
Electrochemically customized assembly of a hybrid xerogel material via combined covalent and non-covalent conjugation chemistry: an approach for boosting the cycling performance of pseudocapacitors†
Taniya
Purkait
,
Dimple
,
Navpreet
Kamboj
,
Manisha
Das
,
Subhajit
Sarkar
,
Abir
De Sarkar
 and
Ramendra Sundar
Dey
*
and
Ramendra Sundar
Dey
*
Institute of Nano Science and Technology (INST), Mohali-160064, Punjab, India. E-mail: rsdey@inst.ac.in
First published on 17th March 2020
Abstract
Organic quinones conjugated with a conductive support like graphene via a covalent and/or non-covalent approach are emerging as low-cost and sustainable alternatives to conventional pseudocapacitive materials because of their fast and reversible redox kinetics. Herein, for the first time, reduced graphene oxide (rGO) networks functionalized with the eco-friendly dopamine (DA) moiety in a combined covalent and non-covalent manner have been explored by a facile hydrothermal synthesis method as high-performance pseudocapacitive materials. Further, a unique in situ electrochemical polymerization approach has been undertaken in an attempt to boost the overall storage capacity as well as cycling stability. Electrochemical tuning of the active material can result in an enhancement in the specific capacitance via dual improvement in faradaic and capacitive current as compared to the initial xerogel material, highlighting the significance of the in situ process. The electrode material shows a highest specific capacitance (CSP) of 348 F g−1 at a current density of 0.5 A g−1 in 1 M H2SO4 while retaining 60% of its initial capacitance even at a very high current density of 200 A g−1. A metal-free all-solid-state symmetric supercapacitor was developed with the in situ electropolymerized active material and the as-fabricated device exhibits an excellent CSP of 218 F g−1 at 0.325 A g−1 in 1 M PVA/H2SO4 gel electrolyte with a maximum energy density of 30.3 W h kg−1 and power density up to 13 kW kg−1. Most importantly, the material exhibited an extraordinary stability of 53![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 charge–discharge cycles while retaining 94% of its initial capacitance, while the device also shows an excellent capacitance retention of 92% even after 10000 continuous cycles. Simultaneously, the DFT study was amended to understand the covalent and non-covalent interaction of the redox species, its charge storage mechanism and charge density distribution as well as to calculate the density of states.
000 charge–discharge cycles while retaining 94% of its initial capacitance, while the device also shows an excellent capacitance retention of 92% even after 10000 continuous cycles. Simultaneously, the DFT study was amended to understand the covalent and non-covalent interaction of the redox species, its charge storage mechanism and charge density distribution as well as to calculate the density of states.
Introduction
Nano-engineered electro-active species are well-known pseudocapacitive materials, but often suffer from poor cycling stability, restricted rate performance and consequently very low power density.1 Their poor electrical conductivity, and limited resources of expensive inorganic oxides along with complex material engineering of nanostructures hinder these materials from being used for large-scale industrialization.2,3 Recently, as a new class of electrode materials, organic molecules with electrochemical reversibility4–8 have emerged as low-cost, renewable and environmentally benign pseudocapacitive species. They are light-weight, easy to synthetically design, and possess large ion-storage capacities beyond those of conventional electroactive metal oxides via multi-electron faradaic reactions in a low-molecular-weight charge-storage unit.9 Among the electroactive organic materials, the quinone species is known to show a fast reversible redox reaction with protons via tautomerism in aqueous electrolytes.10 However, just like other pseudocapacitive materials, poor electroconductivity, slow redox kinetics and poor solubility in common electrolytes also impede the electrochemical performance of these quinone molecules when they are used alone. Therefore, in order to address this problem, organic redox centres have been usually anchored/grafted onto conductive substrates, particularly on carbonaceous materials, via a covalent approach11–15 or non-covalent approach4,6–9,16 and/or by dissolution in the experimental electrolyte.17,18 Unlike transition metal oxides or conducting polymers, these types of hybrid materials often have an interconnected three-dimensional (3D) macroporous xerogel type of structure with stretchability and a low modulus of elasticity. The amount of buffer space available in these highly porous materials can easily alleviate the stress caused by the volume change in redox materials during continuous charge–discharge cycles, leading to an excellent cycling stability.19,20Recently, many researchers have explored non-covalently attaching organic species on a conductive host like graphene as an advanced way to produce electroactive supercapacitors.4,6–9,16 However, most of the findings have failed to achieve a high cycle lifespan of pseudocapacitors. The non-covalently attached redox species are often loosely bound via π–π conjugation, leading to their facile dissolution in the working electrolyte during long service hours and this ultimately results in poor cycling performance.14 On the other hand, grafting or covalent modification of the conductive substrate may offer enhanced energy storage performance as well as improved stability via building well-integrated electronic transport channels, but it evidently decreases the conductivity of the host material. Therefore, to achieve an overall high performance pseudocapacitor with high cycle life, optimization of synthesis engineering is a key step to look into along with the careful selection of electroactive species.
In this work, for the very first time, a combined covalent and non-covalent functionalization approach of reduced graphene oxide (rGO) networks with the eco-friendly dopamine (DA) moiety has been explored via a facile hydrothermal synthesis method. The selection of electroactive species here plays a dynamic role in determining whether a covalent or non-covalent functionalization of graphene will take place. The availability of two facile electron donating head and tail groups, i.e., hydroxyl and alkyl amine functionalities at the para positions in the DA molecule endorses its easy covalent interaction with the functional groups present on GO at the edge-site during the hydrothermal process, whereas the aromatic π cloud helps in non-covalent interaction with molecular level binding. Covalent functionalization provides electron transport channels with faster charge-transport kinetics, leading to a three-dimensional (3D) interconnected xerogel architecture, while non-covalent functionalization helps in retaining the inherent conductivity of the carbon skeleton. The formation of cross-linkage with interconnected 3D macroporous xerogel architecture has not only been confirmed by various physical characterization experiments, but also supported via the first-principles Density Functional Theory (DFT) study. This type of combined interaction helps in retaining the inherent conductivity of the graphene base as the dried dopamine-graphene xerogel (DA-rGO) shows superior electronic conductivity while providing continuous electron transport channels for excellent rate performance. Additionally, DA acts as an efficient spacer element via covalent integration, effectively preventing the agglomeration of the rGO sheet, caused by strong π–π interactions.
Furthermore, in this study, we have explored the well-known polymerization tendency of DA to form polydopamine (pDA) at room temperature via potential cycling of the xerogel material in the experimental electrolyte and generating the in situ electropolymerized pDA-rGO (EpDA-rGO) active material. Potential cycling helps in converting only the non-covalently attached DA moiety into EpDA while the cross-linked DA species remain intact providing channels for electron transport. The in situ formation of EpDA-rGO improves the overall active redox sites covering the surface of the carbonaceous base; therefore, proves to be beneficial in terms of achieving enhanced specific capacitance and excellent rate capability, as well as unprecedented cycling stability (53000 continuous charge–discharge cycles). Additionally, a novel metal-free and self-supported in-plane symmetric solid-state supercapacitor is designed with the EpDA-rGO material, which shows excellent energy density as well as cycling stability (10000 continuous cycles at a high current density of 10 A g−1) to promote its application in commercialization. The combined output from both the theoretical study and experimental findings helps us to understand that the careful selection of the organic moiety ensures optimum interaction with the conductive substrate which in turn helps in addressing the stability of pseudocapacitors, which is the most critical issue of a commercial supercapacitor.
Experimental
Synthesis of the DA-rGO xerogel
Graphene oxide (GO) was synthesized using a modified Hummers' method, reported elsewhere21 and described in the ESI (see the Experimental section in the ESI†). Next, the functionalization of the GO and dopamine (DA) moiety was done in a single step via an economical hydrothermal method, as presented in Fig. 1a. Briefly, a mixture of as-received DA (Sigma-Aldrich) and GO suspension (1 mg mL−1) was stirred for about 10 minutes to get a homogeneous mixture and then transferred to a Teflon-lined autoclave for 6 hours at 160 °C. The autoclave was cooled to obtain the DA-rGO hydrogel, which was further subjected to lyophilization to get the xerogel architecture. For comparison, gel electrodes composed of polydopamine integrated on rGO sheets (pDA-rGO) were also synthesized using the same experimental conditions. A series of DA-rGO gels were prepared by varying the weight percentages of DA (x) to GO (y) and were labelled according to their initial weight ratios [x:y = 1:1, 1:2, 1:5, 1:10 and, 1:20]. To understand the role of the DA moiety, only the GO suspension (1 mg mL−1) was subjected to hydrothermal treatment to obtain the rGO hydrogel.
