
Azobenzene-decorated cellulose nanocrystals as photo-switchable chiral solutes in nematic liquid crystals†
Barış
Sezgin
ab and
Torsten
Hegmann
 *bcd
*bcd
aDepartment of Chemistry, Süleyman Demirel University, 32260 Isparta, Cunur, Turkey
bAdvanced Materials and Liquid Crystal Institute, Kent State University, Kent, OH 44242-001, USA. E-mail: thegmann@kent.edu
cDepartment of Chemistry and Biochemistry, Kent State University, Kent, OH 44242-001, USA
dMaterials Science Graduate Program, Kent State University, Kent, OH 44242-001, USA
First published on 17th November 2022
Abstract
In this work, we validate that cellulose nanocrystals (CNCs), surface-functionalized with pro-mesogenic azobenzene pendants, can act as photoswitchable chiral solutes in a nematic liquid crystal (N-LC). Upon UV illumination, the helical pitch and thus the helical twisting power (βw) can be altered based on the trans–cis photoisomerization of the azobenzene pendants. This approach shows that CNCs—biorenewable chiral additives with a commensurate, rod-like shape to N-LC molecules—can be adapted just like common small molecule organic chiral solutes in LC phases based on simple surface modifications.
1. Introduction
To examine how the geometry of a chiral object affects chirality transfer to a surrounding medium, earlier approaches focused on the transmission and amplification of chirality from colloidal nanomaterials commonly involving a metal (gold) core differing in shape and overall dimensions.1 An efficient strategy takes advantage of a suitable reporter medium that allows one to quantify the transmission efficacy of chiral information from a chiral nano-object to its surrounding.2–8 Our research has shown that to acquire this knowledge, an induced chiral nematic LC (N*-LC) phase, as a responsive and birefringent medium, can be used quite effectively since the induced helical arrangement of the spatial orientation of the constituent building blocks (molecules, molecular assemblies, or anisometric particles) caused by the addition of chiral nanoscale additives translates to the bulk, thereby facilitating the detection, visualization, and measurement of chirality on length scales easily accessible by optical microscopy between crossed polarizers.9 A combination of non-correlated experimental and geometrically derived data revealed how the chirality transfer efficacy depends on the geometry of chiral gold nanoshapes. The underlying mechanism is based on the scalar product of an established pseudoscalar chirality indicator and a scalar geometric shape compatibility factor based on the 2-D isoperimetric ratio of the nanoshape solute and the used N-LC molecules.1,10Previously studied gold nanoshapes differing in size, shape, and aspect ratio were surface functionalized using various types of chiral ligands as chiral additives to study chirality amplification with an achiral N-LC host serving as the reporter. In this context, all gold nanoshape solutes were composed of a molecularly and morphologically achiral core capped with a monolayer of ‘passive’ chiral molecules. In those studies, we established that chiral molecule-decorated rod-like nanoshapes with 2-D isoperimetric quotients matching those of the N-LC host molecules are among the most proficient in their ability to transfer chirality to an N-LC host phase, exhibiting high helical twisting power values βw (βw = (p·c·r)−1, where p is the helical pitch of the induced N*-LC phase, c is the weight fraction of chiral solutes (wt%), and r is the enantiomeric purity).1 The values of βw and a molar helical twisting power (βmol) were one to two orders of magnitude higher than those of the free organic chiral ligands or some of the strongest common organic chiral additives.10
Aside from a few exceptions of engineered, helically structured inorganic films11 or inverse helical organic filament networks,12 nanomaterials with sole or additional core-chirality have only very recently been studied for their ability to transfer chirality.13 While potential contributions to the overall chirality by chiral field effects, those generated by a chiral organic ligand shell,14 or by a chiral adsorption pattern on such plasmonic nanoshapes with high polarizability cannot be overlooked,15 and key questions arising from these prior studies were if and to what extent a potentially chiral core contributes to the overall, highly potent chirality transfer to the surrounding N-LC reporter medium.
To elucidate the role of core chirality, we recently reported on the chirality transfer by nanostructures with an intrinsically chiral core, i.e., cellulose nanocrystals (CNCs) that were further covalently modified with either chiral or achiral molecules.16,17
We found that among such CNCs, those functionalized with achiral moieties structurally related to the N-LC showed better N-LC solubility and thus serving as efficient chiral inducers with βw values from −35 μm−1 for neat to −54 μm−1 for cholesterol-modified CNCs. Here, functionalization with chiral molecules rather than just mesogenic moieties that were structurally alike to the N-LC host only slightly enhanced the efficacy of helical distortion in the host N-LC, indicating the high propensity of CNCs to transfer chirality from an inherently chiral core.16
As for the CNCs inherent core chirality, during acid hydrolysis, the amorphous regions of the cellulose fibers are cleaved, yielding negatively charged rod-like CNCs of high crystallinity with dimensions ranging from 5 to 20 nm in width and 100 to 500 nm in length.19 The final nanocrystals are composed of chiral β(1 → 4) D-glucose subunits, which render CNCs structurally chiral with a twist in both their internal structure and their outer morphology, either on at individual NC or at the formed bundle level.20–30 Their core chirality together with their rod-like shape enable CNCs to self-assemble into left-handed chiral nematic liquid crystal (N*-LC) phases, when dispersed in various solvents (commonly water) or after solvent evaporation.31–40
To further enhance the versatility of hydrophobic CNCs as chiral solutes inducing N*-LC phases, the next logical step seemed to decorate CNCs with photoswitchable molecules that allow for the ‘active’ control (switching) of p, thus βw, by altering the wavelength of light irradiation.41–43 Azobenzene derivatives with aliphatic carboxylic acid end groups that allow for CNC surface modification appeared as the most logical initial choice.
The reversible photoisomerization of azobenzenes has been and still is used in an extensive array of materials systems and for applications ranging from catalysis44–46 over energy harvesting47 and actuators48 to countless optical applications.49,50 The latter presumably receiving the bulk of attention because of the robust and well-understood trans–cis photoisomerization of azobenzene derivatives that can be switched by a combination of light at various wavelengths and temperatures.51
The concept of grafting two structurally purposefully different azobenzene derivatives to CNCs is schematically shown in Fig. 1. CNC-AC features a more polar azobenzene with a terminal cyano-group,52 CNC-AB, in contrast, a more apolar terminal aliphatic hydrocarbon chain,53 thus making this CNC's surface more compatible with the N-LC used in these studies (Felix-2900-03, Fig. 2), which is also flanked by two aliphatic side chains.
