W. Tan and L. Zeng, Anal. Chem., 2009, 81, 10013–10018 CrossRef CAS PubMed.
- Y. Khudyakov and P. Pumpens, Viral Nanotechnology, CRC Press, Boca Raton, FL, USA, 2016 Search PubMed.
- P. van Rijn and R. Schirhagl, Adv. Healthcare Mater., 2016, 5, 1386–1400 CrossRef CAS PubMed.
- A. M. When and N. F. Steinmetz, Chem. Soc. Rev., 2016, 45, 4074–4126 RSC.
- K. Matsuura, Chem. Commun., 2018, 54, 8944–8959 RSC.
- Q. Luo, C. Hou, Y. Bai, R. Wang and J. Liu, Chem. Rev., 2016, 116, 13571–13632 CrossRef CAS PubMed.
- S. L. Kuan, F. R. G. Bergamini and T. Weil, Chem. Soc. Rev., 2018, 47, 9069–9105 RSC.
- S. Lou, X. Wang, Z. Yu and L. Shi, Adv. Sci., 2019, 6, 1802043 CrossRef PubMed.
- M. Cao, Z. Zhang, X. Zhang, Y. Wang, J. Wu, Z. Liu, L. Sun, D. Wang, T. Yue, Y. Han, Y. Wang, Y. Wang and M. Wang, J. Colloid Interface Sci., 2022, 615, 395–407 CrossRef CAS PubMed.
- J. E. Noble, E. De Santis, J. Ravi, B. Lamarre, V. Castelletto, J. Mantell, S. Ray and M. G. Ryadnov, J. Am. Chem. Soc., 2016, 138, 12202–12210 CrossRef CAS PubMed.
- J. Kong, Y. Wang, J. Zhang, W. Qi, R. Su and Z. He, Angew. Chem., Int. Ed., 2018, 57, 14032–14036 CrossRef CAS PubMed.
- Y. Azuma, T. G. W. Edwardson, N. Terasaka and D. Hilvert, J. Am. Chem. Soc., 2018, 140, 566–569 CrossRef CAS PubMed.
- N. Terasaka, Y. Azuma and D. Hilvert, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 5432–5437 CrossRef CAS PubMed.
- S. Tetter, N. Terasaka, A. Steinauer, R. J. Bingham, S. Clark, A. J. P. Scott, N. Patel, M. Leibundgut, E. Wroblewski, N. Ban, P. G. Stockley, R. Twarock and D. Hilvert, Science, 2021, 372, 1220–1224 CrossRef CAS PubMed.
- K. Matsuura, K. Watanabe, T. Matsuzaki, K. Sakurai and N. Kimizuka, Angew. Chem., Int. Ed., 2010, 49, 9662–9665 CrossRef CAS PubMed.
- K. Matsuura, K. Watanabe, Y. Matsushita and N. Kimizuka, Polym. J., 2013, 45, 529–534 CrossRef CAS.
- Y. Nakamura, Y. Sato, H. Inaba, T. Iwasaki and K. Matsuura, Appl. Sci., 2020, 10, 1–10 Search PubMed.
- K. Matsuura, T. Nakamura, K. Watanabe, T. Noguchi, K. Minamihata, N. Kamiya and N. Kimizuka, Org. Biomol. Chem., 2016, 14, 7869–7874 RSC.
- K. Sakamoto, H. Furukawa, J. V. V. Arafiles, M. Imanishi, K. Matsuura and S. Futaki, Bioconjugate Chem., 2022, 33, 311–320 CrossRef CAS PubMed.
- K. Matsuura, G. Ueno and S. Fujita, Polym. J., 2015, 47, 146–151 CrossRef CAS.
- S. Fujita and K. Matsuura, Org. Biomol. Chem., 2017, 15, 5070–5077 RSC.
- Y. Nakamura, S. Yamada, S. Nishikawa and K. Matsuura, J. Pept. Sci., 2017, 23, 636–643 CrossRef CAS PubMed.
- K. Matsuura and T. Honjo, Bioconjugate Chem., 2019, 30, 1636–1641 CrossRef CAS PubMed.
- K. Matsuura, J. Ota, S. Fujita, Y. Shiomi and H. Inaba, J. Org. Chem., 2020, 85, 1668–1673 CrossRef CAS PubMed.
- Y. Liang, H. Furukawa, K. Sakamoto, H. Inaba and K. Matsuura, ChemBioChem, 2022, 23, e202200220 CrossRef CAS PubMed.
- E. M. Molyneux, R. Rochford, B. Griffin, R. Newton, G. Jackson, G. Menon, C. J. Harrison, T. Israels and S. Bailey, Lancet, 2012, 379, 1234–1244 CrossRef PubMed.
- X. Yan, F. Kuipers, L. M. Havekes, R. Havinga, B. Dontje, K. Poelstra, G. L. Scherphof and J. A. A. M. Kamps, Biochem. Biophys. Res. Commun., 2005, 328, 57–62 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3tb00169e |
| This journal is © The Royal Society of Chemistry 2023 |