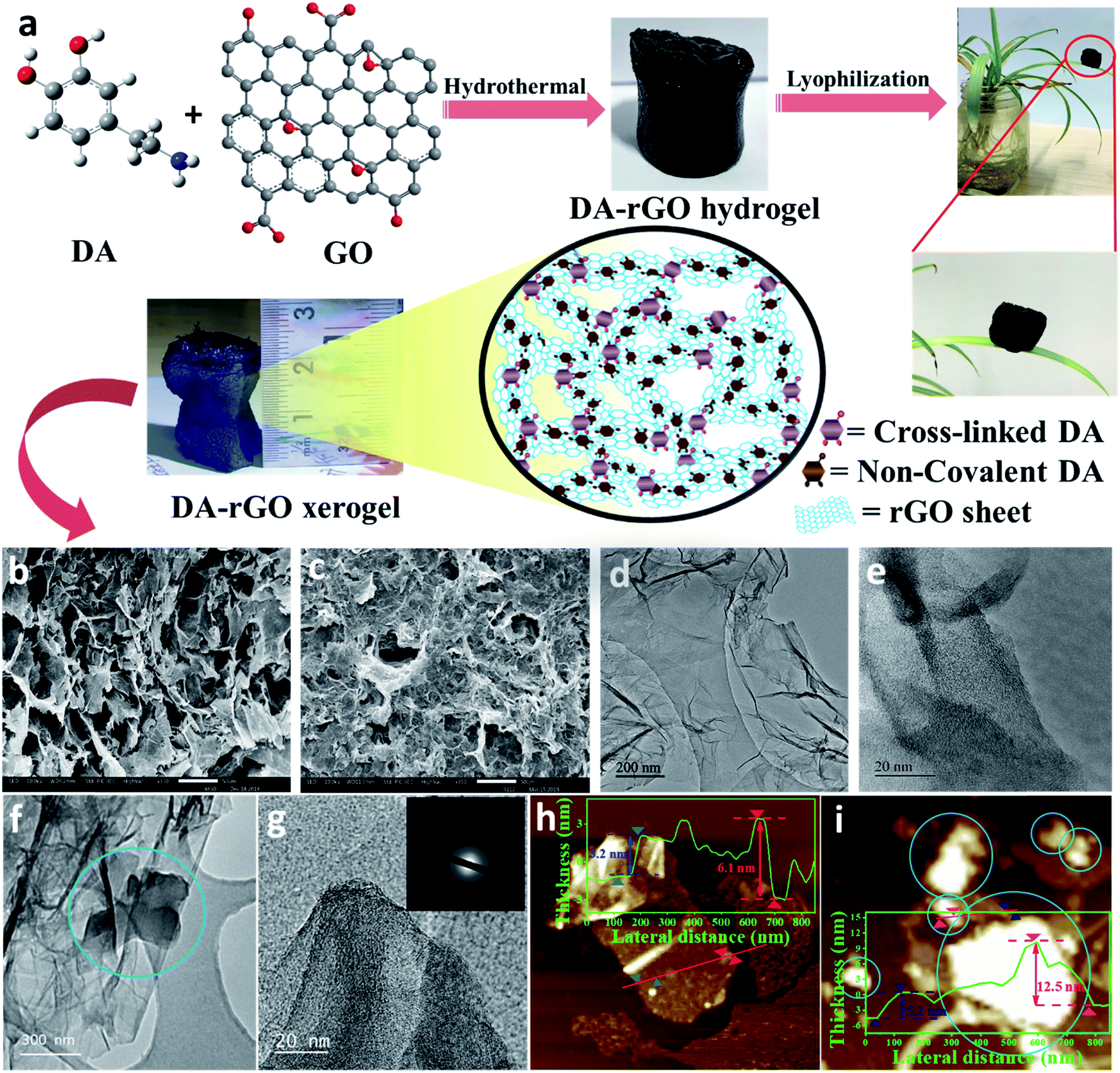 | ||
| Fig. 1 (a) Schematic representation of the hydrothermal synthesis of the dopamine (DA) based reduced graphene oxide (rGO) xerogel (DA-rGO). Surface analysis of the optimized xerogel material, DA-rGO1–2 as well as the in situ electropolymerized active material, EpDA-rGO by various microscopic techniques; (b) SEM image of DA-rGO showing the interconnected 3D porous architecture. (c) SEM micrographs after electropolymerization revealing increased surface coverage via π–π interactions. (d) and (e) TEM and corresponding HRTEM images of the xerogel, DA-rGO. (f) TEM analysis of EpDA-rGO clearly showing large chunks of dark patches (marked by blue circles) revealing the presence of the polymer on the rGO matrix. (g) Related HRTEM image, the inset showing the SAED pattern. (h) AFM study at different positions of a single DA-rGO xerogel flake, the inset showing how the thickness profile varies with the presence of DA (bright spots) on the rGO sheet. (i) AFM analysis confirming the surface coverage of rGO by EpDA with increased bright spots and the inset shows an enhanced thickness of the sheets. | ||
Electrochemical in situ conversion of the DA-rGO xerogel to EpDA-rGO
The as-synthesized DA-rGO sample was used as a conductive ink to be drop-cast on a glassy carbon (GC) electrode to test its electrochemical performance as a storage material. In order to obtain the optimized electro-active material, however, potential cycling was performed at a scan rate of 100 mV s−1 for 100 cycles in the working electrolyte of 1 M H2SO4. The in situ conversion of non-covalently attached DA species to pDA via electrochemical oxidation and rearrangement was observed and the final active material, EpDA-rGO, was further tested for its electrochemical behaviour. On the other hand, another control sample was prepared by taking the aforementioned hydrothermally reduced GO (rGO) and DA in solution (in a similar weight percentage of the hydrothermally optimized xerogel) and drop-casting it on a glassy-carbon electrode in an attempt to electrochemically cross-link the DA moiety with the rGO skeleton. The sample was denoted as rGO/DA.Results and discussion
Physical characterization
DA-rGO was prepared by the functionalization of graphene oxide (GO) via the dopamine (DA) moiety, in a single step via an economical hydrothermal method, as presented in Fig. 1a (for details, please see the Experimental section). Simultaneously, in this work, we introduced a unique in situ electropolymerization technique to obtain the optimized electro-active material. The as-synthesized DA-rGO sample was subjected to potential cycling at a scan rate of 100 mV s−1 for 100 cycles in the working electrolyte of 1 M H2SO4 to obtain the final active material, EpDA-rGO (Experimental section).Surface morphological characterization of the obtained DA-rGO xerogels was performed using a scanning electron microscope (SEM) (Fig. 1b and S1a–h, ESI†). As shown in Fig. 1b, the freeze-dried DA-rGO1–2 has an interconnected 3D macroporous hierarchical framework with pore sizes ranging from sub-micrometers to several micrometers. The SEM images of the other synthesized xerogels are also presented in Fig. S1a–h, ESI,† which also support the 3D porous interconnected morphology of the xerogel. The surface morphological inspections were further extended to study the effect of in situ electropolymerization in the experimental electrolyte on the optimized xerogel structure. Fig. 1c exhibits the SEM micrographs of the EpDA-rGO surface, which shows a similar interconnected architecture to that of DA-rGO, but with more surface coverage. The intensity of the non-covalent π–π interaction may have increased several fold after electropolymerization as compared to only the xerogel material, resulting in more surface coverage of the rGO matrix by EpDA. Transmission electron microscopy (TEM) was adopted to further study the surface interaction of the DA molecule with the overlapping graphene sheets. Interconnected graphene sheets (Fig. 1d) with wrinkles outlining the sheets by DA are clearly visible in the high-resolution TEM (HRTEM) images, as exhibited in Fig. 1e. This kind of open-structured hybrid network provides easy accessibility to the electrolyte ions from both sides of the sheets for rapid charge transport through the electronic channels provided by the amide cross-linkage of DA molecules. Interestingly, in the TEM analysis (Fig. 1f and g) of the EpDA-rGO material, intense dark patches are observed, which are highlighted by blue circles, confirming the increased non-covalent π–π interaction between the polymer and the rGO matrix. The selected area electron diffraction (SAED) analysis (Fig. 1g, inset) in TEM measurements reveals the absence of any characteristic lattice points corresponding to the hexagonal lattice of crystalline graphene, indicating that crystallinity is substantially decreased after inclusion of the DA moiety in the rGO matrix. After in situ polymerization, which substantially improves the coverage of the rGO surface, the crystallinity of the rGO matrix is further reduced. Surface topographic analysis by atomic force microscopy (AFM) further confirms the formation of the cross-linked xerogel structure. As displayed in Fig. 1h, a single DA-rGO flake with DA at the edge as well as on the rGO backbone can be seen. The height profile reveals the sheets with an average height of 3–6 nm (inset of Fig. 1h), which is much higher as compared to that of a single GO sheet (0.9 nm), as exhibited in Fig. S2a and b, ESI.† Detailed surface topological characterization of the EpDA-rGO material by AFM (Fig. 1i) also ensures in situ polymerization with an enhanced presence of bright spots (EpDA) almost covering the rGO sheets beneath. The AFM height profile at various positions on the EpDA-rGO surface, displayed in the inset of Fig. 1i, reveals that the average sheet height increased to around 5–12 nm, as compared to only DA-rGO (Fig. 1h, inset); confirming the in situ electropolymerization.
To investigate the successful reduction of the precursor (GO) in the xerogel material (DA-rGO), X-ray diffraction (XRD) analysis was employed and the results were compared to those of pristine hydrothermally reduced GO (rGO) without incorporating DA. Further inspection of the xerogel structure after its in situ electropolymerization was also performed to follow any change in its graphitic domain. As can be seen in Fig. 2a, the diffraction peak at 2θ = 11.96° corresponding to the (100) graphitic plane of GO completely disappears in the XRD spectra of rGO and DA-rGO as well as EpDA-rGO materials, suggesting that GO is completely reduced in the xerogel material during hydrothermal treatment and the structure is retained even after in situ electropolymerization in the experimental electrolyte. The presence of characteristic diffraction peaks at 2θ = 30.2°, corresponding to the (002) graphitic plane and 2θ = 43°, corresponding to the (110) graphitic plane ensures the removal of most of the oxygen functionalities present in GO in rGO, DA-rGO and EpDA-rGO, respectively.
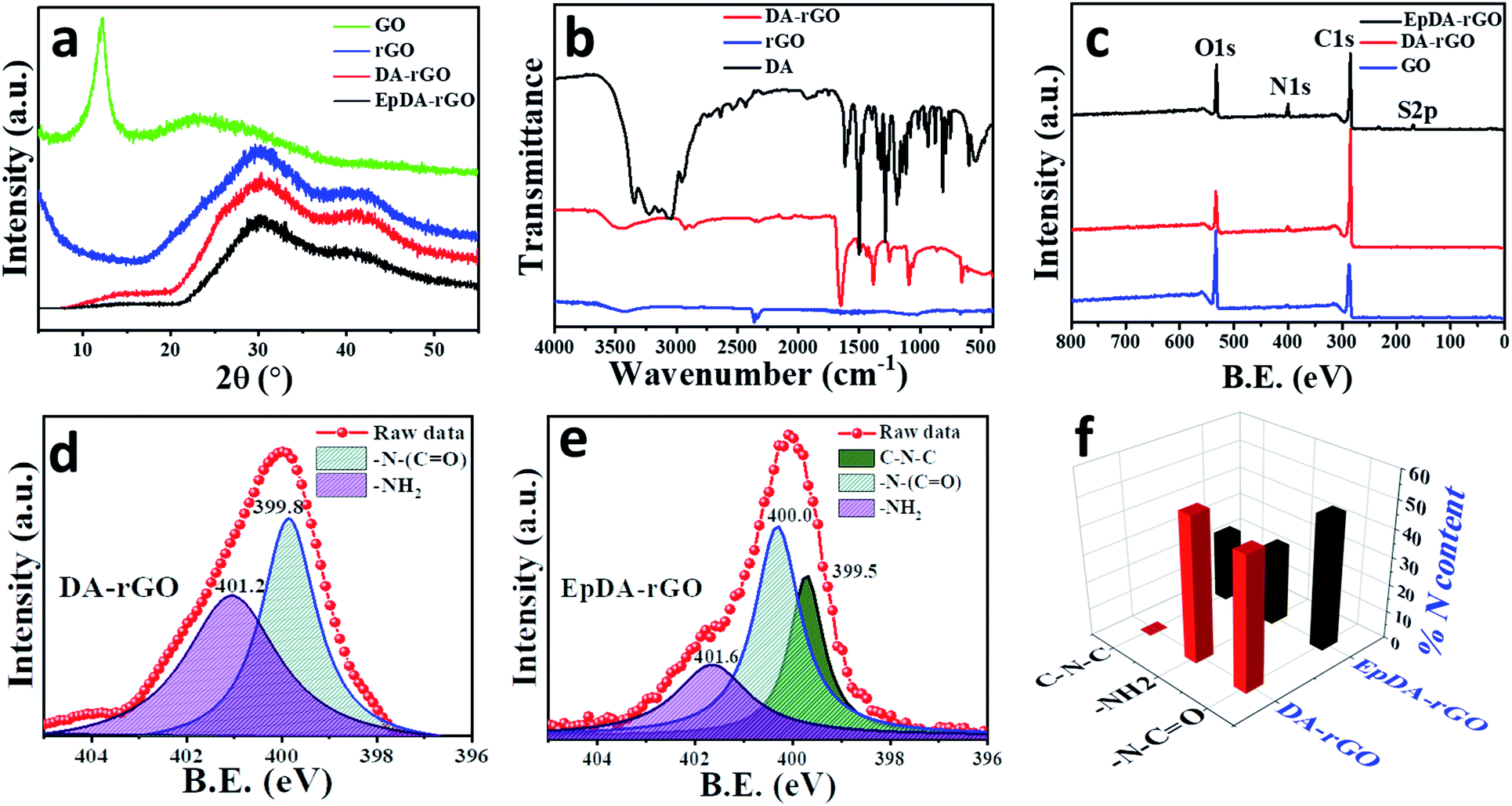 | ||
| Fig. 2 (a) XRD patterns of the xerogel before and after its in situ electropolymerization as compared to GO and rGO. (b) Comparative FTIR analysis of the optimized xerogel with respect to DA and rGO, showing successful incorporation of the DA moiety into the rGO matrix. (c) Relative XPS analysis of EpDA-rGO and DA-rGO as compared to GO. Deconvoluted N1s spectra obtained from high-resolution XPS analysis of (d) DA-rGO and (e) EpDA-rGO. (f) Different types of nitrogen environments in the DA-rGO and EpDA-rGO samples and their relative percentage highlighting the in situ electropolymerization process. | ||
Fourier transform-infrared (FT-IR) spectroscopic analysis was carried out via the attenuated total reflection (ATR) technique to confirm the grafting of DA on the rGO sheet via covalent functionalization. All the FTIR-ATR peaks of DA, rGO and, DA-rGO are identified and assigned to the corresponding characteristic bands, as summarized in Table S1.† As displayed in Fig. 2b, the characteristic bands of the quinonic backbone (1252 and 863 cm−1) is clearly visible in the xerogel structure.22 A broad peak at 3460 cm−1 appears due to the O–H vibration.23 Additionally, the appearance of a peak at 1727 cm−1 corresponding to the stretching frequency of the carbonyl functional group along with shifting in the N–H bending vibration (from 1612 cm−1 to 1652 cm−1) from the pure DA moiety24 ensures the formation of an amide linkage in DA-rGO. The additional C–H stretching vibration, assigned to the peaks obtained at 2930 cm−1 and 2854 cm−1 corresponds to the aromatic and alkane C–H stretching frequency of the linkage group as well as the conjugated rGO architecture at the base of the xerogel.25