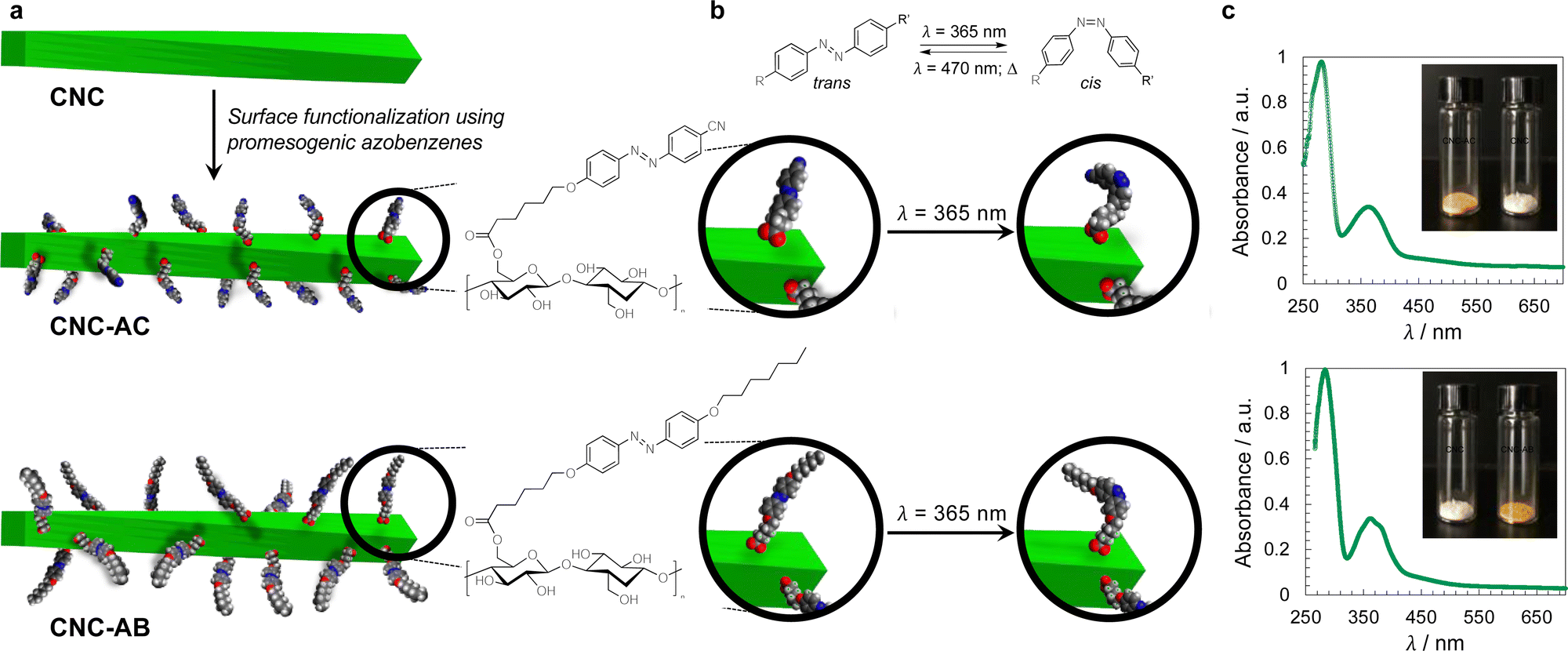 | ||
| Fig. 1 (a) Concept of CNC surface modification with azobenzene derivatives (AC or AB) and (b) depiction of the trans–cis photoisomerization of an azobenzene moiety – principle (top) and on the surface of the functionalized CNCs. (c) UV-vis spectra of CNC-AC and CNC-AB (DMF, 20 °C)—the major peak at λ ∼ 285 nm corresponds to acid-treated CNCs,18 the peak centered at λ ∼ 370 nm corresponds to trans-azobenzenes and the small shoulder at λ ∼ 480 nm corresponds to cis-azobenzenes (insets show photographs of neat and surface-modified CNCs). | ||
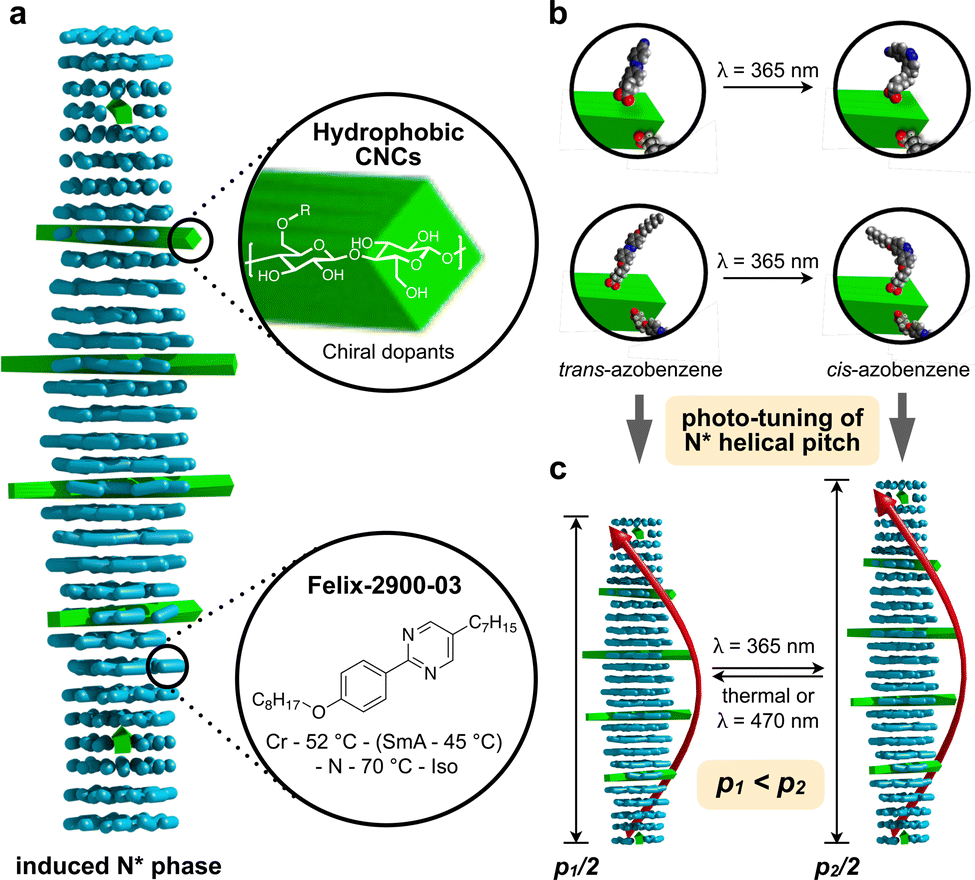 | ||
| Fig. 2 (a) Schematic representation of the N*-LC phase induced by the addition of minute amounts of the functionalized CNCs in the N-LC host, Felix-2300-03. (b) The trans–cis photoisomerization of the azobenzene pendants (AC or AB) induced by illumination with UV light (at λ = 365 nm) to prompt (c) an increase in the helical pitch (p) when the azobenzenes are in their cis configuration. | ||
2. Experimental
2.1 Materials
All reagents used were purchased from Sigma-Aldrich and used without further purification. Cellulose nanocrystals (CNCs) were obtained from CelluForce in the form of the sulphate sodium salt. The two azobenzene pendant molecules in their avid form were prepared as outlined in the ESI† (Section S1, Schemes S1, S2 and Fig. S1–S7).2.2 Methods
1H NMR spectra were recorded on a Bruker DMX 400 MHz spectrometer. The cross-polarization magic angle spinning (CP-MAS) 13C NMR spectra were recorded on a Bruker Avance III (100.6 MHz) 400 NMR spectrometer. 4 mm Zirconia rotors with a stirring speed of 5 kHz were used with 5000 to 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 scans. FT-IR spectra were recorded on a Bruker FT-IR Tensor27 system equipped with a Pike miracle ATR (attenuated total reflectance) accessory. Thermogravimetric analysis (TGA) was performed using a TGA Q500 system (TA Instruments). Differential scanning calorimetry (DSC) experiments were performed using a PerkinElmer Pyris 1 DSC at a heating/cooling rate of 10 °C min−1, reporting the data from the first heating and cooling cycle, respectively (temperatures calibrated with In and Zn standards). UV irradiation experiments were carried out at λ = 365 nm using a UVCureMXLED LED light curing spot gun (P = 6 W). Polarized optical microscopy (POM) observations of the induced N*-LC phases were recorded and photographed using an Olympus BX-53 polarizing microscope equipped