
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePolymer brush-grafted cellulose nanocrystals for the synthesis of porous carbon-coated titania nanocomposites†
Yen Theng
Cheng
ac,
Qingbo
Xia
bc,
Hongwei
Liu
d,
Marcello B.
Solomon
 b,
Chris D.
Ling
bc and
Markus
Müllner
*ac
b,
Chris D.
Ling
bc and
Markus
Müllner
*ac
aKey Centre for Polymers and Colloids, School of Chemistry, The University of Sydney, Sydney, NSW 2006, Australia. E-mail: markus.muellner@sydney.edu.au
bSchool of Chemistry, The University of Sydney, Sydney, NSW 2006, Australia
cThe University of Sydney Nano Institute (Sydney Nano), Sydney, NSW 2006, Australia
dSydney Microscopy & Microanalysis, the University of Sydney node of Microscopy Australia, Sydney, NSW 2006, Australia
First published on 11th April 2023
Abstract
Cellulose nanocrystals (CNCs) are a renewable nanomaterials platform. Their characteristic one-dimensional morphology and surface chemistry offer opportunities for the templated synthesis of functional nanorods and nanotubes. Here we report the template-directed fabrication of mesoporous carbon-coated anatase TiO2 nanotubes using polymer brush-grafted CNCs as nanoreactors. The nanoreactors consisted of a CNC core grafted with a poly(2-(dimethylamino)ethyl methacrylate) (PDMAEMA) shell made by surface-initiated atom transfer radical polymerisation (SI-ATRP). A water-soluble TiO2 precursor, titanium(IV) bis(ammonium lactato)dihydroxide (TALH), was selectively loaded into the nanoreactor shell via ionic interactions. Pyrolysis of the CNC-g-PDMAEMA/TALH hybrids led to the carbonisation of the polymer template, the removal of the CNC core and the crystallisation of the TALH to produce carbon-coated nanocrystalline anatase. The resulting TiO2/C nanotubes retained ∼50 wt% of TiO2 and ∼13% wt% of carbon, and exhibited a specific surface area of ∼111 m2 g−1. The simultaneously forming carbon framework suppressed the growth of titania crystallites to <5 nm. We further demonstrate the potential of our TiO2/C nanomaterials to produce titania anode materials for lithium-ion batteries.
Introduction
Nanocelluloses (i.e. cellulose nanocrystals (CNCs), cellulose nanofibers (CNFs) and bacterial nanocelluloses) are a powerful and versatile class of renewable resources for the fabrication of advanced functional materials.1–4 Their exceptional mechanical properties, ease of modification, and distinct physical properties give them exceedingly diverse applications, such as additives for (nano)composites5 and films,6 modifiers to tune viscosity7 and barrier8,9 properties, and components for producing gels and foams,10,11 among others. Increasing interest in “green” resources means the use of cellulose-based materials is expected to expand rapidly in areas of global importance, such as health and energy.12–14 In this context, CNCs have received growing attention because their inherent one-dimensional (1D) morphology, crystallinity, rheological and mechanical properties offer advantages in materials design; especially for applications where porosity, interconnectedness and hierarchical structure are important considerations.Pristine and modified CNCs have been used as structural and functional components in batteries, supercapacitors, electronics and sensors,14–16 where the CNCs are typically employed as fillers or structural guides in conjunction with functional (nano)materials to fabricate nanocomposites. While pristine nanocelluloses can be readily employed, for example as liquid crystal matrices, surface modifications have been shown to dramatically enhance material processing by introducing better control over interface energy and component compatibility. The use of CNCs as a nanoreactor for functional materials synthesis, however, has not been studied in detail. The intrinsic 1D morphology and surface functionality of CNCs lend themselves to use as structure-guiding agents in the synthesis of inorganic or hybrid nanorods and nanotubes, where 1D soft templating strategies remain limited to cylindrical micelles and unimolecular polymers.17,18 In particular, the surface-initiated growth of polymer chains to afford dense polymer brushes19 produces 1D core–shell particles that can function as nanoreactors.20
Nanostructured and nanoscale metal oxides are desired for a range of applications, including sensing, catalysis and energy storage.21 A metal oxide of growing interest in battery research is titania, due to its elemental abundance and safety profile;22 however, it suffers from relatively low capacity and poor conductivity. Nanostructuring may overcome these limitations.22 Controlling the morphology and porosity of metal oxides at the nanoscale typically enhances their electronic properties. In parallel, CNCs have been used to generate porous carbon scaffolds (e.g. via carbonising CNC aerogels).23,24 Developing an approach that uses CNCs as nanoreactors might therefore combine the benefits of soft templating of metal oxides with the opportunity to introduce a favourable carbon framework.
Herein, we demonstrate the synthesis of porous carbon-coated nanocrystalline anatase (TiO2/C) via a soft templating route. A core–shell nanoreactor (CNC-g-PDMAEMA) was prepared via the surface-initiated atom transfer radical polymerisation (SI-ATRP) of DMAEMA from a CNC-based polyinitiator, CNC-Br (Scheme 1, i). A water-soluble anatase precursor, titanium(IV) bis(ammonium lactate)dihydroxide (TALH), was then complexed into the PDMAEMA polyelectrolyte shell (Scheme 1, ii) for the uniform build-up of hybrid materials. During pyrolysis, two simultaneous processes yielded a well-defined TiO2/C nanocomposite: CNC-g-PDMAEMA transforms into a carbon framework which, in turn, restricts the growth of the anatase crystallites (Scheme 1, iii). The resulting TiO2/C nanotubes were subsequently tested as negative electrode (anode) components in lithium-ion batteries (LIB), and their performance compared to that of their calcined counterparts (Scheme 1, iv).
