
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDihydropyrimidinones: efficient one-pot green synthesis using Montmorillonite-KSF and evaluation of their cytotoxic activity†
Saleem Farooq *ab,
Fahad A. Alharthic,
Ali Alsalmec,
Aashiq Hussaind,
Bashir A. Dare,
Abid Hamid*df and
S. Koulb
*ab,
Fahad A. Alharthic,
Ali Alsalmec,
Aashiq Hussaind,
Bashir A. Dare,
Abid Hamid*df and
S. Koulb
aDepartment of Higher Education, Department of Chemistry, Government Degree College for Boys, Baramulla, 193101, J&K, India. E-mail: farooqprince100@yahoo.in; Fax: +91-1952-234214; Tel: +91-1952-234214
bBioorganic Chemistry Division, CSIR-Indian Institute of Integrative Medicine, Canal Road Jammu, 180001, J&K, India
cDepartment of Chemistry, College of Science, King Saud University, P.O. Box 2455, Riyadh, 11451, Saudi Arabia
dCancer Pharmacology Division, CSIR-Indian Institute of Integrative Medicine, Canal Road Jammu, 180001, J&K, India
eDepartment of Higher Education, Department of Chemistry, Govt. Degree College Sopore, Baramulla, 193201, J&K, India
fDepartment of Biotechnology, Central University of Kashmir, Ganderbal, 191201, J&K, India
First published on 23rd November 2020
Abstract
A simple, efficient, cost-effective, recyclable and green approach has been developed for the synthesis of new dihydropyrimidinone analogs via the Biginelli reaction. The methodology involves a multicomponent reaction catalyzed by “HPA-Montmorillonite-KSF” as a reusable and heterogeneous catalyst. This method gives an efficient and much improved modification of the original Biginelli reaction, in terms of yield and short reaction times under solvent free conditions. All the derivatives were subjected to cytotoxicity screening against a panel of four different human cancer cell lines viz. colon (Colo-205), prostate (PC-3), leukemia (THP-1) and lung (A549) to check their effect on percentage growth. MTT [3-(4,5-dimethylthiazol-yl)-diphenyl tetrazoliumbromide] cytotoxicity assay was employed to check IC50 values. Of the synthesized analogs, 16a showed the best activity with IC50 of 7.1 ± 0.8, 13.1 ± 1.4, 13.8 ± 0.9 and 14.7 ± 1.1 μM against lung (A549), leukemia (THP-1), prostate (PC-3) and colon (Colo-205) cancer lines, respectively. The 16a analog was further checked for its effect on cancer cell properties through clonogenic (colony formation) and scratch motility (wound healing) assays and thereby was found that it reduced both the colony formation and migratory properties of the lung cancer cell line (A549). Further, molecular docking studies were performed with 16a to show its binding mode.
1. Introduction
In modern methods of drug discovery processes, design and synthesis of drugs based on biological targets is of the great interest to medicinal chemists. Additionally, semi-synthetic processes for new compounds, obtained by molecular modification of the functional groups of lead compounds, are able to generate structural analogs with greater pharmacological activity and with fewer side effects.1 A constant enrichment in the science of organic synthesis through improvement of the synthetic methodologies is observed, driven by the needs to improve the capability to synthesize molecules in more facile, efficient and in economical ways.2 The paradigms of organic synthesis have shifted from the traditional concept of efficiency in terms of chemical yield to one that also considers economic and ecological values. The significance of a particular reaction can be judged on its capability to form products with dimensions of high yield, chemo-, regio-, stereo- or enantio-selectivity.3 In last one decade, there has been an incredible increase on the accessibility of Multi-Component Reactions (MCRs), and much still remains to be accomplished.4–9 MCRs are important cornerstones