To obtain a more clear understanding about the nature of chemical bonding between DA and rGO and the subsequent changes occurred to the xerogel structure after in situ polymerization, X-ray photoelectron spectroscopic (XPS) analysis was adopted. The overall spectra of DA-rGO in Fig. 2c ensure the presence of the requisite elements, C, N and O with an atomic percentage of 83%, 4% and 13% respectively in the DA-rGO xerogel material, while the elemental N percentage dramatically improves to 8.4% in the EpDA-rGO structure. Compared to the precursor material, GO, the atomic percentage ratio of carbon to oxygen increases in DA-rGO, confirming the reduction of GO to rGO, as revealed in Fig. 2c. However, EpDA-rGO exhibits a slightly higher O content as compared to DA-rGO and an additional peak for S, which may arise due to the continuous CV cycling in the experimental electrolyte of 1 M H2SO4. The high-resolution deconvoluted C1s spectra of the DA-rGO xerogel material, shown in Fig. S3a,† disclose three characteristic peaks at 284.5 eV, 286.2 eV and 288.9 eV corresponding to the C–C, C–N and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds, respectively.26 Similarly, as expected, the deconvoluted C1s spectra of EpDA-rGO, displayed in Fig. S3b,† reveal three different binding energies, peaking at 284.6 eV, 286.2 eV and 288.1 eV, with only a slight shift in the peak positions, corresponding to the C–C, C–N and O–CO bonds, respectively.26 The deconvoluted N1s spectra (Fig. S3b† and 2e) were instrumental in determining the binding preference in the hydrothermally obtained xerogel structure and the in situ electropolymerized active material, respectively. The spectra corresponding to DA-rGO (Fig. S3b†) were fitted to two peaks at 399.9 eV and 401.2 eV, respectively. The peak at 399.9 eV was assigned to the amide groups (–N–CO) which confirms the occurrence of covalent cross-linking in the hybrid structure,27 while the peak assigned to 401.2 eV indicates the presence of primary amine (R-NH2), which ensures the presence of non-covalently attached DA species.28 On the other hand, N1s spectra (Fig. 2e) of EpDA-rGO were fitted to three distinct characteristic peaks at 399.5, 400.0 and 401.2 eV corresponding to substituted amines (C–N–C), amide groups (–N–CO), and free –NH2 groups.25 As expected, the peaks corresponding to –N–CO and R–NH2 groups in DA-rGO were slightly shifted or remained constant after electropolymerization. However, a significant change is observed in the N 1s core level spectrum via the appearance of C–N–C binding energy at 399.5 eV due to the rearrangement of the non-covalently attached free amine groups into a 5-membered heterocyclic ring (leucodopaminechrome or dopaminechrome species) during the electropolymerization process.25,29 To quantify the relative percentage of the (i) covalent and non-covalent attachment of dopamine to the hybrid material and (ii) amount of DA which has been transformed to EpDA during the polymerization process, we have extracted the %N content from the deconvoluted N1s spectra and presented in Fig. 2f. The %N content from the covalently attached (–N–CO) amide linkage remains almost similar before (∼48% for DA-rGO) and after (∼47.7% for EpDA-rGO) the polymerization process. However, the %N content in non-covalently attached free –NH2 groups present in DA-rGO (∼52%) gets split into C–N–C (∼24.5%) and –NH2 (∼27.8%) binding energies in EpDA-rGO. This confirms the in situ electropolymerization process as well as quantifies the percentage of DA-rGO still present and EpDA-rGO formed after the electropolymerization process.
O bonds, respectively.26 Similarly, as expected, the deconvoluted C1s spectra of EpDA-rGO, displayed in Fig. S3b,† reveal three different binding energies, peaking at 284.6 eV, 286.2 eV and 288.1 eV, with only a slight shift in the peak positions, corresponding to the C–C, C–N and O–CO bonds, respectively.26 The deconvoluted N1s spectra (Fig. S3b† and 2e) were instrumental in determining the binding preference in the hydrothermally obtained xerogel structure and the in situ electropolymerized active material, respectively. The spectra corresponding to DA-rGO (Fig. S3b†) were fitted to two peaks at 399.9 eV and 401.2 eV, respectively. The peak at 399.9 eV was assigned to the amide groups (–N–CO) which confirms the occurrence of covalent cross-linking in the hybrid structure,27 while the peak assigned to 401.2 eV indicates the presence of primary amine (R-NH2), which ensures the presence of non-covalently attached DA species.28 On the other hand, N1s spectra (Fig. 2e) of EpDA-rGO were fitted to three distinct characteristic peaks at 399.5, 400.0 and 401.2 eV corresponding to substituted amines (C–N–C), amide groups (–N–CO), and free –NH2 groups.25 As expected, the peaks corresponding to –N–CO and R–NH2 groups in DA-rGO were slightly shifted or remained constant after electropolymerization. However, a significant change is observed in the N 1s core level spectrum via the appearance of C–N–C binding energy at 399.5 eV due to the rearrangement of the non-covalently attached free amine groups into a 5-membered heterocyclic ring (leucodopaminechrome or dopaminechrome species) during the electropolymerization process.25,29 To quantify the relative percentage of the (i) covalent and non-covalent attachment of dopamine to the hybrid material and (ii) amount of DA which has been transformed to EpDA during the polymerization process, we have extracted the %N content from the deconvoluted N1s spectra and presented in Fig. 2f. The %N content from the covalently attached (–N–CO) amide linkage remains almost similar before (∼48% for DA-rGO) and after (∼47.7% for EpDA-rGO) the polymerization process. However, the %N content in non-covalently attached free –NH2 groups present in DA-rGO (∼52%) gets split into C–N–C (∼24.5%) and –NH2 (∼27.8%) binding energies in EpDA-rGO. This confirms the in situ electropolymerization process as well as quantifies the percentage of DA-rGO still present and EpDA-rGO formed after the electropolymerization process.
Raman spectral analysis was conducted to investigate the structural transformation during hydrothermal treatment as well as after electropolymerization. Fig. S4† displays the characteristic D and G bands of graphitic carbon, which appears at ∼1348 and ∼1589 cm−1, respectively for GO, rGO, DA-rGO and EpDA-rGO structures. As expected, the intensity ratio of ID/IG decreases from GO (1.21) to rGO (1.1) because of the restoration of the graphitic domain. However, in the xerogel structure, the intensity ratio once again increases to 1.25, indicating expansion of the conjugated backbone via covalent functionalization with DA.14 The ID/IG ratio remains almost intact at 1.26 in the EpDA-rGO xerogel material, indicating retention of the graphitic structure even after polymerization.
The N2 adsorption/desorption analysis (Fig. S5†) was carried out to study the specific surface area (SSA), and pore size distribution as well as pore volume. The sorption isotherm displays typical type-IV behaviour with a specific surface area of 105 m2 g−1. The obvious hysteresis loop in the range of P/P0 = 0.4–0.9 and the sharp increase of the adsorption amount at P/P0 > 0.9 indicate the existence of a large number of mesopores in DA-rGO. As expected, an abundance of micropores (∼2 nm) along with mesopores (6–12 nm) can be observed in the inset of Fig. S5,† as calculated by the Barrett–Joyner–Halenda (BJH) pore size distribution method. The total pore volume of 1.28 cm3 g−1 clearly demonstrates the high mass loading of DA on the rGO sheets, a prerequisite for obtaining a high gravimetric capacitance of pseudocapacitive electrodes.
To determine the extent of DA distribution in the DA-rGO xerogel and the distribution of EpDA in the sample after polymerization, EpDA-rGO on the porous rGO network, elemental mapping was carried out via energy dispersive X-ray spectroscopy (EDS). Uniform distribution of all the elements, i.e., carbon, oxygen and, nitrogen, can be clearly observed in Fig. S6a† as well as in Fig. S6b.† Individual elemental contributions are exhibited in Fig. S6c–h.† A close look at Fig. S5a† indicates the abundance of elemental nitrogen on the periphery of the interconnected pores, which highly supports our finding that DA acts as a cross-linker here. The elemental distribution once again substantiates the adequate incorporation of the DA moiety into the DA-rGO structure. On the other hand, the increased percentage of elemental N can be clearly observed because of the enhanced non-covalent interaction with the polymer (Fig. S6b†), which is in close accordance with our XPS results.
Thermogravimetric analysis (TGA) was carried out (Fig. S7†) to compare the thermal stability of different electrode materials in an air atmosphere at a heating rate of 10 °C min−1. GO is thermally unstable and rapid mass loss occurs in two steps. The 15% mass loss observed for GO at ∼100 °C is ascribed to the loss of adsorbed water molecules or sublimation whereas the 40% mass loss at ∼200 °C can be due to the loss of labile oxygen-containing functional groups.30 In contrast, rGO withstands much higher temperature because of the removal of the most of the functional groups. It shows a very gradual decrease in the weight loss at varying temperatures, consistent with previous reports.30 Comparatively, DA-rGO and EpDA-rGO show much better thermal stability than rGO and GO in the temperature range of 40–550 °C. As can be seen in the inset of Fig. S7,† EpDA-rGO and DA-rGO show almost 96% and 92% mass retention, while GO and rGO can retain only about 36% and 84% of the initial sample mass at 400 °C. DA is a synthetic mimic of a mussel adhesive protein and it shows almost no decomposition up to ∼230 °C; then starts to degrade a little up to 300 °C due to the loss of the catechol moiety. The second thermal decomposition occurs in the range of 300–500 °C, most likely due to the decomposition of the alkyl spacer and the amide group.31 This indicates that DA contains necessary thermal stability for incorporation into the composite xerogel material, DA-rGO. It was observed that the composite material, DA-rGO follows a similar decomposition path as well but with higher decomposition temperature. This once again may be attributed to the synergistic thermal stability of both rGO and DA in the composite xerogel material due to the stronger π–π interaction. The first decomposition occurs in the range of 350–550 °C while the 2nd thermal degradation occurs in the range of 550–680 °C. After in situ polymerization, the EpDA-rGO expectedly follows a similar thermal decomposition pathway, however the % mass retention significantly improves to 41% from 16% in DA-rGO after complete decomposition of the functional moieties takes place at ∼680 °C. This once again indicates that the in situ polymerization of the xerogel moiety in the experimental electrolyte greatly improves the thermal stability of the electrode material.
Furthermore, in order to promote the xerogel material as a potential option to be employed for energy storage, the electrical conductivity of all the active electrode materials was determined with a source meter via a two-probe method, while GO shows a very small conductivity of 0.77 mS cm−1; after removal of functional groups, the conductivity in hydrothermally derived rGO improves to 5.35 S cm−1. Interestingly, after the xerogel formation, DA-rGO exhibits an excellent electrical conductivity of around 81 S cm−1, which was slightly improved in EpDA-rGO (82.6 S cm−1) after in situ polymerization.