with a Linkam LTS420E heating/cooling stage. UV-vis data were acquired using an OLIS spectrophotometer (solution data; quartz cuvettes, 1 cm path length). Transmission electron microscopy (TEM) analysis was performed using an FEI Tecnai TF20 TEM instrument at an accelerating voltage of 200 kV. XPS spectra were recorded using a PHI 5000 Versaprobe XPS using a monochromatic Al Kα source (1486.6 eV). Survey spectra were collected from 1100 to 0 eV at a pass energy of 93.90 eV. High resolution C 1s spectra were collected at a pass energy of 11.75 eV with a scan step of 0.1 eV. The Phi MultiPack software was used for data processing. High-resolution XPS spectra were fitted using linear combinations of 70:30 Gauss-Lorentzian functions on the Shirley background-subtracted spectra. Binding energy scale was calibrated using an aliphatic C 1s component, set at 285.0 eV.
000 scans. FT-IR spectra were recorded on a Bruker FT-IR Tensor27 system equipped with a Pike miracle ATR (attenuated total reflectance) accessory. Thermogravimetric analysis (TGA) was performed using a TGA Q500 system (TA Instruments). Differential scanning calorimetry (DSC) experiments were performed using a PerkinElmer Pyris 1 DSC at a heating/cooling rate of 10 °C min−1, reporting the data from the first heating and cooling cycle, respectively (temperatures calibrated with In and Zn standards). UV irradiation experiments were carried out at λ = 365 nm using a UVCureMXLED LED light curing spot gun (P = 6 W). Polarized optical microscopy (POM) observations of the induced N*-LC phases were recorded and photographed using an Olympus BX-53 polarizing microscope equipped with a Linkam LTS420E heating/cooling stage. UV-vis data were acquired using an OLIS spectrophotometer (solution data; quartz cuvettes, 1 cm path length). Transmission electron microscopy (TEM) analysis was performed using an FEI Tecnai TF20 TEM instrument at an accelerating voltage of 200 kV. XPS spectra were recorded using a PHI 5000 Versaprobe XPS using a monochromatic Al Kα source (1486.6 eV). Survey spectra were collected from 1100 to 0 eV at a pass energy of 93.90 eV. High resolution C 1s spectra were collected at a pass energy of 11.75 eV with a scan step of 0.1 eV. The Phi MultiPack software was used for data processing. High-resolution XPS spectra were fitted using linear combinations of 70:30 Gauss-Lorentzian functions on the Shirley background-subtracted spectra. Binding energy scale was calibrated using an aliphatic C 1s component, set at 285.0 eV.
