
The advent and development of organophotoredox catalysis
Tommaso
Bortolato
 ,
Sara
Cuadros
,
Gianluca
Simionato
and
Luca
Dell’Amico
*
,
Sara
Cuadros
,
Gianluca
Simionato
and
Luca
Dell’Amico
*
Department of Chemical Sciences, University of Padova, Via Marzolo 1, Padova, 35131, Italy. E-mail: luca.dellamico@unipd.it
First published on 10th December 2021
Abstract
In the last decade, photoredox catalysis has unlocked unprecedented reactivities in synthetic organic chemistry. Seminal advancements in the field have involved the use of well-studied metal complexes as photoredox catalysts (PCs). More recently, the synthetic community, looking for more sustainable approaches, has been moving towards the use of purely organic molecules. Organic PCs are generally cheaper and less toxic, while allowing their rational modification to an increased generality. Furthermore, organic PCs have allowed reactivities that are inaccessible by using common metal complexes. Likewise, in synthetic catalysis, the field of photocatalysis is now experiencing a green evolution moving from metal catalysis to organocatalysis. In this feature article, we discuss and critically comment on the scientific reasons for this ongoing evolution in the field of photoredox catalysis, showing how and when organic PCs can efficiently replace their metal counterparts.
 From right to left: Tommaso Bortolato, Gianluca Simionato, Luca Dell'Amico and Sara Cuadros | Tommaso Bortolato graduated in Chemistry in 2019 from the University of Padova (Italy). In 2020, he started his PhD in Luca Dell’Amico's group focusing on the development of novel sustainable light-driven methods. Gianluca Simionato completed his MSc in Industrial Chemistry in 2021 at the University of Padova under the supervision of Dr Luca Dell’Amico. His current research deals with the design and study of new heterocyclic organophotoredox catalysts. Luca Dell’Amico completed his PhD at the University of Parma (Italy), under the supervision of Prof. Franca Zanardi, in 2014. He spent a research period with Prof. Karl Anker Jørgensen at Århus University (Denmark), where he was introduced to the field of organocatalysis. From 2014 to 2016, he was a Marie Curie fellow in Prof. Paolo Melchiorre's group at ICIQ (Spain). In 2017, he started his independent career at the University of Padova. His research targets the development and mechanistic investigation of novel photochemical processes. Sara Cuadros graduated from the University of Alicante in 2014. She obtained her MSc and PhD degrees in 2019, under the supervision of Prof. Paolo Melchiorre at ICIQ (Spain). She is currently a postdoctoral researcher at the University of Padova. Her research focuses on the synthesis of novel PET radioligands for functional imaging. |
1 From photochemistry to synthetic photoredox catalysis
Photochemistry is a branch of chemistry that deals with the chemical effects of light.1 Upon light-irradiation, organic molecules can reach an electronically excited state whose reactivity and physicochemical properties are substantially different from those in the ground state. We can track back the first observation of the effect of light on organic molecules in 1834 when the German chemist Johann Bartholomew Trommsdorff discovered that Santonin, an anthelmintic sesquiterpene lactone, photoisomerises upon irradiation with blue and violet light.2 However, it is since the 20th century, thanks to the work of Ciamician and Silber, that photochemistry has been considered a major branch of chemistry worth systematic investigations.3The earliest examples of photochemical transformations involved the direct irradiation of substrates. Nowadays, the most general and rapidly evolving branch of synthetic photochemistry is photoredox catalysis.
Photoredox catalysis makes use of a light-absorbing molecule, the photoredox catalyst (PC): an organometallic complex, an organic molecule or a material which harvests light reaching an electronically excited state (Fig. 1). The S1 singlet excited state can either decay through fluorescence or relax to a triplet T1 state with lower energy through intersystem crossing (ISC). The T1 state can then decay through phosphorescence, which, however, is a spin-forbidden process. Thus, the T1 state is typically longer-lived than the S1 state. The PC in the excited state (S1 or T1) is a stronger oxidant and reductant than in the ground state. The redox potentials in the excited state are determined by the ground-state redox potentials and the energy of the involved excited state E0,0 (Fig. 1).4
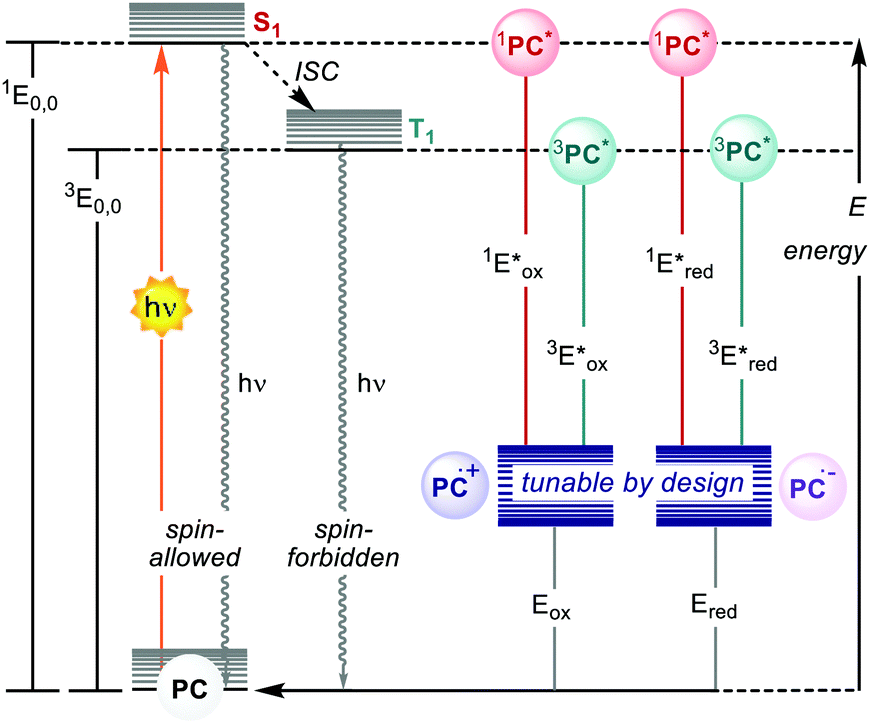 | ||
| Fig. 1 Schematic representation of the ground- and excited-states and electrochemical properties of PCs. | ||
The interaction between the excited-state PC (PC*) and an electron-donor (D) or electron-acceptor (A) leads to a redox event known as photoinduced electron transfer (PET). Its nature depends on the thermodynamic and kinetic feasibility.
Classically, a PET in which the PC acts as the reductant getting oxidised is called oxidative quenching (left pathway, Scheme 1).
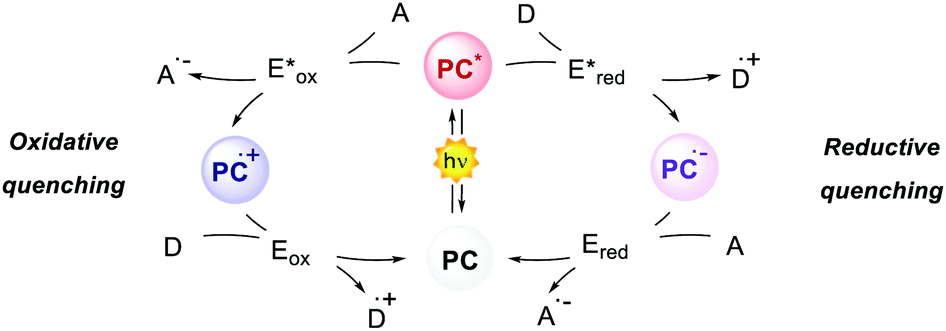 | ||
| Scheme 1 General mechanisms of oxidative and reductive quenching. D = donor; A = acceptor. | ||
On the other hand, we consider a reductive quenching if the PC gets reduced (right pathway, Scheme 1). Through a PET process, it is possible to generate a radical species under mild conditions, using visible light as the only (or the main) energy source.
The earliest example of photoredox catalysis in organic synthesis dates back to 1978 when Kellogg observed that the reduction of phenacyl sulfonium salts by 1,4-dihydropyridines was greatly accelerated under visible light irradiation in the presence of a catalytic amount of Ru(bpy)3Cl2 or eosin.5a Subsequent studies in the 1980s5b–e and early 1990s5f–h contributed to establishing the foundations of modern photoredox catalysis. Between 2008 and 2009, the pioneering reports by Yoon's,6 MacMillan's7 and Stephenson's8 groups fully disclosed the tremendous potential of photoredox catalysis when applied to organic synthesis. Since then, the field has been steadily growing year by year (Fig. 2).9
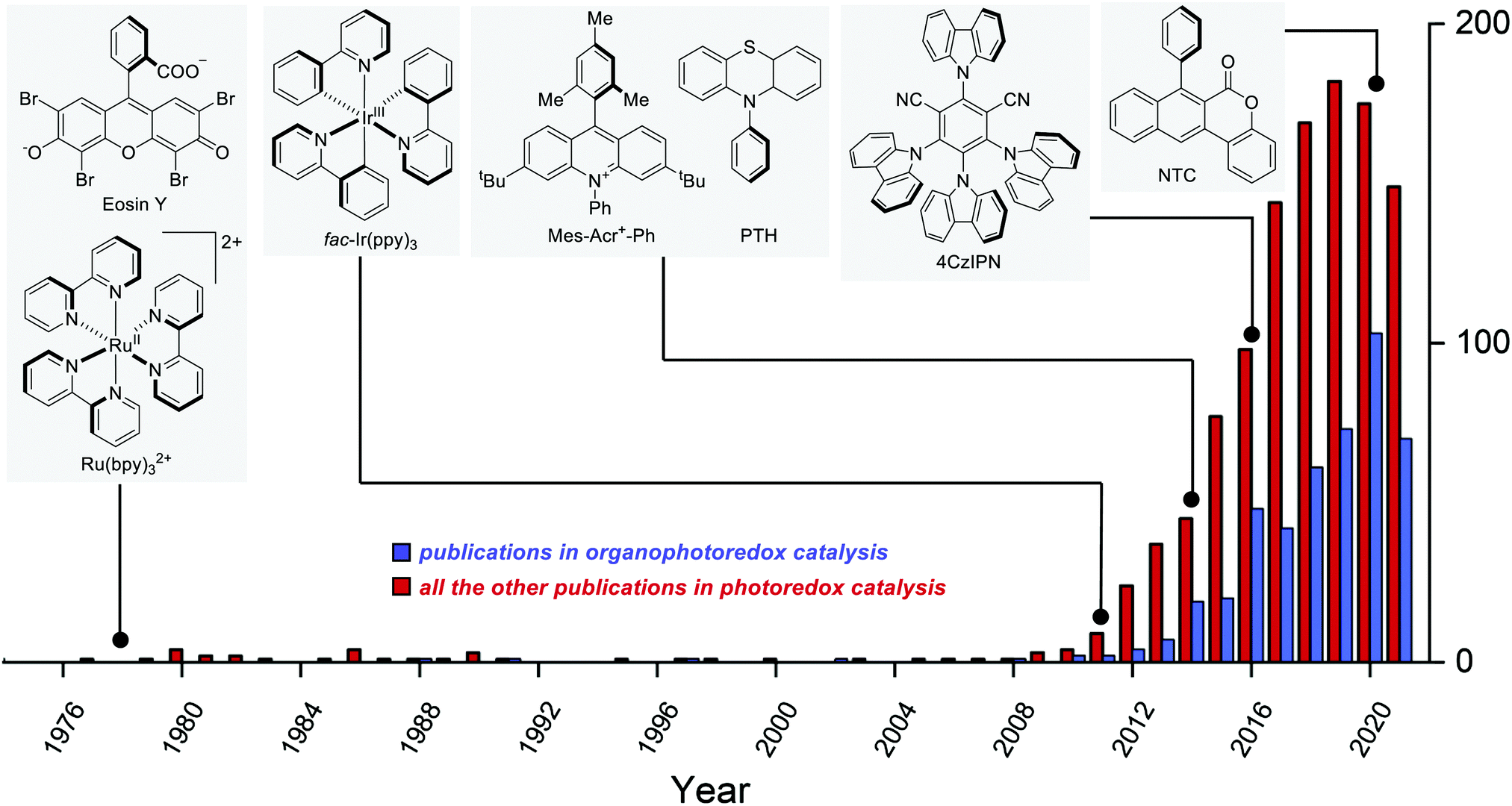 | ||
| Fig. 2 Graphical depiction of the number of articles published each year in the fields of synthetic photoredox catalysis. The blue columns depict the contribution of organophotoredox catalysis to the total, while the red columns depict the rest of the published papers. The structures of selected PCs are linked to the year of their first use in synthetic chemistry. | ||
2 Metal-based photoredox catalysis
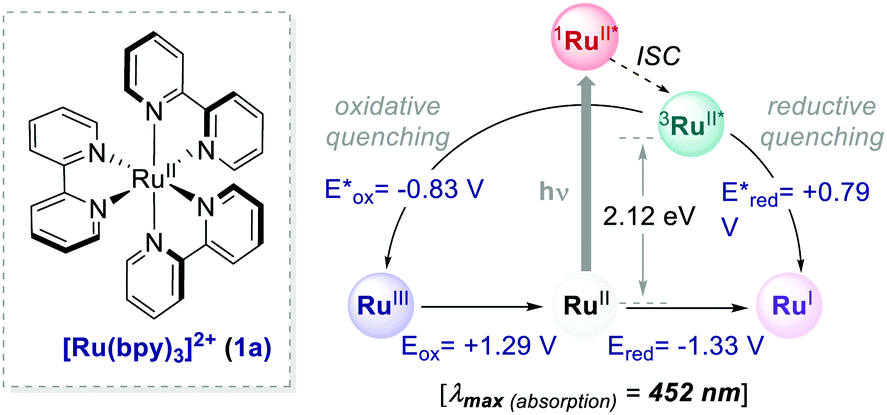
Until recently, Ru- and Ir-based PCs have dominated the field of synthetic photoredox catalysis (Scheme 2). Ru(bpy)32+1a has the longest history as a PC. It has been largely used for applications such as water splitting10 and carbon dioxide reduction.11 The balanced excited-redox potentials of 1a make it able to engage in both oxidative and reductive quenching processes.12 Similarly, fac-Ir(ppy)32a is another transition metal-based PC that has been extensively used in photoredox catalysis. It is a strongly reducing PC,13 with long-standing emission, ideal to be used in organic light emitting diodes (OLEDs).14 The excited state of 2a is about 500 mV less oxidising and 900 mV more reducing than 1a (Scheme 2).15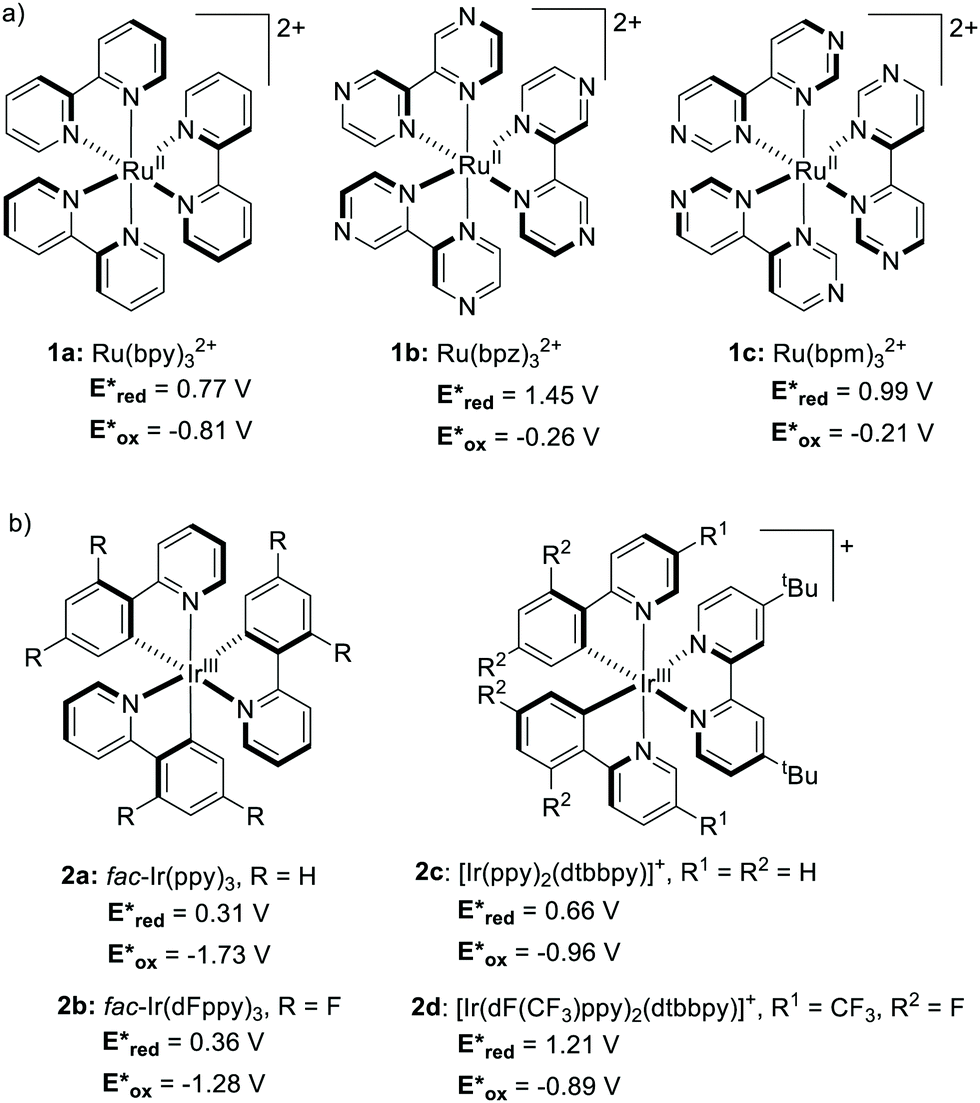 | ||
| Scheme 2 Structures and excited-state redox potentials (vs. SCE in MeCN) of the selected (a) Ru and (b) Ir complexes (ref. 15). | ||
Upon light absorption in the blue region, 1a and 2a get promoted to the S1 state. The S1 state efficiently undergoes ISC to the lower T1 state.15 Both states are involved in an intramolecular metal-to-ligand charge transfer (MLCT) from an electron-donating ligand to the electron-accepting metal centre. The S1 and T1 states are thus named 1MLCT and 3MLCT, respectively.
The popularity of Ru- and Ir-based complexes can be ascribed to a combination of two main factors. First, the photophysical properties of some Ru- and Ir-based complexes have been known for decades. Hence, the synthetic community has benefited from an extensive set of physicochemical data already available. Second, Ru- and Ir-based PCs undergo an efficient ISC to the T1 state. The population of the T1 state produces a long-lived excited state further extended by the MLCT. Such a long-lived excited state (in the order of the μs) facilitates the interaction with the substrate, thus favouring the PET.
Metal complexes have a complex set of excited states.15 Their relative energetic ordering and their photophysics depend on the intrinsic properties of metal and ligands. As a result, the rational fine-tuning of the redox properties of Ru and Ir PCs by ligand modification is particularly complex.16,17 A simple variation on the electronics of the ligand(s) affects the photophysics of the PC (Scheme 2).18 Additionally, the introduction of electron-donating groups into the ligand(s) has little or no effect on the redox potentials.19,20
Transition metal PCs are not the ideal options from a sustainable perspective. Both ruthenium and iridium have an abundance of around 0.001 ppm on the Earth's crust and are thus among the rarest metals.21 This issue also impacts their high prices, hampering their massive use in the future.22
For this reason, the community has evaluated the possibility of using more abundant metals, such as iron. However, the longest-lived excited state for a FeII complex is in the order of 100 ps, while the typical lifetimes of Ru- and Ir-based PCs reach several μs.23 This is because the coordination of a metal with the ligands provides an energy splitting (ΔE) between the ground state and the triplet metal-centred 3MC state. If the ΔE is small enough, the excited state that gets populated is the 3MC state (Fig. 3a). This 3MC state is non-emissive and gets rapidly deactivated by nonradiative decay, being thus unable to engage in PET. Conversely, a large ΔE brings the 3MC state so high in energy that the 3MLCT level becomes populated (Fig. 3b).14 Since the second- and third-row transition metals typically have a larger ΔE compared to their first-row counterparts, the development of abundant first-row metal complexes as PCs is much more challenging.24a On the other hand, Cu-complexes have given promising results.24b,c
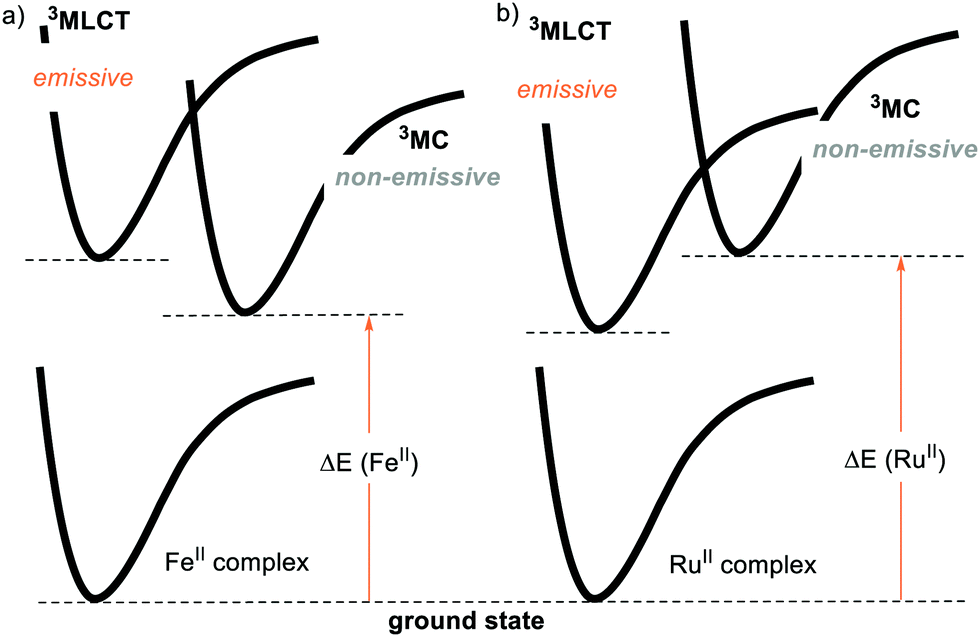 | ||
| Fig. 3 Schematic representation of the metal-to-ligand-charge transfer (3MLCT), metal-centred (3MC) and ground states (GS) energy levels for (a) FeII and (b) RuII complexes. | ||
3 Organic photoredox catalysts
Over the last few years, the development and use of purely organic photoredox catalysts have seen a rapid increase (Fig. 2), as witnessed by the increasing number of papers and reviews on the topic.25 Many organic dyes have been known for a long time, but their synthetic potential has been disclosed only recently.The need for more sustainable catalytic systems and predictable PC properties has prompted recent research towards organic molecules. In contrast to metal complexes, the structure–property relationships and rational design of organic PCs are highly simplified.26
Upon light irradiation, an organic PC is promoted to its excited S1 state, generally through a π–π* transition. The S1 state has a lifetime in the order of ns, and it is usually the state involved in the redox event. However, in some cases, a T1 state is reached by ISC.27 In the S1 state, the redox potential window is dictated by the 1E0,0 with none or minor energy loss (refer to Fig. 1).4 Similarly, the redox potential window of the T1 state is dictated by the 3E0,0, which is lower than 1E0,0 due to the energy lost through the ISC. The main advantage of the T1 state is its longer lifetime, kinetically desirable for PET processes. While Ru- and Ir-based PCs always populate the T1 state with high efficiency, organic PCs can be rationally designed to operate through the S1 or the T1 state.
Analogously to the MLCT character in metal-based PCs, in organic PCs, a charge-transfer (CT) character in the excited state is accessed when the HOMO and LUMO are spatially separated.26b The CT state is useful to increase the excited state lifetime.28 The effect of the CT character on the lifetime is often dramatic. For instance, considering 9,10-dicyanoanthracene (3a) and 4CzTPN, 3b. The first has a lifetime of 14.9 ns, while the second experiences a >100-fold increase, due to the strong CT character of its excited state (Fig. 4).25l
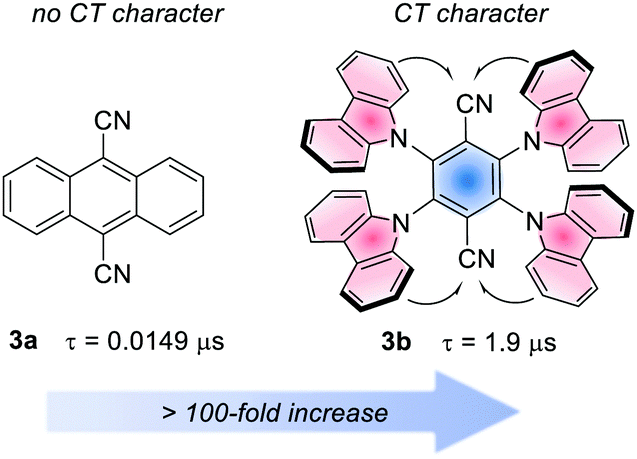 | ||
| Fig. 4 Example of the effect of the CT character for the PC lifetime (τ). | ||
Another advantage of having spatially separated HOMO and LUMO is the possibility to independently alter the energy of the two levels by introducing electron-donating or electron-withdrawing substituents in a specific position of the molecule. This strategy allows tailoring the ground-state and excited-state redox potentials of the PC, in an almost independent manner.26 In this way, it is possible to rationally design new organic PCs with the desired characteristics.