Computational methodology and studies
In order to shed light on the preferred orientation of the redox moiety, DA and its conjugation chemistry with the graphene sheet, DFT calculations have also been performed. The charge density distribution in the preferred orientations as well as the role of density of states (DOS) in the charge storage mechanism was investigated in detail for an atomic-scale insight. Therefore, to unveil the mechanism underlying the conjugation, binding energy has been calculated, at first, for different possible conformations or orientations of the redox moiety at different adsorption sites of DA, including non-covalent binding to the graphene sheet (both in vertical and parallel conformations). The possibility for adsorption in a cross-linking conformation (i.e., covalent interaction during charge-storage) has also been explored. The structure of the DA molecule was optimized first and then it was adsorbed on rGO in various conformations for further structural optimizations of the combined system, DA-rGO. The total energy calculations for the DA molecule were performed with DFT as implemented in the Gaussian16 package.32,33 Fig. S8† shows the relaxed structure of the DA molecule, which is in good agreement with the earlier reports.34In the next step, the binding energies of the DA molecule to the rGO sheet were calculated at different possible sites (shown in Fig. S8†). The calculated binding energy (Eb) is defined as follows:
| Eb = E(rGO+DA) − (ErGO + EDA) |
| Cross-linking | P_1 | P_2 | V_1 | V_2 | V_3 | |
|---|---|---|---|---|---|---|
| B.E. of DA onto rGO (eV) | 3.39 | 0.3519 | 0.3166 | 0.2058 | 0.2256 | 0.34 |
However, interestingly, when DA was chosen as a cross-linker in binding adjacent rGO sheets, the binding energy of the covalently linked structure was found to be even higher (Table 1). Fig. 3a and b show the top and side view of the assembly of rGO sheets via the amide linkage with the DA molecule. Here, the dopamine (DA) molecule acts as a linker with a high binding energy of −3.39 eV and plays an important role in assembling the rGO sheets. One end of DA forms the amide (–CO–NH–) linkage with the –COOH of the rGO whereas its other end connects via O-atoms (by eliminating the 2H2O/DA molecule) to the C-atom of rGO. This further substantiates our proposed mechanism of DA serving as a cross-linker in forming the 3D interconnected macroporous structure.
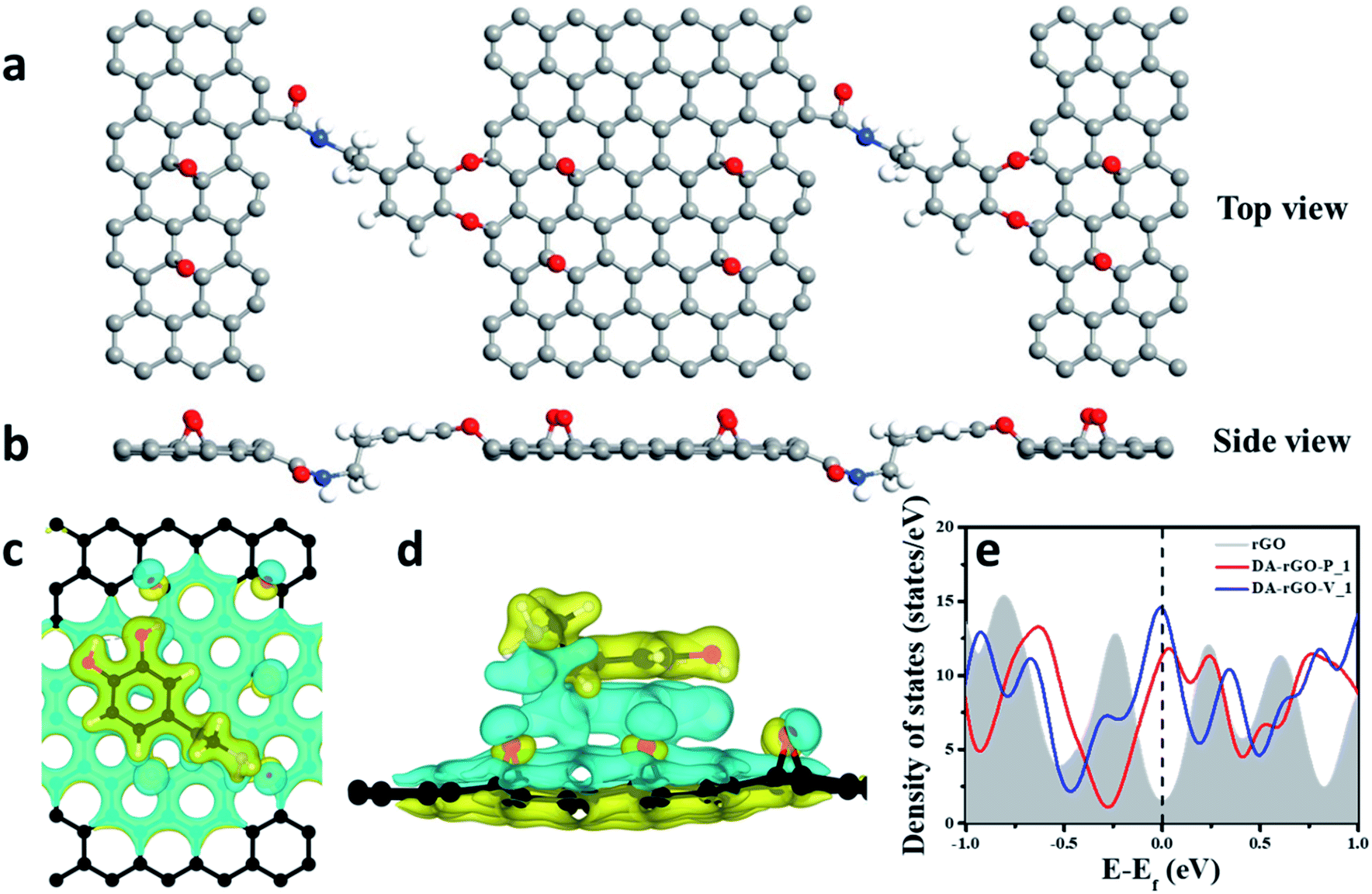 | ||
| Fig. 3 (a) Top and (b) side view of the assembly of rGO sheets via the amide cross-linkage with DA molecules calculated by DFT calculations. (c) Top and (d) side view of the charge density difference plot of the DA molecule over the rGO sheet (at the highest binding-energy site assigned as P_2). Charge accumulation and depletion are represented by the yellow and cyan regions, respectively. The iso-surface level is ±0.07 e Bohr−3. (e) Total density of states (DOS) for pristine rGO and DA-rGO in the parallel as well as vertical stacked position. | ||
Furthermore, we attempted to ascertain the origin of the larger strength of interaction for DA molecules on the graphene surface in the parallel conformation, when both the moieties are super-imposed on each other (P_1) (Fig. 3c and d). A larger charge re-distribution on rGO is caused by DA in the P_1 position on account of the perfect alignment of the π-cloud of rGO on DA due to the availability of oxygen atoms in rGO with the primary amine group of DA as shown in Fig. 3c and d, leading to larger charge distribution.
In addition, the total DOS for pristine rGO and DA-rGO in both parallel and vertical orientations has been calculated (Fig. 3e). The DOS profile shows a large increase in the density of states in DA-rGO with respect to rGO around the Fermi level. These electronic states will take part in current conduction upon the application of bias voltage and thereby contribute to an enhanced conductivity relative to rGO. Compared to previously reported redox-based graphene systems,6 the peaks observed in the present case lie much higher than the Fermi level. These empty bands help in providing more capacitance to store charges. This clearly implies that the pseudocapacitance mainly originates from the carbonyl groups of the hybrid structure, which is consistent with the proposed mechanism.
Electrochemical characterization
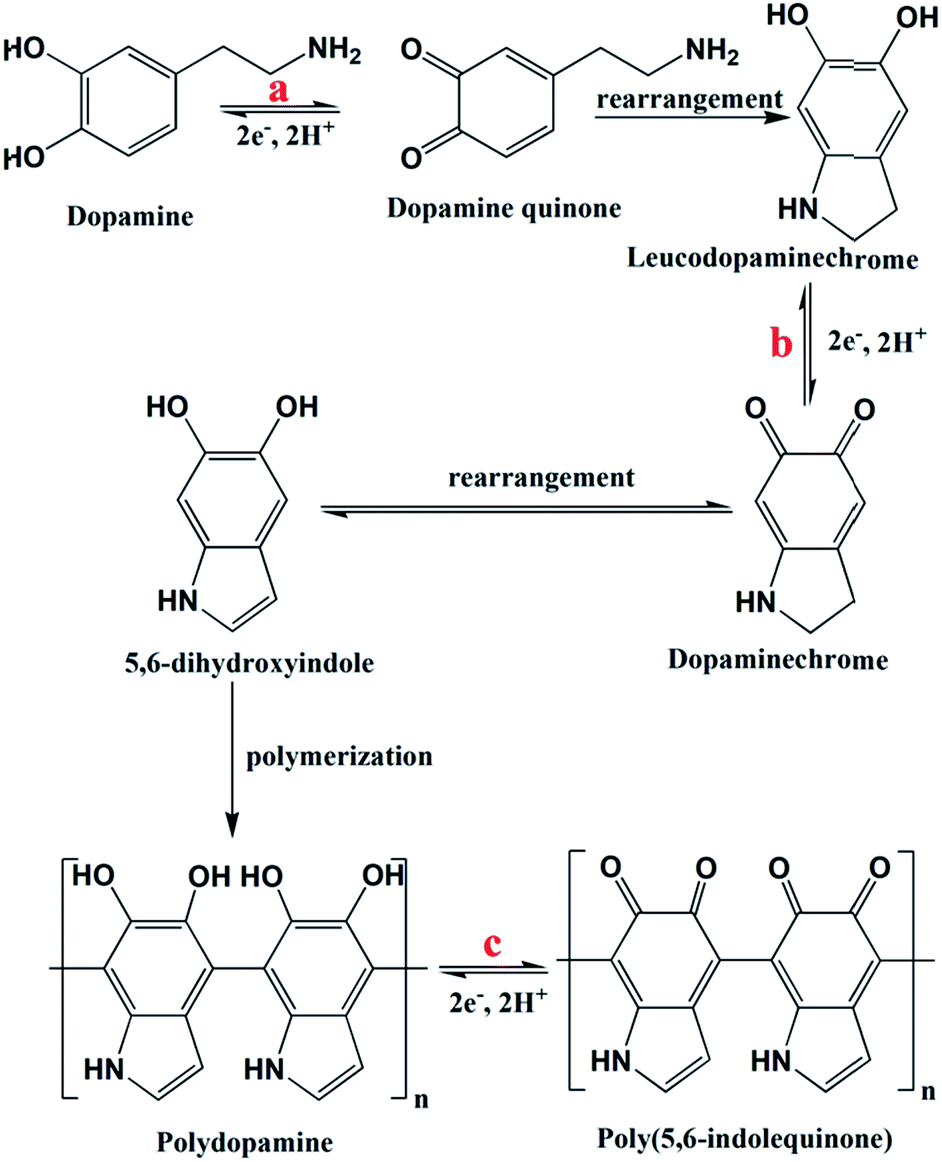 | ||
| Fig. 4 Proposed reaction mechanism of DA-rGO to in situ EpDA-rGO during potential cycling. | ||
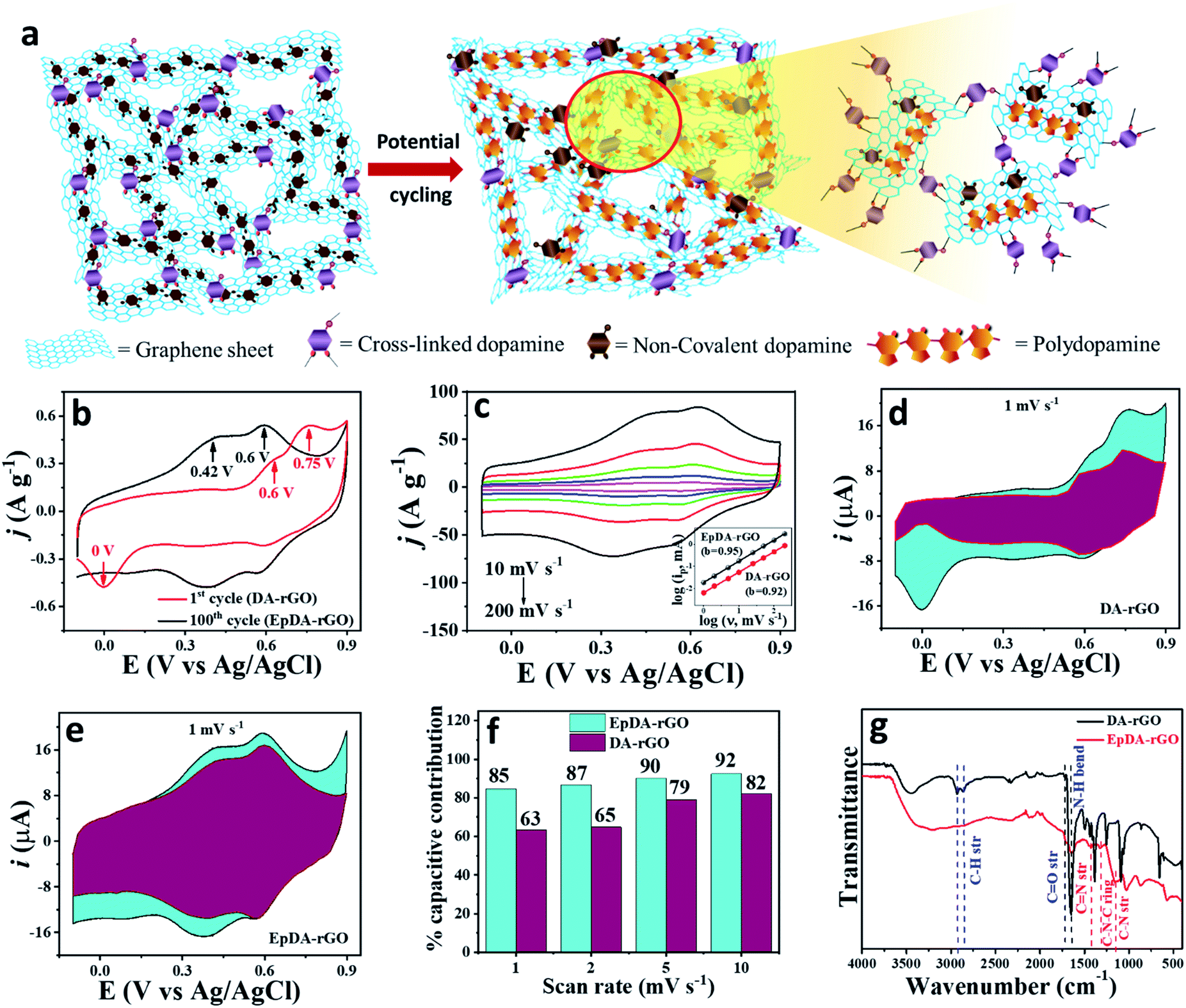 | ||
| Fig. 5 (a) Schematic demonstration of the 3D DA-rGO cross-linked xerogel and its subsequent polymerization to EpDA-rGO after potential cycling; magnified image showing the in situ EpDA-rGO sheets through both covalent and non-covalent linkage. (b) CV at a scan rate of 1 mV s−1 of DA-rGO showing contribution to faradaic capacitance (red curve) caused by loading of DA, and improvement in overall capacitance (black curve) caused by a simple in situ electrochemical conversion to EpDA. (c) Highly reversible cyclic voltammograms of EpDA-rGO at various scan rates; the inset showing the logi vs. logν plot of the anodic peak currents at 0.6 V for both DA-rGO and EpDA-rGO. (d)–(f) Quantitative capacitive analysis of the charge storage process of both DA-rGO and EpDA-rGO electrodes and their comparative study; the capacitive (purple) and faradaic (bright cyan) contribution of (d) DA-rGO and (e) EpDA-rGO at a sweep rate of 1 mV s−1. (f) The ratio of capacitive contribution to the overall capacitance at various scan rates. (g) FTIR-ATR spectra showing generation of characteristic peaks of EpDA after electrochemical cycling. | ||