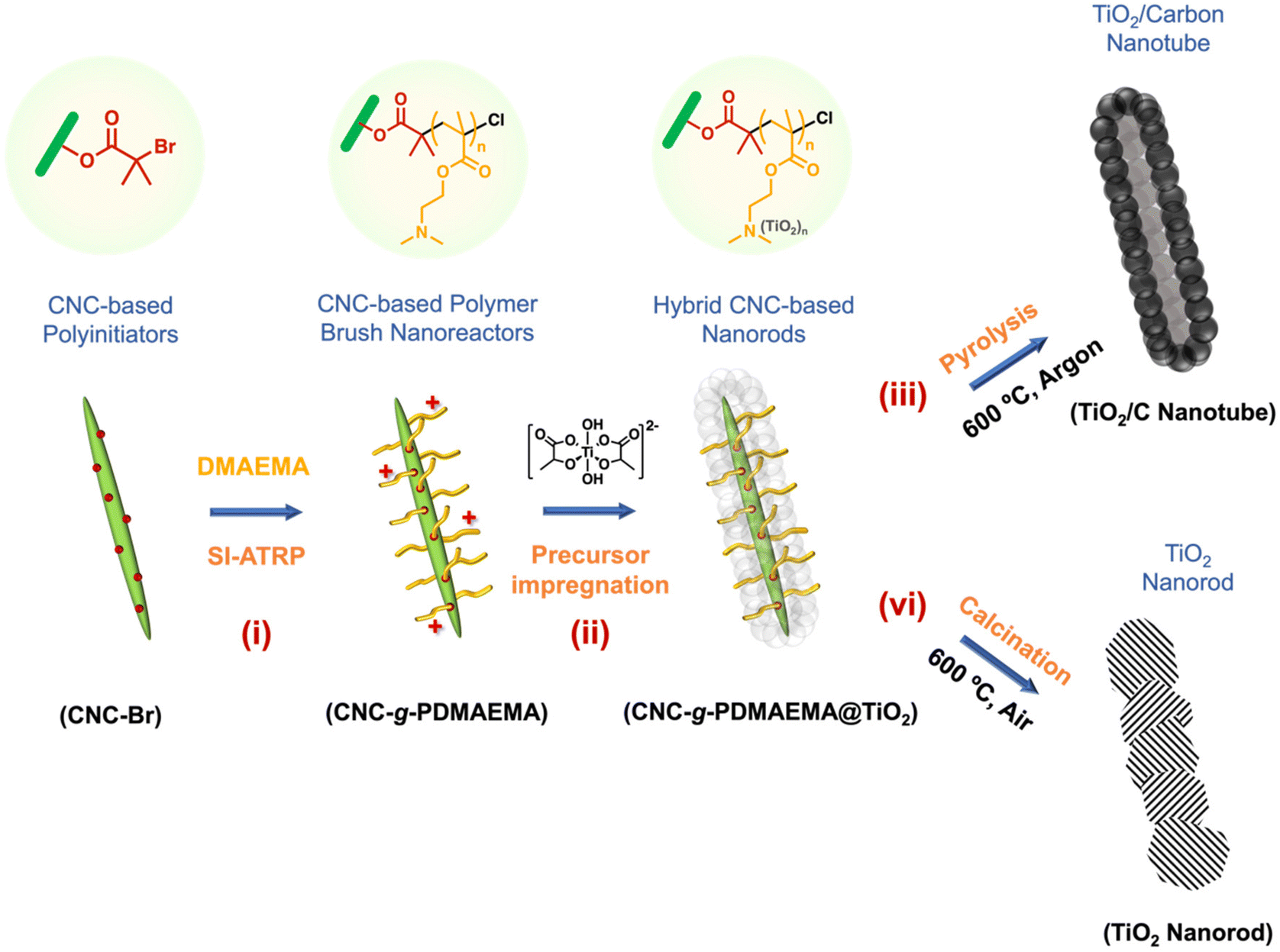 | ||
| Scheme 1 Template-directed formation of anatase TiO2/carbon nanotubes using CNC-based polymer nanoreactors. | ||
Experimental section
Extraction of pristine CNC
Whatman no. 1 filter paper (15 g) was ground into a powder using a coffee grinder. The ground cellulose powder was hydrolysed using sulfuric acid (64 wt%, 260 mL) at 45 °C for 45 min. The acid hydrolysis was quenched by diluting the cellulose suspension with deionised water (2.6 L). The suspension was left to sediment overnight. The acidic supernatant was removed. The cellulose sediment was redispersed in water and centrifuged (6000 rpm) for 20 min. The acidic supernatant was removed. The same redispersion–centrifugation and supernatant disposal processes were repeated. The residual cellulose suspension was dialysed (MWCO = 14.5 kDa) against deionised water for three days with changes of dialysis water regularly until a pH of 5–6 (pH of deionised water) was reached. The cellulose suspension was left to sediment for three days and the supernatant (CNC suspension) was freeze-dried from water to yield pristine CNC (9 g, 55 mmol of glucose units) as a white, fluffy powder.Preparation of the CNC-Br polyinitiators
Pristine CNC (0.300 g, 1.85 mmol) was dispersed in DMF (300 mL) to a homogenous suspension, aided by sonication in a Soniclean 120T sonicator (240 V, 50/60 Hz) for 10 min. To the CNC suspension, triethylamine (TEA, 0.562 g, 5.55 mmol) and DMAP (22.6 mg, 1.85 × 10−1 mmol) were added. The CNC suspension was cooled to 0 °C in an ice bath. After stirring for 30 min, α-bromoisobutyryl bromide (α-BiBB, 1.28 g, 5.55 mmol) was then introduced to the cold CNC suspension in a drop-wise manner. The esterification reaction was left to proceed overnight. The reaction containing CNC-Br was purified by several cycles of centrifugation and redispersion in DMF (3 × 30 mL) and water (1 × 30 mL). The CNC-Br was freeze-dried from water. Br content by mass (Br wt%) was determined by elemental analysis.Preparation of the CNC-g-PDMAEMA nanoreactors
The general CNC-g-PDMAEMA grafting-from SI-ATRP is as follows: in a Schlenk flask containing CNC-Br (50 mg, n(Br) = 4.1 × 10−2 mmol) in DMF, DMAEMA (3.84 mL, 20.5 mmol) and HMTETA (9.5 mg, 4.1 × 10−2 mmol) were added. The reaction mixture was degassed by four freeze–pump–thaw cycles with N2. Following this, CuCl (4.1 mg, 4.1 × 10−2 mmol) was then introduced to the frozen reaction mixture under a gentle N2 flow. After the final freeze–pump–thaw cycle, the SI-ATRP reaction was initiated at 65 °C. After 2 h, the polymerisation was terminated by exposure to atmospheric air and cooling to room temperature. The product was purified by several cycles of centrifugation and redispersion in DMF (3 × 30 mL) and water (1 × 30 mL). The purified CNC-g-PDMAEMA brushes were freeze-dried from water. Monomer conversion was 22% by determining from changes in characteristic 1H NMR integrals and the degree of polymerisation (DP) was calculated to be PDMAEMA112 assuming all initiation sites were initiated during polymerisation. To assess polymerisation control, the same polymerisation was run using a sacrificial initiator ethyl α-bromoisobutyrate (EBiB, 8.0 mg, 4.1 × 10−2 mmol).Preparation of the linear PDMAEMA
DMAEMA (648 mL, 3.9 mmol) and HMTETA (12.0 mg, 5.1 × 10−2 mmol) were added to a Schlenk flask containing EBiB (5.0 mg, 2.6 × 10−2 mmol) in THF. The reaction mixture was degassed by four freeze–pump–thaw cycles with N2. CuCl (3.0 mg, 3.0 × 10−2 mmol) was then introduced to the frozen reaction mixture under a gentle N2 flow. After the final freeze–pump–thaw cycle, the polymersation was initiated at 25 °C. After 2.5 h, the polymerisation was terminated by exposure to atmospheric air and cooling with liquid nitrogen. Copper was removed by passing the mixture through an aluminium oxide column. The product was purified by three cycles of precipitation and redissolving in cold diethyl ether and THF, respectively. The residual solvent was removed from the purified PDMAEMA under high vacuum at room temperature. Monomer conversion was 28% by determining from changes in characteristic 1H NMR integrals, resulting in a DP of 42 DMAEMA units.Formation of the TiO2/carbon and TiO2 nanomaterials