A recent classification of organic PCs is based on their ability to activate the substrate either towards oxidation or reduction. We can thus talk about (i) strongly oxidising, (ii) strongly reducing and (iii) bimodal organic PCs (Fig. 5). Bimodal PCs have a balanced distribution of their redox power which allows their use in both oxidative and reductive quenching manifolds.
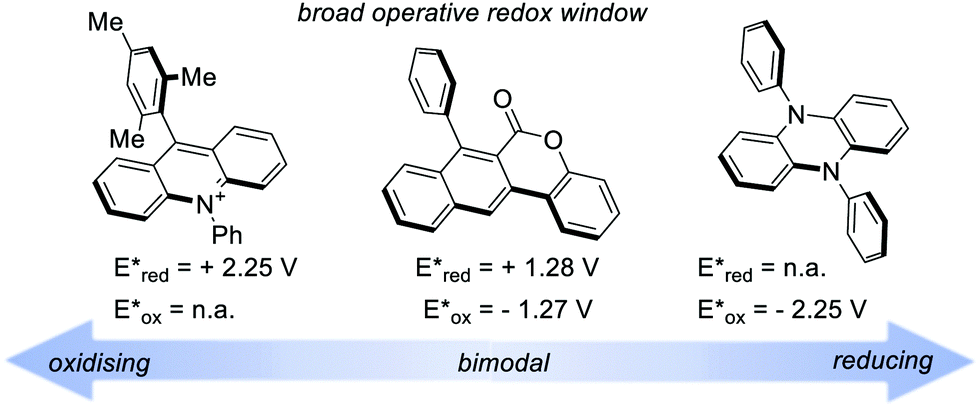 | ||
| Fig. 5 Selected examples of extremely oxidising and reducing PCs. Potentials vs. SCE in MeCN. | ||
4 Unveiling the potential of organic PCs in photoredox catalysis – a comparative analysis
4.1 Replacement of a Ru-complex for an organic PC
Ru(bpy)32+1a absorbs visible photons (λmax = 452 nm), giving access to a long-lived triplet 3MLCT state (τ = 1.1 μs).15 Thus, this metal-based PC can participate in both oxidative (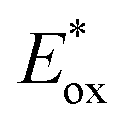 = −0.81 V vs. SCE in MeCN) and reductive quenching processes (
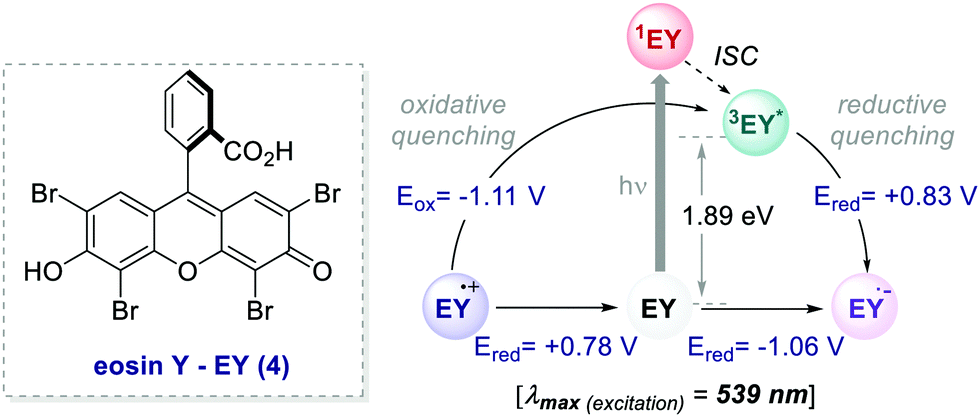
= −0.81 V vs. SCE in MeCN) and reductive quenching processes (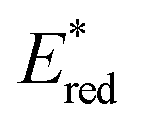 = +0.77 V vs. SCE in MeCN) (Scheme 3).11 Nevertheless, its relatively narrow redox potential window has hampered the use of this catalyst for the activation of substrates with high thermodynamic requirements. Likewise, eosin Y 4 (EY, Scheme 4) exhibits a narrow redox window and similar redox properties.25b
= +0.77 V vs. SCE in MeCN) (Scheme 3).11 Nevertheless, its relatively narrow redox potential window has hampered the use of this catalyst for the activation of substrates with high thermodynamic requirements. Likewise, eosin Y 4 (EY, Scheme 4) exhibits a narrow redox window and similar redox properties.25b
 | ||
| Scheme 3 Photophysical properties and redox potentials (vs. SCE in MeCN) of Ru(bpy)32+ (ref. 15). | ||
 | ||
Scheme 4 Photophysical properties and redox potentials (vs. SCE in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 MeCN:H2O) of eosin Y (ref. 25b and 29). :1 MeCN:H2O) of eosin Y (ref. 25b and 29). | ||
Thus, the commercially available and cheaper EY can be effectively used to replace Ru(bpy)32+, especially for reactions proceeding through a reductive quenching mechanism.30 Upon irradiation (λmax = 539 nm), EY reaches the S1 state that undergoes a rapid ISC to the lowest T1 state, which has a lifetime of 24 μs.31 Additionally, this well-known organic dye31–34 allows the use of low-energy green light, representing a milder alternative to trigger photoredox processes.
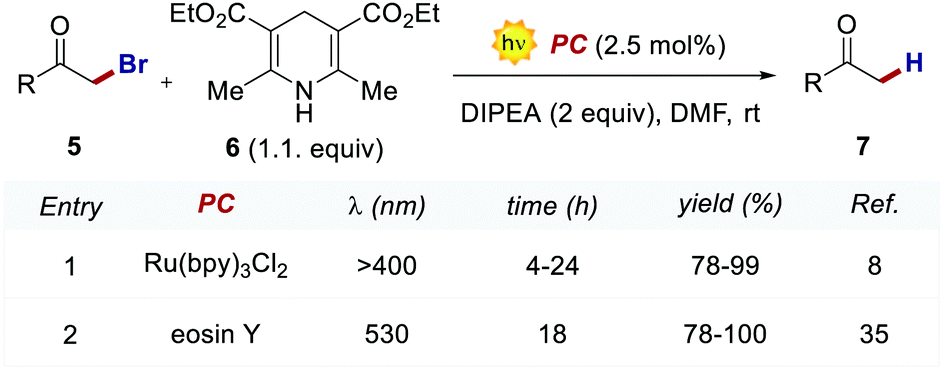
In 2011, König and Zeitler's groups reported a comparative study between the performances of Ru(bpy)32+ and EY, in two photoredox reactions.35 The first reaction tested was the dehalogenation of α-haloacetophenones 5 (Scheme 5).8 Here, EY outperformed the metal-based PC with better yields of the dehalogenated products 7 (Scheme 5, entries 1 vs. 2).
 | ||
| Scheme 5 Photocatalytic reductive dehalogenation. Comparison between the performances of Ru(bpy)32+ and eosin Y. | ||
Importantly, when using EY, it was possible to use a less energetic 530 nm light which avoids the degradation of this photosensitizer while providing a clean and high yielding dehalogenation process. The authors also tested this organic PC in the photochemical asymmetric α-alkylation of aldehydes 8 (Scheme 6).7 In this case, the use of 0.5 mol% of EY allowed the formation of the alkylated products 11 in comparable yields and enantioselectivities (Scheme 6a).
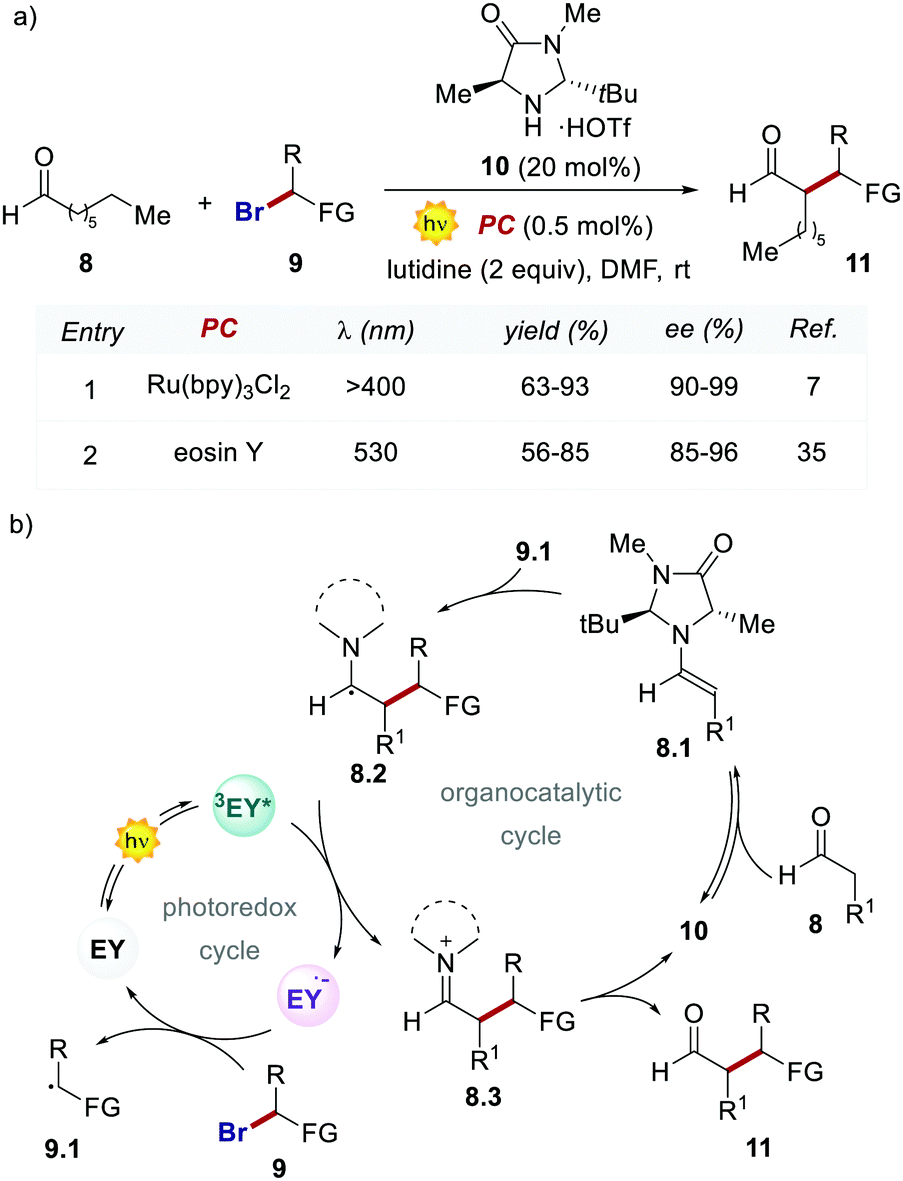 | ||
| Scheme 6 (a) Photo-organocatalytic enantioselective α-alkylation/perfluoroalkylation of aldehydes. Comparison between the performances of Ru(bpy)32+ and eosin Y. (b) The proposed mechanism for the photochemical asymmetric α-alkylation of aldehydes with eosin Y. | ||
Following the working hypothesis of MacMillan,7 the authors proposed the two cooperative catalytic cycles depicted in Scheme 6, where the EY orchestrates this process similarly to Ru(bpy)32+ (Scheme 6a, entries 1 vs. 2). However, the highly different quantum yields obtained (φ = 0.075 for EY versus φ = 18 for Ru(bpy)32+) revealed the operation of the two alternative mechanistic pathways.36
In the following work, Zeitler's group proved the possibility of merging EY with other activation modes, such as hydrogen-bonding catalysis. Specifically, the authors revisited the reductive cyclisation of (aryl)bisenones 12 under protic conditions, previously reported by Yoon's group with Ru(bpy)32+.37 In this case, the use of 20 mol% of thiourea 14 in combination with EY afforded the cyclised products 13 with complete trans-diastereoselectivity, in comparable yields to the Ru-catalysed process (Scheme 7a, entries 1 vs. 2).38
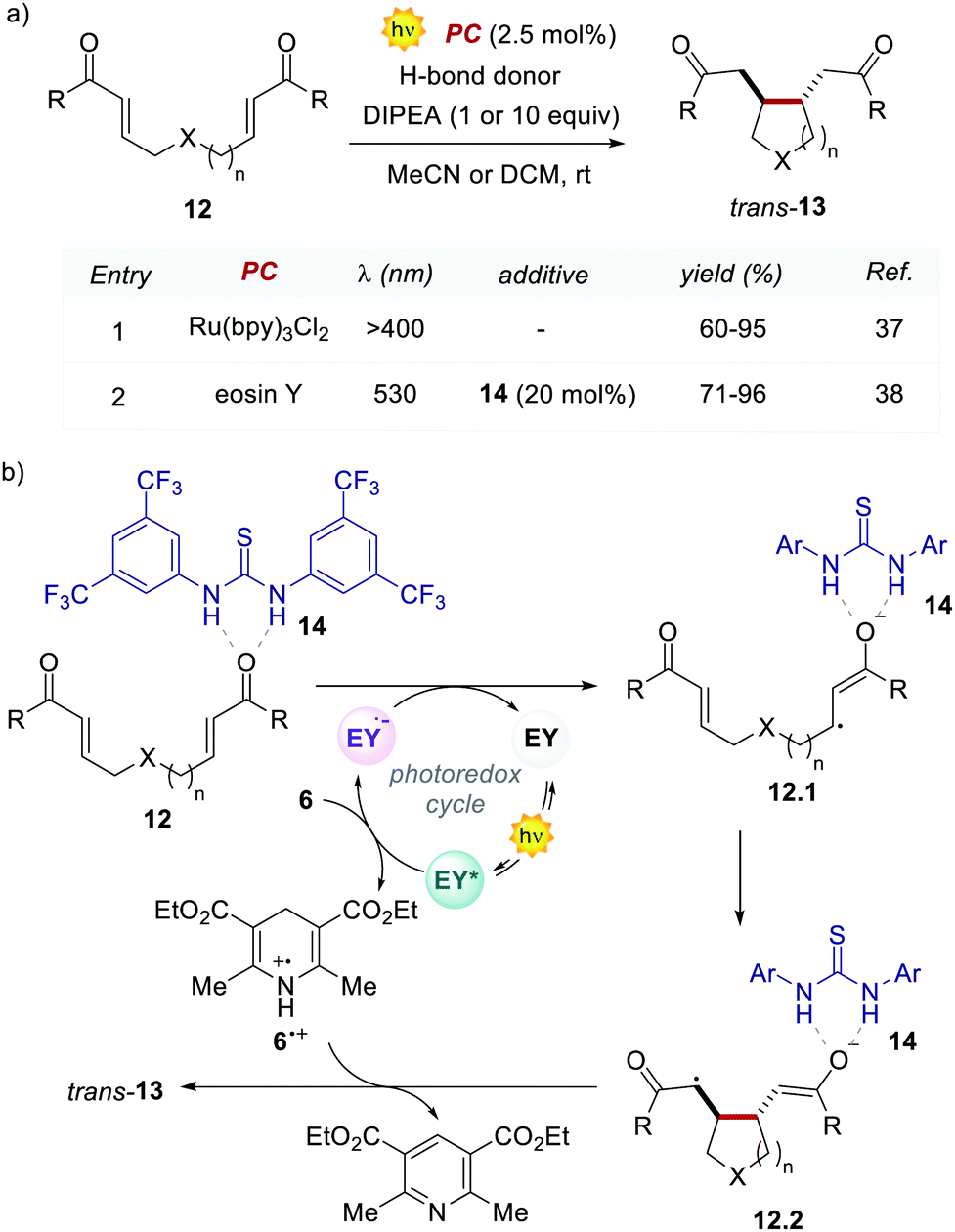 | ||
| Scheme 7 (a) Photo-reductive cyclisation of (bis)enones. Comparison between the performances of Ru(bpy)32+ and eosin Y. (b) Mechanism of the reductive cyclisation of (aryl)bisenones promoted by eosin Y. | ||
Interestingly, the method implemented by Zeitler's group avoids the use of over-stoichiometric amounts of Lewis acids. Instead, this organocatalytic reaction relies on the ability of thiourea 14 to activate 12 through H-bond interactions (Scheme 7b), thus making the coordinated enone 12 more reactive towards the PET process with the EY˙−. In this regard, the reductive power of the EY˙− (Ered = −1.06 V vs. SCE in MeCN) proved to be sufficient to reduce a variety of enones 12.
In 2013, Scaiano's group unveiled the potential of methylene blue 15 (MB) to serve as a purely organic PC (Scheme 8).39 MB is characterised by a maximum absorption set at λ = 650 nm (red-light), and a triplet state (3MB*) with a τ = 32 μs, which can participate in both oxidative and reductive quenching events.40
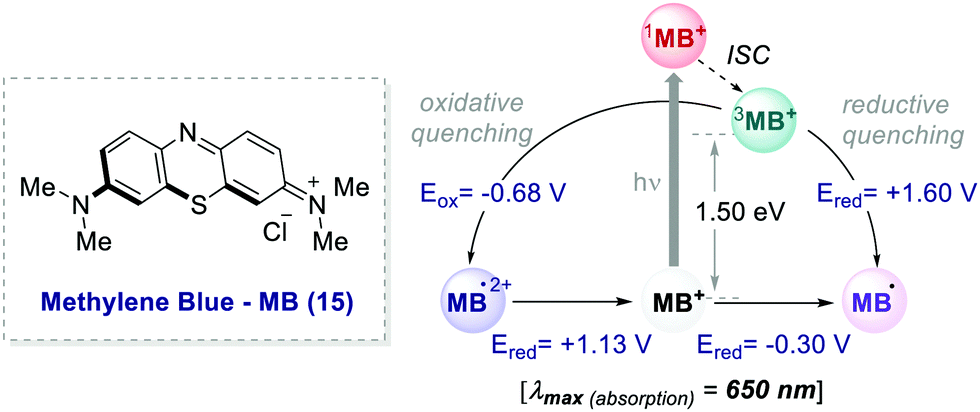 | ||
| Scheme 8 Photophysical properties and redox potentials (vs. SCE in MeCN) of methylene blue (ref. 40c and d). | ||
The use of this PC was previously limited to producing singlet oxygen for applications in the fields of chemistry and biology.41
In the work, Scaiano's group developed a metal-free variant of the photocatalytic oxidative hydroxylation of aryl boronic acids 16 with molecular oxygen (Scheme 9). Such transformation was previously reported by Xiao and Jørgensen's groups employing Ru(bpy)32+ as a PC, but it suffered from prolonged reaction times (up to 72 h).42 A direct comparison between Ru(bpy)3Cl2 and MB at a given reaction time (7 h) showed the superior performance of MB (Scheme 9a, entries 1 vs. 2).
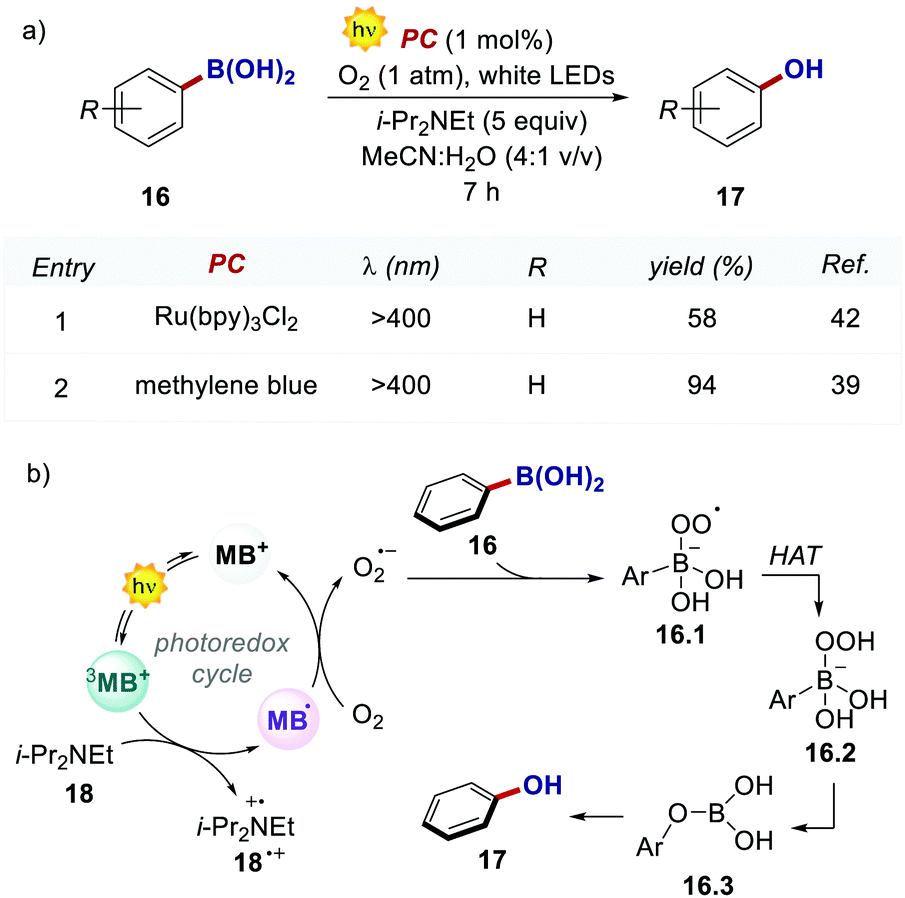 | ||
| Scheme 9 (a) Photocatalytic oxidative hydroxylation of aryl boronic acids. Comparison between the performances of Ru(bpy)32+ and methylene blue. (b) Mechanistic proposal for the oxidative hydroxylation of aryl boronic acids with MB as PC. | ||
The authors concluded that one of the key events in the photocatalytic cycle is the reductive quenching of the MB triplet state (3MB+) via a SET process from diisopropylethylamine (DIPEA 18, Scheme 9b). In fact, kinetic investigations with laser flash photolysis revealed that 3MB+* is quenched nearly 40 times faster than Ru(bpy)32+*.
Collectively, these reports emphasise the potential of already established organic dyes as suitable alternatives to transition metal chromophores and have encouraged the discovery of new families of versatile organic molecules serving as PCs. The search for new organic PCs capable to replace metal complexes is still an active research area. In this regard, one of the latest examples has been documented by Gianetti's group.43a They have successfully applied N,N′-di-n-propyl-1,13-dimethoxyquinacridinium (nPr-DMQA+) tetrafluoroborate salt 19 (Scheme 10) as an efficient PC in a variety of known photoredox reactions. This PC can be synthesized following a high-yielding multi-step procedure.43b,c One remarkable aspect of this PC is that it allows the use of red light (λ = 600–700 nm), thus offering a milder option for the commonly used white, blue or even green light mediated photoreactions. Furthermore, this low-energy irradiation avoids side reactions, has fewer health risks and it is naturally abundant from sunlight.