The change in the electrode active material structure was also confirmed by the FTIR-ATR analysis (Fig. 5g). The disappearance of the alkane C–H stretching frequency (2860 cm−1)23,25 of DA-rGO in the EpDA-rGO structure clearly indicates the formation of the 5-membered ring in 5,6-dihydroxyindole via rearrangement, as described in Fig. 4. An intense peak corresponding to the N–H bending39 frequency of the π-conjugated free DA species on the rGO sheet in DA-rGO almost vanished in the EpDA-rGO structure, confirming the polymerization process. On the other hand, several important characteristic stretching frequencies appear in the FTIR-ATR analysis of the EpDA structure, for example –CN stretching (1427 cm−1),41,42 C–N stretching in secondary amine (1152 cm−1),41,43 C–N–C ring vibration (1368 cm−1),25etc. which confirms the formation of in situ EpDA during potential cycling. The in situ polymerization not only helps in better π–π conjugation and contributes to higher overall capacitance as compared to only the DA-rGO xerogel material, but is also responsible for improved cycling stability by preventing the loss of loosely bound DA species from the hybrid structure. In order to get the composition-dependent charge-storage capacity, the cyclic voltammogram of the EpDA-rGO electrode was compared with those of pristine GO and rGO modified electrodes at a scan rate of 100 mV s−1, as shown in Fig. S12a.† EpDA-rGO electrodes show much-expanded CVs (150 times higher CSP than GO and 10 times higher CSP than rGO, respectively) with pronounced redox peaks due to the in situ electrochemical conversion after potential cycling. To understand the shift in the characteristic redox peaks in EpDA-rGO as compared to DA-rGO, chemically synthesized (similar hydrothermal treatment) pDA anchored on the rGO sheet (denoted as pDA-rGO) was also studied for its capacitive behaviour. Although similar oxidation peaks (centered at 0.42 V and 0.6 V) are observed in the pDA-rGO electrode, the decreased area under the CV curve (1.5 times lesser CSP) clearly demonstrates the advantage of the in situ electrochemical conversion process. Another electrode was also modified with rGO and DA aqueous solution (denoted as rGO/DA) to see whether the DA-rGO hybrid can be achieved electrochemically. But as exhibited in Fig. S12a,† there is an insubstantial current response in the rGO/DA electrode, which may be due to the presence of the loosely absorbed DA moiety only at the rGO surface and clearly an absence of any potential interaction between DA and rGO. This observation promotes the advantage of the economical hydrothermal method followed by potential cycling. In order to further establish the advantage of our facile hydrothermal synthesis method followed by novel in situ electropolymerization, another pure EpDA-rGO electrode was fabricated, following methods reported in the literature. Typically, the hydrothermally derived rGO material was subjected to potential CV cycling in the experimental electrolyte containing 1 mg mL−1 DA for obtaining the pure EpDA-rGO electrode. As can be seen in Fig. S12b† (magnified version of Fig. S12a†), the as-obtained pure EpDA-rGO electrode material exhibits specific capacitance (CSP = 33 F g−1) in the range of that of the hydrothermally obtained rGO electrode (CSP = 27 F g−1); whereas our optimized EpDA-rGO electrode (CSP = 263 F g−1) shows much higher specific capacitance. This once again substantiates the importance of the facile hydrothermal synthesis method that helps in covalently linking the DA species onto the rGO skeleton and helps in improving the overall capacitance of the composite material drastically.
To demonstrate the effect of DA loading on the capacitance, a comparative CV study was carried out by synthesizing various DA-rGO xerogels with different ratios (w/w) of DA to GO via hydrothermal treatment followed by potential cycling and the results are presented in Fig. S12c.† A strong dependence between the initial DA loading in the hybrid material and capacitance was observed. The change in specific capacitance as a function of the DA weight ratio, at a scan rate of 100 mV s−1, is shown in a bar diagram in Fig. S12d.† The highest CSP (263 F g−1) is observed with EpDA-rGO1–2 mass loading. Therefore, a detailed electrochemical analysis of the optimized EpDA-rGO1–2 material was carried out. Fig. S12e† demonstrates the effect of varying DA concentration on both the CSP and rate capability of the electrodes at a sweep rate of 10–1000 mV s−1. EpDA-rGO1–2 not only shows the highest CSP, the rate capability of this particular composition is also the best. Even at a high scan rate of 1000 mV s−1, it can retain 74% of its initial capacitance at 10 mV s−1, demonstrating the excellent rate capability of the optimized material.
Galvanostatic charge–discharge (GCD) analysis was further carried out as the best practice method44 to assess the advantage of the in situ potential cycling more precisely. The charge–discharge profile of the optimized EpDA-rGO1–2 (in a wide range of current densities; from 0.5 A g−1 to 500 A g−1), as depicted in Fig. 6a, also corroborated the results obtained from CV. A highest CSP of 348 F g−1 was achieved at a current density of 0.5 A g−1. The shape of the discharge curve also suggests the pseudocapacitive contribution to the overall capacitance, with varying slopes from 0.9 V to 0.6 V (capacitive contribution) and 0.6 V to 0 V (faradaic contribution). Aiming for high energy density without compromising on rate capability was another aspect of our study, which was satisfied when a highest energy density of 48.26 W h kg−1 was also registered with EpDA-rGO1–2 at a current density of 0.5 A g−1. The comparative Ragone plots of all the various compositions are collectively exhibited in Fig. S12f† that clearly shows the superior performance of EpDA-rGO1–2 in terms of both energy and power density.