3. Results and discussion
3.1 Surface-modified CNC synthesis and characterization
To prepare azobenzene-functionalized CNCs, we esterified the surface of the CNCs (specifically the primary hydroxyl group at carbon C6) with two promesogenic azobenzene derivatives: one with a terminal cyano group (AC) and one with a terminal aliphatic hydrocarbon chain (AB) as shown in Fig. 1. Prior to this step, the sodium sulfonate groups, despite the overall low degree of sulfonation, were converted into sulfonic acid groups to render the CNCs more soluble in organic solvents.Functionalization of the CNCs with the corresponding carboxylic acids was performed using Steglich esterification (EDC/DMAP) (ESI,† Section S1.2).54 FT-IR analysis shows that the CNCs’ primary hydroxyl groups were successfully converted, judging from the presence of peaks at about 1700 cm−1 and 1600 cm−1, which correspond to the ester C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretches and aromatic CC skeletal vibrations, respectively (ESI,† Fig. S8).42 Moreover, the solid-state cross-polarization magic angle spinning (CP-MAS) 13C NMR experiments confirmed that each of the CNCs was indeed functionalized with the appearance of the carbonyl-C peaks at around 175 to 160 ppm, weak aromatic-C peaks between 140 and 120 ppm, and aliphatic carbon signals at around 50 to 10 ppm. The area of the peak corresponding to the C6 amorphous region, labeled as C6 (and small shoulder on the right in Fig. S9, ESI†), appears to decrease for the functionalized CNC products when compared to the untreated CNCs,16 and thus indicates the esterification of the C6 hydroxyl groups in the amorphous region.55 CP-MAS solid state 13C NMR analysis also demonstrated that the structural crystallinity was preserved.56 X-ray photoelectron spectroscopy (XPS) analysis57 (ESI,† Fig. S10 and S11) afforded estimations of the degree of surface functionalization (DSF) for the functionalized CNCs, which were calculated to be 0.10 for CNC-AC and 0.17 for CNC-AB (see the ESI,† Section S2.3).58 The thermogravimetric analysis (TGA) of all samples further supported the functionalization by showing altered weight loss curves (ESI,† Fig. S12).16 Transmission electron microscopy (TEM) micrographs obtained for the functionalized CNCs corroborate the 13C NMR data by showing that neither crystallinity nor morphology were affected by the surface modifications when compared to the TEM images collected for the unmodified CNCs (ESI,† Fig. S13).59 The average width was determined to be 20 nm and the length varied from about 100 to 150 nm, giving an average aspect ratio of 6.25.
O stretches and aromatic CC skeletal vibrations, respectively (ESI,† Fig. S8).42 Moreover, the solid-state cross-polarization magic angle spinning (CP-MAS) 13C NMR experiments confirmed that each of the CNCs was indeed functionalized with the appearance of the carbonyl-C peaks at around 175 to 160 ppm, weak aromatic-C peaks between 140 and 120 ppm, and aliphatic carbon signals at around 50 to 10 ppm. The area of the peak corresponding to the C6 amorphous region, labeled as C6 (and small shoulder on the right in Fig. S9, ESI†), appears to decrease for the functionalized CNC products when compared to the untreated CNCs,16 and thus indicates the esterification of the C6 hydroxyl groups in the amorphous region.55 CP-MAS solid state 13C NMR analysis also demonstrated that the structural crystallinity was preserved.56 X-ray photoelectron spectroscopy (XPS) analysis57 (ESI,† Fig. S10 and S11) afforded estimations of the degree of surface functionalization (DSF) for the functionalized CNCs, which were calculated to be 0.10 for CNC-AC and 0.17 for CNC-AB (see the ESI,† Section S2.3).58 The thermogravimetric analysis (TGA) of all samples further supported the functionalization by showing altered weight loss curves (ESI,† Fig. S12).16 Transmission electron microscopy (TEM) micrographs obtained for the functionalized CNCs corroborate the 13C NMR data by showing that neither crystallinity nor morphology were affected by the surface modifications when compared to the TEM images collected for the unmodified CNCs (ESI,† Fig. S13).59 The average width was determined to be 20 nm and the length varied from about 100 to 150 nm, giving an average aspect ratio of 6.25.
3.2 Mixtures of the N-LC host doped with CNC solutes
To examine how efficiently the surface-modified CNCs can induce a helical distortion of guest molecules of an achiral N-LC host medium, we prepared and examined binary mixtures at various concentrations of the functionalized CNCs in the N-LC phase of Felix-2900-03. Minute amounts of the chiral CNC additives were added at a temperature just above the isotropic-nematic phase transition (TIso-N) of the N-LC to formulate mixtures, ranging from 0.05 to 2.0 wt% CNCs, which were rigorously sonicated and then slowly cooled down until a phase transition to the induced N*-LC was observed. The textural characteristics of the induced N*-LC phase would thereby evidence chirality transfer from the CNC chiral core to the achiral N-LC bulk.Polarized optical microscopy (POM) studies of the CNC-doped samples prepared between untreated glass slides show the formation of the characteristic fingerprint textures, resulting from the induction of an N*-LC phase with values of p ranging from about 6 to 8 μm at 0.05 wt% and 3 to 4 μm at 0.2 wt% prior to illumination with 365 nm UV light (Fig. 3). As expected, the values decrease with an increasing concentration of either CNC, but lower values are observed throughout each series for CNC-AB featuring azobenzene pendants that more closely resemble the N-LC host (either one is flanked by two aliphatic side chains).