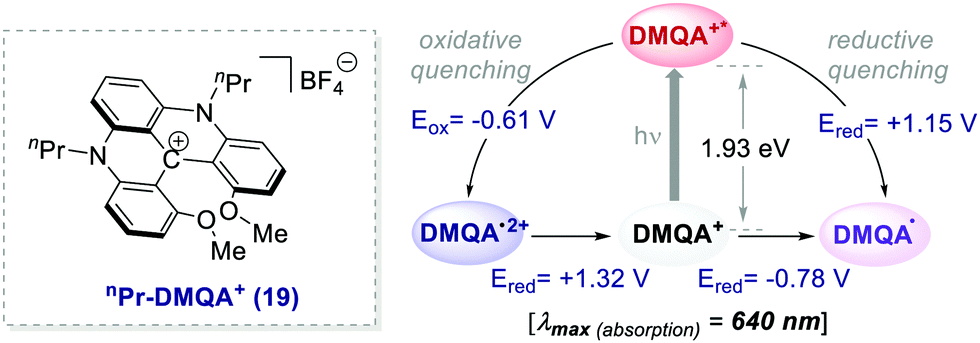 | ||
| Scheme 10 Photophysical properties and redox potentials (vs. SCE in MeCN) of nPr-DMQA+ (ref. 43a). | ||
The authors evaluated the performances of this new PC in the dual photoredox/Pd-catalysed C(sp2)–H arylation of substituted arenes 20, which was first documented by Sanford's group, using Ru(bpy)32+ in the presence of a 26 W compact fluorescent light (CFL, Scheme 11).44
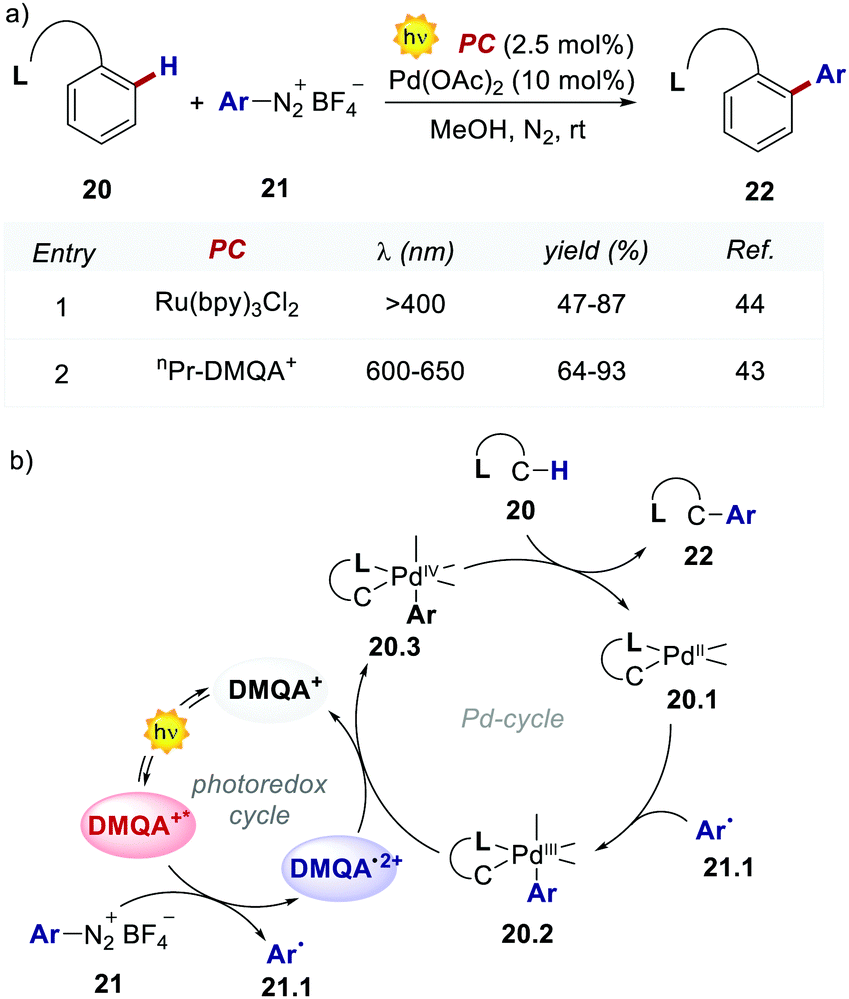 | ||
| Scheme 11 (a) Dual photoredox/Pd-catalysed C–H arylation of substituted arenes. Comparison between the performances of Ru(bpy)32+ and nPr-DMQA+. (b) The proposed mechanism for the photoredox/Pd-catalysed C–H arylation of arenes, depicted with DMQA+ as PC. | ||
The replacement of Ru-PC for nPr-DMQA+ provided enhanced results for the formation of the arylated product 22, with the additional advantage of using low-energy red-light (entries 1 vs. 2, Scheme 11a). In the original light-driven process, the excited Ru(bpy)32+* reduces ArN2+21 to the aryl radical 20.1 (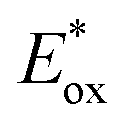 (RuIII/RuII*) = −0.81 V; Ered (PhN2+/Ph˙) = −0.10 V vs. SCE in MeCN) (Scheme 11b). Then, the one-electron oxidation of the PdIII-complex 20.2 by Ru(bpy)33+ regenerates the Ru(bpy)32+ (Ered (RuIII/RuII) = +1.29 V vs. SCE in MeCN). Following a similar mechanistic pathway, the excited DMQA+* undergoes an unconventional oxidative quenching with 21, to deliver the required aryl radical 21.1 (
(RuIII/RuII*) = −0.81 V; Ered (PhN2+/Ph˙) = −0.10 V vs. SCE in MeCN) (Scheme 11b). Then, the one-electron oxidation of the PdIII-complex 20.2 by Ru(bpy)33+ regenerates the Ru(bpy)32+ (Ered (RuIII/RuII) = +1.29 V vs. SCE in MeCN). Following a similar mechanistic pathway, the excited DMQA+* undergoes an unconventional oxidative quenching with 21, to deliver the required aryl radical 21.1 (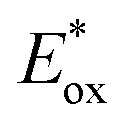 (DMQA˙++/DMQA+*) = −0.62 V vs. SCE in MeCN). The catalyst regeneration occurs through the reduction of DMQA˙++ by the Pd-intermediate 20.2 (Ered (DMQA˙++/DMQA+) = +1.32 V vs. SCE in MeCN). The photoinduced aerobic oxidative hydroxylation of arylboronic acids 16 (refer to Scheme 9) was also successfully promoted by DMQA+ PC, using red-light irradiation. Under the new photochemical conditions, the benchmark product 17 (Scheme 9, with R = H) was obtained in 83% yield, thus outcompeting the performance of the Ru-complex PC (entry 1, Scheme 9a).
(DMQA˙++/DMQA+*) = −0.62 V vs. SCE in MeCN). The catalyst regeneration occurs through the reduction of DMQA˙++ by the Pd-intermediate 20.2 (Ered (DMQA˙++/DMQA+) = +1.32 V vs. SCE in MeCN). The photoinduced aerobic oxidative hydroxylation of arylboronic acids 16 (refer to Scheme 9) was also successfully promoted by DMQA+ PC, using red-light irradiation. Under the new photochemical conditions, the benchmark product 17 (Scheme 9, with R = H) was obtained in 83% yield, thus outcompeting the performance of the Ru-complex PC (entry 1, Scheme 9a).
In 2020, Dell’Amico's group reported the synthesis, photophysical characterisation and use of naphthochromenones (NTCs), a novel class of bimodal PCs (Scheme 12).45 The NTC scaffolds were assembled through a microfluidic photoenolization/Diels–Alder sequence from benzophenones,46 followed by water elimination and aromatisation (Scheme 12a). This strategy allowed the synthesis of the six photoactive scaffolds 25a–f (Scheme 12b). Further manipulation of 25e provided another six additional structures (products 25g–l).
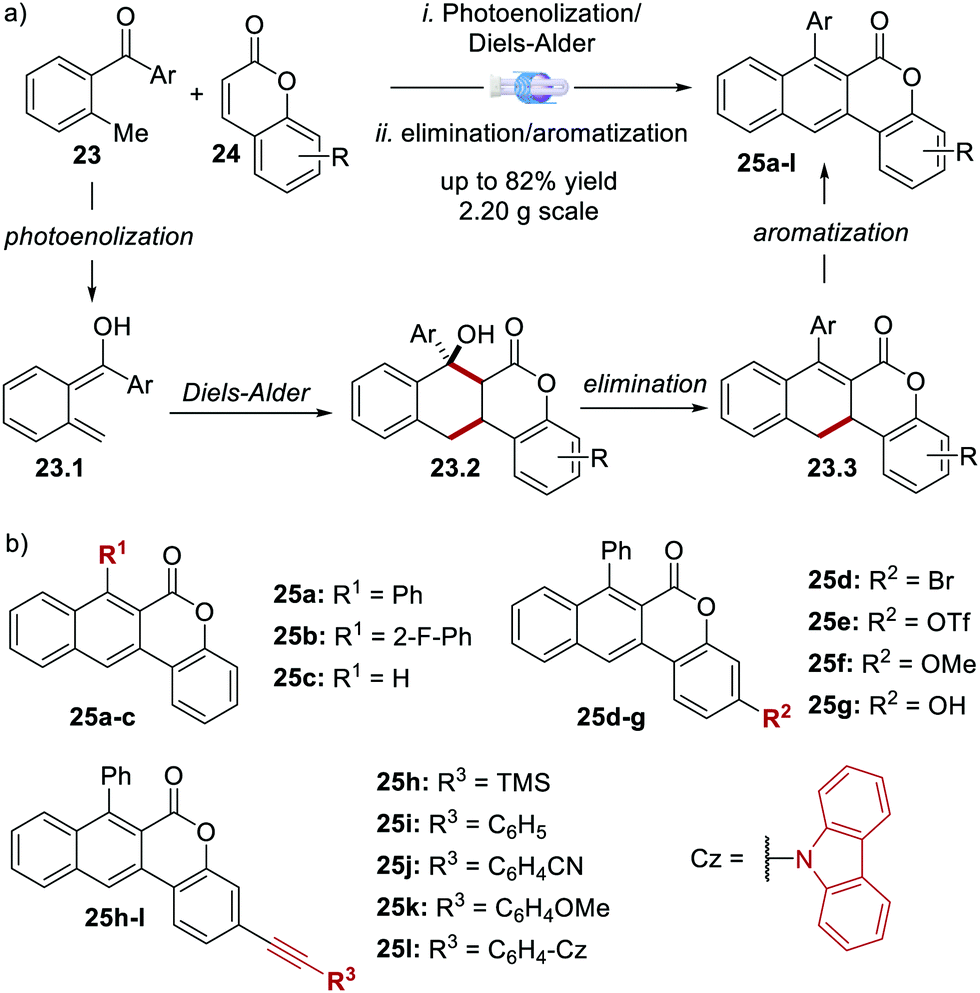 | ||
| Scheme 12 (a) Synthetic strategy for the assembly of the naphthochromenone scaffold. (b) Structures of the NTC photocatalysts (ref. 45). | ||
The 12 NTCs absorb in the visible region towards the higher-energy region of the spectrum (400–420 nm). This feature allows the use of common LEDs for their excitation, but at the same time, guaranteeing access to a highly energetic excited state (up to 3.22 eV). Additionally, their balanced and broad redox windows (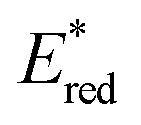 up to 1.65 V,
up to 1.65 V, 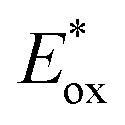 up to −1.77 V vs. SCE in MeCN) make them able to engage in both reductive and oxidative quenching processes (Scheme 13).
up to −1.77 V vs. SCE in MeCN) make them able to engage in both reductive and oxidative quenching processes (Scheme 13).
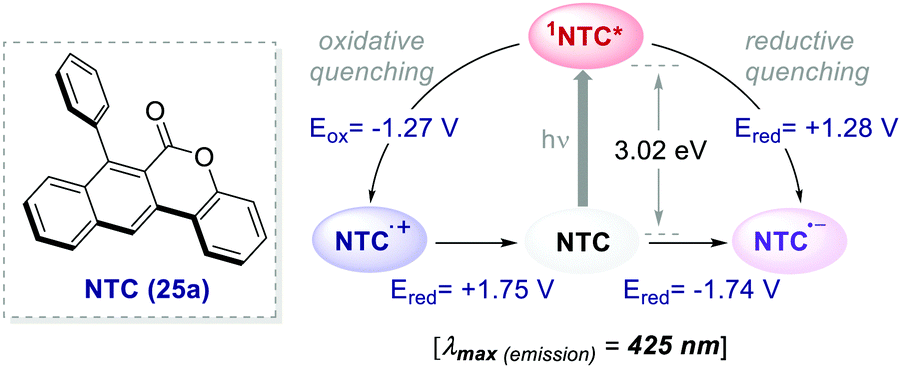 | ||
| Scheme 13 Photophysical properties and redox potentials (vs. SCE in MeCN) of NTC 25a (ref. 45). | ||
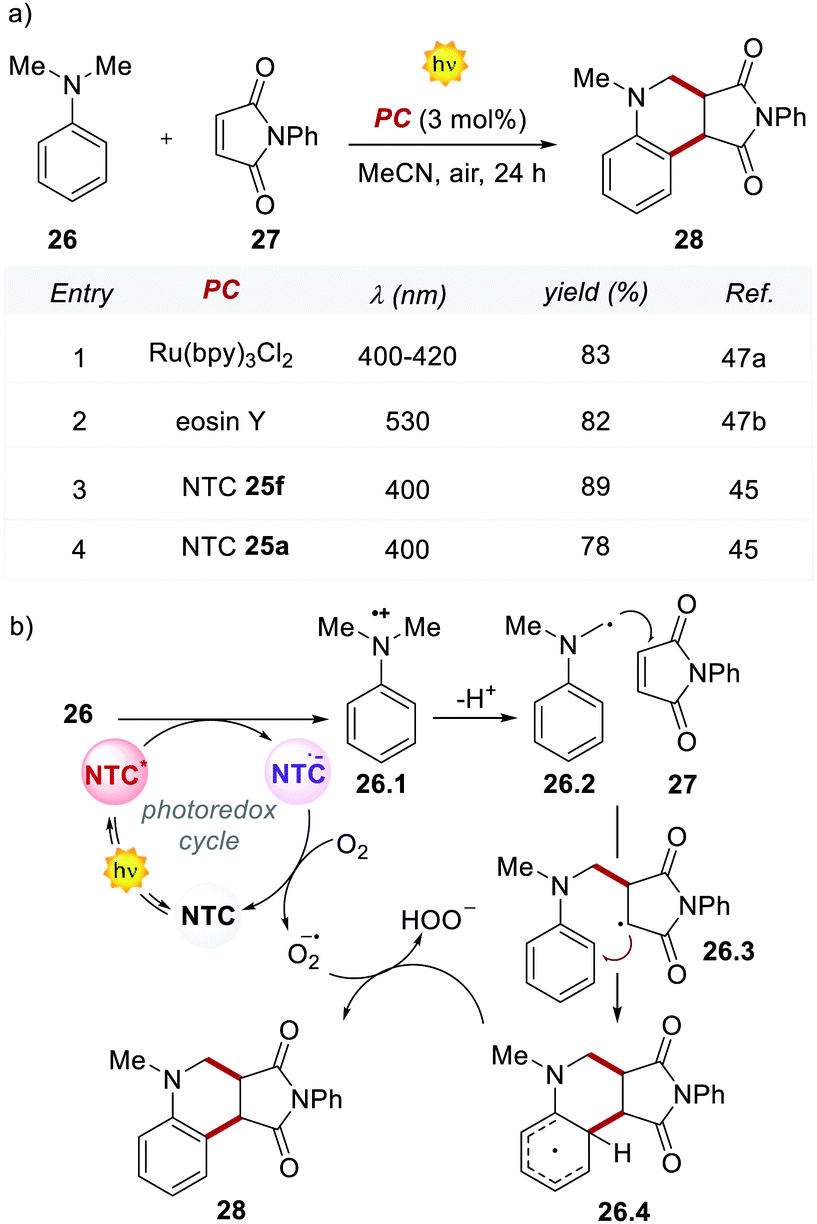
The performances of the 12 NTCs were first evaluated in the Povarov-type addition of N,N-dimethylaniline 26 to phenylmaleimide 27 (Scheme 14). Interestingly, the methoxy-substituted NTC 25f outperformed the previously reported yields achieved with Ru(bpy)32+ and EY, which afforded product 28 in 83% and 82% yields, respectively (Scheme 14, entries 1 and 2 vs. 3). It is worth noting that the structurally simpler NTC 25a provided the product in comparable yield to Ru(bpy)32+ and EY (Scheme 14, entries 1 and 2 vs. 4).
 | ||
| Scheme 14 (a) Povarov-type addition of N,N-dimethylaniline to phenylmaleimide. Comparison between the performance of Ru(bpy)32+ and NTC 25f. (b) The reported mechanism for the Povarov-type addition of N,N-dimethylaniline to phenylmaleimide, depicted with NTCs as PCs. | ||
Based on the reported mechanism,47 the oxidation of aniline 26 occurs from the excited NTC*, generating the radical cation 26.1 (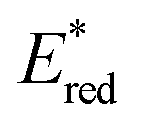 (25f*/25f˙−) = 1.21 V; Eox (26˙+/26) = +0.80 V vs. SCE in MeCN). The resulting NTC 25f˙− is then oxidised by O2, thus closing the photoredox cycle (Ered (25f/25f˙−) = −1.76 V; Ered (O2/O2˙−) = −0.64 V vs. SCE in MeCN). The formation of the product occurs through the addition of the α-aminoradical 26.2 to 27, leading to 26.3. A second radical cyclization event, followed by the oxidation of the resulting intermediate 26.4, delivers product 28.
(25f*/25f˙−) = 1.21 V; Eox (26˙+/26) = +0.80 V vs. SCE in MeCN). The resulting NTC 25f˙− is then oxidised by O2, thus closing the photoredox cycle (Ered (25f/25f˙−) = −1.76 V; Ered (O2/O2˙−) = −0.64 V vs. SCE in MeCN). The formation of the product occurs through the addition of the α-aminoradical 26.2 to 27, leading to 26.3. A second radical cyclization event, followed by the oxidation of the resulting intermediate 26.4, delivers product 28.
4.2 Replacement of an Ir-complex for an organic PC
Ir-based PCs have also found extensive use in photoredox catalysis. The complex fac-Ir(ppy)32a absorbs visible photons (λmax = 375 nm), giving access to a long-lived triplet 3MLCT state (τ = 1.9 μs).14 It is a strong reductant in the excited state (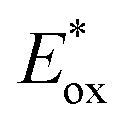 = −1.79 V vs. SCE in MeCN), and a relatively poor oxidant (
= −1.79 V vs. SCE in MeCN), and a relatively poor oxidant (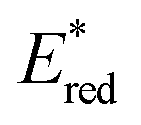 = 0.31 V vs. SCE in MeCN) (Scheme 15).
= 0.31 V vs. SCE in MeCN) (Scheme 15).
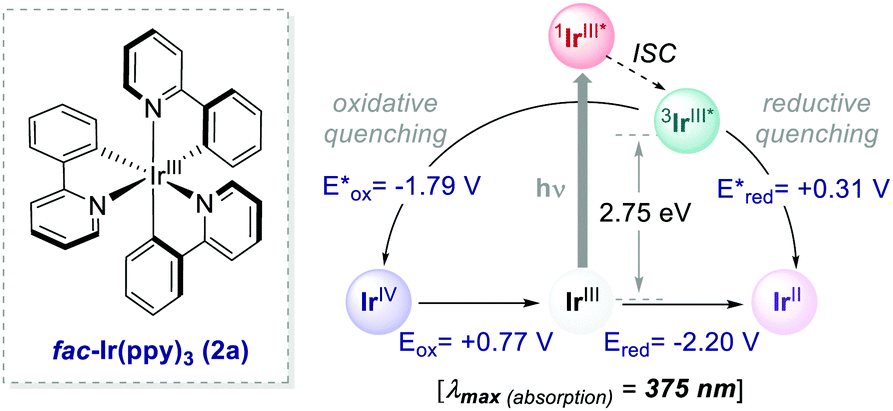 | ||
| Scheme 15 Photophysical properties and redox potentials (vs. SCE in MeCN) of fac-Ir(ppy)3 (ref. 14). | ||
Research towards the identification of suitable organic alternatives to this strongly reducing Ir complex initially began in the field of polymer chemistry, specifically in atom transfer radical polymerisation (ATRP). Controlled radical polymerisations (including ATRP) are synthetic processes that allow obtaining polymers with low dispersity, precise molecular weight and controlled composition.48
The use of purely organic PCs in polymerisation reactions is essential because the metal-traces contamination in a polymeric material prevents any possible applications.49
In 2011, Choi's group presented the first method of photo-induced radical polymerisation of methyl methacrylate 30.50 In 2012, Hawker's group proposed the first visible light controlled ATRP of 30 with fac-Ir(ppy)3, which allowed an improved control on the chain growth.51 The PC operates through an oxidative quenching mechanism with the growing polymer chain (Scheme 16). The continuous activation and deactivation of the chain limit the number of active radicals present at the same time in the system. This mechanism also results in superior control over molecular weight dispersity.
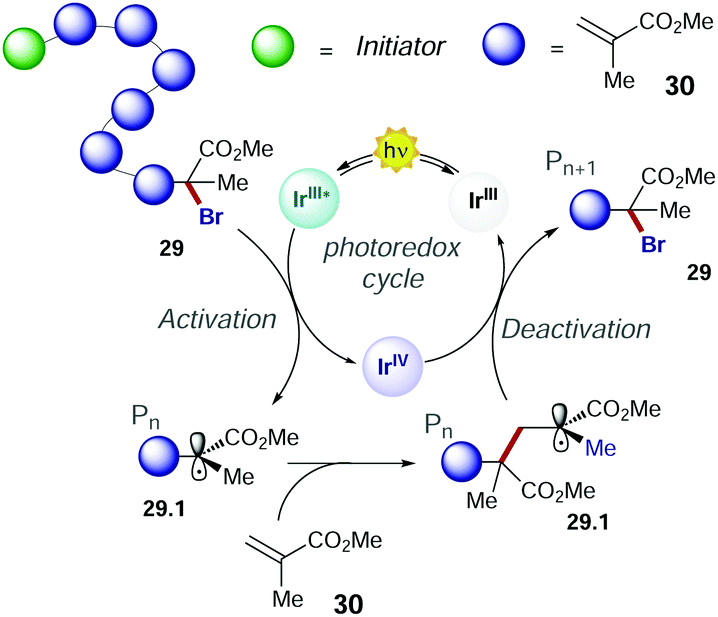 | ||
| Scheme 16 Mechanism of polymerisation of methyl methacrylate with fac-Ir(ppy)3. | ||
Consequently, the main challenge for replacing the fac-Ir(ppy)3 with an organic PC is to develop a strong photoreductant with appropriate redox potentials to promote the reduction of the alkyl halide, while maintaining the ideal equilibrium between the activation and deactivation events (Scheme 16).
The earliest examples of photoinduced organic ATRP (O-ATRP) dates back to 2014, when Hawker's and Miyake's groups independently reported a metal-free method for the polymerisation of 30.52 Hawker successfully proposed the use of 10-phenyl phenothiazine 31 (PTH) as an organic PC (Scheme 17), whereas Miyake employed perylene. PTH was synthesized by the authors from commercially available phenothiazine and chlorobenzene in a one-step procedure through routine C–N cross-coupling reactions.
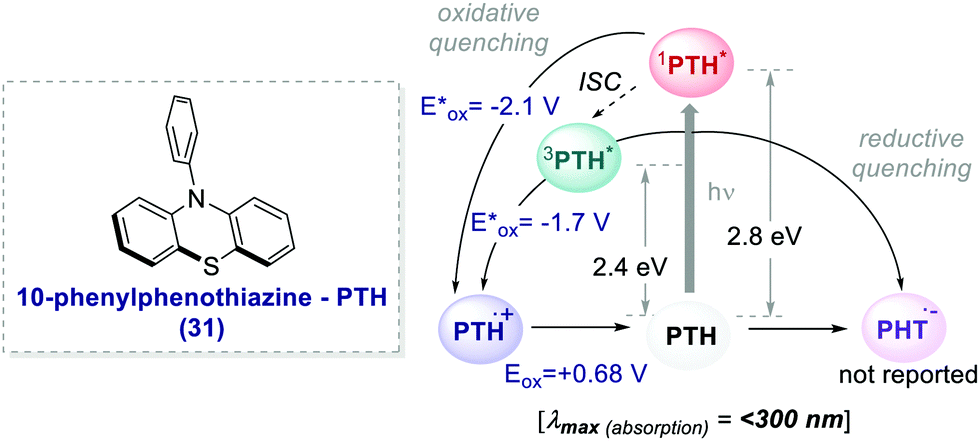 | ||
| Scheme 17 Photophysical properties and redox potentials (vs. SCE in MeCN) of PTH (ref. 25c). | ||
In both cases, the PCs operate through an oxidative quenching mechanism. They showed excellent results in terms of dispersion and retention of the chain end functionality.
PTH gave also access to the polymerisation of monomers that were inaccessible with Ir-based PCs. The polymerisation of dimethylaminoethyl methacrylate 32 was reported (Scheme 18) following the same oxidative quenching mechanism. PTH achieved a much lower level of dispersion in comparison with fac-Ir(ppy)3, showing a better control on the growth of the chain (Scheme 18a, entry 1 vs. entry 2).