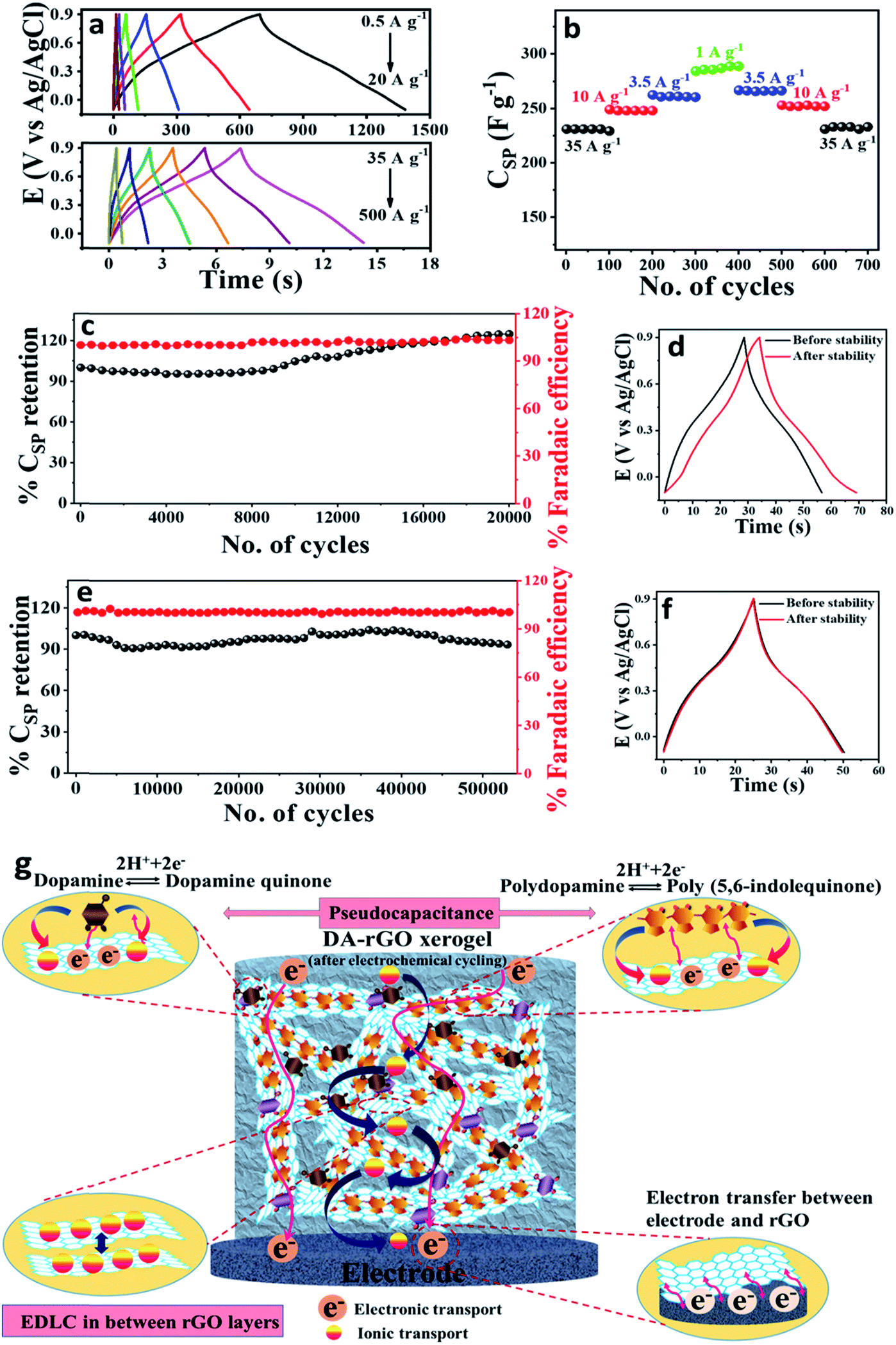 | ||
| Fig. 6 Electrochemical characterization of the electrochemically in situ polymerized EpDA-rGO1–2 samples in 1 M H2SO4 aqueous electrolyte. (a) Galvanostatic charge–discharge (GCD) profile at various current densities. (b) Continuous cycling stability study at various current densities for 100 charge–discharge cycles each to indicate rate capability and cycling reversibility. Cycling stability and retention of faradaic efficiency study at a current density of (c) 10 A g−1 for 20000 cycles with an initial capacitance retention of 125% and (e) 35 A g−1 for 53000 cycles with an initial capacitance retention of 94%. (d) and (f) show GCD analysis before and after the stability study. (g) Schematic illustration of the ionic and electronic transport taking place in the 3D network of the EpDA-rGO1–2 structure. | ||
The conductivity and ease of ionic transport of the EpDA-rGO1–2 electrode are further characterized by electrochemical impedance spectroscopy (EIS). As observed in the high frequency region of the Nyquist plot (Fig. S13a†), the small semi-circular region corresponding to the EpDA-rGO1–2 may stem from a negligible interfacial charge transfer resistance.45 This confirms the high conductivity of the hybrid structure even after the incorporation of the DA moiety. Similarly, in the low frequency range, EpDA-rGO1–2 shows slight deviation from the vertical line, as compared to the pristine rGO electrode, which may be ascribed to the increase of Warburg resistance due to the presence of the pseudocapacitive DA molecule between the rGO sheets. The dependence of the phase angle on the applied frequency was evaluated in the corresponding Bode plot, as exhibited in Fig. S13b.† The frequency at which the phase angle reaches 45° is known as the knee frequency (f0), where the capacitive and resistive impedances are equal. The relaxation time (τ0 = 1/f0) denotes the minimum time required to discharge all the energy from the storage system with an efficiency of greater than 50%.46 The relaxation time for the EpDA-rGO1–2 hybrid electrode (157 ms) was far less than that of the pristine rGO electrode (257 ms), indicating faster ion-transport indicating faster ion-transport indicating faster ion-transport kinetics and good electrical conductivity.
000 GCD cycles (Fig. 6c), which exhibits around 125% of the initial capacitance retention. A slight enhancement in the specific capacitance can be observed after GCD cycling measurements (Fig. 6d) as a result of the increase in the active redox sites of the electrode surface. Fig. 6c also shows the retention of coulombic or faradaic efficiency around 103.2% indicating a slight involvement of faradaic processes during the GCD cycling test. Therefore, instigated by the excellent capacitance retention at 10 A g−1, the cycling durability of the EpDA-rGO1–2 electrode was investigated at further lower current densities (1 and 3.5 A g−1) for various galvanostatic charge–discharge cycles (500 and 2000 cycles, respectively) (Fig. S14†). The inset of all the stability plots in Fig. S14† represents the charge–discharge scans before and after the stability study. On the other hand, the rate capability and power delivery of a supercapacitor system are always best evaluated by its performance at higher current densities, where the charge–discharge process is very fast. Fig. 6d shows that 94% of the initial capacitance is retained by the EpDA-rGO1–2 electrode even after 53000 continuous GCD cycles at a very high current density of 35 A g−1. Interestingly, the EpDA-rGO1–2 electrode shows more than 93% retention of the initial capacitance at all the above-mentioned current densities, promoting it as the most versatile pseudocapacitors reported till date, for commercial use. It exceeds the cycling stability of various xerogel materials, for example – DMQ@rGO (99% CSP retention after 25000 CV cycles at 5 mV s−1),6 PDA-rGO (99% CSP retention after 10000 GCD cycles at 2 A g−1)47 or AQS@rGO (89.1% after 10000 GCD cycles at 10 A g−1).7 It even surpasses the cycling stability of a recently reported carbon-CP composite material, PVA/G/PAni-4C/G, showing 82% capacitance retention after 50000 continuous GCD cycles19 (Table 2). The impressive cycling stability may be attributed to a number of reasons: (a) the unique 3D interconnected porous architecture with uniform distribution of the DA moiety that ensured a continuous conductive pathway for rapid charge-transfer kinetics without the depletion of the electrode material in the electrolyte; (b) covalent interaction of the DA moiety to the rGO sheet via the amide linkage, which not only provides higher charge accumulation and a depletion region as compared to only π–π interactions, but also prevents the aggregation and degradation of the organic molecule during rigorous charge–discharge cycling; (c) in situ electropolymerization also plays a dynamic role in enhancing the charge-distribution on the rGO surface via the formation of EpDA therefore effectively preventing the parasitic side reactions and enhancing the cycle-life; (d) the basal plane of the rGO sheets provides excellent mechanical stability and conductivity that ensured continuous electronic channels even at high current density for stable electrochemical performances. GCD curves before and after 53000 cycles are exhibited in Fig. 6d that clearly shows that the shape of the curve remains intact with negligible decay in the discharge time, indicating excellent integration of the active electrode material onto the substrate surface. Fig. 6d also shows the retention of coulombic or faradaic efficiency around 100.4% indicating an absence of any side reactions or faradaic processes during the GCD cycling test.
| Material | Electrolyte | % Retention (cycle number) | Scan-rate, current density | References |
|---|---|---|---|---|
| a EpDA – electropolymerized dopamine; rGO – reduced graphene oxide; PQ – 9,10-phenanthrenequinone; OLC – onion-like carbon; DMQ – 2,5-dimethoxy-1,4-benzoquinone; AQS – anthraquinone-2-sulfonate; Th – thionine; GA – graphene aerogel; PVA – polyvinyl alcohol; G – graphene; PANI – polyaniline; PPy – polypyrrole; Ex-GF – exfoliated graphite foil; NDS – 1,5-naphthalene disulphonate; GNS – graphene nanosheets. | ||||
| EpDA-rGO | H2SO4 | 94% (53000) |
35 A g−1 | This work |
| PQ-OLC | H2SO4 | 97% (10000) |
1.3 A g−1 | 50 |
| DMQ@rGO | H2SO4 | 99% (25000) |
50 mV s−1 | 6 |
| AQS@rGO | H2SO4 | 89.1% (10000) |
10 A g−1 | 7 |
| Th-GA | H2SO4 | 91% (10000) |
10 A g−1 | 8 |
| PVA/G/PAni-4C/G | H2SO4 | 82% (50000) |
50 mA | 19 |
| Ex-GF/PPy-NDS | KCl | 82% (1000) | 5 A g−1 | 54 |
| Co3O4/GNS | KOH | 70% (4000) | 0.2 A g−1 | 53 |
020 W kg−1 was achieved even while maintaining the specific energy at 9.04 W h kg−1. A highest volumetric energy density of 70.7 W h L−1 with a highest power density of 30.57 W L−1 was measured based on the thickness of the material in the device. In order to further demonstrate the excellent storage performance as well as instant power delivery capacity of our as-fabricated device, Ragone plot comparison has been drawn with some previously reported graphene hydrogel/aerogel based solid state devices and is depicted in Fig. S15e.† It shows much superior energy as well as power density as compared to LS-GHs (13.8 W h kg−1, 5000 W kg−1),49 BN-GAs (8.7 h kg−1, 1650 W kg−1)50 and OMC-GAs (6.2 W h kg−1, 3545 W kg−1).51 A detailed comparative electrochemical performance analysis to contemporary composite pseudocapacitors including CPs, metal-oxides and similar organic redox centre-based xerogel materials as well as devices is provided in Table S2.† The EpDA-rGO1–2 electrode material exhibits a highest energy density of 48.26 W h kg−1 and the EpDA-rGO//EpDA-rGO symmetric solid-state device shows a highest energy density of 30.3 W h kg−1, much higher than those of most of the reported organic electro-active xerogel pseudocapacitors, for example – AZ-SHGs (18.2 W h kg−1),5 PQ-OLCs (4.5 W h kg−1),52 AQ@CFs (19.3 W h kg−1),53 PPD-C-DCNTs (19.1 W h kg−1),14etc. It even shows superior or almost comparable energy density to those of mostly reported carbon-CP composites, for example – PANI-graphene foam (43.64 W h kg−1)54 or carbon-TMO composite structures, like Co3O4/GNS (21.9 W h kg−1),55etc. Although there have been some reports mentioning higher energy density, for instance – DMQ@rGO xerogel (113.4 W h kg−1),6 or composites of carbon-CPs, like – Ex-GF/PPy-NDS (82.4 W h kg−1)56 or PAG80 (66.67 W h kg−1),57 the solid-state symmetric device shows an excellent power delivery of 13.02 kW kg−1, which is better than those of most of the reported two-electrode systems. For example – AZ-SHGs (0.7 kW kg−1),5 PPD-C-DCNTs (0.8 kW kg−1),14 TH-GAs (8.7 kW kg−1),8 or Ex-GF/PPy-NDS (13 kW kg−1)56 or Co3O4/GNS (2.19 kW kg−1).55 Our EpDA-rGO1-2 electrode demonstrates excellent electrochemical activity due to an enhanced ionic and electronic conductivity coming from a novel synthesis strategy of both covalent and non-covalent interactions.