 | ||
| Fig. 3 POM photomicrographs (crossed polarizer, P, and analyzer, A) taken on cooling from the isotropic liquid phase at a T of 45 °C, showing the fingerprint textures obtained from the samples prepared between two untreated glass slides of Felix-2900-03 doped with CNC-AC or CNC-AB before and after illumination with UV light at a wavelength of = 365 nm at concentrations of the CNCs ranging from 0.05 to 0.2 wt% as indicated above each column of images. Images on the right of each set show the contact preparations with cholesteryl oleyl carbonate (COC) for CNC-AC (taken at T = 45 °C on cooling) or cholesteryl nonanoate (ChN) for CNC-AB (taken at T = 65 °C on cooling), and in each case, no discontinuation was seen in contact with the two known left-handed N*-LCs (handedness is indicated by light blue circular arrows). Scale bars = 50 μm. | ||
Noteworthily, neither of the two azobenzene-decorated CNCs reported here is as effective with respect to inducing a tighter pitch (leading to higher values for βw) as the previously tested CNCs that were decorated with mesogenic pendants that either mimicked the host N-LC or were chiral nematic LCs such as the cholesterol-decorated CNCs.16 However, illumination with UV light at λ = 365 nm resulted in the expected pitch increase of the induced N*-LC phase60–67 for concentrations of each of the CNCs not exceeding 0.15 wt% (Fig. 4). A further increase of the CNC concentration to 0.2 wt% for both CNC-AC and CNC-AB led to an increase in p after UV illumination at λ = 365 nm (Fig. 4) that is surely linked to the increasing incompatibility (lower miscibility)5 of the CNCs with the ensuing cis-configuration (Fig. 5) of the two azobenzene pendants on the CNC surfaces in the N-LC as reported previously in numerous studies.60–67
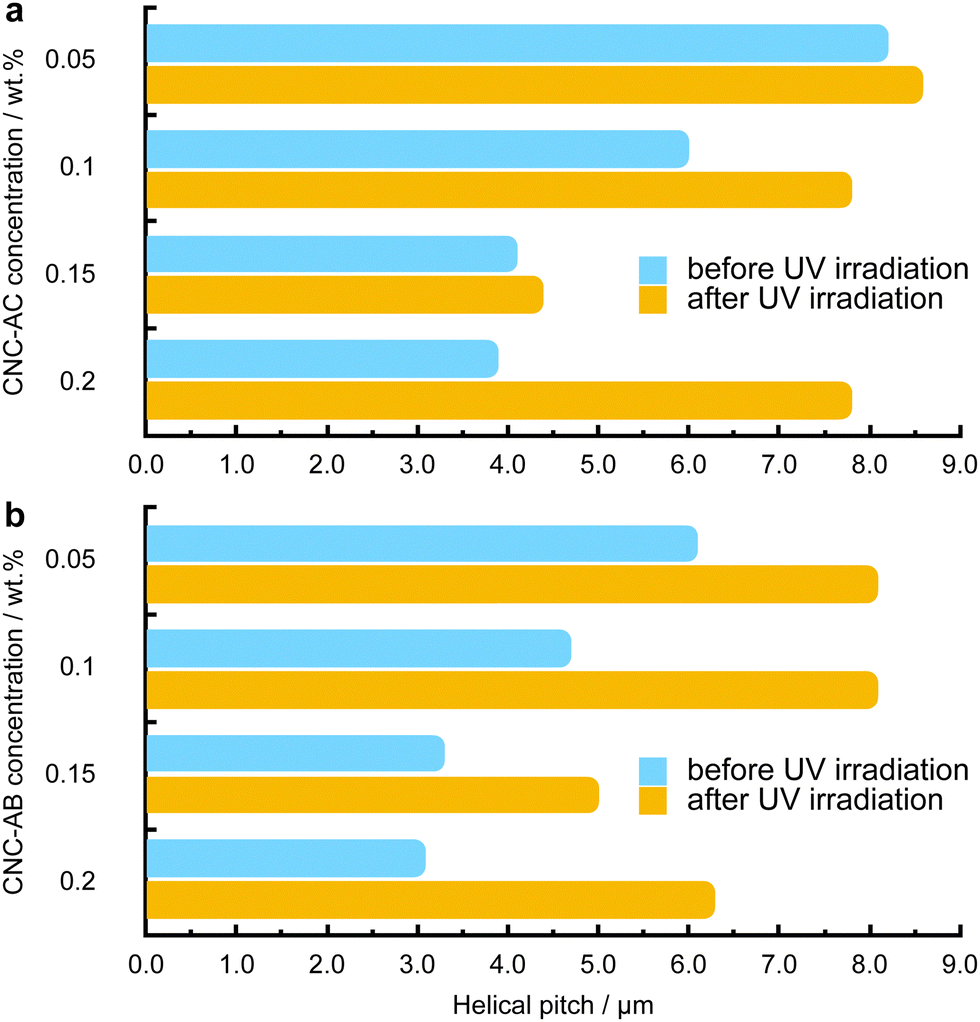 | ||
| Fig. 4 Plots of the measured helical pitch (p) at 5 °C above the N-SmA phase transition on cooling from the isotropic liquid phase versus the concentration of (a) CNC-AC and (b) CNC-AB. Light blue upper data bars in each plot and for each concentration are samples prior to illumination with UV light and yellow lower bars after UV illumination. | ||
 | ||
| Fig. 5 UV-vis spectra of the CNCs in DMF (20 °C) before and after illumination with UV light at λ = 365 nm for a time period of t = 10 s: (a) CNC-AC and (b) CNC-AB. Longer exposure, up to t = 60 s, did not lead to a discernible change in the spectra, and thus, all azobenzene pendants are already in the cis configuration in solution after a t = 10 s exposure to UV light (ESI,† Fig. S16). | ||
The handedness of the induced N*-LC phase was analyzed using contact preparations with cholesteryl oleyl carbonate (COC) or cholesterol nonanoate (ChN), each known to form left-handed N*-LC phases.68,69 Since no discontinuation (i.e., no achiral N-LC phase) was seen in the contact zone with COC (ChN), this reveals that the CNCs induced a left-handed N*-LC phase just like the previous CNC chiral solutes in this N-LC host16 or their native handedness in an aqueous suspension.34 The values of βw derived from the measured p data are comparatively lower than that of the previous mesogen-crafted CNCs,16 here βw = −2.14 μm−1 for CNC-AB and −1.70 μm−1 for CNC-AC prior to UV exposure and −1.39 μm−1 and −1.50 μm−1 (“−” meaning left-handed) after UV illumination (ESI,† Fig. S14), which we attribute to a more limited compatibility of the azobenzene moieties with the used N-LC. In fact, both azobenzene-decorated CNCs described here appear less soluble in Felix-2900-03, which we relate to two factors: (1) neither of the azobenzene pendants is liquid crystalline at the temperature range of the studied N-LC host. Only AC is monotropic nematic with a phase sequence of Iso 202 N 162 Cr on cooling (with some degrees of decomposition after the first heating);52 AB, however, is non-mesogenic.53 (2) Small fractions of the azobenzene pendants are already in the cis-configuration even before UV illumination, which can be seen in the UV-vis spectra shown in Fig. 1c. The deconvolution of the peaks in the UV spectra prior to UV illumination indicates that for CNC-AC 27% of the AC units and for CNC-AB 26% of the AB units, respectively, are in the cis configuration (ESI,† Fig. S15).