In a typical hybrid formation procedure, CNC-g-PDMAEMA (0.300 g, 1.79 mmol DMAEMA) was dispersed in water, aided by sonication in a Soniclean 120T sonicator (240 V, 50/60 Hz) for 10 min, to yield a homogenous suspension (0.5 mg mL−1). TALH (2.7 g, 8.9 mmol) was added dropwise to the suspension under stirring. The pH of the suspension was adjusted to pH 2 using HCl (1.0 M). The TALH infiltration of the CNC-g-PDMAEMA was continuously stirred overnight. The resulting CNC-g-PDMAEMA/TALH hybrids were purified via several cycles of centrifugation (4200 rpm for 15 min) and redispersion in water (3 × 30 mL). Specifically, unreacted/free TALH dissolved in the water was removed by decanting the supernatant layer consecutively. The purified CNC-g-PDMAEMA/TALH hybrids were then heated to 95 °C overnight, according to a procedure by Caruso et al.25 Finally, the hybrids were either pyrolysed in argon or calcined in air (600 °C, 2 h, heating rate: 3 °C min−1) to produce TiO2/C nanotubes or pure TiO2 nanomaterials, respectively.Results and discussion
The stepwise build-up of core–shell CNC-g-PDMAEMA polymer brush nanoreactors is schematically summarised in Scheme 1. Firstly, pristine CNCs (with lengths ranging between 30–100 nm) were extracted from filter paper with reference to the protocol reported by Edgar et al.26 Their morphology and dimensions were verified via transmission electron microscopy (TEM) (Fig. S1-1†). Pristine CNC with cellulose I structure was also identified using powder X-ray diffraction (PXRD) (Fig. S2†). Hydroxyl groups on the CNCs surface were then converted using α-BiBB to install ATRP initiator sites on the surface of the CNCs, yielding a 1D polyinitiator (CNC-Br). The successful introduction of ATRP initiators to the surface of CNCs was confirmed via Fourier-transform infrared (FTIR) spectroscopy (Fig. 1a). Compared to pristine CNCs (Fig. 1a, black), the presence of a carbonyl stretch at 1748 cm−1 for the CNC-Br polyinitiator indicated the attachment of α-BiBB. The esterification reaction was also verified using PXRD, in which a change in the crystallinity of CNC was observed (Fig. S2†). The bromine content on the polyinitiator was determined to be 6.66 wt% by elemental analysis, which is equivalent to a degree of substitution of ∼40% of surface hydroxyl groups (detailed calculation is provided in the ESI, section S3†).27,28 Next, PDMAEMA112 polymer sidechains were grafted via SI-ATRP. Note, DMAEMA itself has been shown to serve as a reducing agent/ligand in ATRP.29 For our work, we opted for HMTETA as the ligand as it is a commonly used when grafting from CNC surfaces.30–32 Moreover, the sacrificial initiator approach indicated a controlled polymerisation with first-order polymerisation kinetics (Fig. S3†) and the resultant sacrificial PDMAEMA has a dispersity (Đ) of 1.18 (Fig. S4†). The 1H NMR spectrum of CNC-g-PDMAEMA112 with peak assignment is shown in Fig. 1b. A more pronounced carbonyl stretch and a new methyl signal at 1442 cm−1 on the corresponding FTIR spectrum further confirmed the successful grafting of the PDMAEMA sidechains (Fig. 1a). In our previous study on CNC-g-PDMAEMA, we could use atomic force microscopy phase imaging to visualise the core–shell character and nanoparticle-like morphology of these nanoreactors.20 In addition, differences in the thermal degradation profiles of the pristine, CNC-Br and CNC-g-PDMAEMA (Fig. 1c) further underscored the successful modifications on the CNC surface. Thermogravimetric analysis (TGA) showed an onset degradation temperature of 150–170 °C for pristine CNCs, which is consistent with the value in literature reported for CNC with a cellulose I structure.33 The onset degradation temperature was increased once ATRP initiators and polymer sidechains were installed. The increased thermal stability of CNC-Br may be attributed to the desulfation of CNC surface when the surface sulfate groups are substituted with 2-bromoisobutyryl groups.34,35 Additionally, both desulfation and the grafted PDMAEMA sidechains further contributed to the thermal stability of CNC-g-PDMAEMA.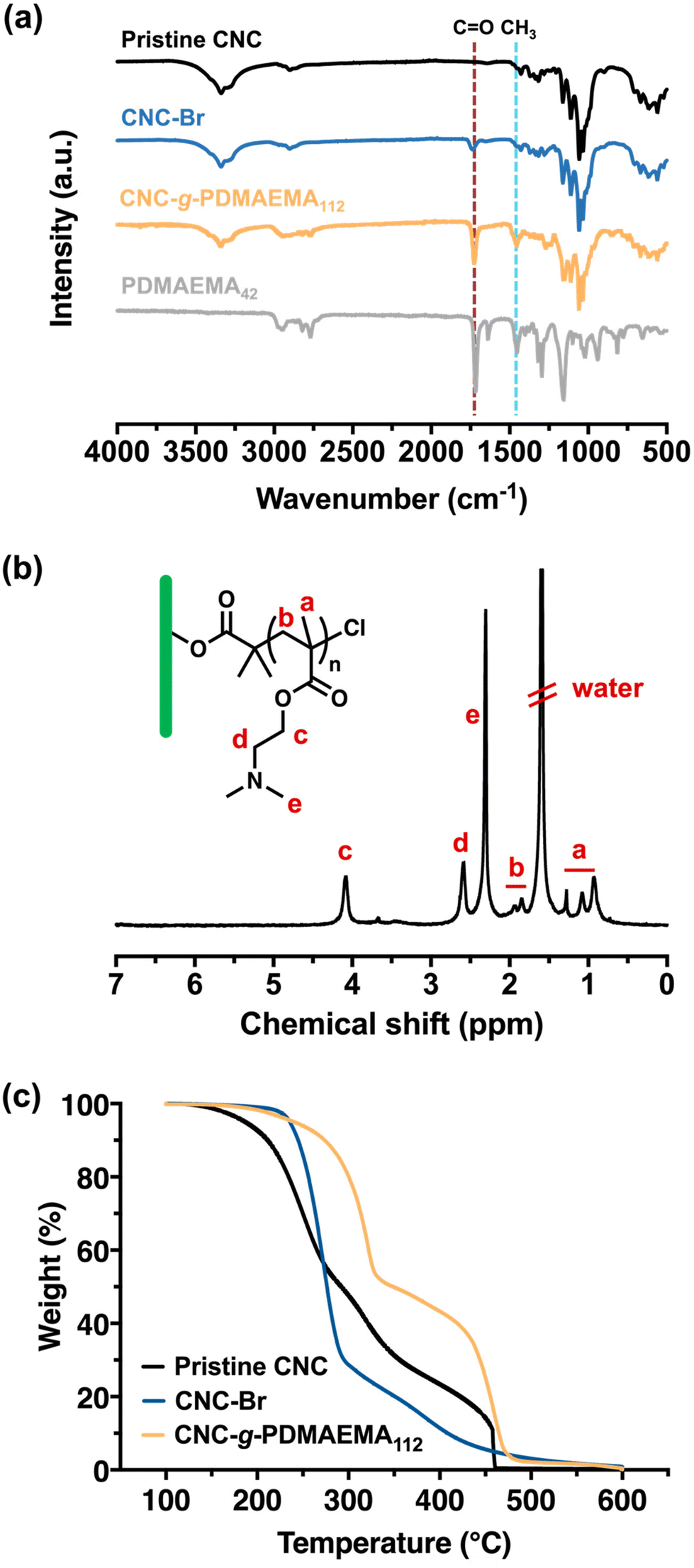 | ||