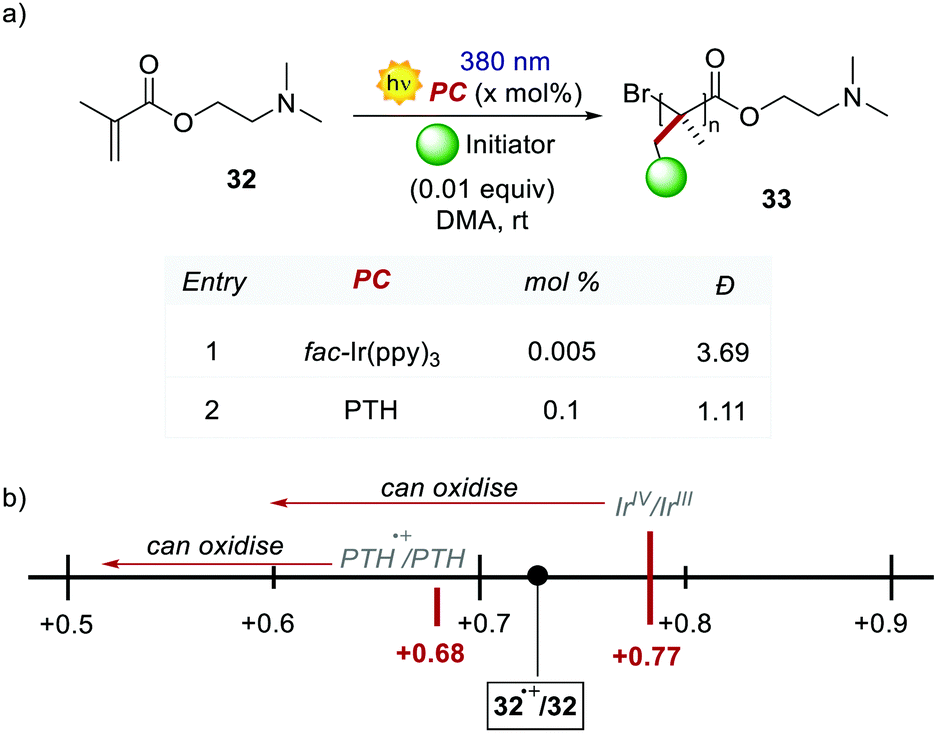 | ||
| Scheme 18 (a) Polymerisation of dimethylaminoethyl methyl methacrylate. Comparison between the performances of PTH and fac-Ir(ppy)3. (b) Graphical depiction of the oxidation potentials (vs. SCE in MeCN) of 32, PTH and fac-Ir(ppy)3. | ||
The authors suggested that the reason was the difference in the Eox of PTH and fac-Ir(ppy)3, respectively +0.68 V and +0.77 V vs. SCE in MeCN. The higher ground-state oxidation potential of fac-Ir(ppy)3 causes the oxidation of the amino group of the monomer, and this decreases the control over the growth of the polymer chain.
Shortly after, these highly reducing organic dyes started to find applications as photocatalysts for small molecule activation. In 2015, Hawker's group, inspirited by the results of O-ATRP, proposed the use of PTH as a replacement for fac-Ir(ppy)3 in the dehalogenation of alkyl halides, following the report by Stephenson with fac-Ir(ppy)3.53,54 Employing PTH, the authors extended the application of this protocol not only to aryl iodides but also to difficult-to-activate substrates such as aryl bromides and chlorides (Scheme 19a). PTH was used in higher loading than fac-Ir(ppy)3. However, the authors pointed out that 1 mg of PTH is more than 7000 times cheaper than 1 mg of fac-Ir(ppy)3. The reaction mechanism, originally proposed by Stephenson, begins with the reduction of aryl halide 34 and the subsequent formation of the radical 34.1 upon the homolytic cleavage of the carbon–halogen bond. Product 35 is finally obtained after a H-atom abstraction event. The oxidation of tributyl amine 36 closes the photocatalytic cycle restoring the ground-state PC. Dehalogenation of phenyl bromide 34b performed with PTH resulted in 85% yield in 72 hours while no conversion was observed with fac-Ir(ppy)3. This result is a consequence of the higher 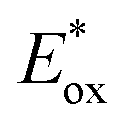 of the organic photocatalyst. Contrary to fac-Ir(ppy)3, with PTH, both the S1 and T1 states can take part in the reduction of the carbon–halogen bond.
of the organic photocatalyst. Contrary to fac-Ir(ppy)3, with PTH, both the S1 and T1 states can take part in the reduction of the carbon–halogen bond.
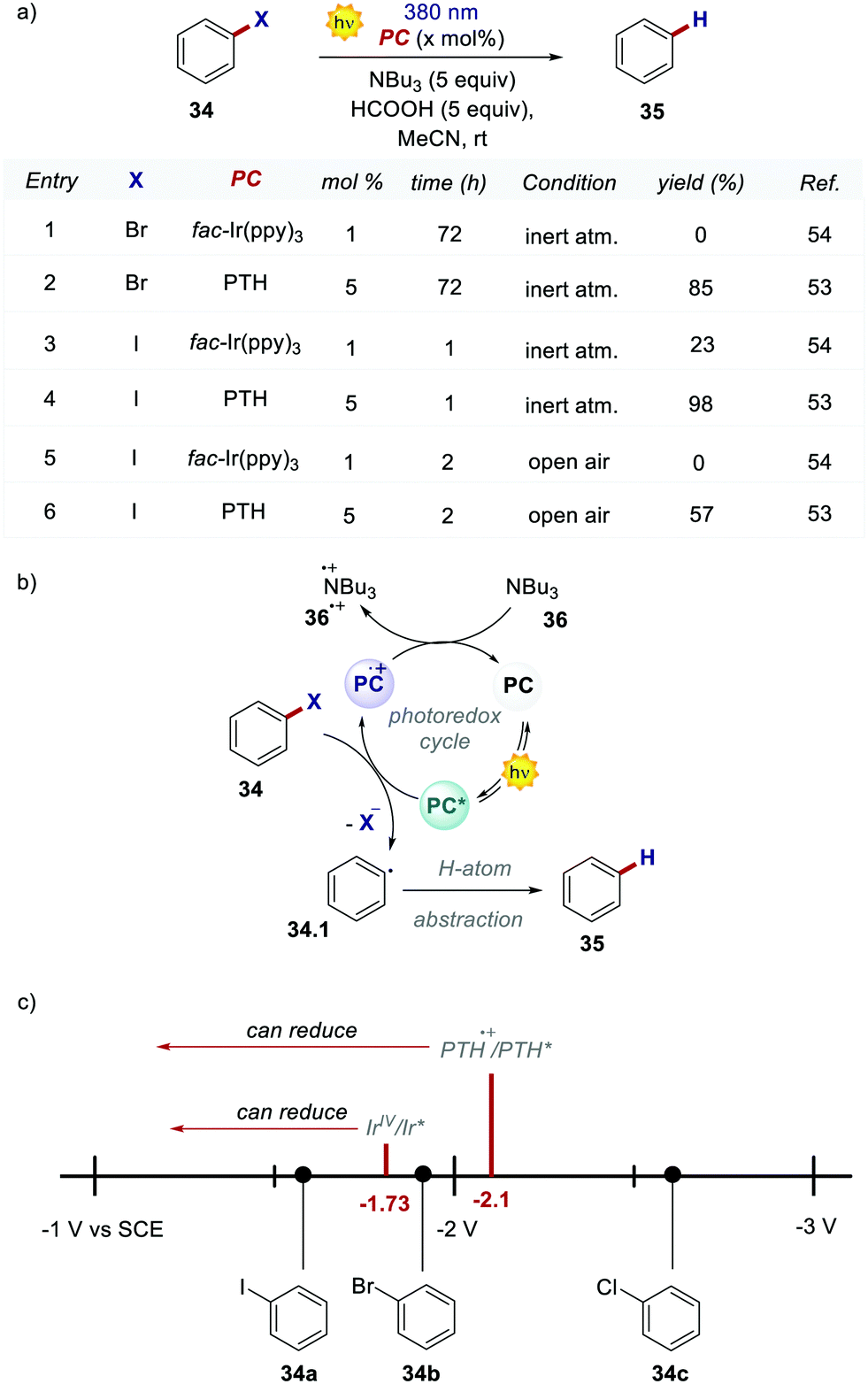 | ||
Scheme 19 (a) Reductive dehalogenation of aryl halides. Comparison between the performances of PTH and fac-Ir(ppy)3. (b) Mechanism of the dehalogenation reaction. (c) Graphical depiction of the reduction potentials (vs. SCE in MeCN) of the selected aryl halides and 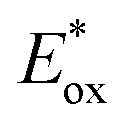 of PTH and fac-Ir(ppy)3. of PTH and fac-Ir(ppy)3. | ||
Despite the fact that the S1 state is dominant due to its longer lifetime, the reduction of unactivated aryl bromides or chlorides is achieved only by the more reducing S1 state. This feature was confirmed by carrying out a control reaction open to air in the presence of oxygen, which is a well-known triplet quencher.55 The tolerance to oxygen was tested with different substrates and in every case, no reaction was observed with iridium. PTH was tested also in a higher-scale reaction with excellent results. Notably, during the purification of the product, PTH was also isolated and reused with unvaried reactivity.
In 2020, Dell’Amico's group chose this dehalogenation reaction to evaluate the performances of NTCs under oxidative quenching.45 Thus, following the same mechanistic scheme, the reduction of methyl 4-iodobenzoate 37 was examined (Scheme 19). Interestingly, NTC 25a performed well, achieving the formation of 34 in 86% yield. The more reducing NTC 25i delivered 38 in 96% yield, providing a valuable metal-free alternative to fac-Ir(ppy)3.54 It is worth noting how NTC 25a could be successfully employed under both oxidative (Scheme 20) and reductive quenching (Scheme 14), thus being a competent metal-free alternative to both Ru(bpy)32+ and fac-Ir(ppy)3.45
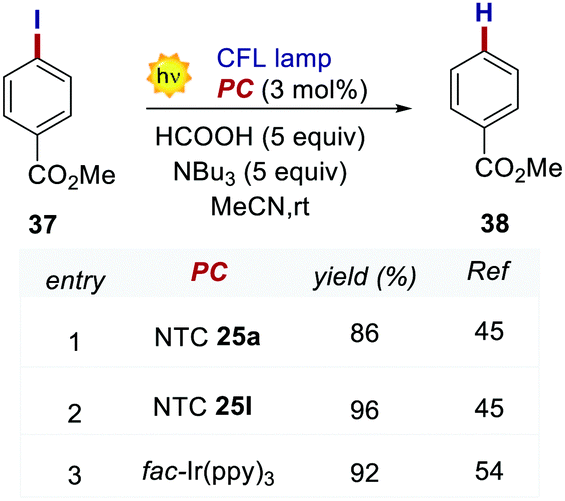 | ||
| Scheme 20 Reductive dehalogenation of methyl 4-iodobenzoate. Comparison between the performances of NTCs and fac-Ir(ppy)3. | ||
In 2017, Ooi's group proposed the use of 3,6-dimethoxy-9H-thioxanthen-9-one 39 (TX, Scheme 21), a fully organic PC synthesized in three steps and high overall yield, to replace fac-Ir(ppy)3 for the light-promoted imidation of arenes (Scheme 22).56,57 The mechanism proceeds via the generation of the nitrogen-centred radical 40.1 upon the oxidative quenching of the PC* by the redox-active ester 41, and the subsequent formation of radical 41.1 (Scheme 22b). The Wheland intermediate 41.2 is then formed upon the reoxidation of 41.1 by PC˙+.
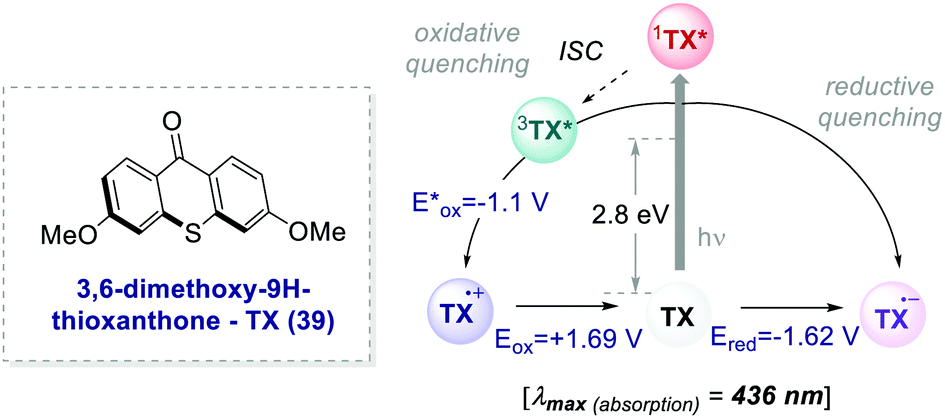 | ||
| Scheme 21 Photophysical properties and redox potentials (vs. SCE in MeCN) of TX (ref. 25c). | ||
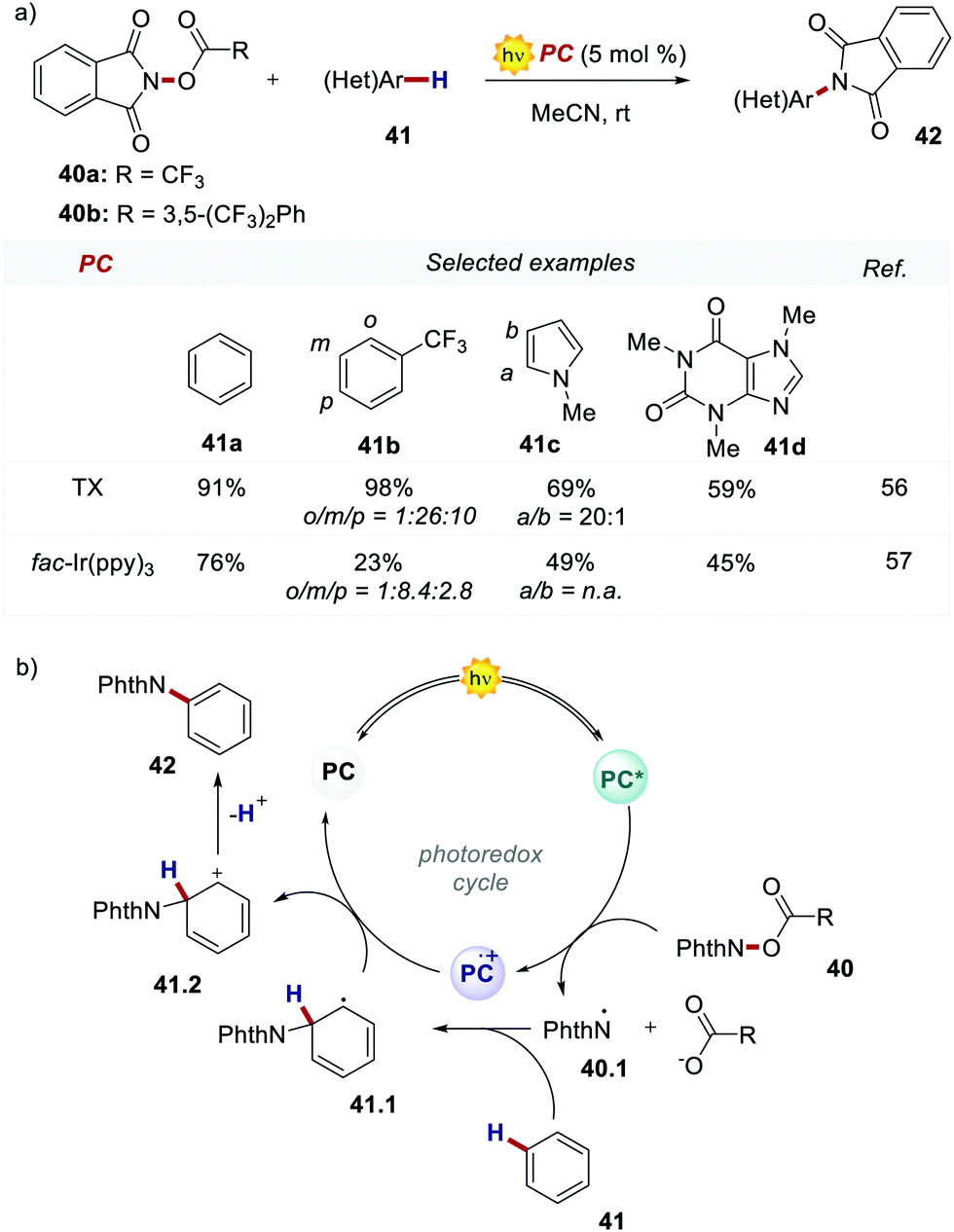 | ||
| Scheme 22 (a) Light-promoted imidation of arenes. Comparison between the performances of TX and fac-Ir(ppy)3. (b) Mechanism of the light-promoted imidation of arenes. | ||
Ooi's group replaced fac-Ir(ppy)3 with TX 39 and the reaction was optimised with the redox-active ester 40b. The organic PC outperformed fac-Ir(ppy)3 with various substrates including caffeine.
With appropriate substituents, Ir-based PCs can also operate under reductive quenching mechanisms. In 2014, McMillan's group reported the use of the iridium complex 2d (Scheme 23) as the photooxidant in a decarboxylative Giese-type radical addition (Scheme 24a, entry 4).58
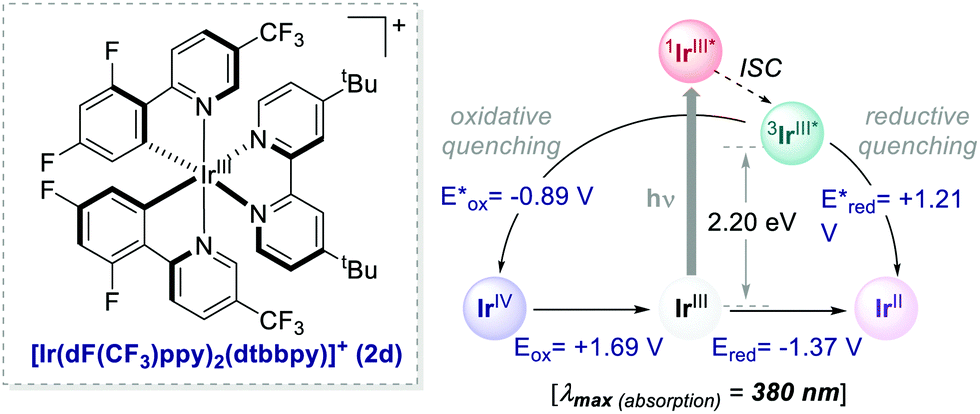 | ||
| Scheme 23 Photophysical properties and redox potentials (in MeCN) of [Ir[dF(CF3)ppy]2(dtbbpy)]+ (ref. 15). | ||
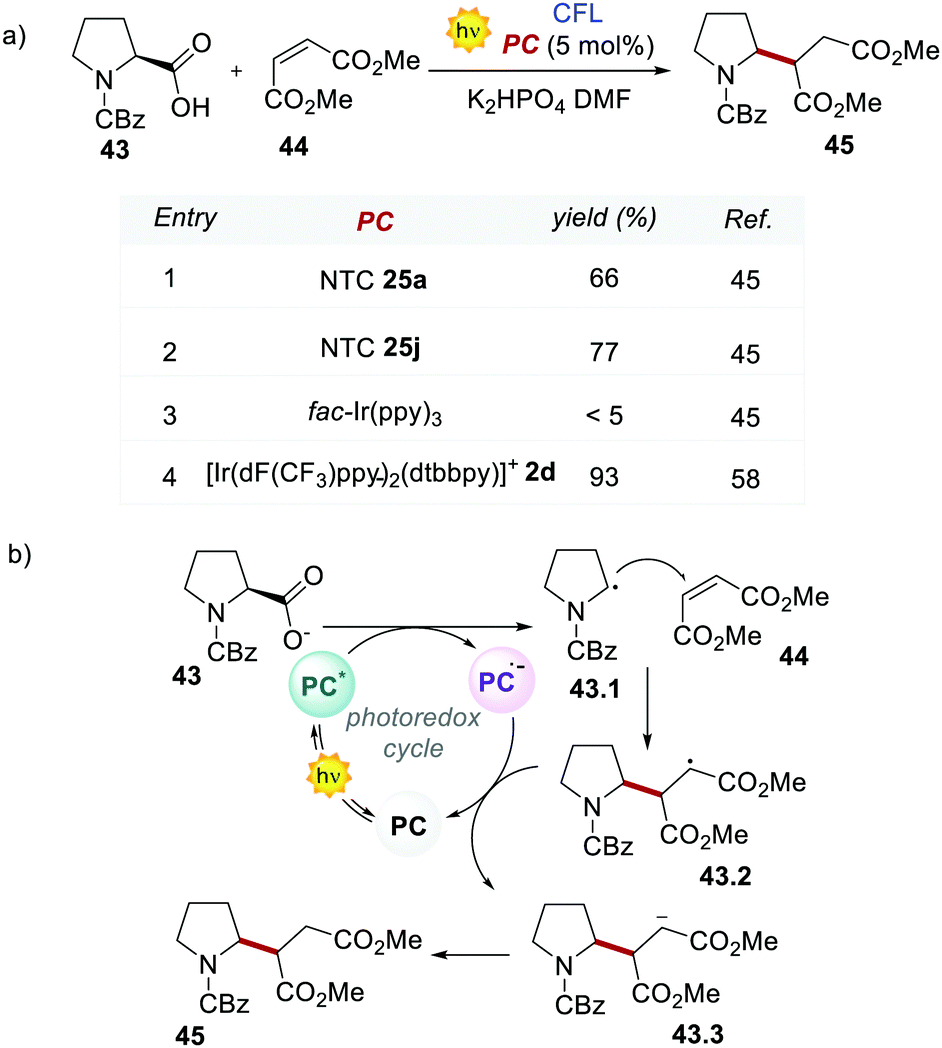 | ||
| Scheme 24 (a) Decarboxylative Giese-type addition of 43 and dimethyl maleate 44. Comparison between the performances of NTCs and Ir-based PCs. (b) Mechanism of the decarboxylative Giese-type addition. | ||
This challenging reaction was used also by Dell’Amico's group to further evaluate the performance of NTCs under reductive quenching. The mechanism depicted in Scheme 24b follows a reductive quenching mechanism. The first step is the oxidation of 43 by the excited photocatalyst and the subsequent formation of the α-amino radical 43.1. This reacts with the electron-poor olefin 44 generating the radical intermediate 43.2 which is then reduced by PC˙− closing the photoredox cycle. NTC 25a gave a yield of 66% but the best result was delivered by NTC 25j with a yield of 77% (Scheme 24a, entries 1 and 2). Conversely, fac-Ir(ppy)3 was ineffective in this reaction (Scheme 24a, entry 3).
Despite the fact that NTCs performed slightly worse than 2d, it is relevant to notice that NTC 25a was applied successfully in various photochemical transformations under both oxidative and reductive quenching (Schemes 14, 20 and 24).45
5 Pushing the boundaries of photoredox catalysis with organic PCs
As mentioned, organic PCs are not only cheaper and sustainable alternatives to metal complexes, but they have also allowed the activation of previously unreactive targets, exceeding the redox limits of their metal counterparts. This significant feature has allowed the discovery of unprecedented reactivity and the development of previously inaccessible chemical transformations. In this section, we highlight that privileged organic PCs can perform thermodynamically demanding photoredox cycles.5.1 Highly oxidising organic PCs
One of the most used highly oxidising PC classes is acridinium salts (46a–c, Scheme 25). Their scaffold was introduced by Fukuzumi in 2001,59 and later was widely used and modified by Nicewicz among others.60 The ground-state reduction of acridinium ions occurs at moderate potentials (around −0.5 V vs. SCE). In contrast, their excited state is characterised by a very high oxidative power, reaching redox potentials [Ered (Acr+*/Acr˙)] up to +2.32 eV. Structural modifications of the acridinium core have allowed improving their chemical stability towards (i) N-dealkylation reactions and (ii) nucleophilic or radical additions at the 2,7 or 3,6-positions.26a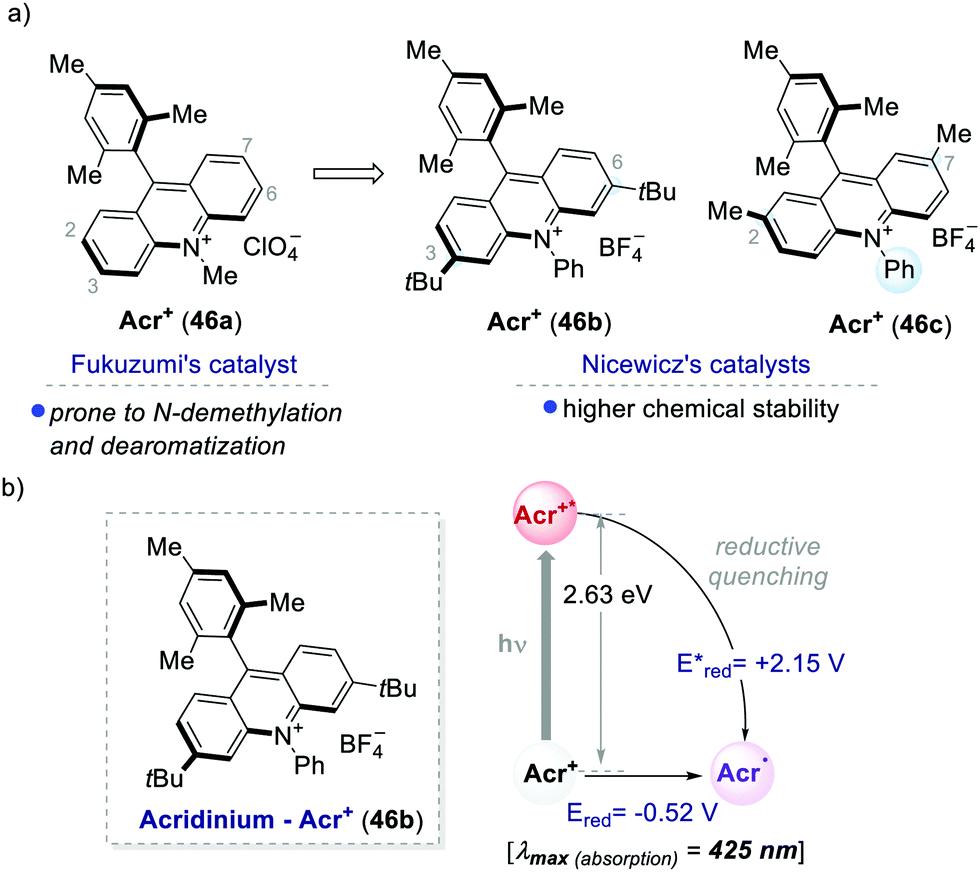 | ||
| Scheme 25 (a) Representative acridinium catalysts developed by Fukuzumi's and Nicewicz's groups. (b) Photophysical properties and redox potentials (in MeCN) of Acr+46b (ref. 25c). | ||
Acridinium ions, synthesized through multi-step procedures,60 have been efficiently used as PCs under reductive quenching for a wide variety of thermodynamically challenging redox processes, including the oxidation of alkenes.61,62 In this regard, Nicewicz's group has reported various photochemical methods for the intra- and intermolecular anti-Markovnikov addition of different heteronucleophiles, using Acr+46a and a redox active hydrogen atom donor (Scheme 26a).60a,63 The general postulated mechanism for these transformations involves the initial formation of the alkene radical cation 49.1 by single-electron oxidation (Scheme 26b, inter-molecular manifold shown), followed by nucleophilic addition with a nucleophile (Nu in Scheme 26b) with anti-Markovnikov regioselectivity. The desired hydro-functionalised product 50 is then formed after a hydrogen atom transfer (HAT) event. Regeneration of the catalyst Acr+46a occurs by another SET process between the thiyl radical 51.1 and the acridinium radical Acr˙ 46a. Considering the reduction potentials of this substrate couple (Ered (46.1/46.2) = +0.45 V vs. SCE64 and Eox (Acr+46a/Acr˙ 46a) = −0.57 V vs. SCE59,65), this latter step is thermodynamically favoured. Notably, this photochemical method allows the use of alkenes having oxidation potentials up to 2.10 V. Such values are well beyond the redox potentials of the common Ir- and Ru-based PCs.