The change in p under λ = 365 nm illumination is reasonably fast (seconds), but the thermal cis-to-trans back relaxation of the azobenzene units is significantly slower (several tens of minutes or more). However, by heating the sample back to the isotropic liquid phase (i.e., to higher temperatures), samples recover the tighter pitch values seen prior to UV illumination (ESI,† Fig. S17).
4. Conclusions
CNCs may not be ideal chiral additives for some applications, but their origin from a wide range of renewable resources70 coupled with facile opportunities for surface functionalization render them a new opportunity in the realm of chiral solutes. Here, we verified that surface modification with an active photoswitch in the form of azobenzene pendants leads to CNCs as chiral solutes that can induce N*-LC phases with a phototunable helical pitch. According to the predictions, and following the earlier trend,16 CNC-AB, which is decorated with azobenzene pendants that are structurally more alike to the N-LC host, shows both higher values of βw and more effective photomodulation of p as verified by comparing the values of Δβw (0.75 μm−1 for CNC-AB and 0.20 μm−1 for CNC-AC, respectively). This may be further exacerbated by the higher DSF value of CNC-AB in comparison to that of CNC-AC. In CNC-AC, only one of every ten D-glucose subunits is functionalized with an AC pendant; for CNC-AB, this increases to one AB pendant in almost every six D-glucose subunits, thus rendering CNC-AB not only more compatible (structurally) but also more miscible.Further opportunities to optimize this system are available via the use of more effective (higher βw), better soluble (miscible), room temperature mesogenic and photoswitchable pendant molecules61,65 or by selecting (preparing) CNC aspect ratios71 that would provide an improved match to the N-LC host molecule aspect ratio based on our earlier work on matching the 2-D isoperimetric ratio of nanoscale chiral solutes in N-LCs,1 which is the work that is currently ongoing in our laboratory. Ultimately, photoresponsive (photoswitchable) CNCs may find applications requiring stimuli-responsive48,72 and smart nanoarchitectonic73–77 materials for chiral photonics,78–80 soft actuators, and other adaptive materials81 based on their renewable nature and high versatility.
Author contributions
B. S. performed the synthesis and characterization of the materials, the sample preparation, and all measurements. B. S. and T. H. analyzed the data and co-wrote the manuscript. T. H. directed the study.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Scientific and Technological Research Council of Turkey (TUBITAK) – 2214-A – International Research Fellowship Program (1059B142100482, B. S.), the U.S. National Science Foundation (NSF, DMR-1904091; T. H.), and the Ohio Third Frontier (OTF) program for Ohio Research Scholars “Research Cluster on Surfaces in Advanced Materials”, which also supports the Materials Characterization and Imaging Facility at the Advanced Materials and Liquid Crystal Institute (AMLCI) at Kent State University, where the current TEM data were acquired. We thank the Swagelok Center for the Surface Analysis of Materials at Case Western Reserve University and Dr T. K. J. Kim for access to and assistance with the XPS experiments and data analysis. Finally, we acknowledge the help of Dr M. Gangoda for CP-MAS 13C NMR spectroscopy experiments.References
- A. Nemati, L. Querciagrossa, C. Callison, S. Shadpour, D. P. N. Gonçalves, T. Mori, X. Cui, R. Ai, J. Wang, C. Zannoni and T. Hegmann, Sci. Adv., 2022, 8, eabl4385 CrossRef CAS.
- A. Sharma, T. Mori, H.-C. Lee, M. Worden, E. Bidwell and T. Hegmann, ACS Nano, 2014, 8, 11966–11976 CrossRef CAS PubMed.
- T. Mori, A. Sharma and T. Hegmann, ACS Nano, 2016, 10, 1552–1564 CrossRef CAS PubMed.
- R. K. Shukla, A. Sharma, T. Mori, T. Hegmann and W. Haase, Liq. Cryst., 2016, 43, 695–703 CrossRef CAS.
- A. Nemati, S. Shadpour, L. Querciagrossa, L. Li, T. Mori, M. Gao, C. Zannoni and T. Hegmann, Nat. Commun., 2018, 9, 3908 CrossRef PubMed.
- S. Shadpour, J. P. Vanegas, A. Nemati and T. Hegmann, ACS Omega, 2019, 4, 1662–1668 CrossRef CAS PubMed.
- A. Nemati, S. Shadpour, L. Querciagrossa, T. Mori, C. Zannoni and T. Hegmann, ACS Nano, 2019, 13, 10312–10326 CrossRef CAS PubMed.
- A. Poryvai, M. Šmahel, M. Švecová, A. Nemati, S. Shadpour, P. Ulbrich, T. Ogolla, J. Liu, V. Novotná, M. Veverka, J. Vejpravová, T. Hegmann and M. Kohout, ACS Nano, 2022, 16, 11833–11841 CrossRef CAS PubMed.
- D. P. N. Gonçalves, M. E. Prévôt, Ş. Üstünel, T. Ogolla, A. Nemati, S. Shadpour and T. Hegmann, Liq. Cryst. Rev., 2021, 9, 1–34 CrossRef.
- A. Sharma, T. Mori, A. Nemati, D. P. N. Gonçalves, L. Querciagrossa, C. Zannoni and T. Hegmann, Mater. Adv., 2022, 3, 3346–3354 RSC.
- K. Robbie, D. J. Broer and M. J. Brett, Nature, 1999, 399, 764–766 CrossRef CAS.
- J.-J. Lee, B.-C. Kim, H.-J. Choi, S. Bae, F. Araoka and S.-W. Choi, ACS Nano, 2020, 14, 5243–5250 CrossRef CAS PubMed.
- X. Zhang, Y. Xu, C. Valenzuela, X. Zhang, L. Wang, W. Feng and Q. Li, Light: Sci. Appl., 2022, 11, 223 CrossRef CAS.
- S. O. Pour, L. Rocks, K. Faulds, D. Graham, V. Parchaňský, P. Bouř and E. W. Blanch, Nat. Chem., 2015, 7, 591–596 CrossRef.