| Fig. 1 (a) FTIR spectra of pristine CNC (black), CNC-Br (blue) and CNC-g-PDMAEMA112 (yellow) and linear PDMAEMA42 reference (grey). The appearance of a carbonyl peak at 1748 cm−1 confirmed the introduction of ATRP initiator on CNC-Br. The intensifying peaks at 1748 cm−1 and 1442 cm−1 highlighted the presence of the PDMAEMA sidechains. (b) 1H NMR spectral assignment of CNC-g-PDMAEMA112, measured in CDCl3. (c) TGA data of the thermal degradation of pristine CNC (black), CNC-Br (blue) and CNC-g-PDMAEMA112 (yellow) in air. | ||
With a polyelectrolyte shell, the CNC-g-PDMAEMA nanoreactors could be dispersed into acidic water to be used as uniform and shape-anisotropic templates for the fabrication of organic–inorganic hybrid materials. At neutral and low pH, PDMAEMA is positively charged and can complex the oppositely charged and water-soluble titania precursor (TALH). The benefits of using a stable water-soluble titania precursor with controllable hydrolysis, like TALH, has found increasing interest in recent years, including by our group.36–41,56 Complexing the CNC-g-PDMAEMA with TALH yielded uniform rod-like hybrids (denoted as CNC/TALH hereinafter), as verified by scanning electron microscopy (SEM) and TEM (Fig. 2a and b). TALH exclusively infiltrated the polymer shell, while the CNC core ensured the morphology preservation but remained inaccessible. This could be visualised via a greyscale analysis of TEM bright field images of the hybrid nanorods (Fig. 2c, inset), whereby the lower contrast in the middle of each nanorod indicated the CNC core. No crystallinity was observed using PXRD of the CNC/TALH hybrids (Fig. 3b, black plot, Table 1).
 | ||
| Fig. 2 SEM and TEM micrographs of the (a–c) CNC/TALH hybrids, (d–f) TiO2/C nanotubes and (g–i) TiO2 nanorods. The insets in (e/h) are the corresponding HRTEM images, see Fig. S5† for the full figures. The insets in (c/f/i) show the grey-scale analysis of the diameter across the corresponding materials. | ||
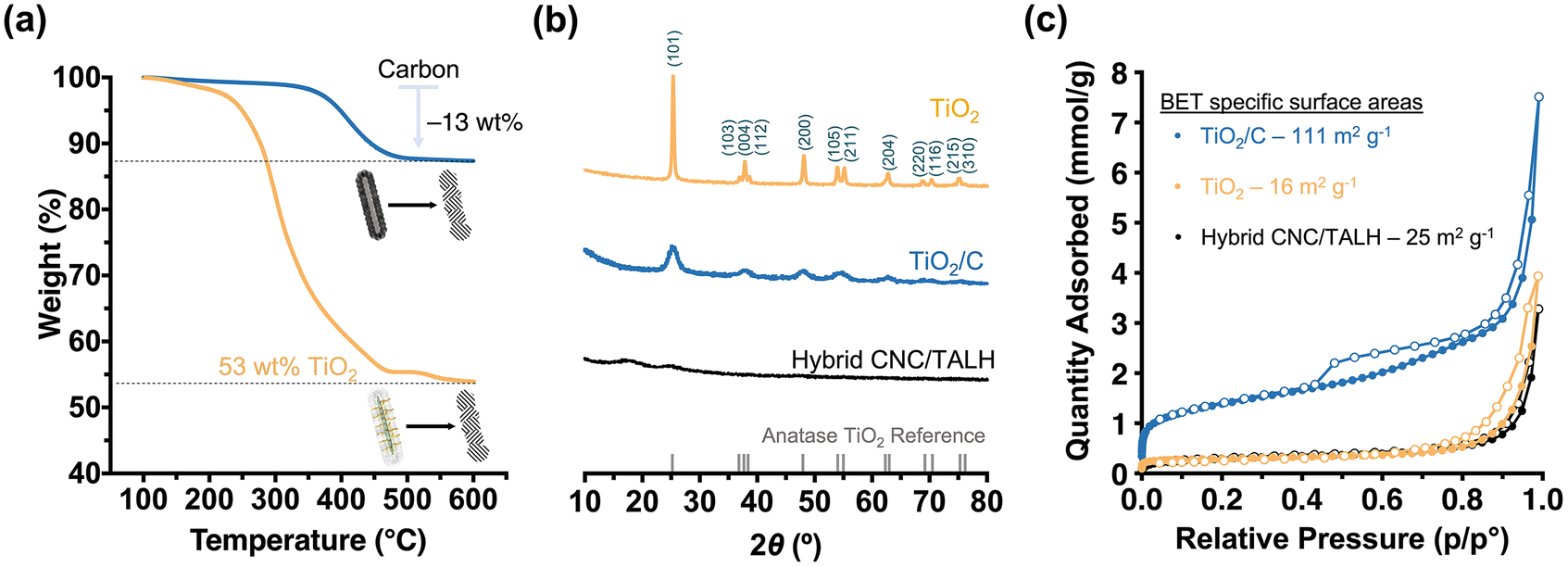 | ||
| Fig. 3 (a) TGA data of hybrid nanorods (yellow) and TiO2/C nanotubes (blue) in air. The residual weight for the hybrids corresponds to the TiO2 content of the CNC-g-PDMAEMA template while weight loss recorded for TiO2/C corresponds to the carbon content. (b) PXRD data (Cu Kα, λ = 1.5406 Å) for the hybrids (black), TiO2/C (blue) and TiO2 (yellow) nanomaterials. The grey vertical ticks designate the Bragg diffraction peaks of anatase TiO2. (c) N2-physisorption isotherms at 77 K of hybrid (black), TiO2/C (blue) and TiO2 (yellow) nanomaterials, determined by applying the Brunauer–Emmett–Teller (BET) theory. Filled and open symbols indicate adsorption and desorption isotherms, respectively. | ||
| Samples | Crystallite sizea (nm) |
S
BET![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) b (m2 g−1) b (m2 g−1) |
Pore volumeb (cm3 g−1) | Capacityc (mA h g−1) | Capacity lossd (%) |
|---|---|---|---|---|---|
| a Average crystallite size calculated from PXRD using the Debye–Scherrer equation. b S BET: specific surface areas and pore volumes estimated based on the BET theory and DFT models, respectively. c Discharge capacity based on the fifth cycle. d Capacity loss as determined by the discharge capacity loss at the 100th cycle since the 5th cycle. | |||||
| CNC/TALH | n/a | 25 | 0.010 | — | — |
| TiO2/C | 4.6 | 111 | 0.046 | 341 | 5 |
| TiO2 | 36.4 | 16 | 0.016 | 189 | 40 |
Next, we pyrolysed our hybrid materials under argon. We hypothesised that pyrolysis would convert polymers into carbon while TALH begins to crystallise and form anatase. However, the forming carbon scaffold should limit the overall anatase crystallite growth and prevent sintering. As TEM showed, pyrolysis of the hybrid nanorods preserved the overall 1D morphology, albeit introducing a level of roughness to the surface of the formed TiO2/C nanotubes (Fig. 2e and f). Nevertheless, the overall core–shell structure was preserved, with the lower contrast core still visible in TEM (Fig. 2f, inset). Instead of a cellulose core, TiO2/C now consisted of a pyrolysed hollow core (Fig. 2e and f), which can be further shown through N2-physisorption measurements (Fig. S6†). At the same time, an amorphous carbon layer coating the TiO2/C nanomaterials was introduced, as can be seen by high-resolution TEM (HR-TEM) image (Fig. 