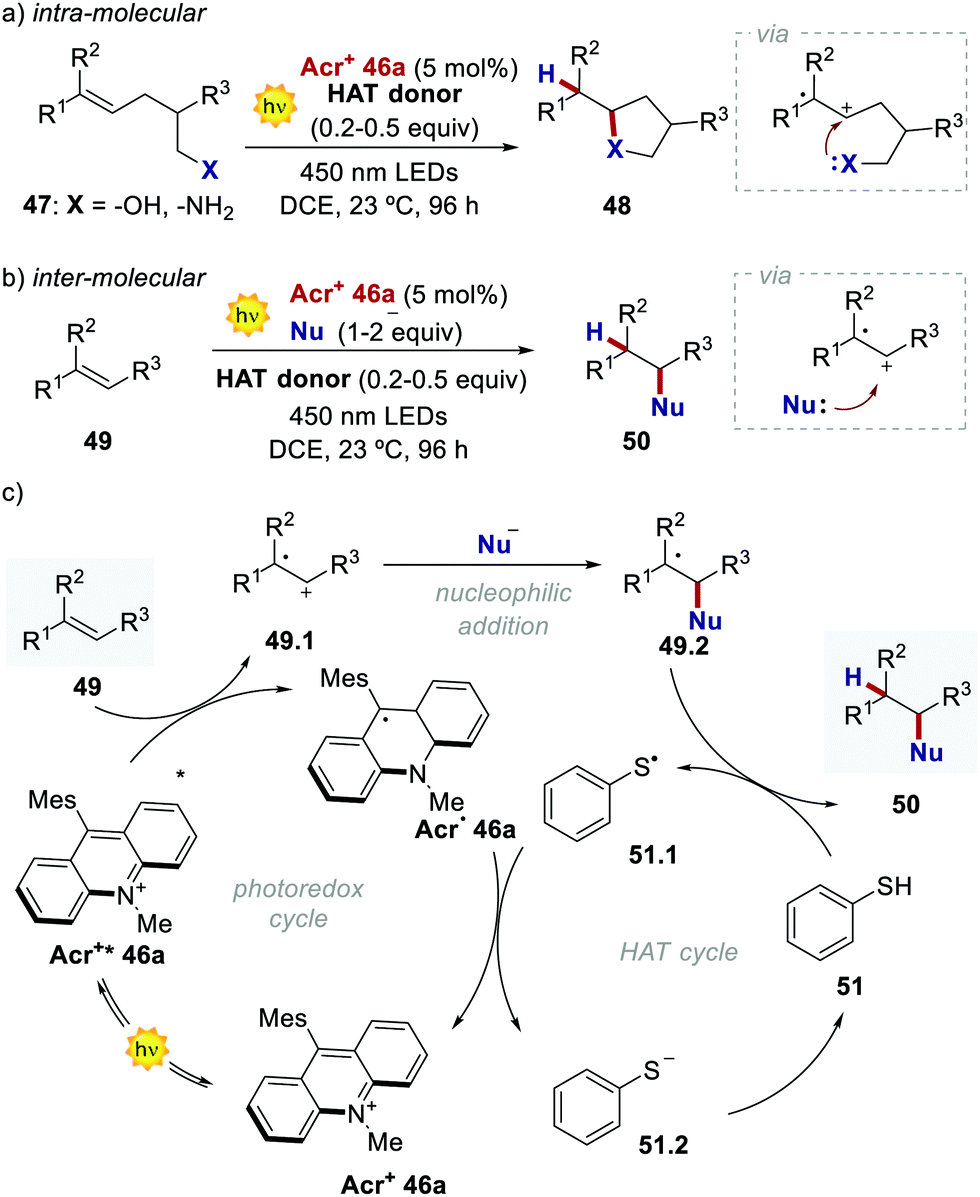 | ||
| Scheme 26 (a) Catalytic intra- and inter-molecular anti-Markonikov hydrofunctionalisation of alkenes enabled by the photocatalytic activity of acridinium salts. (b) General mechanism (for simplicity, only the inter-molecular reactivity is shown). Nu: nucleophile. HAT: hydrogen atom transfer. | ||
In the following studies, Nicewicz's group has showcased the potential of acridinium PCs to provide new metal-free strategies for the C–H functionalisation of arenes.66 In 2015, they used the more robust catalyst Acr+46b (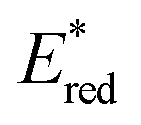 = +2.25 V) to develop a site-selective C–H amination of substituted aromatic compounds (Scheme 27a).60b This transformation exploits the ability of the photoexcited acridinium Acr+* 46b to generate arene radical cations 47.1 through a single electron oxidation pathway (Scheme 27b). Then, the nucleophilic addition of amine 53 on the intermediate 52.1, followed by the oxidation of the resulting species 52.3, delivers the C–N-coupled arene adduct in high yields (up to 88%) and with good to excellent levels of regioselectivity (up to >20:1). During the optimization studies, degradation of the acridinium catalyst was observed, as well as of the starting arene derivative 52 (where R1 = –OMe). Indeed, both compounds are prone to decompose in the presence of oxygen-centred radicals that may be formed during the reaction, under the presence of O2.60c Such a degradation pathway was mitigated by the addition of 20 mol% of TEMPO 55, thus improving the yield of the process.
= +2.25 V) to develop a site-selective C–H amination of substituted aromatic compounds (Scheme 27a).60b This transformation exploits the ability of the photoexcited acridinium Acr+* 46b to generate arene radical cations 47.1 through a single electron oxidation pathway (Scheme 27b). Then, the nucleophilic addition of amine 53 on the intermediate 52.1, followed by the oxidation of the resulting species 52.3, delivers the C–N-coupled arene adduct in high yields (up to 88%) and with good to excellent levels of regioselectivity (up to >20:1). During the optimization studies, degradation of the acridinium catalyst was observed, as well as of the starting arene derivative 52 (where R1 = –OMe). Indeed, both compounds are prone to decompose in the presence of oxygen-centred radicals that may be formed during the reaction, under the presence of O2.60c Such a degradation pathway was mitigated by the addition of 20 mol% of TEMPO 55, thus improving the yield of the process.
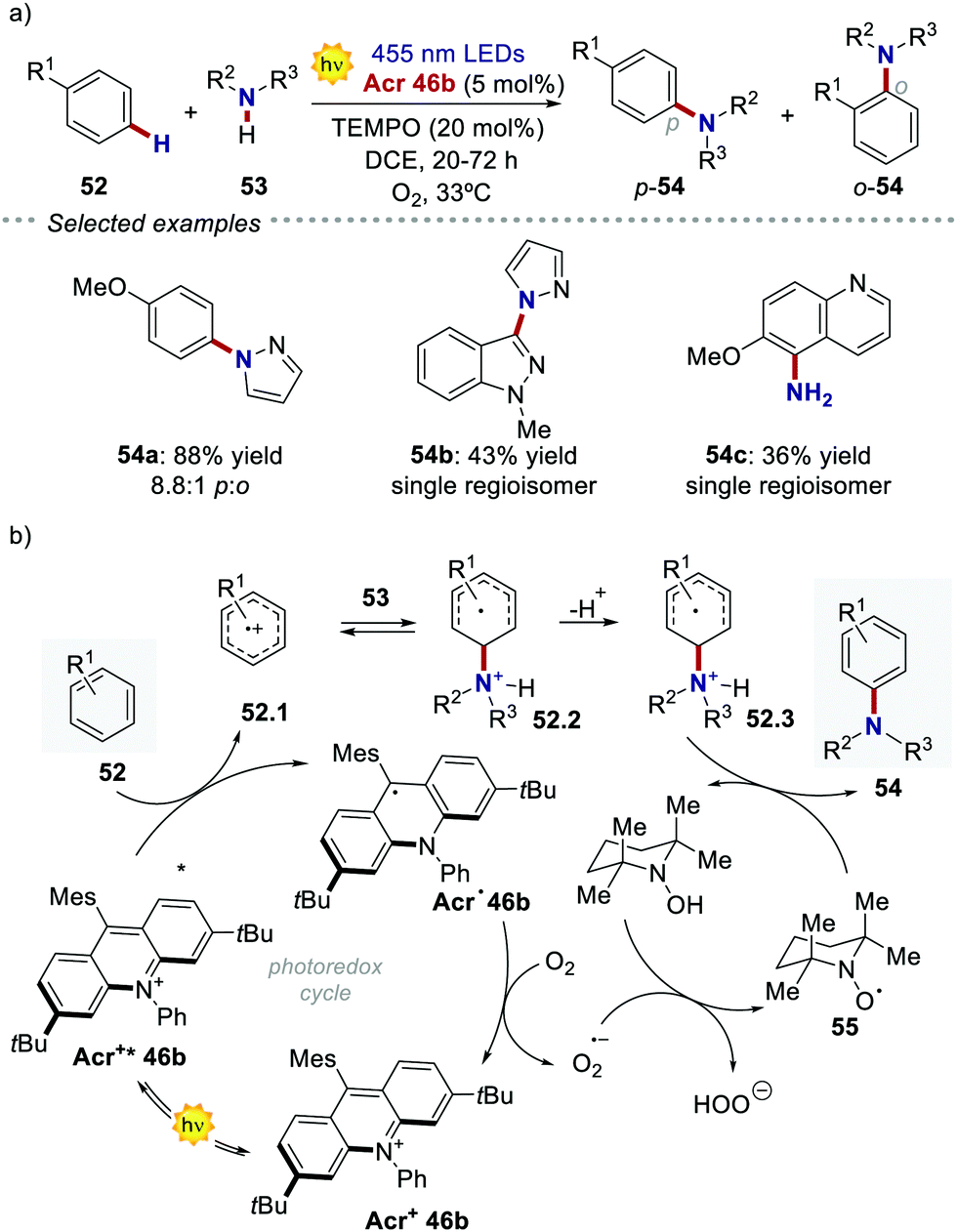 | ||
| Scheme 27 (a) Photochemical C–H amination of arenes enabled by acridinium PCs. (b) One of the plausible mechanisms [ref. 60b]. | ||
Recently, new libraries of acridinium salts with diverse photophysical and electrochemical properties were reported, facilitating a rational selection of the most adequate acridinium PC for a given photoredox transformation.67 Interestingly, Sparr's group has recently documented three protocols for the synthesis of libraries of symmetrical 3,6-diamino- and 1,8-dimethoxy-substituted PCs (56 and 57, respectively, in Scheme 28a), as well as unsymmetrical acridiniums (58).67a,b,f
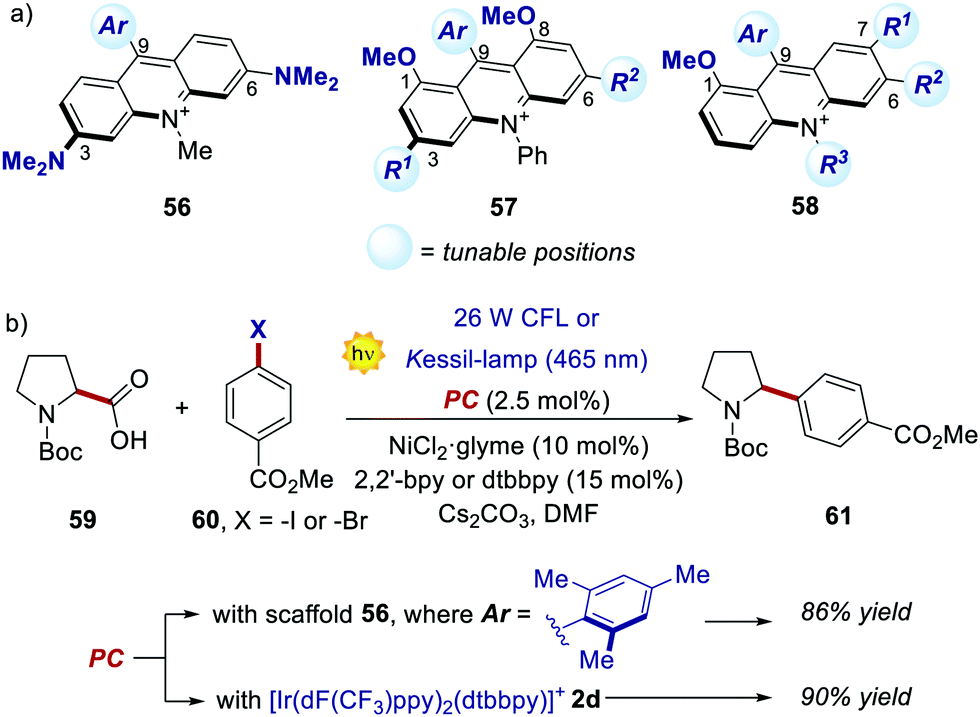 | ||
| Scheme 28 (a) Emerging acridinium libraries. (b) Comparison of the performances between an acridinium derivative 56 and an Ir–PC, in a photoredox-Ni catalysed decarboxylative cross-coupling process. | ||
Interestingly, the performance of some of these new PCs was tested in various photoreactions, showing comparable or even enhanced results relative to the ones offered by the corresponding Ir- or Ru-PCs. For instance, acridiniums 56 exhibited redox potentials similar to those of iridium complex 2d (Ered (IrIII/IrII) = −1.37 V; Ered (IrIII*/IrII) = +1.21 V vs. SCE).67a This feature allowed the use of these new PCs in the metallaphotoredox-nickel catalysed decarboxylative cross-coupling between aminoacids 59 and aryl halides 60 (Scheme 28b). This photochemical process was originally developed by MacMillan and co-workers in 2014, using 2d as the optimal PC.68 In Sparr's work, the mesityl-substituted catalyst 56 (where Ar = 2,4,6-trimethylphenyl) could promote the same transformation with very similar results (86% yield with 56vs. 90% yield with 2d), thus highlighting the potential of these new families of PCs for replacing their metal counterparts.
5.2 Highly reducing organic PCs
In the last few years, research on strongly reducing organic PCs has rapidly evolved. Many new PCs were introduced to replace metal complexes. Also, in this case, most of them were first applied in O-ATRP. In 2016, Miyake's group reported the synthesis and use of 5,10-dihydrophenazines 62 and N-aryl phenoxazine 63. More recently, the same group reported the successful use of dihydroacridines 64 and N-alkyl phenoxazines 65 (Scheme 29).69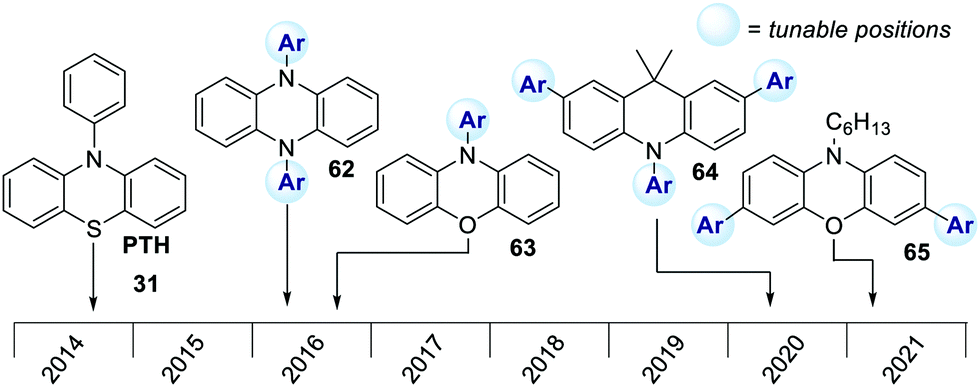 | ||
| Scheme 29 Main classes of organic PCs employed in O-ATRP. | ||
Despite their similar structures, PC 63 displayed enhanced abilities in the O-ATRP of methyl methacrylate 30 in comparison with PTH. Due to the bigger size of the sulfur atom, PTH adopts a bent conformation in the ground state, whereas PC 63 is planar. Nevertheless, they both assume a planar conformation in the excited state. Consequently, the former has a higher reorganisation energy that decreases the rate of electron transfer during the reduction of PTH˙+ to restore the ground-state PTH. In 2018, Miyake's group performed structure–property relationship studies on phenoxazine-based photocatalysts.26b This study was made possible by the high synthetic versatility and availability phenoxazine scaffold. C–N coupling was exploited to functionalize the nitrogen atom. The functionalization of the main core was accomplished in a two-step process: (i) radical bromination, followed by (ii) a cross-coupling reaction.
Different substituents in the core of the molecule and on the N-aryl moiety can dramatically alter the redox potentials and the absorption spectrum. The result was the generation of a library of 19 different PCs whose triplet  covers a wide window ranging from −2.11 to −1.40 V vs. SCE.
covers a wide window ranging from −2.11 to −1.40 V vs. SCE.
One of the most promising candidates is 3,7-4(biphenyl)-1-naphthalene-10-phenoxazine 66 (POx) represented in Scheme 30.
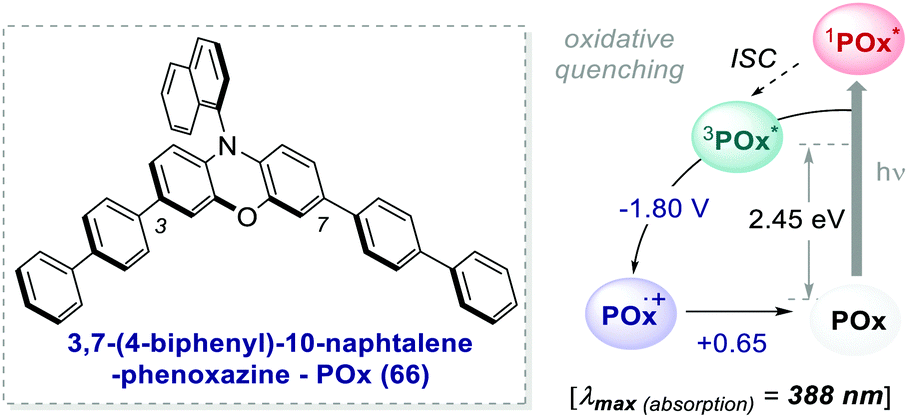 | ||
| Scheme 30 Photophysical properties and redox potentials (vs. SCE, in MeCN) of POx (ref. 25d). | ||
The addition of biphenyl substituents at positions 3 and 7 of the phenoxazine core doubles the molar absorptivity and redshifts the maximum wavelength of absorption by 65 nm compared to the unsubstituted counterpart. Therefore, the absorption profile reaches the visible region, contrary to PC 63. The naphthyl moiety on the nitrogen atom instead allows the access to a CT excited state from the electron-rich core.26b
The capability to work under visible-light and the exceptionally long triplet lifetime of 480 μs25d made this PC suitable for various photochemical reactions.
In 2018, König's group used 66 in a Barbier-type reaction for the allylation of aldehydes and ketones (Scheme 31).70 The mechanism (Scheme 31b) follows an oxidative quenching cycle and involves the reduction of benzaldehyde 67 with the formation of a ketyl radical 67.1. Remarkably, product 69a is obtained in higher yields and with less formation of the homocoupling side products 69b and 69c, by using POx 66 instead of the Ir catalyst 2d.
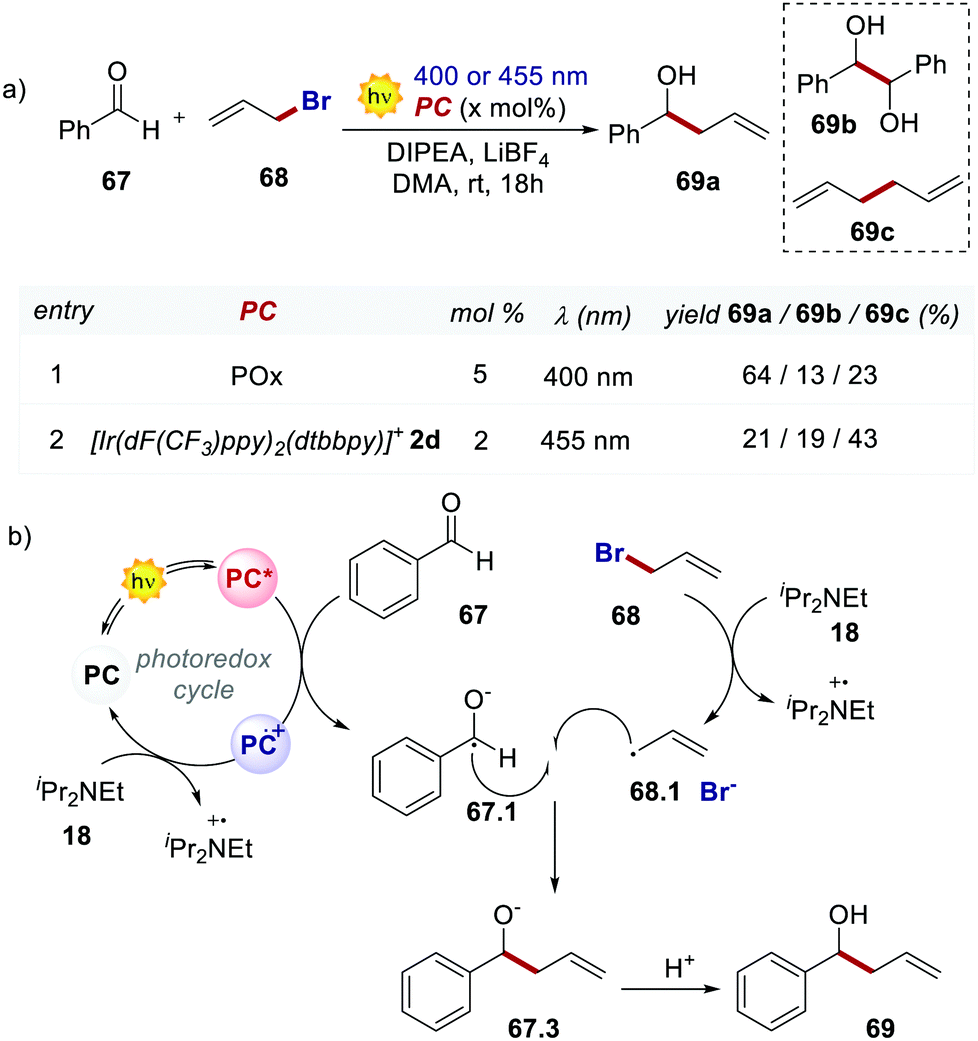 | ||
| Scheme 31 (a) Barbier-type reaction between benzaldehyde and allyl bromide. Comparison between the performances of POx and [Ir(dF(CF3)ppy)2(dtbbpy)]+. (b) Mechanism of the Barbier-type reaction. | ||
POx 66 was used also by Jui's group in 2019 for the functionalisation of the C–F bond of unactivated trifluoromethyl arenes.71 At temperatures higher than 50 °C, POx performed better than fac-Ir(ppy)3 and PTH (Scheme 32a). The higher conversion achieved was a result of the longer excited state lifetime of POx compared to the other PCs.