in the diversity-oriented construction of molecular complexity due to their ability to incorporate, in a fast and efficient manner, three or more components into a single product.10–14 Reactions that build up carbon–carbon bonds and at the same time introduce nitrogen-containing functionalities into the structural framework are especially attractive for the rapid construction of organic molecules. As MCRs are one-pot processes with simpler experimental conditions that do not require the isolation of intermediates, they are perfect candidates for combinatorial, and generation of products in a single synthetic operation15,16 and are of increasing importance in organic and medicinal chemistry.17–19 Since rapidity and diversity are considered as key factors in modern drug discovery, MCR strategies offer significant advantages over conventional linear-type syntheses, owing to their exceptional synthetic efficiency.20 MCRs contribute to the requirements of an environmentally benign process by reducing the number of synthetic steps, energy consumption and waste production.21 Functionalized nitrogen-heterocycles play a prominent role in medicinal chemistry and therefore they have been intensively used as scaffolds in the field of drug development.22–26 Despite several reports on fused heterocycles, there is a continuing demand for development of new methods for synthesis of novel fused heterocycles due to their plethora of medicinal applications.27In this context pyrimidine derivatives are of particular interest because of their interesting pharmacological profile.28–30 Dihydropyrimidin-2(1H)-ones (DHPMs), also known as Biginelli compounds are easily accessible via a multicomponent condensation process, first reported more than a century ago.31 The synthesis of 3,4-dihydropyrimidin-2(1H)-ones (Biginelli compounds) and their derivatives is of great importance due to the biological investigation of these molecules via molecular manipulation which have shown several activities such as calcium channel blockers,32–35 α1a-adrenergic receptor antagonists,35 mitotic kinesin inhibitors35 along with anti-bacterial and anti-tubercular activities.36,37 Monastrol, a structurally simple DHPM has been found as a novel cell-permeable molecule that blocks normal bipolar mitotic spindle assembly in mammalian cells by specifically inhibiting the motor activity of the mitotic kinesin Eg5, a motor protein required for spindle bipolarity, and therefore, causes cell cycle arrest.38 There are only a limited number of cell-permeable molecules currently known to specifically inhibit mitotic kinesin Eg5 and such molecules having DHPM scaffolds can therefore be considered as leads for the development of new anticancer drugs.
Based on the above cited findings and inspiration from the potential anticancer activity of DHPMs,39–44 we directed this work towards the synthesis of a diverse series of novel DHPM derivatives of biological interest using a simple, efficient, cost-effective, recyclable and green approach via Biginelli reaction involving the one-pot multicomponent reaction catalyzed by novel “HPA-Montmorillonite-KSF” as a reusable and heterogeneous catalyst. All the newly synthesized compounds were subjected to MTT cytotoxicity screening against a panel of four different human cancer cell lines viz. colon (Colo-205), prostate (PC-3), leukemia (THP-1) and lung (A549) along with normal epithelial cell line (fR-2) followed by clonogenic (colony formation) and scratch motility (wound healing) assays of most potent molecule to analyze any effect on certain cancer properties. Molecular docking studies were carried out to establish the binding capabilities of the same derivative.
2. Results and discussion
2.1. Chemistry
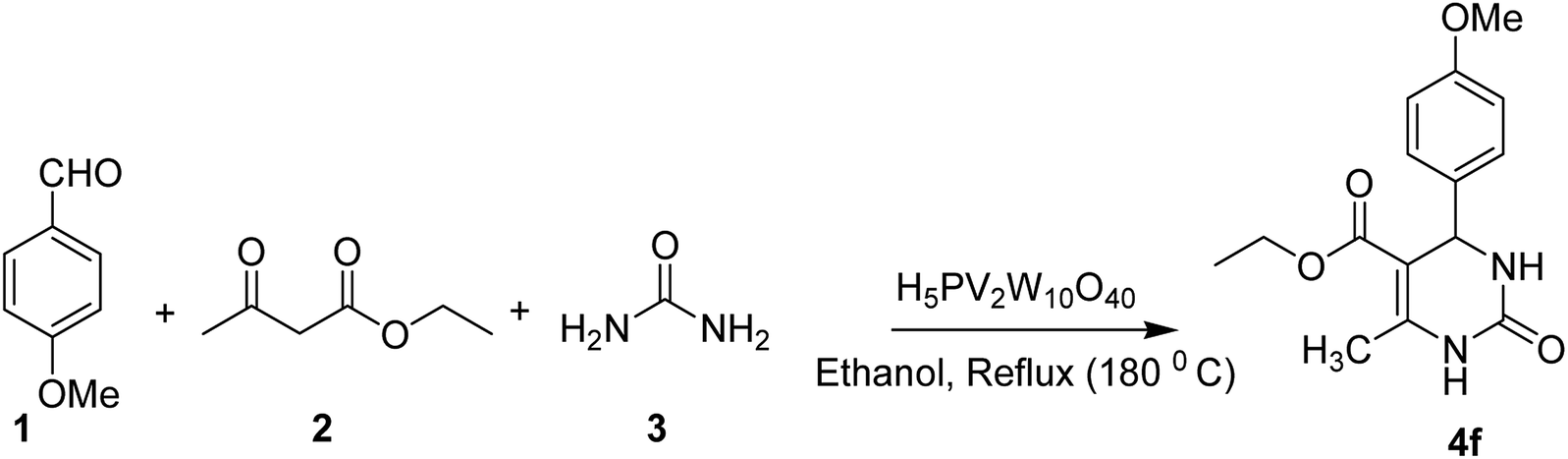