2e, inset). TGA measurements revealed the retention of carbon (13 wt%) and TiO2 (53 wt%) in the TiO2/C nanotubes (Fig. 3a). Moreover, PXRD confirmed the exclusive formation of anatase from TALH (Fig. 3) with all peaks fully indexed to the anatase reference. Peak broadening suggested the presence of small TiO2 crystallites. By applying the Debye–Scherrer equation,42 the average crystallite size in TiO2/C was estimated to be 4.6 nm (Fig. 3b, blue plot, Table 1), consistent with HR-TEM (Fig. 2e, inset). The carbon structure of TiO2/C was further investigated using Raman spectroscopy, identifying two peaks corresponding to disordered (D-band) and graphitic (G-band) carbon (Fig. S7†). The peak intensity ratio of D-band and G-band (ID/IG) was found to be 1.24, which implied amorphous carbon is the dominant carbon in this material. Similarly, characteristic signals of TiO2 were identified in the TiO2/C sample but with weakened intensity due to the carbon scaffold encasing the TiO2 crystallites (Fig. S7†).
In stark contrast, calcination in air removed all organic materials, and no carbon scaffold was preserved (Fig. 2b and Fig. S7†). While the overall rod-shape of the CNC template can still be seen (Fig. 2g), the formation of larger TiO2 crystallites due to sintering indicated a loss of control in the templating process (Fig. 2h and i). For this reason, the internal core structure becomes less obvious and progressively inaccessible. The average crystallite size in the TiO2 nanomaterial, as calculated via Debye–Scherrer equation, was ∼36 nm, which is eight-fold larger than that of TiO2/C. This underscored the contribution and benefit of the carbon scaffold in minimising crystallite growth.
The preservation of carbon on the material also had a pronounced effect on the specific surface area of the nanomaterials. N2-physisorption measurements were performed on all three nanomaterials (hybrid, TiO2/C and TiO2) in order to measure the BET43 specific surface areas and pore size distributions based on density functional theory (DFT)44 (Fig. 3c, Table 1 and Fig. S6†). As expected, the organic–inorganic hybrids showed minimal meso-porosity in the system with a relatively small specific surface area (25 m2 g−1) and pore volume (0.010 cm3 g−1). After pyrolysis, however, the TiO2/C nanotubes demonstrated a significant increase in accessible surface area (111 m2 g−1) and pore volume (0.046 cm3 g−1), especially due to the formation of new micro- and mesopores (mostly within 1–12 nm pore width) (Fig. S6b†). As discussed above, calcination led to a less pronounced templating effect, crystallite sintering and the absence of a carbon scaffold in the TiO2 nanomaterials. Together, this limited the specific surface area (16 m2 g−1) and pore volume (0.016 cm3 g−1) due to the loss of micropores and meso-porosity in the process (Fig. 3c and Fig. S6c†).
We established that our CNC-based templating route affords 1D nanomaterials with high specific surface areas, while a carbon scaffold can minimise the crystallite growth of anatase. We had targeted anatase in our study as it is a promising semiconductor with great potential in batteries.22 Considering the desired size of <7 nm for anatase in lithiation processes,45,46 we then assessed the viability and performance of our nanomaterials in LIBs. Several studies had further demonstrated that the presence of a carbon scaffold may offer additional stability not only to the nanomaterials but also to their use in electronic applications.22,47–49 Our focus was, therefore, on the TiO2/C nanomaterials, but also prepared LIBs from the calcined nanomaterials (Fig. S8-1†) and commercial, non-templated TiO2 nanoparticles (Fig. S8-2†) for comparison. Our TiO2/C nanotubes exhibited a promising electrochemical performance as LIB anodes. The performance was studied under galvanostatic conditions over the potential window 2.5–0.01 V vs. Li/Li+ (Fig. 4). Table 1 provides an overview of the discharge capacities and the capacity retention over 100 cycles of the templated TiO2/C and TiO2 nanomaterials tested at the current density of 0.1 C. From their initial discharge voltage curves (Fig. S8-3†), it became evident that these two materials behave very differently, as we discuss below.