- W. Ma, L. Xu, A. F. de Moura, X. Wu, H. Kuang, C. Xu and N. A. Kotov, Chem. Rev., 2017, 117, 8041–8093 CrossRef CAS PubMed.
- D. P. N. Gonçalves and T. Hegmann, Angew. Chem., Int. Ed., 2021, 60, 17344–17349 CrossRef PubMed.
- D. P. N. Gonçalves, T. Ogolla and T. Hegmann, ChemPhysChem, 2022 DOI:10.1002/cphc.202200685.
- S. K. Shukla, G. C. Dubey, A. Tiwari and A. Bharadvaja, Adv. Mater. Lett., 2013, 4, 714–719 CrossRef CAS.
- T. Abitbol, A. Rivkin, Y. Cao, Y. Nevo, E. Abraham, T. Ben-Shalom, S. Lapidot and O. Shoseyov, Curr. Opin. Biotechnol, 2016, 39, 76–88 CrossRef CAS PubMed.
- W. J. Orts, L. Godbout, R. H. Marchessault and J.-F. Revol, Macromolecules, 1998, 31, 5717–5725 CrossRef CAS.
- J. Araki and S. Kuga, Langmuir, 2001, 17, 4493–4496 CrossRef CAS.
- S. Elazzouzi-Hafraoui, Y. Nishiyama, J.-L. Putaux, F. F. Dubreuil and C. Rochas, Biomacromolecules, 2008, 9, 57–65 CrossRef CAS PubMed.
- I. Usov, G. Nyström, J. Adamclik, S. Handschin, C. Schütz, A. Fall, L. Bergström and R. Mezzenga, Nat. Commun., 2015, 6, 7564–7575 CrossRef CAS PubMed.
- K. Conley, L. Godbout, M. A. Whitehead and T. G. M. Van de Ven, Carbohydr. Polym., 2016, 135, 285–299 CrossRef CAS PubMed.
- Y. Ogawa, Nanoscale, 2019, 11, 21767–21774 RSC.
- G. Delepierre, S. Eyley, W. Thielemans, C. Weder, E. D. Cranston and J. O. Zoppe, Nanoscale, 2020, 12, 17480–17493 RSC.
- S. Gim, G. Fittolani, Y. Nishiyama, P. H. Seeberger, Y. Ogawa and M. Delbianco, Angew. Chem., Int. Ed., 2020, 59, 22577–22583 CrossRef CAS PubMed.
- K. Uetani, T. Uto and N. Suzuki, Sci. Rep., 2021, 11, 790 CrossRef CAS PubMed.
- T. Willhammar, K. Daicho, D. N. Johnstone, K. Kobayashi, Y. Liu, P. A. Midgley, L. Bergström and T. Saito, ACS Nano, 2021, 15, 2730–2737 CrossRef CAS.
- T. G. Parton, R. M. Parker, G. T. van de Kerkhof, A. Narkevicius, J. S. Haataja, B. Frka-Petesic and S. Vignolini, Nat. Commun., 2022, 13, 2657 CrossRef CAS.
- Y. Habibi, L. A. Lucia and O. J. Rojas, Chem. Rev., 2010, 110, 3479–3500 CrossRef CAS PubMed.
- J. Majoinen, E. Kontturi, O. Ikkala and D. G. Gray, Cellulose, 2012, 19, 1599–1605 CrossRef CAS.
- J. A. Kelly, A. M. Shukaliak, C. C. Y. Cheung, K. E. Shopsowitz, W. Y. Hamad and M. J. MacLachlan, Angew. Chem., Int. Ed., 2013, 52, 8912–8916 CrossRef CAS PubMed.
- J. P. F. Lagerwall, C. Schütz, M. Salajkova, J. Noh, J. H. Park, G. Scalia and L. Bergström, NPG Asia, 2014, 6, e80 CrossRef CAS.
- P.-X. Wang, W. Y. Hamad and M. J. MacLachlan, Nat. Commun., 2016, 7, 11515 CrossRef PubMed.
- Y. Li, J. Jun-Yan Suen, E. Prince, E. M. Larin, A. Klinkova, H. Thérien-Aubin, S. Zhu, B. Yang, A. S. Helmy, O. D. Lavrentovich and E. Kumacheva, Nat. Commun., 2016, 7, 12520 CrossRef CAS.
- O. Liu and I. I. Smalyukh, Sci. Adv., 2017, 3, e1700981 CrossRef PubMed.
- B. Vollick, P.-Y. Kuo, H. Thérien-Aubin, N. Yan and E. Kumacheva, Chem. Mater., 2017, 29, 789–795 CrossRef CAS.
- O. Kose, A. Tran, L. Lewis, W. Y. Hamad and M. J. MacLachlan, Nat. Commun., 2019, 10, 510 CrossRef CAS PubMed.
- R. Kádár, S. Spirk and T. Nypelö, ACS Nano, 2021, 15, 7931–7945 CrossRef.
- X. Liu, M. Li, X. Zheng, E. Retulainen and S. Fu, Materials, 2018, 11, 1725 CrossRef.
- I. Otsuka and C. J. Barrett, Cellulose, 2019, 26, 6903–6915 CrossRef CAS.
- Z. Xu, S. Peng, G. Zhou and X. Xu, J. Compos. Sci., 2020, 4, 186 CrossRef CAS.
- M. V. Peters, R. S. Stoll, A. Kühn and S. Hecht, Angew. Chem., Int. Ed., 2008, 47, 5968–5972 CrossRef CAS.
- T. Imahori, R. Yamaguchi and S. Kurihara, Chem. – Eur. J., 2012, 18, 10802–10807 CrossRef CAS.
- L. Osorio-Planes, C. Rodríguez-Escrich and M. A. Pericas, Org. Lett., 2014, 16, 1704–1707 CrossRef CAS PubMed.
- B. Zhang, Y. Feng and W. Feng, Nano-Micro Lett., 2022, 14, 138 CrossRef CAS PubMed.
- For a recent review, see: X. Pang, J. Lv, L. Qin and Y. Yu, Adv. Mater., 2019, 31, 1904224 CrossRef CAS PubMed.