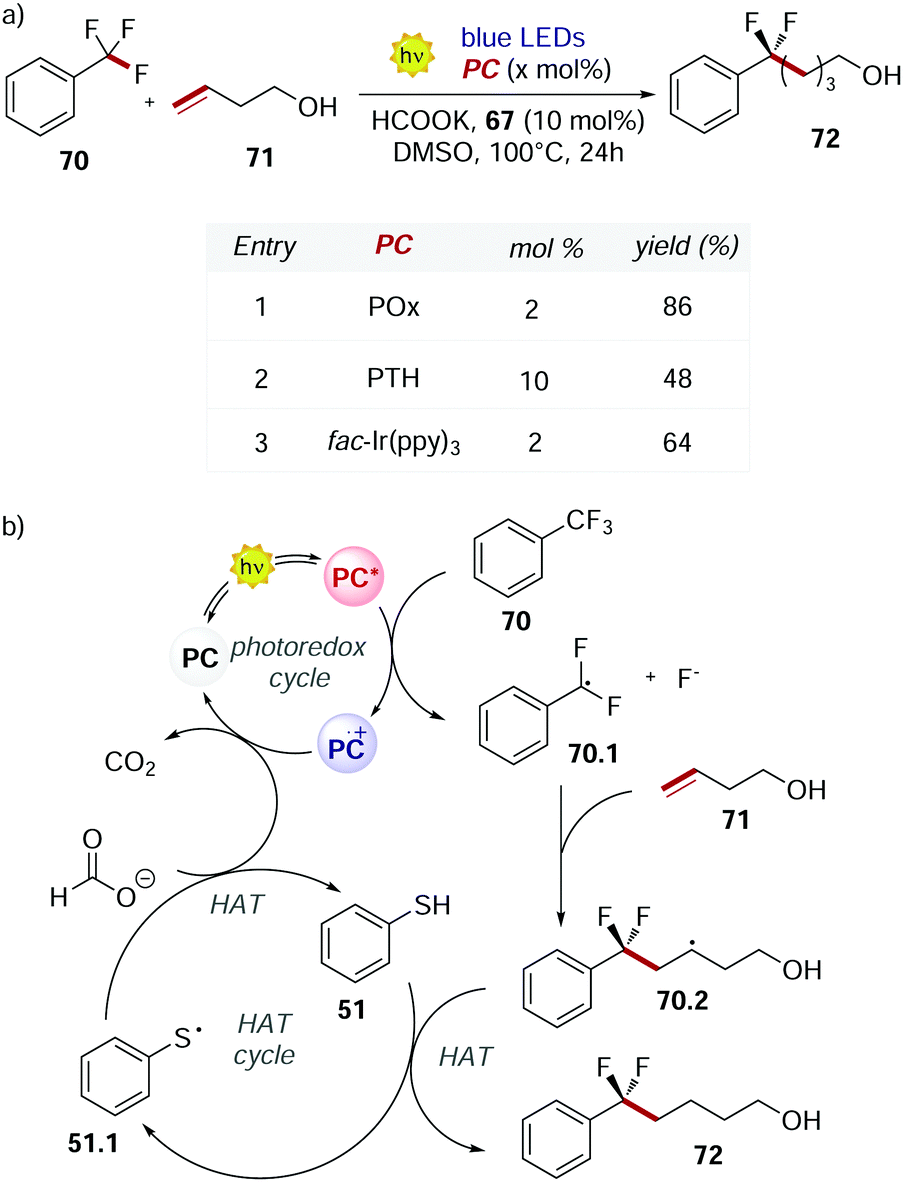 | ||
| Scheme 32 (a) Selective C–F functionalisation of unactivated trifluoromethyl arenes. Comparison between the performances of POx, PTH and fac-Ir(ppy)3. (b) Mechanism of the C–F functionalisation. | ||
This mechanism (Scheme 32b) is an example of dual photocatalysis where a photoredox cycle is merged with a second catalytic cycle with a different activation mode.72
The excited PC reduces 70 and, upon the cleavage of the C–F bond, radical 70.1 is formed. This radical undergoes intermolecular addiction with olefin 71. Finally, a HAT event between the alkyl radical 70.2 and thiophenol 51 provides product 72. Potassium formate is needed in stoichiometric amounts to close both the photoredox cycle and the HAT cycle restoring the PC and 51, liberating CO2 as a traceless side product.
5.3 Bimodal organic PCs
The light-driven acylation of methyl crotonate 74 by acetyl silane 73 was performed using the developed NTCs (Scheme 33). According to the reported reaction mechanism, the PC needs to exceed an 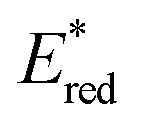 of 1.46 V to engage in reductive quenching with acetyl silane 73.73 Giese-type addition of 73.2 to methyl crotonate 74 yields the intermediate 74.1, which is reduced by PC˙−. In this step, the thermodynamic requirement is that the Eox < −0.6 V.74 NTC 25j (
of 1.46 V to engage in reductive quenching with acetyl silane 73.73 Giese-type addition of 73.2 to methyl crotonate 74 yields the intermediate 74.1, which is reduced by PC˙−. In this step, the thermodynamic requirement is that the Eox < −0.6 V.74 NTC 25j (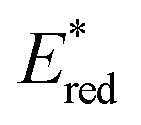 = 1.56 V and Eox = 1.45 V) delivered the product in 88% yield. The simpler NTC 25a also provided the product in synthetically useful yield. Importantly, under the same experimental conditions, product 75 was not obtained using Ru(bpy)32+ or fac-Ir(ppy)3.
= 1.56 V and Eox = 1.45 V) delivered the product in 88% yield. The simpler NTC 25a also provided the product in synthetically useful yield. Importantly, under the same experimental conditions, product 75 was not obtained using Ru(bpy)32+ or fac-Ir(ppy)3.
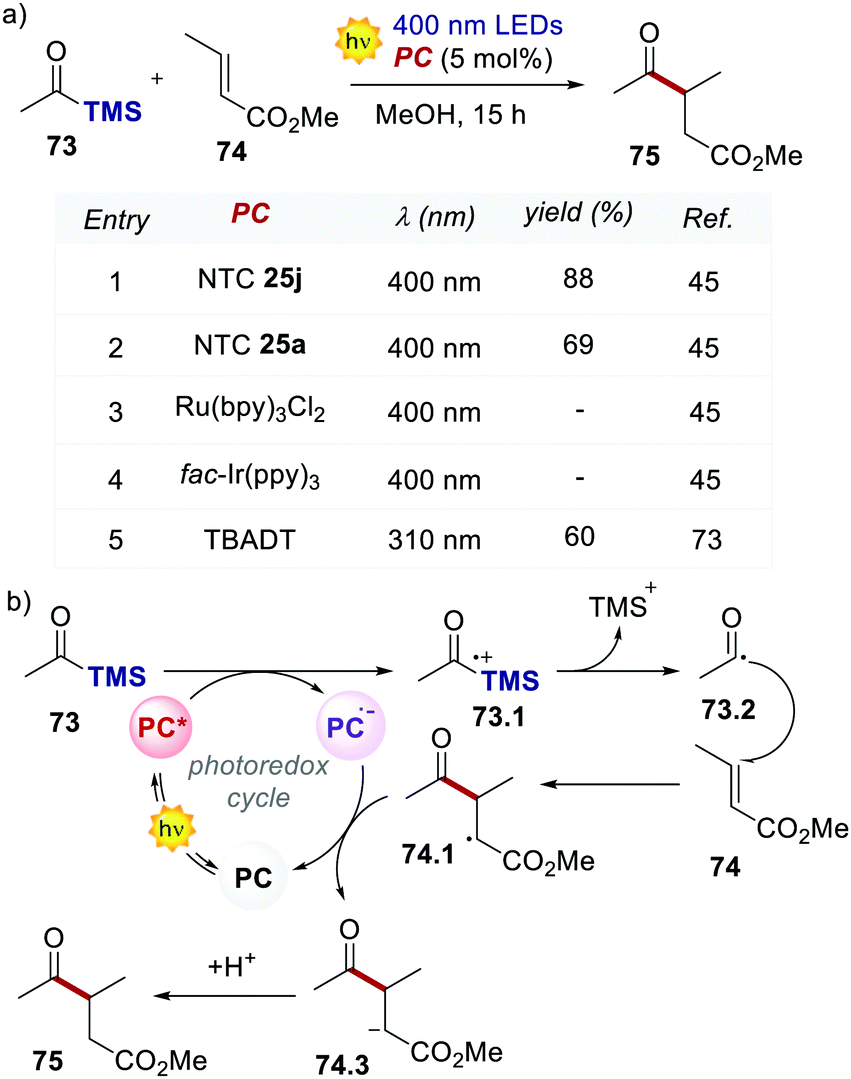 | ||
| Scheme 33 (a) Light-driven Giese-type acylation of methyl crotonate. Comparison between the performances of various PCs. (b) The proposed mechanism for the light-driven acylation of methyl crotonate. | ||
The reductive dehalogenation of 4-cyanobenzyl halides 76 was also investigated with NTCs to benchmark their performances in an oxidative quenching cycle (Scheme 34). When using NTC 25k, product 77 was obtained in 51% yield. The use of highly reducing fac-Ir(ppy)3 only led to traces of the desired product, while fac-Ir(dFppy)3 gave the product in only 19% yield. Ru(bpy)32+ was not a competent catalyst for the transformation, and no product was detected.
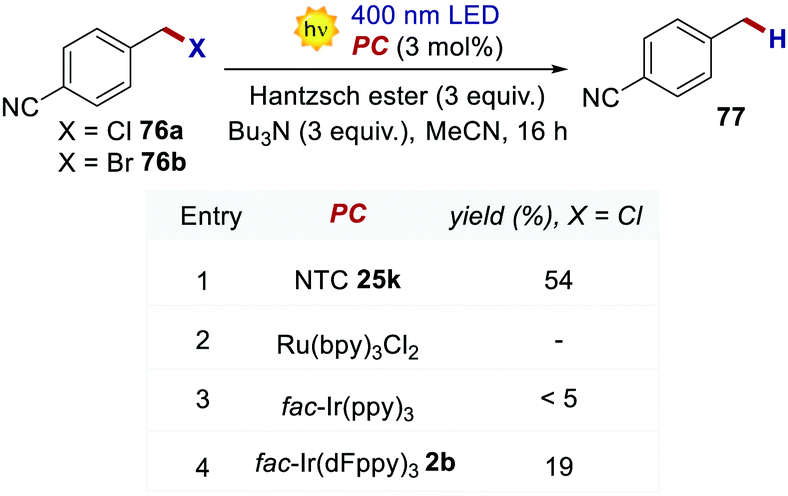 | ||
| Scheme 34 Light-driven reduction of 4-cyanobenzyl halides. Comparison between the performances of various PCs. | ||
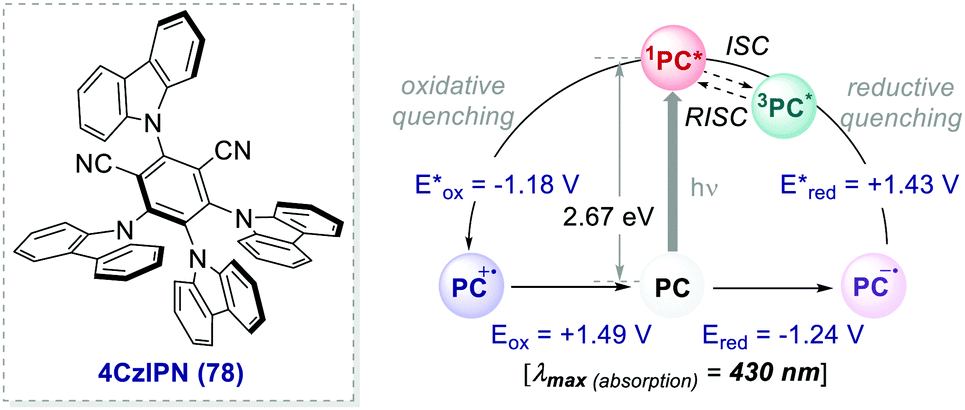 | ||
| Scheme 35 Photophysical properties and redox potentials (vs. SCE, in MeCN) of 4CzIPN (ref. 26c). | ||
In 2016, Zhang and Luo reported the first synthetic use of 4CzIPN in a dual photoredox/Ni cross-coupling as a cost-effective alternative to iridium complex 2d.76 In 2018, Zeitler's group expanded the class of cyanoarenes derived from isophthalonitrile (IPN) and benzonitrile (BN), showing how the rational design of photocatalytic scaffolds can deliver PCs able to engage in both oxidative and reductive quenching cycles.26c
The redox potentials of the prototypical 4CzIPN are close to those of iridium complex 2d, and 4CzIPN is often used as its metal-free alternative. It is however worth pointing out the main advantage of cyanoarenes. In 4CzIPN and its analogues, the HOMO and LUMO are spatially separated on the donor substituents and the acceptor core, respectively. Hence, it is possible to build a wide library of PCs with fine-tailored redox potentials in straightforward single-step syntheses (Fig. 6).26c,76 The evaluation of the thermodynamic requirements of each reaction guides the rational selection of the ad hoc derivative.
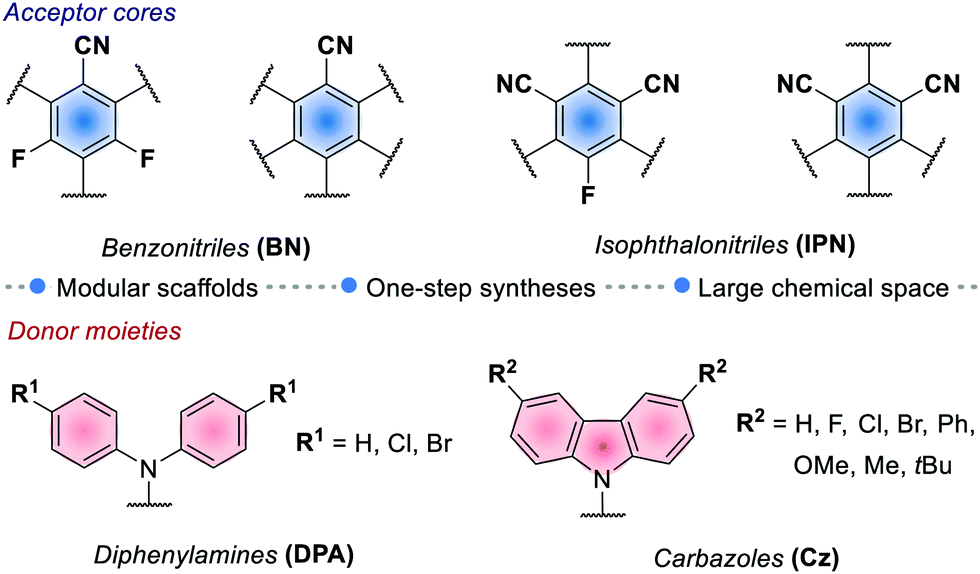 | ||
| Fig. 6 Common acceptor and donor moieties for cyanoarenes synthesis. | ||
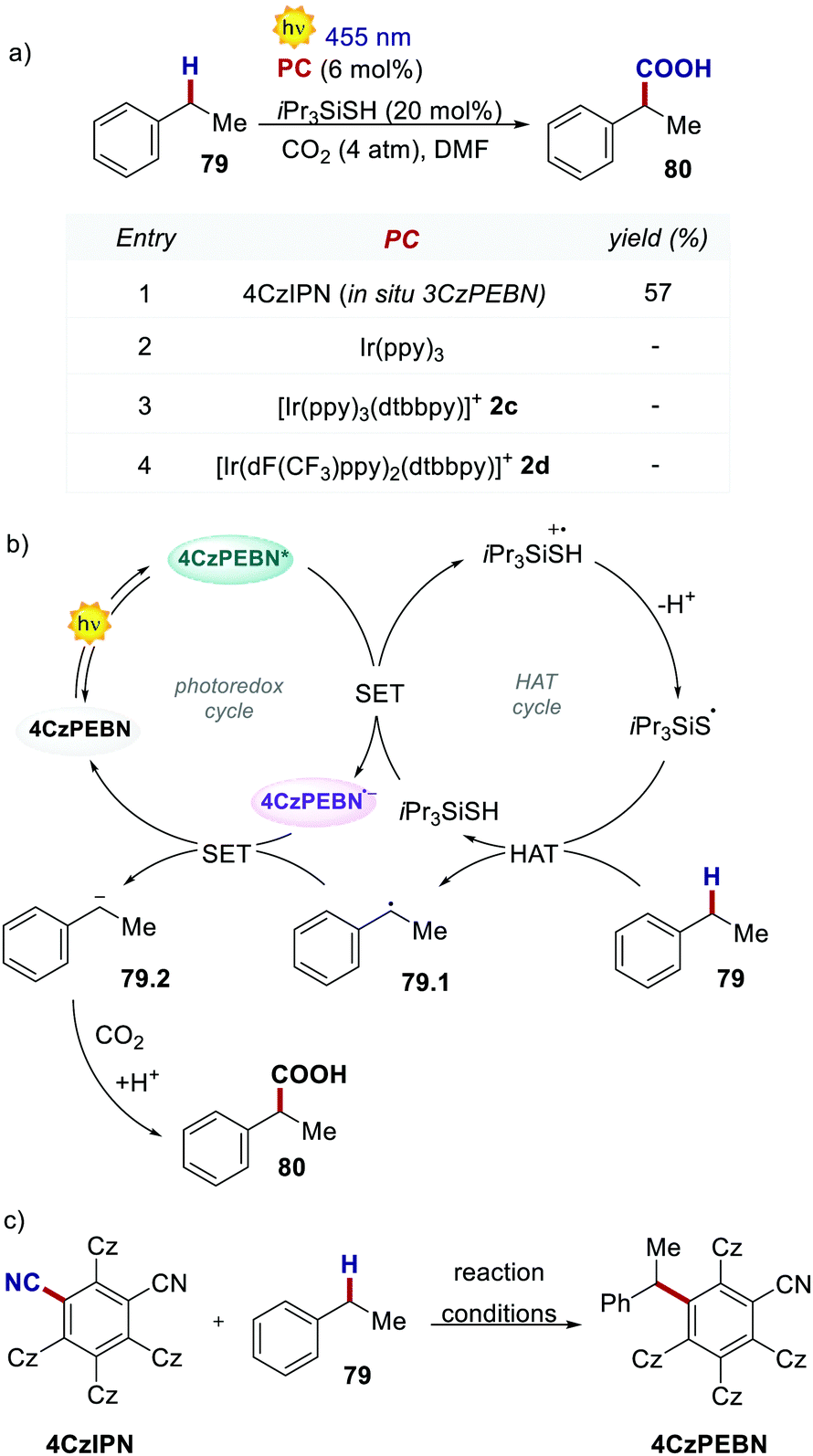
The molecular structure of 4CzIPN is essential to its success in a recent example of dual photoredox/HAT catalysis. In 2019, König's group reported the redox-neutral photocarboxylation of benzylic C–H bonds (Scheme 36).77 During the initial screening of the PCs, 4CzIPN provided product 79 in 14% yield, while fac-Ir(ppy)3 and iridium complexes 2c and 2d were completely ineffective. Further optimisation with 4CzIPN increased the yield to 57%. Under reaction conditions, 4CzIPN is converted into its benzylated derivative 3CzPEBN. Its radical anion 3CzPEBN˙− (Ered = −1.69 V for 3CzPEBN) is a much stronger reductant compared to 4CzIPN˙− (Ered = −1.24 V for 4CzIPN) and is thus able to reduce the benzylic radical 79.1 (Ered = −1.60 V vs. SCE) to the anion 79.2. 3CzPEBN is the active catalyst in the transformation, which also explains the lack of reactivity observed with the Ir-based PCs.
 | ||
| Scheme 36 (a) Dual photoredox/HAT catalysis for the carboxylation of C–H benzylic bonds. Comparison between the performances of various PCs. (b) Proposed reaction mechanism for the carboxylation of C–H benzylic bonds. (c) In situ formation of 3CzPEBN. | ||
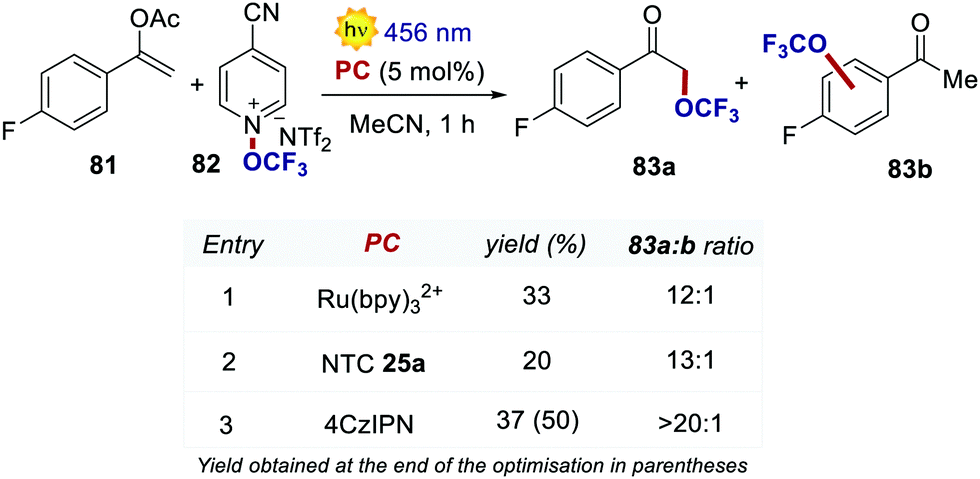
Very recently, Dell’Amico, Magnier and Togni's groups reported a protocol for the radical α-trifluoromethoxylation of ketones under photoredox conditions (Scheme 37).78 The selection of an organic PC was key for increasing the regioselectivity of the reaction. During the optimisation, the use of Ru(bpy)32+ afforded a regioselectivity of 12:1 between 83a and 83b, with significant trifluoromethoxylation on the aromatic ring.79 With NTC 25a, the ratio was improved to 13:1, albeit with a lower reactivity. When using 4CzIPN, the trifluoromethoxylation took place with full selectivity on the alpha position of the ketone (3:4 ratio >20:1) and with increased yield.
 | ||
| Scheme 37 Radical α-trifluoromethoxylation of ketones. | ||
6 Consecutive photoinduced electron transfer: overcoming the thermodynamic limits in photoredox catalysis
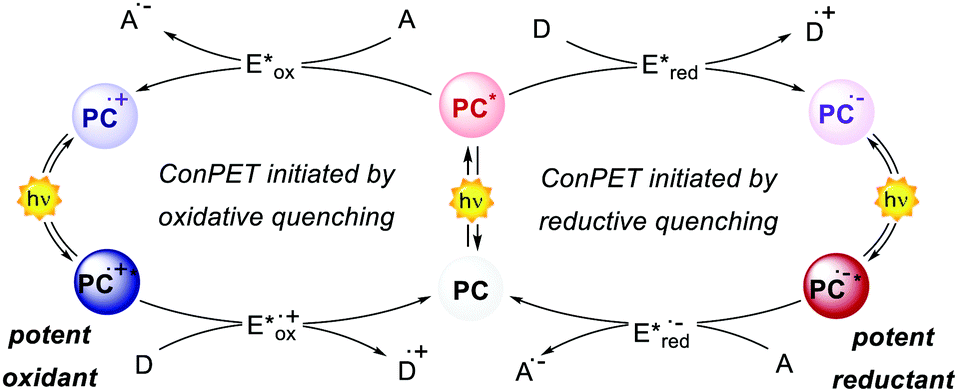
According to the Rehm–Weller theory, the excited state energy E0,0 of a PC sets the maximum redox potential window achievable under oxidative or reductive quenching.4 For a PC absorbing in the visible region (>400 nm), the maximum E0,0 approaches 3 eV. However, energy losses due to ISC or nonradiative processes further decrease this value. This issue poses a lower thermodynamic limit for PET and for several synthetically valuable transformations that can be carried out.Consecutive photoinduced electron transfer (ConPET) is a catalytic solution to this limitation, first reported by König's group in 2014 using perylene diimide (PDI) as an organic PC.80 Through a ConPET, it is possible to overcome the thermodynamic limitations of PCs by expanding their redox borders.
The general concept of ConPET involves (i) the PC excitation and (ii) the subsequent quenching by a suitable donor or acceptor, followed by (iii) the light-excitation of the resulting radical anion (PC˙−) or radical cation (PC˙+, Scheme 38).
 | ||
| Scheme 38 General catalytic manifolds of ConPET. | ||
The requirement is that PC˙− or PC˙+ to be further photoexcited must be sufficiently stable and long-lived, and the corresponding excited species are sufficiently long-lived (>1 ns) to allow the ET to happen.