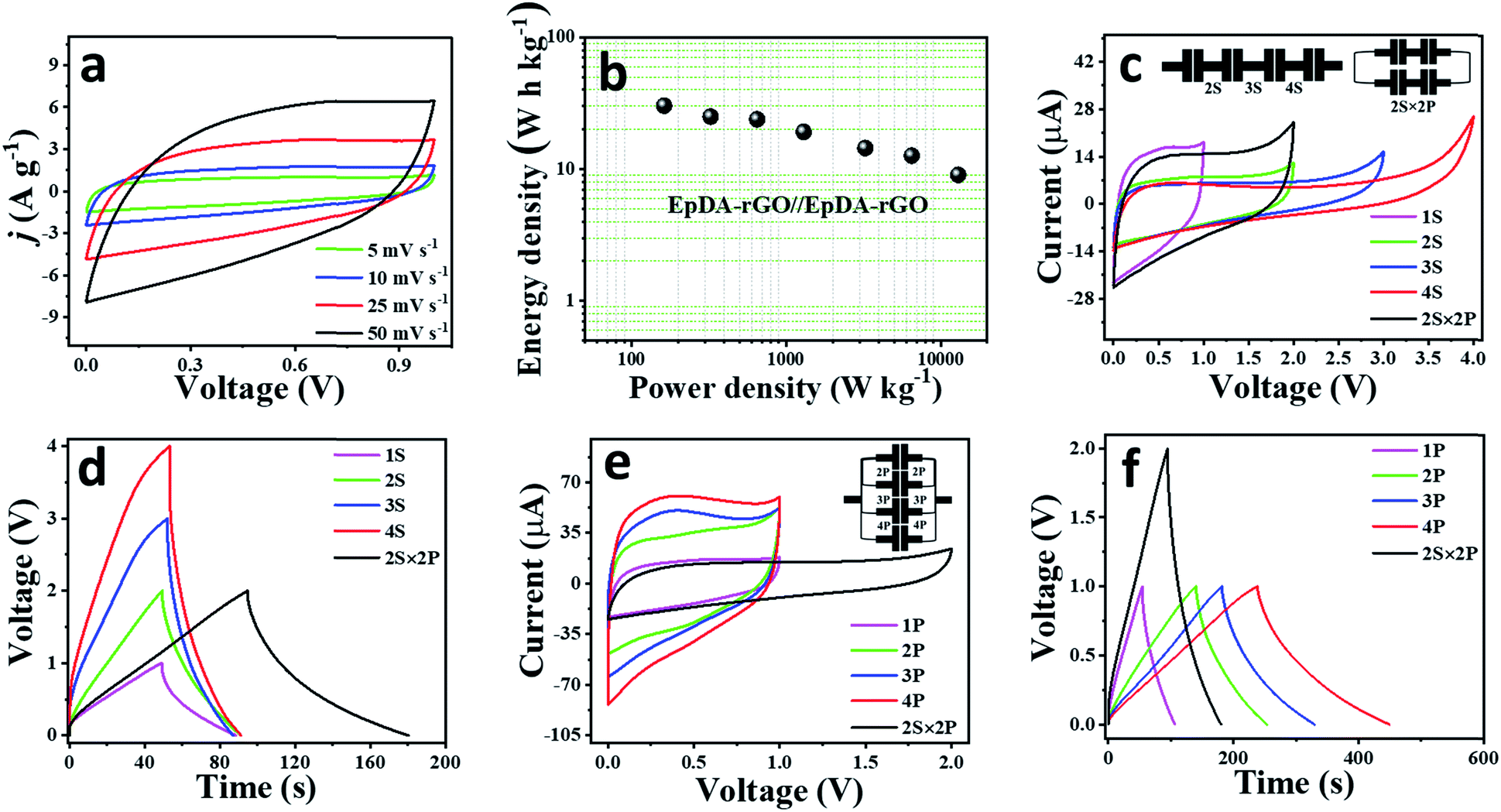 | ||
| Fig. 7 (a) Cyclic voltammograms at various scan rates of the symmetric all-solid-state planar device, EpDA-rGO//EpDA-rGO in 1 M PVA/H2SO4 solid state gel electrolyte. (b) Ragone plot of the device. (c)–(e) Integration of such planar devices in various series and parallel combinations and their electrochemical response. (c) CV curves at a scan rate of 10 mV s−1 and (d) GCD profiles at a current density of 2.6 A g−1 obtained from series (1S to 4S) as well as the 2S × 2P configuration, the inset of (c) showing the schematic representation of the linear devices arranged in series and 2S × 2P configurations. Similarly (e) and (f) show CV and GCD responses of the devices respectively, arranged in parallel (1P to 4P), compared with the 2S × 2P configuration, the inset of (e) showing the schematic representation of the linear devices arranged in the parallel configuration. | ||
The fact that the specific energy and power delivery capability of a single device are often not sufficient to run commercial devices, it is generally preferred to package the cells in various series and parallel arrangements or a combination of both to meet the energy and power requirements.58 To meet the demand of high voltage, four as-fabricated solid-state linear devices were arranged in series, while to improve the current response and capacitance, they were connected in a parallel configuration. The electrochemical performance of the tandem devices was investigated in terms of their CV and GCD responses. As can be observed in Fig. 7c and d, the series combination of four devices exhibited an enhanced potential window of 4 V as compared to 1 V of a single device. The inset of Fig. 7c schematically shows the series configuration of the four devices. Similarly, when arranged in a parallel fashion, the tandem configuration shows an almost 4-fold increase in the current response as compared to a single device both in CV (Fig. 7e) and GCD (Fig. 7f) profiles. The inset of Fig. 7e schematically shows the parallel configuration of the four devices. It is highly required that by the tandem configuration of the individual cells, the highest current response is attained in the highest operating potential window, leading to minimum energy loss. Therefore, the performances of the four devices were also evaluated by arranging two of them in series and then connecting the two series devices in a parallel fashion (2S × 2P configuration). As expected, the 2S × 2P tandem configuration shows an enhanced potential window of 2 V while improving the current response to 2-fold as compared to two devices in series (Fig. 7c–f).
One of the most routine features of any commercial device is to have a long service-life. Based on the unprecedented cycling performance of our active electrode material; it was highly anticipated that the symmetric solid-state device, EpDA-rGO//EpDA-rGO will eventually show excellent cycling stability. However, in a solid-state gel electrolyte, the increased resistance at the electrolyte–electrode interface hampers the rate performance and eventually the device performance suffers with continuous charge–discharge cycles. In spite of this, in Fig. 8a, the device shows a remarkable capacitance retention of 92% even after 10000 continuous GCD cycles at a very high current density of 10 A g−1, which is much superior to those of most of the gel-electrolyte based pseudocapacitors reported to date, for example – LS-GHs (84% (10000) @ 2 A g−1)49 or rGO/MoO3 (80% (5000) @ 2 A g−1)59 (Table S2†). Fig. 8a also shows the retention of coulombic efficiency around 100% indicating an absence of any side reactions during the GCD cycling test. The inset of Fig. 8a shows the first and last four cycles revealing almost unchanged GCD profiles, indicating negligible ohmic drop in the device.
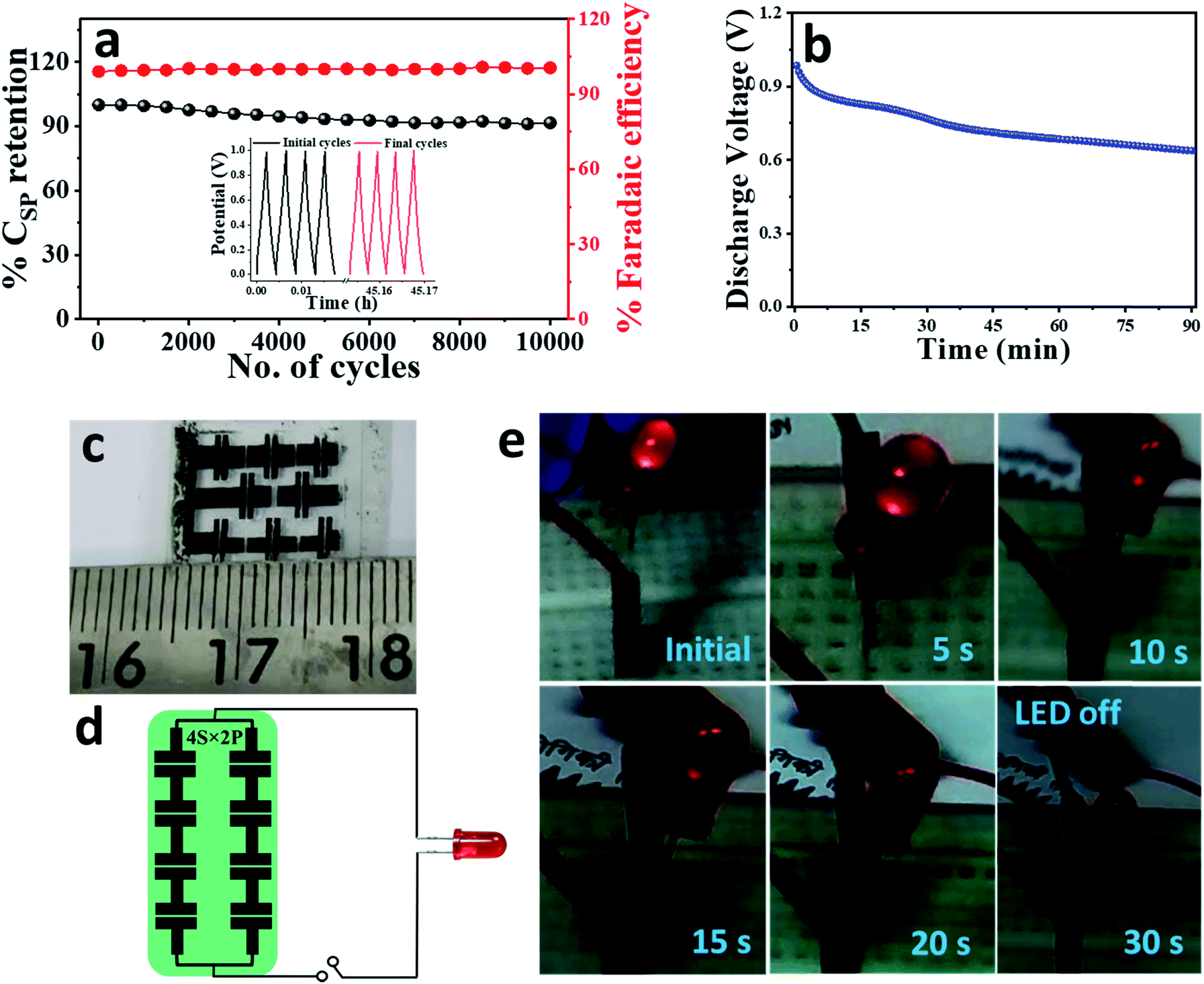 | ||
| Fig. 8 (a) Percentage capacitance retention (92%) as well as faradaic efficiency of the solid-state device, EpDA-rGO//EpDA-rGO after 10000 continuous cycles at a high current density of 10 A g−1, the inset showing GCD profiles of the first and last four cycles. (b) Self-discharge study of a single charged supercapacitor device. (c) Digital photograph of the laser-patterned linear devices. (d) Integration of the devices in a 4S × 2P array exhibited schematically for powering up an LED. (e) Digital images showing the lighting of a single LED device for 30 s after charging the module in (d) at 4 V for 1 minute. | ||
To endorse the development of self-supported metal free light-weight supercapacitors for portable electronic applications, a number of studies were performed with the EpDA-rGO//EpDA-rGO device as a power source. At first, the discharging performance of a single device was evaluated by charging it at 3 V for 3 minutes using a commercial DC power source. Self-discharge is a valuable performance metric and of major practical significance for pseudocapacitors. After 3 minutes, the device was able to sustain almost 70% of the cell voltage even after 1.5 h. Fig. 8b shows the discharge voltage profile of the device as a function of time. The device performance was also validated by its ability to power up a commercial 1.6 V LED system. The digital image of the as-patterned in-plane devices is shown in Fig. 8c. The devices were arranged in a 4S × 2P tandem configuration as schematically shown in Fig. 8d and then charged at 4 V for 1 minute. As can be seen in Fig. 8e, after charging, the tandem system was able to power up the LED for 30 seconds continuously, demonstrating the practical viability of the EpDA-rGO//EpDA-rGO symmetric device.
The overall material as well as device performance as a pseudocapacitor promotes EpDA-rGO as a potential future storage material, ready to be employed in commercial supercapacitors.