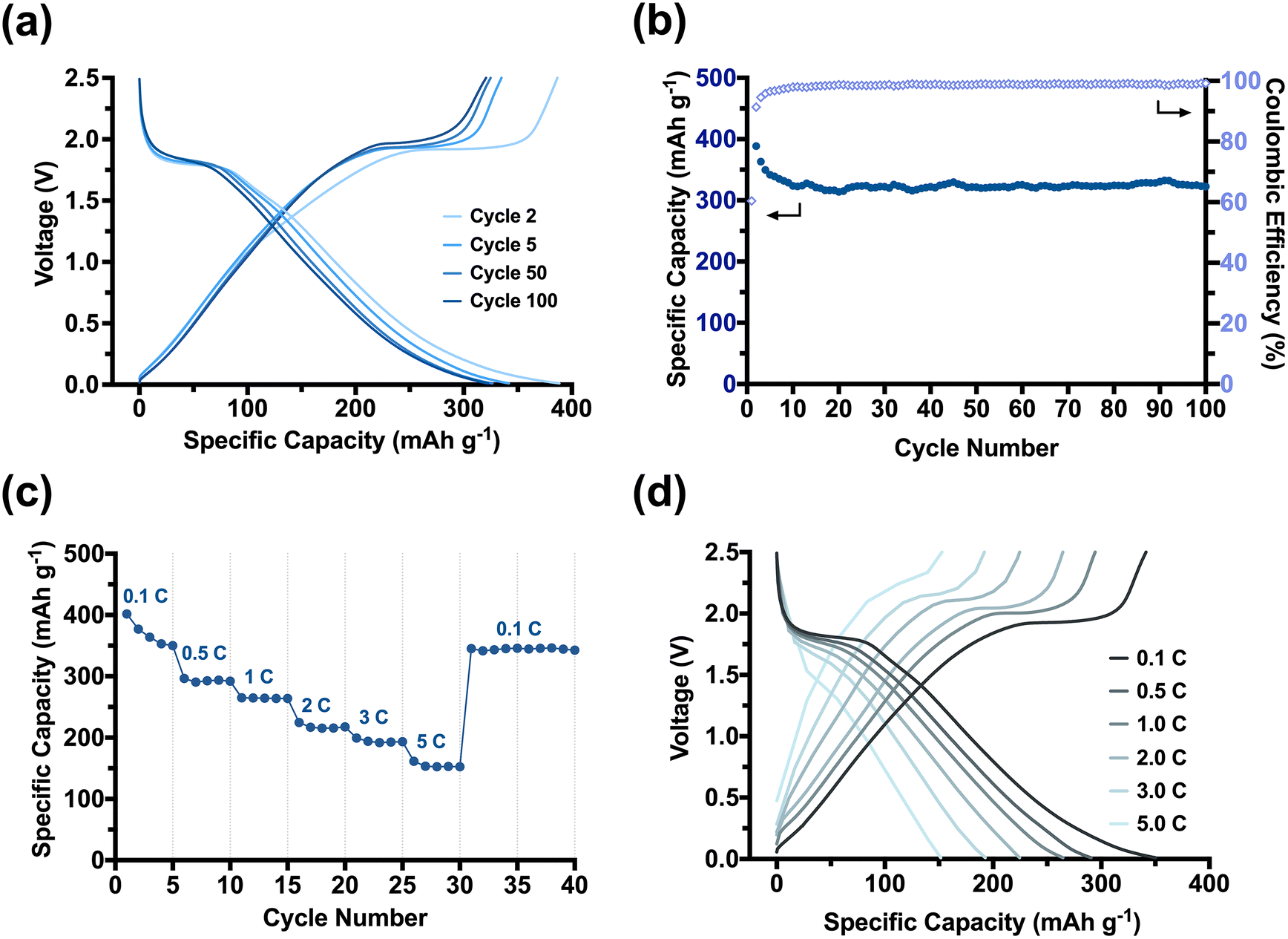 | ||
| Fig. 4 (a) The electrochemical performance of TiO2/C nanotubes as LIB anodes: (a) galvanostatic charge/discharge voltage profiles in cycles 2, 5, 50 and 100 at 0.1 C, (b) cycling performance over 100 cycles at 0.1 C (the plot fluctuation observed in the specific capacity in (b) is caused by the changes in room temperature during battery cycling). (c) Rate performance and (d) galvanostatic charge/discharge curves at various C-rates. | ||
The TiO2/C nanotubes achieved a reversible high discharge and charge capacities of 388 mA h g−1 and 386 mA h g−1 in the second cycle, respectively (Fig. 4a). The resulting capacities were larger than the theoretical capacity of anatase (335 mA h g−1),50 which we attributed to the in situ installed porous carbon scaffold within our pyrolysed nanomaterial. Porous carbons can, apart from Li-intercalation of crystalline graphitic nanodomains, facilitate additional lithiation mechanisms, such as the surface absorption and Li-deposition into nano-pores.51,52 The TiO2/C nanotubes experienced a significant but gradual capacity loss of ∼50 mA h g−1 in the first five cycles until reaching a stable capacity of (341 mA h g−1). The loss can be ascribed to a stabilisation process between the porous anode and the electrolyte. Electrolyte decomposition is a dominant side reaction that typically occurs at the beginning of battery cycling, leading to the formation of a passivation layer known as the solid-electrolyte interphase (SEI) layer on the surfaces of the anode electrodes.53,54
As TiO2/C nanotubes exhibited a high specific surface area (111 m2 g−1), the electrolyte decomposition on them is expected to be significant. Consequently, an extremely high discharge capacity of 640 mA h g−1 was only achieved in their initial discharge cycle (Fig. S8-3†). Moreover, for our porous material, micropores (<2 nm) may become blocked by the decomposing electrolyte, reducing Li-deposition in the early cycles. It takes several initial cycles for the SEI layer to be chemically and morphologically stabilised on the electrode surfaces (especially for materials with high surface areas), from which point our batteries stabilised and minimised further capacity loss between the 5th to 50th cycles (∼4 mA h g−1, 1%). The Coulombic efficiency rapidly reached ∼100% confirming a reversible lithiation–delithiation process in the subsequent cycling of our system (Fig. 4b). The average reversible capacity of TiO2/C nanotubes for cycles between 50 and 100 was 317 mA h g−1, which is comparable to or even higher than that achieved from other anatase-carbon nanomaterials previously reported.55 In comparison to the calcined TiO2 nanorods (Fig. S8-1a and S8-1b†) and commercial nano-sized anatase (Fig. S8-2a and S8-2b†), the TiO2/C nanotubes showed a much improved and stable cycling performance. Moreover, the rate capability (Fig. 4c and d) of the TiO2/C was strongly enhanced compared to the calcined nanomaterials (Fig. S8-1c and S8-1d†). A high capacity of 193 mA h g−1, ∼55% of the at 0.1 C, was achieved when the current density increased by 50 times to 5.0 C. Switching the current density back from 5.0 C to 0.1 C, the maximum capacity of 350 mA h g−1 was immediately recovered, demonstrating a robust electrochemical stability and fast lithiation–delithiation kinetics of our TiO2/C nanotube materials. We attribute this excellent rate capability to the synergistic effects stemming from our templating method. The PDMAEMA shell ensured a homogenous distribution of anatase TiO2 around the CNCs and arranged them into a nanorod. The in situ carbon formed during the pyrolysis step retained the 1D morphology and introduced meso-porosity with accessible pores that promote Li+ movement. More importantly, the carbon also restricted the anatase crystallite growth to an advantageous size of <5 nm. The small crystallites enabled our material to better accommodate faster lithiation and delithiation reactions at high current densities (such as 1.0 C–5.0 C). The large surface area provided an increased reaction interface between the electrolyte and electrode for Li+ diffusion into/from the TiO2/C nanotubes, while the small anatase crystallites enable an efficient and short diffusion pathway for Li+ ions and electrons across the anatase bulk.