- Z. Chu, Y. Han, T. Bian, S. De, P. Král and R. Klajn, J. Am. Chem. Soc., 2019, 141, 1949–1960 CrossRef CAS PubMed.
- Q. M. Zhang, X. Li, M. R. Islam, M. Wei and M. J. Serpe, J. Mater. Chem. C, 2014, 2, 6961–6965 RSC.
- M. Quick, A. L. Dobryakov, M. Gerecke, C. Richter, F. Berndt, I. N. Ioffe, A. A. Granovsky, R. Mahrwald, N. P. Ernsting and S. A. Kovalenko, J. Phys. Chem. B, 2014, 118, 8756–8771 CrossRef CAS PubMed.
- G. Mao, J. Wang, S. R. Clingman, C. K. Ober, J. T. Chen and E. L. Thomas, Macromolecules, 1997, 30, 2556–2567 CrossRef CAS.
- M. Matsumoto, K. Tanaka, R. Azumi, Y. Kondo and N. Yoshino, Langmuir, 2004, 20, 8728–8734 CrossRef CAS PubMed.
- Y. Wang, X. Wang, Y. Xie and K. Zhang, Cellulose, 2018, 25, 3703–3731 CrossRef CAS.
- H. Kono, S. Yunoki, T. Shikano, M. Fujiwara, T. Erata and M. Takai, J. Am. Chem. Soc., 2002, 124, 7506–7511 CrossRef CAS PubMed.
- A. Brinkmann, M. Chen, M. Couillard, Z. J. Jakubek, T. Leng and L. J. Johnston, Langmuir, 2016, 32, 6105–6114 CrossRef CAS.
- G. Rodionova, M. Lenes, Ø. Eriksen and Ø. Gregersen, Cellulose, 2011, 18, 127–134 CrossRef CAS.
- Among the many reports for surface functionalization of CNCs by esterification, the degree of surface functionalization, DSF, is typically ≤1 and can be as low as 0.06; see: S. Eyley and W. Thielemans, Nanoscale, 2014, 6, 7764–7779 RSC.
- J. Meija, M. Bushell, M. Couillard, S. Beck, J. Bonevich, K. Cui, J. Foster, J. Will, D. Fox, W. Cho, M. Heidelmann, B. C. Park, Y. C. Park, L. Ren, L. Xu, A. B. Stefaniak, A. K. Knepp, R. Theissmann, H. Purwin, Z. Wang, N. de Val and L. J. Johnston, Anal. Chem., 2020, 92, 13434–13442 CrossRef CAS PubMed.
- Q. Li, L. Green, N. Venkataraman, I. Shiyanovskaya, A. Khan, A. Urbas and J. W. Doane, J. Am. Chem. Soc., 2007, 129, 12908–12909 CrossRef CAS PubMed.
- Y. Wang and Q. Li, Adv. Mater., 2012, 24, 1926–1945 CrossRef CAS PubMed.
- Y. Kim and N. Tamaoki, J. Mater. Chem. C, 2014, 2, 9258–9264 RSC.
- A. Varanytsia and L.-C. Chien, Opt. Lett., 2015, 40, 4392–4395 CrossRef CAS PubMed.
- Y. Kim and N. Tamaoki, ACS Appl. Mater. Interfaces, 2016, 8, 4918–4926 CrossRef CAS PubMed.
- H. K. Bisoyi and Q. Li, Chem. Rev., 2016, 116, 15089–15166 CrossRef CAS PubMed.
- Y. Kim and N. Tamaoki, ChemPhotoChem, 2019, 3, 284–303 CrossRef CAS.
- Y. Kim, N. N. Mafy, S. Maisonneuve, C. Lin, N. Tamaoki and J. Xie, ACS Appl. Mater. Interfaces, 2020, 12, 52146–52155 CrossRef CAS PubMed.
- T. Mori, M. Kyotani and K. Akagi, Chem. Sci., 2011, 2, 1389–1395 RSC.
- K. Akagi and T. Mori, Chem. Rec., 2008, 8, 395–406 CrossRef CAS PubMed.
- J. Gong, J. Li, J. Xu, Z. Xiang and L. Mo, RSC Adv., 2017, 7, 33486–33493 RSC.
- X. Wang, C. H. Chang, J. Jiang, Q. Liu, Y.-P. Liao, L. Lu, L. Li, X. Liu, J. Kim, A. Ahmed, A. E. Nel and T. Xia, Small, 2019, 15, 1901642 CrossRef CAS PubMed.
- R. Nasseri, C. P. Deutschman, L. Han, M. A. Pope and K. C. Tam, Mater. Today Adv., 2020, 5, 100055 CrossRef.
- K. Ariga, Nanoscale Horiz., 2021, 6, 364–378 RSC.
- P. Lv, X. Lu, L. Wang and W. Feng, Adv. Funct. Mater., 2021, 31, 2104991 CrossRef CAS.
- J. Ma, Y. Yang, C. Valenzuela, X. Zhang, L. Wang and W. Feng, Angew. Chem., Int. Ed., 2022, 61, e202116219 CAS.
- L. Wang, A. M. Urbas and Q. Li, Adv. Mater., 2020, 32, 1801335 CrossRef CAS PubMed.
- X. Zhang, Y. Yang, P. Xue, C. Valenzuela, Y. Chen, X. Yang, L. Wang and W. Feng, Angew. Chem., Int. Ed., 2022, 61, e202211030 CAS.
- L. Wang and Q. Li, Chem. Soc. Rev., 2018, 47, 1044–1097 RSC.
- L. Dong, Y. Feng, L. Wang and W. Feng, Chem. Soc. Rev., 2018, 47, 7339–7368 RSC.
- F. Zhai, Y. Feng, K. Zhou, L. Wang, Z. Zheng and W. Feng, J. Mater. Chem. C, 2019, 7, 2146–2171 RSC.
- J. Yang, X. Zhang, X. Zhang, L. Wang, W. Feng and Q. Li, Adv. Mater., 2021, 33, 2004754 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2tc04444g |
| This journal is © The Royal Society of Chemistry 2022 |