Conclusions
In summary, a unique combination of covalent and non-covalent functionalization of rGO via an organic redox species, dopamine (DA) has been explored to develop a high-rate pseudocapacitor with outstanding cycle-life. The availability of two electron donating groups, i.e., hydroxyl and alkyl amine functionalities at the para positions in the DA molecule promotes its easy covalent interaction with rGO, during the hydrothermal reduction process, whereas the aromatic π cloud helps in non-covalent interaction or molecular level binding; which was further confirmed by theoretical DFT analysis. Additionally, in this study, we have also explored the well-known polymerization tendency of DA to form pDA by cycling the xerogel material in the experimental electrolyte and generating the in situ EpDA-rGO xerogel material. Among various tested electrodes, EpDA-rGO1-2 exhibits the highest specific capacitance of 348 F g−1 at a current density of 0.5 A g−1. In situ electro-polymerization induces large charge-distribution over the rGO surface via the formation of EpDA, preventing the parasitic side reactions of the organic species and leading to an outstanding cycling stability of 94% after 53000 continuous GCD cycles at 35 A g−1. Additionally, an all-solid-state symmetric planar supercapacitor device was fabricated using the EpDA-rGO1-2 active electrode material, which delivers an exceptional energy density of 30.3 W h kg−1, one of the highest specific energies delivered by solid-state symmetric graphene hydrogel/aerogel-based pseudocapacitors, reported to date. Even the device shows an exceptional cycling stability of 92% after 10000 GCD cycles at a very high current density of 10 A g−1. The combined output from both the theoretical study and experimental findings helps us to conclude that the careful selection of the organic moiety ensures optimum interaction with the conductive substrate which in turn helps in addressing the cycling performance of pseudocapacitors. This work is a unique attempt to study the mechanism of redox supercapacitors at the molecular level and offers new insights into how to improve the long-standing issue of stability and inferior power output of pseudocapacitors. It may promote future research in the field of organic pseudocapacitors as suitable candidates for commercial applications.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
TP acknowledges DST INSPIRE and CSIR SRF for providing a fellowship and Panjab University for Ph. D. registration. This work was financially supported by DST INSPIRE (DST/INSPIRE/04/2015/000337) and DST SERB EMR (EMR/2016/000040). The authors acknowledge INST, Mohali for instrumental support. Our sincere thanks are extended to CDAC, Pune's supercomputing resources on PARAM YUVA II for theoretical calculations.Notes and references
- M. Kim, C. Lee and J. Jang, Adv. Funct. Mater., 2014, 24, 2489–2499 CrossRef CAS.
- A. Jaffe, A. Saldivar Valdes and H. I. Karunadasa, Chem. Mater., 2015, 27, 3568–3571 CrossRef CAS.
- V. Augustyn, P. Simon and B. Dunn, Energy Environ. Sci., 2014, 7, 1597–1614 RSC.
- L. Madec, A. Bouvrée, P. Blanchard, C. Cougnon, T. Brousse, B. Lestriez, D. Guyomard and J. Gaubicher, Energy Environ. Sci., 2012, 5, 5379–5386 RSC.
- N. An, Y. An, Z. Hu, B. Guo, Y. Yang and Z. Lei, J. Mater. Chem. A, 2015, 3, 22239–22246 RSC.
- M. Boota, C. Chen, M. Bécuwe, L. Miao and Y. Gogotsi, Energy Environ. Sci., 2016, 9, 2586–2594 RSC.
- R. Shi, C. Han, H. Duan, L. Xu, D. Zhou, H. Li, J. Li, F. Kang, B. Li and G. Wang, Adv. Energy Mater., 2018, 8, 1802088 CrossRef.
- Y. Shabangoli, M. S. Rahmanifar, M. F. El-Kady, A. Noori, M. F. Mousavi and R. B. Kaner, Adv. Energy Mater., 2018, 8, 1802869 CrossRef.
- T. Tomai, S. Mitani, D. Komatsu, Y. Kawaguchi and I. Honma, Sci. Rep., 2014, 4, 1–6 Search PubMed.
- A. Yu, A. Davies and Z. Chen, in Electrochemical Technologies for Energy Storage and Conversion, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2012, 1st edn, pp. 317–382 Search PubMed.
- Y. Liang, Z. Tao and J. Chen, Adv. Energy Mater., 2012, 2, 742–769 CrossRef CAS.
- K. Kalinathan, D. P. DesRoches, X. Liu and P. G. Pickup, J. Power Sources, 2008, 181, 182–185 CrossRef CAS.
- G. Pognon, C. Cougnon, D. Mayilukila and D. Bélanger, ACS Appl. Mater. Interfaces, 2012, 4, 3788–3796 CrossRef CAS PubMed.
- Y. He, X. Yang, N. An, X. Wang, Y. Yang and Z.-A. Hu, New J. Chem., 2019, 43, 1688–1698 RSC.
- Q. Wu, Y. Sun, H. Bai and G. Shi, Phys. Chem. Chem. Phys., 2011, 13, 11193–11198 RSC.
- L. Hamenu, A. Madzvamuse, L. Mohammed, M. Hu, J. Park, M. H. Ryou, Y. M. Lee and J. M. Ko, Synth. Met., 2017, 231, 25–33 CrossRef CAS.
- S. Roldán, M. Granda, R. Menéndez, R. Santamaría and C. Blanco, J. Phys. Chem. C, 2011, 115, 17606–17611 CrossRef.
- S. Roldán, C. Blanco, M. Granda, R. Menéndez and R. Santamaría, Angew. Chem., Int. Ed., 2011, 50, 1699–1701 CrossRef PubMed.
- A. Khosrozadeh, G. Singh, Q. Wang, G. Luo and M. Xing, J. Mater. Chem. A, 2018, 6, 21064–21077 RSC.
- T. Purkait, G. Singh, N. Kamboj, M. Das and R. S. Dey, J. Mater. Chem. A, 2018, 6, 22858–22869 RSC.
- R. S. Dey, H. A. Hjuler and Q. Chi, J. Mater. Chem. A, 2015, 3, 6324–6329 RSC.
- L. Q. Xu, W. J. Yang, K. G. Neoh, E. T. Kang and G. D. Fu, Macromolecules, 2010, 43, 8336–8339 CrossRef CAS.
- T. Yadav and V. Mukherjee, J. Mol. Struct., 2018, 1160, 256–270 CrossRef CAS.
- J. E. Carter, J. H. Johnson and D. M. Baaske, Anal. Profiles Drug Subst. Excipients, 1982, 11, 257–272 CAS.
- R. A. Zangmeister, T. A. Morris and M. J. Tarlov, Langmuir, 2013, 29, 8619–8628 CrossRef CAS PubMed.
- X. Jian, H. min Yang, J. gang Li, E. hui Zhang, L. le Cao and Z. hai Liang, Electrochim. Acta, 2017, 228, 483–493 CrossRef CAS.
- E. Desimoni and B. Brunetti, Chemosensors, 2015, 3, 70–117 CrossRef CAS.
- H. Gao, Y. Sun, J. Zhou, R. Xu and H. Duan, ACS Appl. Mater. Interfaces, 2013, 5, 425–432 CrossRef CAS PubMed.
- F. Bernsmann, A. Ponche, C. Ringwald, J. Hemmerlé, J. Raya, B. Bechinger, J.-C. Voegel, P. Schaaf and V. Ball, J. Phys. Chem. C, 2009, 113, 8234–8242 CrossRef CAS.
- R. S. Dey, S. Hajra, R. K. Sahu, C. R. Raj and M. K. Panigrahi, Chem. Commun., 2012, 48, 1787 RSC.
- V. K. Thakur, M. F. Lin, E. J. Tan and P. S. Lee, J. Mater. Chem., 2012, 22, 5951–5959 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts and B. Me, Gaussian 16, Revision B. 01, 2016, 1 Search PubMed.
- R. Dennington, T. A. Keith and J. M. Millam, GaussView, Version 6.1, 2016 Search PubMed.
- D. García-Toral, M. González-Melchor, J. F. Rivas-Silva, E. Meneses-Juárez, J. Cano-Ordaz and G. H. Cocoletzi, J. Phys. Chem. B, 2018, 122, 5885–5896 CrossRef PubMed.
- J. lei Wang, B. chao Li, Z. jun Li, K. feng Ren, L. jiang Jin, S. miao Zhang, H. Chang, Y. xin Sun and J. Ji, Biomaterials, 2014, 35, 7679–7689 CrossRef PubMed.
- M. D. Hawley, S. V. Tatawawadi, S. Piekarski and R. N. Adams, J. Am. Chem. Soc., 1967, 89, 447–450 CrossRef CAS.
- T. C. Liu, W. G. Pell, B. E. Conway and S. L. Roberson, J. Electrochem. Soc., 1998, 145, 1882 CrossRef CAS.
- S. Ardizzone, G. Fregonara and S. Trasatti, Electrochim. Acta, 1990, 35, 263–267 CrossRef CAS.
- V. Augustyn, J. Come, M. A. Lowe, J. W. Kim, P.-L. Taberna, S. H. Tolbert, H. D. Abruña, P. Simon and B. Dunn, Nat. Mater., 2013, 12, 518–522 CrossRef CAS PubMed.
- T. Brousse, D. Bélanger and J. W. Long, J. Electrochem. Soc., 2015, 162, A5185–A5189 CrossRef CAS.
- V. Krishnakumar and R. John Xavier, Spectrochim. Acta, Part A, 2004, 60, 709–714 CrossRef CAS.
- L. E. Clougherty, J. A. Sousa and G. M. Wyman, J. Org. Chem., 1957, 22, 462 CrossRef CAS.
- T. Noguchi, Y. Inoue and X. S. Tang, Biochemistry, 1999, 38, 399–403 CrossRef CAS PubMed.
- M. D. Stoller and R. S. Ruoff, Energy Environ. Sci., 2010, 3, 1294–1301 RSC.
- Q. Wu, Y. Xu, Z. Yao, A. Liu and G. Shi, ACS Nano, 2010, 4, 1963–1970 CrossRef CAS PubMed.
- W. Liu, C. Lu, H. Li, R. Y. Tay, L. Sun, X. Wang, W. L. Chow, X. Wang, B. K. Tay, Z. Chen, J. Yan, K. Feng, G. Lui, R. Tjandra, L. Rasenthiram, G. Chiu and A. Yu, J. Mater. Chem. A, 2016, 4, 3754–3764 RSC.
- S. Dong, Z. Xie, Y. Fang, K. Zhu, Y. Gao, G. Wang, J. Yan, K. Cheng, K. Ye and D. Cao, ChemistrySelect, 2019, 4, 2711–2715 CrossRef CAS.
- Z. Dai, C. Peng, J. H. Chae, K. C. Ng and G. Z. Chen, Sci. Rep., 2015, 5, 9854 CrossRef CAS PubMed.
- F. Li, X. Wang and R. Sun, J. Mater. Chem. A, 2017, 5, 20643–20650 RSC.
- Z. S. Wu, A. Winter, L. Chen, Y. Sun, A. Turchanin, X. Feng and K. Müllen, Adv. Mater., 2012, 24, 5130–5135 CrossRef CAS.
- R. Liu, L. Wan, S. Liu, L. Pan, D. Wu and D. Zhao, Adv. Funct. Mater., 2015, 25, 526–533 CrossRef CAS.
- D. M. Anjos, J. K. McDonough, E. Perre, G. M. Brown, S. H. Overbury, Y. Gogotsi and V. Presser, Nano Energy, 2013, 2, 702–712 CrossRef CAS.
- H. Wang, H. Yi, C. Zhu, X. Wang and H. Jin Fan, Nano Energy, 2015, 13, 658–669 CrossRef CAS.
- Y. Xu, Y. Tao, X. Zheng, H. Ma, J. Luo, F. Kang and Q. H. Yang, Adv. Mater., 2015, 27, 8082–8087 CrossRef CAS.
- Q. Guan, J. Cheng, B. Wang, W. Ni, G. Gu, X. Li, L. Huang, G. Yang and F. Nie, ACS Appl. Mater. Interfaces, 2014, 6, 7626–7632 CrossRef CAS.
- Y. Song, J. L. Xu and X. X. Liu, J. Power Sources, 2014, 249, 48–58 CrossRef CAS.
- K. Zhang, L. L. Zhang, X. S. Zhao and J. Wu, Chem. Mater., 2010, 22, 1392–1401 CrossRef CAS.
- N. Kamboj, T. Purkait, M. Das, S. Sarkar, K. S. Hazra and R. S. Dey, Energy Environ. Sci., 2019, 12, 2507–2517 RSC.
- X. Cao, B. Zheng, W. Shi, J. Yang, Z. Fan, Z. Luo, X. Rui, B. Chen, Q. Yan and H. Zhang, Adv. Mater., 2015, 27, 4695–4701 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedure, formula, SEM, AFM, XPS, Raman, BET, TGA, EDS mapping, computational analysis, Nyquist plot, stability study and other electrochemical data. See DOI: 10.1039/d0ta02477e |
| This journal is © The Royal Society of Chemistry 2020 |