Conclusions
We have presented a facile fabrication method for mesoporous carbon-coated anatase with rod-like morphology starting from a renewable CNC resource. The TiO2 precursor was exclusively incorporated into the PDMAEMA shell of our core–shell template nanorods to yield 1D organic–inorganic hybrid nanomaterials. Pyrolysis of the materials produced TiO2/C nanotubes with high surface area and meso-porosity. Moreover, the CNC-g-PDMAEMA template converted into a carbon scaffold that surrounded the forming anatase crystallites and limited their overall growth to below 5 nm. The synergistic effect of the small TiO2 nanocrystals in a highly porous, carbon-based framework highlight the potential use of our nanomaterials as anodes in LIBs. We anticipate that the construction of in situ carbon-coated metal oxide nanorods/nanotubes through our approach can be applied to other functional oxide materials, particularly for battery electrode materials that suffer from intrinsically low electrical and ionic conductivities. Moreover, the composition and carbon content may be easily tailored via the brush length, while the overall process could be extended to other cellulosic materials, such as cellulose nanofibers.Author contributions
M. M. conceived the idea and supervised the research with C. D. L., Y. T. C. and M. M. planned and developed the experimental setup. Y. T. C. performed all the experiments and analysed the data. Q. X. and Y. T. C. performed the electrochemical experiments and analysed the data. H. L. contributed to the collection of TEM images. Y. T. C. and M. B. S. performed the N2 physisorption measurements and analysed the data. All the authors discussed the results. Y. T. C. and M. M. co-wrote the manuscript with inputs from all the authors.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
M.M. and C. D. L. acknowledge the Australian Research Council for their Future Fellowship (FT200100185, M. M.) and Discovery Projects (DP200100959, C.D.L.; DP220100452, M. M.), respectively. This research was facilitated by access to Sydney Analytical, a core research facility at the University of Sydney. The authors acknowledge the technical and scientific assistance of Sydney Microscopy & Microanalysis, The University of Sydney node of Microscopy Australia. We acknowledge Prof. Deanna D'Alessandro for use of the 3-Flex Gas Adsorption Analyzer and Dr Michelle Wood for performing the Raman measurements. Y. T. C. is a grateful recipient of a University of Sydney International Scholarship (USydIS). Q. X. thanks the Australian Institute of Nuclear Science and Engineering for the Early Career Researcher (ECR) Grant and the Sydney Nano Institute for the ECR Support Fund. M. M. is a grateful recipient of a University of Sydney Research Accelerator (SOAR) Prize.References
- E. Kontturi, P. Laaksonen, M. B. Linder, Nonappa, A. H. Gröschel, O. J. Rojas and O. Ikkala, Adv. Mater., 2018, 30, 1703779 CrossRef.
- K. Heise, E. Kontturi, Y. Allahverdiyeva, T. Tammelin, M. B. Linder, Nonappa and O. Ikkala, Adv. Mater., 2021, 33, 2004349 CrossRef CAS PubMed.
- E. Lizundia, D. Puglia, T. D. Nguyen and I. Armentano, Prog. Mater. Sci., 2020, 112, 100668 CrossRef CAS.
- P. Lv, X. Lu, L. Wang and W. Feng, Adv. Funct. Mater., 2021, 31, 2104991 CrossRef CAS.
- T. Xu, H. Du, H. Liu, W. Liu, X. Zhang, C. Si, P. Liu and K. Zhang, Adv. Mater., 2021, 33, 2101368 CrossRef CAS.
- Z. Fang, G. Hou, C. Chen and L. Hu, Curr. Opin. Solid State Mater. Sci., 2019, 23, 100764 CrossRef CAS.
- S. Shafiei-Sabet, W. Y. Hamad and S. G. Hatzikiriakos, Langmuir, 2012, 28, 17124–17133 CrossRef CAS PubMed.
- Y. Wu, Y. Liang, C. Mei, L. Cai, A. Nadda, Q. Van Le, Y. Peng, S. S. Lam, C. Sonne and C. Xia, Chemosphere, 2022, 286, 131891 CrossRef CAS PubMed.
- S. S. Nair, J. Zhu, Y. Deng and A. J. Ragauskas, Sustainable Chem. Processes, 2014, 2, 1–7 CrossRef.
- K. J. De France, T. Hoare and E. D. Cranston, Chem. Mater., 2017, 29, 4609–4631 CrossRef CAS.
- F. V. Ferreira, C. G. Otoni, K. J. De France, H. S. Barud, L. M. F. Lona, E. D. Cranston and O. J. Rojas, Mater. Today, 2020, 37, 126–141 CrossRef CAS.
- R. G. R. Brenes, M. A. B. Grieco, N. Bojorge and N. Pereira, ChemNanoMat, 2021, 7, 1259–1272 CrossRef CAS.
- S. Peter, N. Lyczko, D. Gopakumar, H. J. Maria, A. Nzihou and S. Thomas, J. Mater. Sci., 2022, 57, 6835–6880 CrossRef CAS.
- R. Calle-Gil, E. Castillo-Martínez and J. Carretero-González, Adv. Sustainable Syst., 2022, 6, 2100395 CrossRef CAS.
- P. A. Nizam, D. A. Gopakumar, Y. B. Pottathara, D. Pasquini, A. Nzihou and S. Thomas, in Nanocellulose Based Composites for Electronics, Elsevier, 2021, pp. 15–29 Search PubMed.
- L. H. Nguyen, S. Naficy, R. Chandrawati and F. Dehghani, Adv. Mater. Interfaces, 2019, 6, 30–33 Search PubMed.
- M. Müllner and A. H. E. Müller, Polymer, 2016, 98, 389–401 CrossRef.
- J. H. Swisher, L. Jibril, S. H. Petrosko and C. A. Mirkin, Nat. Rev. Mater., 2022, 7, 428–448 CrossRef CAS.
- S. Wohlhauser, G. Delepierre, M. Labet, G. Morandi, W. Thielemans, C. Weder and J. O. Zoppe, Macromolecules, 2018, 51, 6157–6189 CrossRef CAS.