Metal-based PCs have found a restricted use in ConPET cycles. Recent reports on the use of Ru and Ir complexes in two-photon photoredox cycles only involve the generation of highly reducing species in aqueous and/or micellar environments, with limited synthetic applicability.81 On the contrary, organic PCs have demonstrated a much broader versatility both for thermodynamically challenging reduction and oxidation.
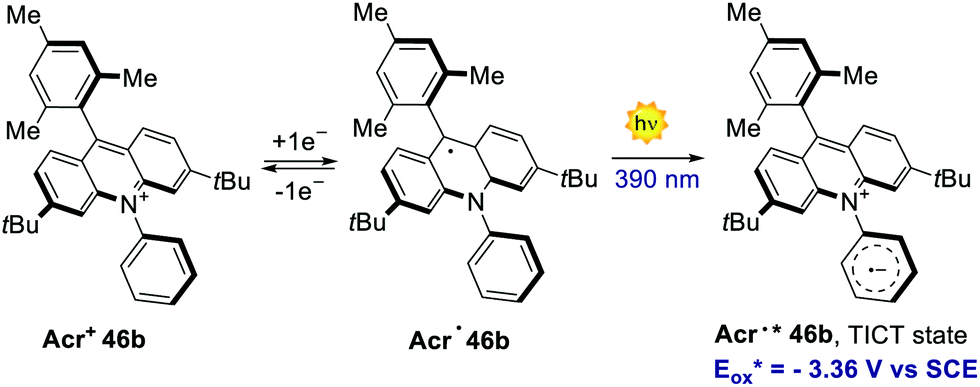
In 2020, Nicewicz's group documented the discovery, characterisation and reactivity of the excited acridine radical Acr˙* 46b (Scheme 39).82 Spectroscopic and computational investigations revealed the formation of two main excited states for Acr˙* 46b, assigned as a lower-energy doublet (D1) and a higher-energy twisted intramolecular charge-transfer state (TICT). The oxidation potentials of these excited states were estimated to be −2.91 V and −3.36 V vs. SCE, respectively, which are values comparable to those of elemental lithium (Eox (Li+/Li0) = −3.29 V). The singly occupied molecular orbital (SOMO) density of Acr˙* 46b was found to be localised on the acridine core, while the LUMO was placed on the N-phenyl ring. Further spectroscopical analysis with time-dependent density functional theory (TD-DFT) supported the formation of a charge-transfer state possessing both the aromatic radical anion and acridinium features, as it is expected for the proposed TICT state.
 | ||
| Scheme 39 The strongly reducing radical Acr˙* 46b, formed upon the excitation of the acridium radical Acr˙ 46b. | ||
The reactivity of the highly reducing acridinium radical Acr˙˙* 46b was then demonstrated by the dehalogenation of aryl halides 84, as well as by the desulfonylation of tosyl amines 85 (Scheme 40a). Both reduced products 86 and 87 were obtained in generally high yields (up to 98%) under optimised conditions. The scope of this process included substrates showing an Ered up to −2.9 V, which are values not accessible to commonly used PCs. The proposed mechanism for the dehalogenation process involves the reductive quenching of the excited acridinium Acr+* 46b by DIPEA 18, generating the acridinium radical Acr˙ 46b (Scheme 40b). This intermediate Acr˙ 46b is then excited with a 390 nm light, populating both the highly reducing Dn and TICT excited states (Acr˙* 46b). A single electron transfer subsequently occurs from Acr˙* 46b to the aryl halide 84, forming the arene radical anion 84.1, while regenerating Acr+46b. The intermediate 84.1 undergoes fragmentation, yielding the aryl radical 84.2. Finally, a HAT event occurs for 84.2 from the amine radical cation 18˙+, which delivers the reduced product 86.
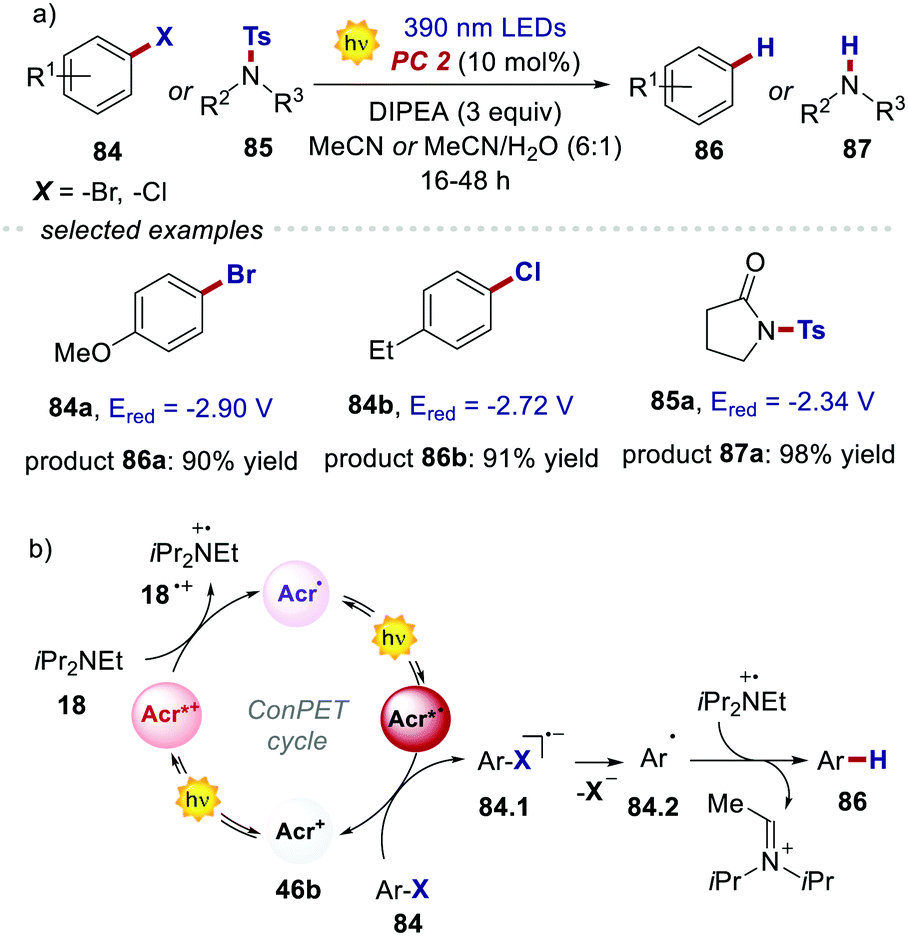 | ||
| Scheme 40 (a) Reductive dehalogenation of aryl halides (81) and reductive detosylation of amines (82) triggered by the excited radical acridinium Acr˙* 41b. (b) The proposed mechanism for the dehalogenation of arenes. | ||
On the other hand, two independent reports have exploited a ConPET manifold with PTH. In 2018, Wagenknecht's group realised the pentafluorosulfanylation/nucleophilic trapping of styrene derivatives through the use of UV and green light (Scheme 41).83
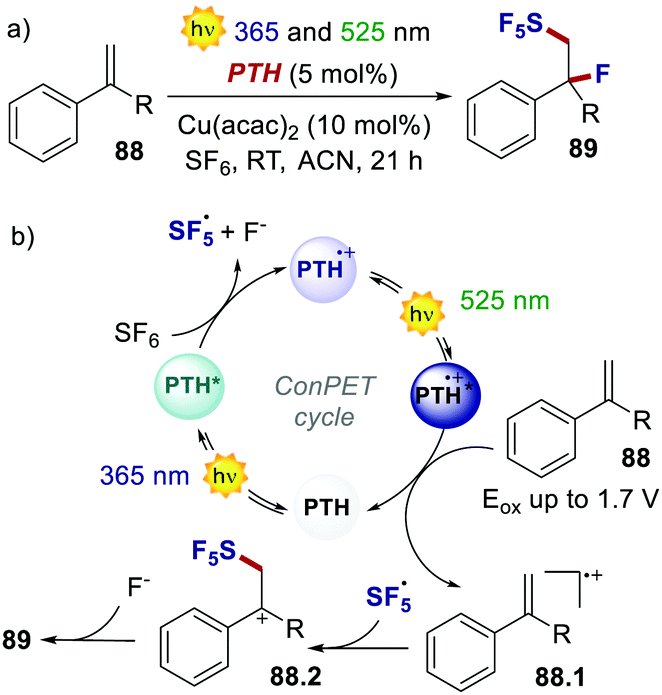 | ||
| Scheme 41 (a) ConPET-enabled pentafluorosulfanylation/nucleophilic trapping of styrene derivatives. (b) The proposed mechanism. | ||
The highly reducing PTH* engages in oxidative quenching with SF6, delivering SF5˙ and fluoride. The excitation of the poorly oxidising PTH˙+ (Eox = 0.68 V) with green light transforms it into a powerful oxidant, which oxidises the thermodynamically demanding styrene derivative 88. Coupling between SF5˙ and 88.1 affords the cationic intermediate 88.2, which is then trapped by the nucleophilic fluoride ion. As a result, PTH acts in the same photoredox cycle as a strong reductant for the reduction and fragmentation of SF6 and as a strong oxidant for the oxidation of the styrene.
In 2021, Wickens's group realised the Nicewicz-type oxidative coupling between arenes 90 and N-heterocyclic nucleophile 91 through a similar ConPET manifold with PTH as the PC (Scheme 42).84 In this case, oxygen serves as the electron acceptor to generate PTH˙+ which, upon photoexcitation, can oxidise arenes with extreme oxidation potentials (Eox = 2.5 V for benzene).
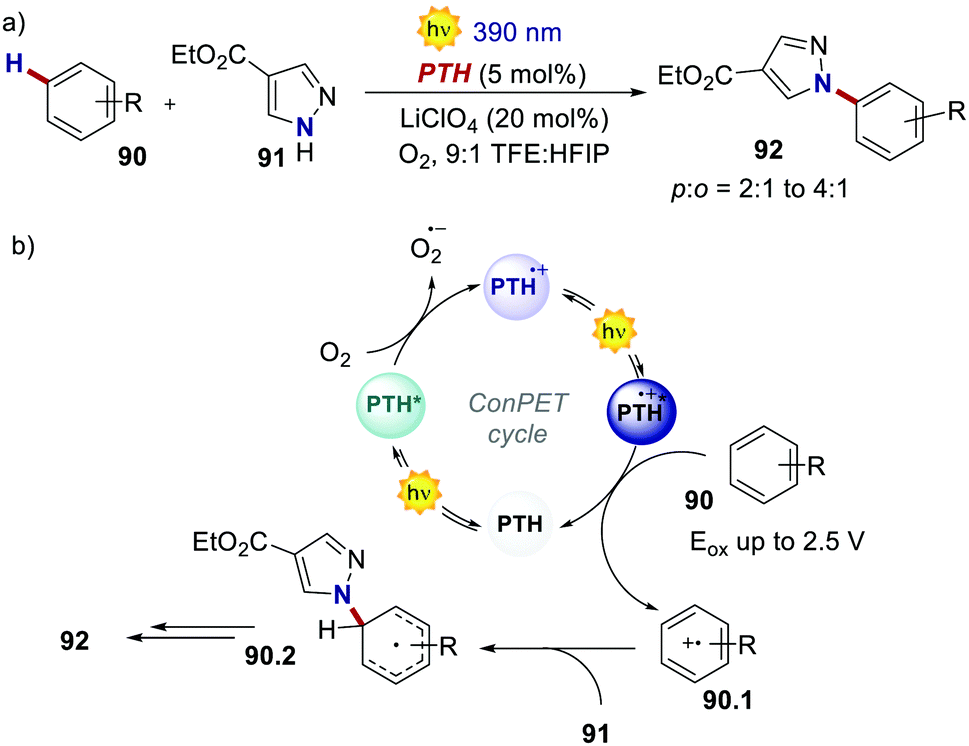 | ||
| Scheme 42 (a) ConPET-enabled oxidative arene/N-heterocyclic nucleophile coupling. (b) The proposed mechanism. | ||
Very recently, also 4CzIPN and its derivatives were used in ConPET cycles. Wickens's group realised the reductive defunctionalisation of electron-rich aryl chlorides 93 with 4DPAIPN and sodium formate as the electron donor.85 Through the ConPET mechanism, the authors were able to reduce extremely challenging aryl chlorides 93 with reduction potentials as low as −3.4 V (Scheme 43).
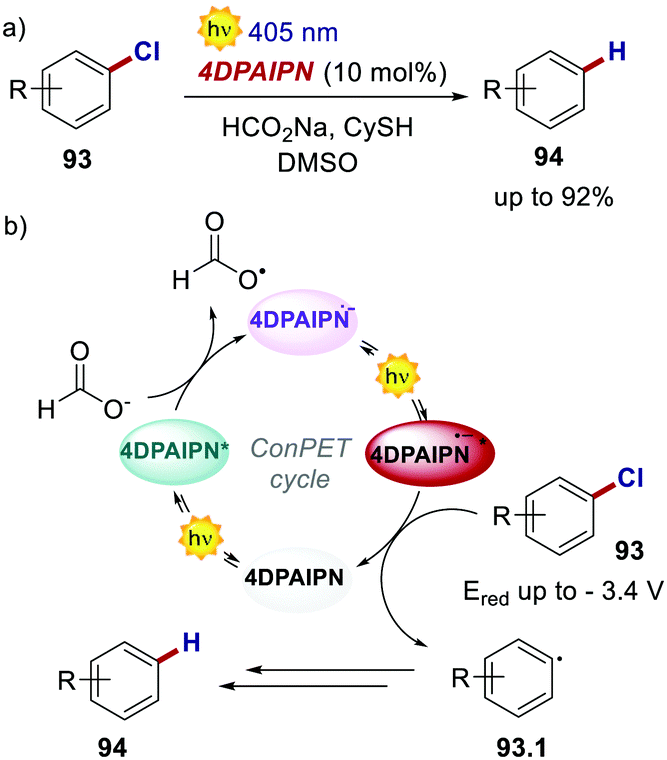 | ||
| Scheme 43 (a) Reductive defunctionalisation of electron-rich aryl chlorides. (b) Proposed reaction mechanism. | ||
Such a redox potential is beyond the thermodynamic limit accessible by a classic photoredox cycle under visible-light irradiation. The authors also exploited the developed catalytic manifold for the phosphonylation, hydroarylation and borylation of aryl chlorides. However, complete suppression of the net reduction product 94 could not be achieved in such cases.
Similarly, Zhou and Wu's groups obtained the reductive borylation and phosphorylation of extremely challenging aryl chlorides through an analogous ConPET mechanism.86 Key for the optimisation of the system was the use of 3CzEPAIPN, a modified derivative of 4CzIPN, as the PC (Scheme 44). Higher yields are attributed to the higher photostability of 3CzEPAIPN and the longer lifetime of its radical anion.
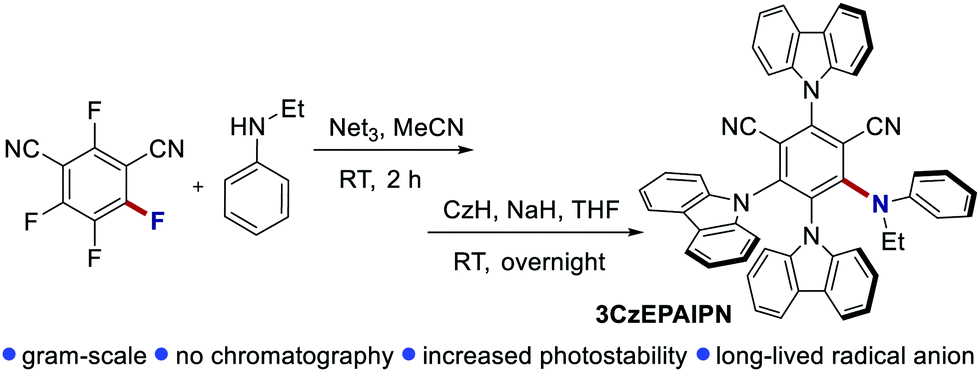 | ||
| Scheme 44 Synthesis and structure of 3CzEPAIPN. | ||
A catalytic platform for the activation of similarly demanding aryl chlorides was developed by Lambert and Lin's groups.87 In this case, the radical anion of 9,10-dicyanoanthracene 3a was produced by electrochemical reduction and was subsequently excited by blue light irradiation to yield a potent reductant (E*˙−red = −3.2 V vs. SCE).
These examples highlight the versatility of organic PCs in non-conventional multi-photon photoredox cycles. Through this strategy, it is possible to perform both thermodynamically challenging oxidation and reduction using the same organic PC.
7 Conclusions and future perspectives
Over the last decade, the field of organic photoredox catalysis has proven to have great potential to provide new synthetic methods under mild reaction conditions. Importantly, organic PCs are generally cheaper, less toxic and easier to modulate than transition metal PCs, thus representing a convenient and more sustainable alternative. In this feature article, we have outlined the evolution of the field of photoredox catalysis from the use of classical Ir- and Ru-complexes towards purely organic PCs. Remarkably, organic PCs can be efficiently used to replace their metal counterparts, while promoting previously inaccessible transformations. Their easy tunability also offers the possibility of building a large variety of structural and electronic variants and adopting a more rational PC selection. Recently, bimodal PCs, such as cyanoarenes (IPN scaffold), have become more and more popular thanks to their CT excited state character that mimics the MLCT of metal complexes, inferring long-lived excited states and a balanced distribution of the redox potentials (Fig. 7). However, the chemical stability of these PCs still represents an issue in the presence of highly nucleophilic radicals. König and co-workers demonstrated that 4CzIPN 78 can be in situ alkylated when benzylic radicals are generated under reaction conditions (Scheme 36).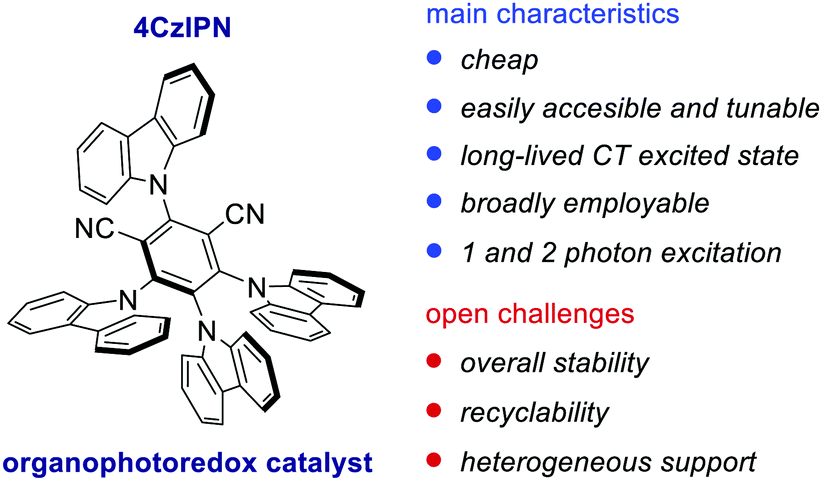 | ||
| Fig. 7 Main advantages and open challenges associated with the use of IPN PCs. | ||
Our group has recently contributed to the field of organophotoredox catalysis by identifying naphtochromenone (NTC) PCs as a new class of bimodal organic PCs, with the ability to participate in both thermodynamically demanding oxidative and reductive photochemical processes. Interestingly, we have evaluated their stability and we were able to recover and reuse the NTC PC up to 5 times, maintaining virtually identical catalytic performances.45 The recyclability of the PC is one important aspect often overlooked in academia, but may become essential towards industrial implementations. An additional open challenge regarding the possibility of supporting organic PCs is generating easily recoverable heterogeneous systems. This represents a complex issue, since solid supports can profoundly alter the ground and excited state properties of PCs, while introducing scattering problems.88
However, more sustainable reusable PCs,89 having tuneable photochemical and redox properties, are needed to respond to the increasing demands of novel reactivity and bioactive molecules. To this end, novel structural variants must be evaluated, possibly unlocking new mechanistic paradigms. In particular, several heterocyclic scaffolds, which have already found applications as TADF emitters in OLEDs, have demonstrated interesting photochemical and redox properties.90
It is worth noting that while metal-based PCs have found a restricted use in only reductive ConPET cycles, organic PCs have instead exhibited a much broader versatility in both reductive and oxidative ConPET processes with PTH 31, cyanoarenes and Acr-Mes+46b playing a central role.82–86 This indicates that purely organic molecules will likely govern the evolution of the field. However, an unmet goal in ConPET processes is the use of the same PC class under both reductive and oxidative quenching. The possibility of using the same PC or PC class under both 1- and 2-photon thermodynamically opposite quenching mechanisms will represent a decisive advancement in terms of generality and versatility. In our group, we are currently evaluating this possibility with the use of a new generation of structurally robust NTC and IPN PCs.
Major discoveries are expected to come in the near future, given the new and broad opportunities that this class of relatively simple purely organic molecules can offer.
Author contributions
T. B., S. C. and G. S. wrote the manuscript and drew the schemes and figures upon coordination by T. B. L. D. selected the topic and conceptualised the overall idea, directed the project, and corrected the text and the figures.Conflicts of interest
There are no conflicts to declare.Acknowledgements
University of Padova P-DiSC#11BIRD2020-UNIPD (L. D.) and CariParo Foundation, Synergy – Progetti di Eccellenza 2018 (L. D.) are acknowledged for financial support.Notes and references
- S. E. Braslavsky, Pure Appl. Chem., 2007, 79, 293–465 CAS.
- H. D. Roth, Angew. Chem., Int. Ed. Engl., 1989, 28, 1193–1207 CrossRef.
- G. Ciamician, Science, 1912, 36, 385–394 CrossRef CAS PubMed.
- (a) D. Rehm and A. Weller, Isr. J. Chem., 1970, 8, 259–271 CrossRef CAS; (b) L. Buzzetti, G. E. M. Crisenza and P. Melchiorre, Angew. Chem., Int. Ed., 2019, 58, 3730–3747 CrossRef CAS PubMed.
- For early examples on synthetic photoredox catalysis, see: (a) D. M. Hedstrand, W. H. Kruizinga and R. M. Kellogg, Tetrahedron Lett., 1978, 19, 1255–1258 CrossRef; (b) H. Cano-Yelo and A. Deronzier, Tetrahedron Lett., 1984, 25, 5517–5520 CrossRef CAS; (c) H. Cano-Yelo and A. Deronzier, J. Chem. Soc., Perkin Trans. 2, 1984, 1093–1098 RSC; (d) H. Cano-Yelo and A. Deronzier, J. Photochem., 1987, 37, 315–321 CrossRef CAS; (e) K. Okada, K. Okamoto and M. Oda, J. Am. Chem. Soc., 1988, 110, 8736–8738 CrossRef CAS; (f) K. Okada, K. Okamoto, N. Morita, K. Okubo and M. Oda, J. Am. Chem. Soc., 1991, 113, 9401–9402 CrossRef CAS; (g) K. Okada, K. Okubo, N. Morita and M. Oda, Tetrahedron Lett., 1992, 33, 7377–7380 CrossRef CAS; (h) K. Okada, K. Okubo, N. Morita and M. Oda, Chem. Lett., 1993, 2021–2024 CrossRef CAS.
- M. A. Ischay, M. E. Anzovino, J. Du and T. P. Yoon, J. Am. Chem. Soc., 2008, 130, 12886–12887 CrossRef CAS PubMed.
- D. A. Nicewicz and D. W. C. MacMillan, Science, 2008, 322, 77–80 CrossRef CAS PubMed.
- J. M. R. Narayanam, J. W. Tucker and C. R. J. Stephenson, J. Am. Chem. Soc., 2009, 131, 8756–8757 CrossRef CAS PubMed.
- The results were obtained from a search on Elsevier Scopus using the keywords “photoredox catalyst” to obtain the total number of articles on the topic and “organic photoredox catalyst” for the number of articles on organophotoredox catalysis. The raw data were then refined to exclude reports that are not dealing with the use of photoredox catalysis for synthetic purposes. Despite this, it is possible that spurious results are still included, but they are expected to account for a non-significant fraction of the total.
- (a) M. Graetzel, Acc. Chem. Res., 1981, 14, 376–384 CrossRef CAS; (b) T. J. Meyer, Acc. Chem. Res., 1989, 22, 163–170 CrossRef CAS.
- H. Takeda and O. Ishitani, Coord. Chem. Rev., 2010, 254, 346–354 CrossRef CAS.
- C. R. Bock, J. A. Connor, A. R. Gutierrez, T. J. Meyer, D. G. Whitten, B. P. Sullivan and J. K. Nagle, J. Am. Chem. Soc., 1979, 101, 4815–4824 CrossRef CAS.
- K. A. King, P. J. Spellane and R. J. Watts, J. Am. Chem. Soc., 1985, 107, 1431–1432 CrossRef CAS.