- M. Morits, V. Hynninen, Nonappa, A. Niederberger, O. Ikkala, A. H. Gröschel and M. Müllner, Polym. Chem., 2018, 9, 1650–1657 RSC.
- R. Wu, S. Shen, G. Xia, F. Zhu, C. Lastoskie and J. Zhang, ACS Appl. Mater. Interfaces, 2016, 8, 19968–19978 CrossRef CAS PubMed.
- S. Liang, X. Wang, Y. J. Cheng, Y. Xia and P. Müller-Buschbaum, Energy Storage Mater., 2022, 45, 201–264 CrossRef.
- Y. Meng, T. M. Young, P. Liu, C. I. Contescu, B. Huang and S. Wang, Cellulose, 2015, 22, 435–447 CrossRef CAS.
- F. D'Acierno, W. Y. Hamad, C. A. Michal and M. J. MacLachlan, Bioresour. Technol. Rep., 2021, 16, 100838 CrossRef.
- K. S. Mayya, D. I. Gittins and F. Caruso, Chem. Mater., 2001, 13, 3833–3836 CrossRef CAS.
- C. D. Edgar and D. G. Gray, Cellulose, 2003, 10, 299–306 CrossRef CAS.
- M. S. Islam, L. Chen, J. Sisler and K. C. Tam, J. Mater. Chem. B, 2018, 6, 864–883 RSC.
- J. Majoinen, A. Walther, J. R. McKee, E. Kontturi, V. Aseyev, J. M. Malho, J. Ruokolainen and O. Ikkala, Biomacromolecules, 2011, 12, 2997–3006 CrossRef CAS PubMed.
- Y. Kwak and K. Matyjaszewski, Polym. Int., 2009, 58, 242–247 CrossRef CAS.
- Q. Xu, J. Yi, X. Zhang and H. Zhang, Eur. Polym. J., 2008, 44, 2830–2837 CrossRef CAS.
- H. Rosilo, J. R. McKee, E. Kontturi, T. Koho, V. P. Hytönen, O. Ikkala and M. A. Kostiainen, Nanoscale, 2014, 6, 11871–11881 RSC.
- J. Yi, Q. Xu, X. Zhang and H. Zhang, Polymer, 2008, 49, 4406–4412 CrossRef CAS.
- P. Dhar, D. Tarafder, A. Kumar and V. Katiyar, RSC Adv., 2015, 5, 60426–60440 RSC.
- X. Zhang, J. Zhang, L. Dong, S. Ren, Q. Wu and T. Lei, Cellulose, 2017, 24, 4189–4203 CrossRef CAS.
- Y. Yin, X. Tian, X. Jiang, H. Wang and W. Gao, Carbohydr. Polym., 2016, 142, 206–212 CrossRef CAS PubMed.
- M. Schöttle, Q. Xia, Y. T. Cheng, N. D. Shepherd, C. D. Ling and M. Müllner, Chem. Mater., 2020, 32, 4716–4723 CrossRef.
- M. Müllner, T. Lunkenbein, N. Miyajima, J. Breu and A. H. E. Müller, Small, 2012, 8, 2636–2640 CrossRef PubMed.
- H. Li, X. Mao, H. Wang, Z. Geng, B. Xiong, L. Zhang, S. Liu, J. Xu and J. Zhu, Macromolecules, 2020, 53, 4214–4223 CrossRef CAS.
- M. Müllner, T. Lunkenbein, M. Schieder, A. H. Gröschel, N. Miyajima, M. Förtsch, J. Breu, F. Caruso and A. H. E. Müller, Macromolecules, 2012, 45, 6981–6988 CrossRef.
- O. F. McRae, Q. Xia, S. Tjaberings, A. H. Gröschel, C. D. Ling and M. Müllner, J. Polym. Sci., Part A: Polym. Chem., 2019, 57, 1890–1896 CrossRef CAS.
- K. Subramanya Mayya, D. I. Gittins and F. Caruso, Chem. Mater., 2001, 3833–3836 CrossRef.
- U. Holzwarth and N. Gibson, Nat. Nanotechnol., 2011, 6, 534–534 CrossRef CAS PubMed.
- S. Brunauer, P. H. Emmett and E. Teller, J. Am. Chem. Soc., 1938, 60, 309–319 CrossRef CAS.
- J. Landers, G. Y. Gor and A. V. Neimark, Colloids Surf., A, 2013, 437, 3–32 CrossRef CAS.
- S. Leijonmarck, A. Cornell, G. Lindbergh and L. Wågberg, J. Mater. Chem. A, 2013, 1, 4671–4677 RSC.
- J. Janek, M. Martin and K. D. Becker, Phys. Chem. Chem. Phys., 2009, 11, 3010 RSC.
- R. Qian, H. Lu, T. Yao, F. Xiao, J.-W. Shi, Y. Cheng and H. Wang, ACS Sustainable Chem. Eng., 2022, 10, 61–70 CrossRef CAS.
- K. Zhu, W. Feng, Z. Xue, M. Sun, S. Wu, Z. Jing and Y. Yu, ACS Sustainable Chem. Eng., 2022, 10, 10955–10965 CrossRef.
- W. J. Zhang, J. Power Sources, 2011, 196, 2962–2970 CrossRef CAS.
- L. Bai, F. Fang, Y. Zhao, Y. Liu, J. Li, G. Huang and H. Sun, RSC Adv., 2014, 4, 43039–43046 RSC.
- G. Wang, M. Yu and X. Feng, Chem. Soc. Rev., 2021, 50, 2388–2443 RSC.
- K. Persson, V. A. Sethuraman, L. J. Hardwick, Y. Hinuma, Y. S. Meng, A. Van Der Ven, V. Srinivasan, R. Kostecki and G. Ceder, J. Phys. Chem. Lett., 2010, 1, 1176–1180 CrossRef CAS.
- A. Wang, S. Kadam, H. Li, S. Shi and Y. Qi, npj Comput. Mater., 2018, 4 Search PubMed.
- S. K. Heiskanen, J. Kim and B. L. Lucht, Joule, 2019, 3, 2322–2333 CrossRef CAS.
- S. Paul, M. A. Rahman, S. Bin Sharif, J.-H. Kim, S.-E.-T. Siddiqui and M. A. M. Hossain, Nanomaterials, 2022, 12, 2034 CrossRef CAS PubMed.
- Y. T. Cheng, Q. Xia, H. Liu, M. B. Solomon, E. R. L. Brisson, L. D. Blackman, C. D. Ling and M. Müllner, ACS Appl. Mater. Interfaces, 2023, 15(9), 12261–12272 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Materials, methods, and supplementary results, including polymer and hybrid characterisation, electron microscopy, physisorption and electrochemical performance studies. See DOI: https://doi.org/10.1039/d3py00194f |
| This journal is © The Royal Society of Chemistry 2023 |