- Selected photophysical studies: (a) W. J. Finkenzeller and H. Yersin, Chem. Phys. Lett., 2003, 377, 299–305 CrossRef CAS; (b) T. Hofbeck and H. Yersin, Inorg. Chem., 2010, 49, 9290–9299 CrossRef CAS PubMed For a comprehensive review, see: ; (c) H. Yersin, A. F. Rausch, R. Czerwieniec, T. Hofbeck and T. Fischer, Coord. Chem. Rev., 2011, 255, 2622–2652 CrossRef CAS.
- For comprehensive reviews on the photophysical properties of iridium and ruthenium complexes, see: (a) L. Flamigni, A. Barbieri, C. Sabatini, B. Ventura and F. Barigelletti, in Photochemistry and Photophysics of Coordination Compounds II, ed. V. Balzani and S. Campagna, Springer, Berlin Heidelberg, 2007, ch. 4, vol. 281, pp. 143–203 For ruthenium, see Search PubMed; (b) S. Campagna, F. Puntoriero, F. Nastasi, G. Bergamini and V. Balzani, in Photochemistry and Photophysics of Coordination Compounds II, ed. V. Balzani and S. Campagna, Springer, Berlin Heidelberg, 2007, ch. 4, vol. 280, pp. 117–214 For reviews on the use of photoactive metal complexes in organic synthesis, see Search PubMed; (c) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed; (d) K. Teegardin, J. I. Day, J. Chan and J. Weaver, Org. Process Res. Dev., 2016, 20, 1156–1163 CrossRef CAS PubMed; (e) J.-H. Shon and T. S. Teets, Comments Inorg. Chem., 2020, 40, 53–85 CrossRef CAS; (f) J. M. R. Narayanam and C. R. J. Stephenson, Chem. Soc. Rev., 2011, 40, 102–113 RSC; (g) M. Reckenthäler and A. G. Griesbeck, Adv. Synth. Catal., 2013, 355, 2727–2744 CrossRef; (h) K. L. Skubi, T. R. Blum and T. P. Yoon, Chem. Rev., 2016, 116, 10035–10074 CrossRef CAS PubMed; (i) J. Twilton, C. Le, P. Zhang, M. H. Shaw, R. W. Evans and D. W. C. MacMillan, Nat. Chem. Rev., 2017, 1, 0052 CrossRef CAS; (j) F. Glaser and O. S. Wenger, Coord. Chem. Rev., 2020, 405, 213129 CrossRef CAS.
- For selected examples on tailored Ir complexes able to operate under reductive quenching, see: (a) D. A. Nagib, M. E. Scott and D. W. C. MacMillan, J. Am. Chem. Soc., 2009, 131, 10875–10877 CrossRef CAS PubMed; (b) A. G. Condie, J. C. González-Gómez and C. R. J. Stephenson, J. Am. Chem. Soc., 2010, 132, 1464–1465 CrossRef CAS PubMed; (c) J. W. Tucker, J. D. Nguyen, J. M. R. Narayanam, S. W. Krabbe and C. R. J. Stephenson, Chem. Commun., 2010, 46, 4985–4987 RSC; (d) E. B. Corcoran, M. T. Pirnot, S. Lin, S. D. Dreher, D. A. DiRocco, I. W. Davies, S. L. Buchwald and D. W. C. MacMillan, Science, 2016, 353, 279–283 CrossRef CAS PubMed.
- For the synthesis of tailored Ir complexes, see: M. S. Lowry, J. I. Goldsmith, J. D. Slinker, R. Rohl, R. A. Pascal, G. G. Malliaras and S. Bernhard, Chem. Mater., 2005, 17, 5712–5719 CrossRef CAS.
- (a) D. P. Rillema, G. Allen, T. J. Meyer and D. Conrad, Inorg. Chem., 1983, 22, 1617–1622 CrossRef CAS; (b) M. Haga, E. S. Dodsworth, G. Eryavec, P. Seymour and A. B. P. Lever, Inorg. Chem., 1985, 24, 1901–1906 CrossRef CAS.
- E. D. Nacsa and D. W. C. MacMillan, J. Am. Chem. Soc., 2018, 140, 3322–3330 CrossRef CAS PubMed.
- K. Dedeian, P. I. Djurovich, F. O. Garces, G. Carlson and R. J. Watts, Inorg. Chem., 1991, 30, 1685–1687 CrossRef CAS.
- W. M. Haynes, CRC Handbook of Chemistry and Physics, CRC Press, Boca Raton, 2014 Search PubMed.
- Nat. Catal., 2019, 2, 735 Search PubMed.
- P. Chábera, K. S. Kjaer, O. Prakash, A. Honarfar, Y. Liu, L. A. Fredin, T. C. B. Harlang, S. Lidin, J. Uhlig, V. Sundström, R. Lomoth, P. Persson and K. Wärnmark, J. Phys. Chem. Lett., 2018, 9, 459–463 CrossRef PubMed.
- (a) B. M. Hockin, C. Li, N. Robertson and E. Zysman-Colman, Catal. Sci. Technol., 2019, 9, 889–915 RSC; (b) S. Paria and O. Reiser, in Visible Light Photocatalysis in Organic Chemistry, ed. C. Stephenson, T. Yoon and D. W. C. MacMillan, Wiley-VCH, Weinheim, 2018, ch. 7, pp. 233–251 Search PubMed; (c) G. Noirbent and F. Dumur, Catalysts, 2020, 10, 953 CrossRef CAS.
- For selected reviews, see: (a) S. Fukuzumi and K. Ohkubo, Org. Biomol. Chem., 2014, 12, 6059–6071 RSC; (b) D. P. Hari and B. König, Chem. Commun., 2014, 50, 6688–6699 RSC; (c) N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS PubMed; (d) Y. Du, R. M. Pearson, C.-H. Lim, S. M. Sartor, M. D. Ryan, H. Yang, N. H. Damrauer and G. M. Miyake, Chem. – Eur. J., 2017, 23, 10962–10968 CrossRef CAS PubMed; (e) T.-Y. Shang, L.-H. Lu, Z. Cao, Y. Liu, W.-M. He and B. Yu, Chem. Commun., 2019, 55, 5408–5419 RSC; (f) S. G. E. Amos, M. Garreau, L. Buzzetti and J. Waser, Beilstein J. Org. Chem., 2020, 16, 1163–1187 CrossRef CAS PubMed; (g) P. P. Singh and V. Srivastava, Org. Biomol. Chem., 2021, 19, 313–321 RSC; (h) Y. Lee and M. S. Kwon, Eur. J. Org. Chem., 2020, 6028–6043 CrossRef CAS; (i) M. V. Bobo, J. J. Kuchta and A. K. Vannucci, Org. Biomol. Chem., 2021, 19, 4816–4834 RSC; (j) M. A. Bryden and E. Zysman-Colman, Chem. Soc. Rev., 2021, 50, 7587–7680 RSC; (k) A. Tlili and S. Lakhdar, Angew. Chem., Int. Ed., 2021, 60, 19526–19549 CrossRef CAS PubMed; (l) Y. Wu, D. Kim and T. S. Teets, Synlett, 2021 DOI:10.1055/a-1390-9065.
- (a) A. Joshi-Pangu, F. Lévesque, H. G. Roth, S. F. Oliver, L.-C. Campeau, D. Nicewicz and D. A. DiRocco, J. Org. Chem., 2016, 81, 7244–7249 CrossRef CAS PubMed; (b) B. G. McCarthy, R. M. Pearson, C.-H. Lim, S. M. Sartor, N. H. Damrauer and G. M. Miyake, J. Am. Chem. Soc., 2018, 140, 5088–5101 CrossRef CAS PubMed; (c) E. Speckmeier, T. G. Fischer and K. Zeitler, J. Am. Chem. Soc., 2018, 140, 15353–15365 CrossRef CAS PubMed; (d) A. Vega-Peñaloza, J. Mateos, X. Companyó, M. Escudero-Casao and L. Dell’Amico, Angew. Chem., Int. Ed., 2021, 60, 1082–1097 CrossRef PubMed.
- F. Strieth-Kalthoff, M. J. James, M. Teders, L. Pitzer and F. Glorius, Chem. Soc. Rev., 2018, 47, 7190–7202 RSC.
- At the same time, however, the stabilisation of the CT state in polar solvents produces a redshift in the emission (Stokes shift), lowering the energy of the excited state and reducing the redox potential window.
- M. V. Bobo, A. M. Arcidiacono, P. J. Ayare, J. C. Reed, M. R. Helton, T. Ngo, K. Hanson and A. K. Vannucci, ChemPhotoChem, 2021, 5, 51–57 CrossRef CAS.
- K. Zeitler, in Visible Light Photocatalysis in Organic Chemistry, ed. C. Stephenson, T. Yoon and D.W.C. MacMillan, Wiley-VCH, Weinheim, 2018, ch. 6, pp. 159–232 Search PubMed.
- (a) A. Penzkofer, A. Beidoun and M. Daiber, J. Lumin., 1992, 51, 297–314 CrossRef CAS; (b) A. Penzkofer and A. Beidoun, Chem. Phys., 1993, 177, 203–216 CrossRef CAS; (c) A. Penzkofer, A. Beidoun and S. Speiser, Chem. Phys., 1993, 170, 139–148 CrossRef CAS.
- M. E. Selsted and H. W. Becker, Anal. Biochem., 1986, 155, 270–274 CrossRef CAS PubMed.
- (a) A. Chisvert and A. Salvador, in Analysis of Cosmetic Products, ed. A. Salvador and A. Chisvert, Elsevier, Amsterdam, 2nd edn, 2018, ch. 5, pp. 85–106 Search PubMed; (b) R. W. Sabnis, Handbook of Biological Dyes and Stains, John Wiley & Sons, Inc., Hoboken, 2010 CrossRef.
- R. W. Sabnis, Handbook of Acid-Base Indicators, CRC Press, Boca Raton, 2007 Search PubMed.
- M. Neumann, S. Füldner, B. König and K. Zeitler, Angew. Chem., Int. Ed., 2011, 50, 951–954 CrossRef CAS PubMed.
- M. A. Cismesia and T. P. Yoon, Chem. Sci., 2015, 6, 5426–5434 RSC.
- J. Du, L. R. Espelt, I. A. Guzei and T. P. Yoon, Chem. Sci., 2011, 2, 2115–2119 RSC.
- M. Neumann and K. Zeitler, Chem. – Eur. J., 2013, 19, 6950–6955 CrossRef CAS PubMed.
- S. P. Pitre, C. D. McTiernan, H. Ismaili and J. C. Scaiano, J. Am. Chem. Soc., 2013, 135, 13286–13289 CrossRef CAS PubMed.
- (a) T. Ohno and N. N. Lichtin, J. Am. Chem. Soc., 1980, 102, 4636–4643 CrossRef CAS; (b) R. H. Kayser and R. H. Young, Photochem. Photobiol., 1976, 24, 395–401 CrossRef CAS; (c) H.-J. Timpe and S. Neuenfeld, J. Chem. Soc., Faraday Trans., 1992, 88, 2329–2336 RSC; (d) T. Shen, Z.-G. Zhao, Q. Yu and H.-J. Xu, J. Photochem. Photobiol., A, 1989, 47, 203–212 CrossRef CAS.
- (a) F. Wilkinson, W. P. Helman and A. B. Ross, J. Phys. Chem. Ref. Data, 1995, 24, 663–677 CrossRef CAS; (b) C. Tanielian and C. Wolff, J. Phys. Chem., 1995, 99, 9831–9837 CrossRef CAS.
- Y. Q. Zou, J. R. Chen, X. P. Liu, L. Q. Lu, R. L. Davis, K. A. Jørgensen and W. J. Xiao, Angew. Chem., Int. Ed., 2012, 51, 784–788 CrossRef CAS PubMed.
- (a) L. Mei, J. M. Veleta and T. L. Gianetti, J. Am. Chem. Soc., 2020, 142, 12056–12061 CrossRef CAS PubMed For the synthesis of the precursors of PC 19, see: ; (b) B. Laursen and F. Krebs, Angew. Chem., Int. Ed., 2000, 39, 3432–3434 CrossRef CAS; (c) J. C. Martin and Russell G. Smith, J. Am. Chem. Soc., 1964, 86, 2252–2256 CrossRef CAS.
- D. Kalyani, K. B. McMurtrey, S. R. Neufeldt and M. S. Sanford, J. Am. Chem. Soc., 2011, 133, 18566–18569 CrossRef CAS PubMed.
- J. Mateos, F. Rigodanza, A. Vega-Peñaloza, A. Sartorel, M. Natali, T. Bortolato, G. Pelosi, X. Companyó, M. Bonchio and L. Dell’Amico, Angew. Chem., Int. Ed., 2020, 59, 1302–1312 CrossRef CAS PubMed.
- J. Mateos, N. Meneghini, M. Bonchio, N. Marino, T. Carofiglio, X. Companyó and L. Dell’Amico, Beilstein J. Org. Chem., 2018, 14, 2418–2424 CrossRef CAS PubMed.
- (a) X. Ju, D. Li, W. Li, W. Yu and F. Bian, Adv. Synth. Catal., 2012, 354, 3561–3567 CrossRef CAS; (b) Z. Liang, S. Xu, W. Tian and R. Zhang, Beilstein J. Org. Chem., 2015, 11, 425–430 CrossRef CAS PubMed.
- K. Matyjaszewski, Macromolecules, 2012, 45, 4015–4039 CrossRef CAS.
- V. K. Singh, C. Yu, S. Badgujar, Y. Kim, Y. Kwon, D. Kim, J. Lee, T. Akhter, G. Thangavel, L. S. Park, J. Lee, P. C. Nandajan, R. Wannemacher, B. Milián-Medina, L. Lüer, K. S. Kim, J. Gierschner and M. S. Kwon, Nat. Catal., 2018, 1, 794–804 CrossRef CAS.
- G. Zhang, Y. Song, K. H. Ahn, T. Park and W. Choi, Macromolecules, 2011, 44, 7594–7599 CrossRef CAS.
- B. P. Fors and C. J. Hawker, Angew. Chem., Int. Ed., 2012, 51, 8850–8853 CrossRef CAS PubMed.
- (a) N. J. Treat, H. Sprafke, J. W. Kramer, P. G. Clark, B. E. Barton, J. Read de Alaniz, B. P. Fors and C. J. Hawker, J. Am. Chem. Soc., 2014, 136, 16096–16101 CrossRef CAS PubMed; (b) G. M. Miyake and J. C. Theriot, Macromolecules, 2014, 47, 8255–8261 CrossRef CAS.
- E. H. Discekici, N. J. Treat, S. O. Poelma, K. M. Mattson, Z. M. Hudson, Y. Luo, C. J. Hawker and J. R. de Alaniz, Chem. Commun., 2015, 51, 11705–11708 RSC.
- J. D. Nguyen, E. M. D’Amato, J. M. R. Narayanam and C. R. J. Stephenson, Nat. Chem., 2012, 4, 854–859 CrossRef CAS PubMed.
- (a) C. D. McTiernan, S. P. Pitre and J. C. Scaiano, ACS Catal., 2014, 4, 4034–4039 CrossRef CAS; (b) E. Arceo, E. Montroni and P. Melchiorre, Angew. Chem., Int. Ed., 2014, 53, 12064–12068 CrossRef CAS PubMed.
- C. B. Tripathi, T. Ohtani, M. T. Corbett and T. Ooi, Chem. Sci., 2017, 8, 5622–5627 RSC.
- L. J. Allen, P. J. Cabrera, M. Lee and M. S. Sanford, J. Am. Chem. Soc., 2014, 136, 5607–5610 CrossRef CAS PubMed.
- L. Chu, C. Ohta, Z. Zuo and D. W. C. MacMillan, J. Am. Chem. Soc., 2014, 136, 10886–10889 CrossRef CAS PubMed.
- S. Fukuzumi, H. Kotani, K. Ohkubo, S. Ogo, N. V. Tkachenko and H. Lemmetyinen, J. Am. Chem. Soc., 2004, 126, 1600–1601 CrossRef CAS PubMed.
- (a) D. S. Hamilton and D. A. Nicewicz, J. Am. Chem. Soc., 2012, 134, 18577–18580 CrossRef CAS PubMed; (b) N. A. Romero, K. A. Margrey, N. E. Tay and D. A. Nicewicz, Science, 2015, 349, 1326–1330 CrossRef CAS PubMed; (c) W. P. Hess and F. P. Tully, J. Phys. Chem., 1989, 93, 1944–1947 CrossRef CAS.
- For selected examples on the reducing ability of acridinium ions, see: (a) M. Xiang, Z.-K. Xin, B. Chen, C.-H. Tung and L.-Z. Wu, Org. Lett., 2017, 19, 3009–3012 CrossRef CAS PubMed; (b) M. Xiang, C. Zhou, X.-L. Yang, B. Chen, C.-H. Tung and L.-Z. Wu, J. Org. Chem., 2020, 85, 9080–9087 CrossRef CAS PubMed.
- H. G. Roth, N. A. Romero and D. A. Nicewicz, Synlett, 2016, 714–723 CAS.
- (a) T. M. Nguyen and D. A. Nicewicz, J. Am. Chem. Soc., 2013, 135, 9588–9591 CrossRef CAS PubMed; (b) A. J. Perkowski and D. A. Nicewicz, J. Am. Chem. Soc., 2013, 135, 10334–10337 CrossRef CAS PubMed; (c) D. J. Wilger, J.-M. M. Grandjean, T. R. Lammert and D. A. Nicewicz, Nat. Chem., 2014, 6, 720–726 CrossRef CAS PubMed; (d) N. A. Romero and D. A. Nicewicz, J. Am. Chem. Soc., 2014, 136, 17024–17035 CrossRef CAS PubMed.
- D. A. Armstrong, Q. Sun and R. H. Schuler, J. Phys. Chem., 1996, 100, 9892–9899 CrossRef CAS.
- K. Ohkubo, K. Mizushima, R. Iwata, K. Souma, N. Suzuki and S. Fukuzumi, Chem. Commun., 2010, 46, 601–603 RSC.
- (a) N. E. S. Tay and D. A. Nicewicz, J. Am. Chem. Soc., 2017, 139, 16100–16104 CrossRef CAS PubMed; (b) W. Chen, Z. Huang, N. E. S. Tay, B. Giglio, M. Wang, H. Wang, Z. Wu, D. A. Nicewicz and Z. Li, Science, 2019, 364, 1170–1174 CrossRef CAS PubMed; (c) N. Holmberg-Douglas, N. P. R. Onuska and D. A. Nicewicz, Angew. Chem., Int. Ed., 2020, 59, 7425–7429 CrossRef CAS PubMed.
- (a) C. Fischer and C. Sparr, Angew. Chem., Int. Ed., 2018, 57, 2436–2440 CrossRef CAS PubMed; (b) C. Fischer and C. Sparr, Tetrahedron, 2018, 74, 5486–5493 CrossRef CAS; (c) B. Zilate, C. Fischer, L. Schneider and C. Sparr, Synthesis, 2019, 4359–4365 CAS; (d) A. Gini, M. Uygur, T. Rigotti, J. Alemán and O. García Mancheño, Chem. – Eur. J., 2018, 24, 12509–12514 CrossRef CAS PubMed; (e) A. R. White, L. Wang and D. A. Nicewicz, Synlett, 2019, 827–832 CAS; (f) C. Fischer, C. Kerzig, B. Zilate, O. S. Wenger and C. Sparr, ACS Catal., 2020, 10, 210–215 CrossRef CAS.
- Z. Zuo, D. T. Ahneman, L. Chu, J. A. Terrett, A. G. Doyle and D. W. C. MacMillan, Science, 2014, 345, 437–440 CrossRef CAS PubMed.
- (a) J. C. Theriot, C. H. Lim, H. Yang, M. D. Ryan, C. B. Musgrave and G. M. Miyake, Science, 2016, 352, 1082–1086 CrossRef CAS PubMed; (b) R. M. Pearson, C. H. Lim, B. G. McCarthy, C. B. Musgrave and G. M. Miyake, J. Am. Chem. Soc., 2016, 138, 11399–11407 CrossRef CAS PubMed; (c) B. L. Buss, C. H. Lim and G. M. Miyake, Angew. Chem., Int. Ed., 2020, 59, 3209–3217 CrossRef CAS PubMed; (d) N. A. Swisher, D. A. Corbin and G. M. Miyake, ACS Macro Lett., 2021, 10, 453–459 CrossRef CAS PubMed.
- A. L. Berger, K. Donabauer and B. König, Chem. Sci., 2018, 9, 7230–7235 RSC.
- D. B. Vogt, C. P. Seath, H. Wang and N. T. Jui, J. Am. Chem. Soc., 2019, 141, 13203–13211 CrossRef CAS PubMed.
- M. H. Shaw, J. Twilton and D. W. C. MacMillan, J. Org. Chem., 2016, 81, 6898–6926 CrossRef CAS PubMed.
- L. Capaldo, R. Riccardi, D. Ravelli and M. Fagnoni, ACS Catal., 2018, 8, 304–309 CrossRef CAS.
- N. Bortolamei, A. A. Isse and A. Gennaro, Electrochim. Acta, 2010, 55, 8312–8318 CrossRef CAS.
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234–238 CrossRef CAS PubMed.
- J. Luo and J. Zhang, ACS Catal., 2016, 6, 873–877 CrossRef CAS.
- Q.-Y. Meng, T. E. Schirmer, A. L. Berger, K. Donabauer and B. König, J. Am. Chem. Soc., 2019, 141, 11393–11397 CrossRef CAS PubMed.
- T. Duhail, T. Bortolato, J. Mateos, E. Anselmi, B. Jelier, A. Togni, E. Magnier, G. Dagousset and L. Dell’Amico, Org. Lett., 2021, 23, 7088–7093 CrossRef CAS PubMed.
- B. J. Jelier, P. F. Tripet, E. Pietrasiak, I. Franzoni, G. Jeschke and A. Togni, Angew. Chem., Int. Ed., 2018, 57, 13784–13789 CrossRef CAS PubMed.
- I. Ghosh, T. Ghosh, J. I. Bardagi and B. König, Science, 2014, 346, 725–728 CrossRef CAS PubMed.
- (a) R. Naumann, F. Lehmann and M. Goez, Chem. – Eur. J., 2018, 24, 13259–13269 CrossRef CAS PubMed; (b) R. Naumann and M. Goez, Chem. – Eur. J., 2018, 24, 9833–9840 CrossRef CAS PubMed; (c) R. Naumann and M. Goez, Chem. – Eur. J., 2018, 24, 17557–17567 CrossRef CAS PubMed; (d) C. Kerzig, X. Guo and O. S. Wenger, J. Am. Chem. Soc., 2019, 141, 2122–2127 CrossRef CAS PubMed; (e) M. Giedyk, R. Narobe, S. Weiß, D. Touraud, W. Kunz and B. König, Nat. Catal., 2020, 3, 40–47 CrossRef CAS.
- I. A. MacKenzie, L. Wang, N. P. R. Onuska, O. F. Williams, K. Begam, A. M. Moran, B. D. Dunietz and D. A. Nicewicz, Nature, 2020, 580, 76–80 CrossRef CAS PubMed.
- D. Rombach and H.-A. Wagenknecht, ChemCatChem, 2018, 10, 2955–2961 CrossRef CAS.
- K. Targos, O. P. Williams and Z. K. Wickens, J. Am. Chem. Soc., 2021, 143, 4125–4132 CrossRef CAS PubMed.
- A. F. Chmiel, O. P. Williams, C. P. Chernowsky, C. S. Yeung and Z. K. Wickens, J. Am. Chem. Soc., 2021, 143, 10882–10889 CrossRef CAS PubMed.
- J. Xu, J. Cao, X. Wu, H. Wang, X. Yang, X. Tang, R. W. Toh, R. Zhou, E. K. L. Yeow and J. Wu, J. Am. Chem. Soc., 2021, 143, 13266–13273 CrossRef PubMed.
- H. Kim, H. Kim, T. H. Lambert and S. Lin, J. Am. Chem. Soc., 2020, 142, 2087–2092 CrossRef CAS PubMed.
- M. Melchionna and P. Fornasiero, ACS Catal., 2020, 10, 5493–5501 CrossRef CAS.
- B. Bartolomei, G. Gentile, C. Rosso, G. Filippini and M. Prato, Chem. – Eur. J., 2021, 27, 16062–16070 CrossRef CAS PubMed.
- (a) T. Serevičius, T. Nakagawa, M.-C. Kuo, S.-H. Cheng, K.-T. Wong, C.-H. Chang, R. C. Kwong, S. Xiae and C. Adachi, Phys. Chem. Chem. Phys., 2013, 15, 15850–15855 RSC; (b) C. Tang, R. Bi, Y. Tao, F. Wang, X. Cao, S. Wang, T. Jiang, C. Zhong, H. Zhang and W. Huang, Chem. Commun., 2015, 51, 1650–1653 RSC.
| This journal is © The Royal Society of Chemistry 2022 |