From the literature scan it is evident that DHPM moiety is pharmacologically important and many researchers are engaged to prepare their libraries by applying various modifications using different synthetic strategies to shorten the reaction time and increase the yield using a variation of different catalysts. Some of the reported catalysts used include Cu(OTf)2/MWI,45 Yb(PFO)3,46 HCl/EtOH,47 H3PO3/Pd-Cat, TMSCl/CAN, silica immobilized Ni(II), Ca(OCl)2, Mg(NO3)2, PPA-SiO2, sulfated tungstate, melamine trisulfonic acid, Cu(NO3)2·3H2O, acidic ionic liquids, organocatalysts, H3PMo12O40, H3PW12O40, H6P2W18O62·24H2O, H4PMo11VO40 (ref. 48) and other catalysts. These chemical methods in spite of their potential utility, involve expensive/toxic reagents and adverse reaction conditions like, strong acids, long reaction times, high temperature, stoichiometric amount of catalysts, environmental concerns and poor yields. Therefore, in order to overcome these limitations, the discovery of a new and efficient catalyst with high catalytic activity, short reaction time, recyclability and simple work-up procedures for the preparation of 3,4-dihydropyrimidin-2(1H)-ones under neutral, mild and practical conditions is of prime interest. Biginelli reaction catalysed by heteropolyacids (HPAs) can be one of the sources for the synthesis of large number of new molecules. HPA catalysts are widely exploited for the production of fine organic chemicals, health care products, pharmaceuticals and agrochemical products.49 HPAs are more reactive catalysts than conventional inorganic and organic acids for reactions in solution50 and have been used in organic transformations, such as synthesis of acylals, tetrahydropyranilation of phenols, thioacetalization and transacetalization reactions. They are also used as industrial catalysts for several liquid-phase reactions, including alcohol dehydration, alkylation and esterification.51 HPAs that contain super acidic properties have found numerous applications during last three decades, as useful and versatile acid catalysts for some acid-catalyzed reactions. They are usually solids that are insoluble in non-polar solvents while as highly soluble in polar ones. They can be used in bulk or in supported forms, in both homogeneous and heterogeneous system. Furthermore, HPAs have several advantages, including high flexibility in modification of the acid strength, ease of handling, environmental compatibility, non-toxicity, and experimental simplicity.52Inspired by above mentioned facts, a novel green methodology has been developed which involves the synthesis of DHPM analogs by Biginelli reaction and their in vitro screening against different human cancer cell lines. The methodology involves the multicomponent reaction catalyzed by HPA-Montmorillonite-KSF (H5PV2W10O40) as a reusable and heterogeneous catalyst. The strategy involves a three-component one-pot Biginelli-type reaction for the condensation of urea, ethylacetoacetate and different aldehydes to corresponding pyrimidinones in presence of a catalytic amount of HPA-Montmorillonite-KSF. The best conditions were achieved under solvent free conditions (neat) using 2 mol% of HPA, 1.5 equivalents of urea/thiourea and ethyl acetoacetate and 1 equivalent of aldehyde under reflux conditions for 1 h, affording the desired product in good yields (Table 1).
| S. no. | Solvent | Time (h) | Amount of catalyst (mol%) | Yield (%) | Temperature (°C) |
|---|---|---|---|---|---|
a Para-Methoxy benzaldehyde![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :urea:ethylacetoacetate in the ratio of 1:1.5:1.5. :urea:ethylacetoacetate in the ratio of 1:1.5:1.5. |
|||||
| 1 | 1,4-Dioxane | 6 h | 10 | 65 | Reflux |
| 2 | Acetonitrile | 5 h | 10 | 77 | Reflux |
| 3 | Toluene | 7 h | 10 | 71 | Reflux |
| 4 | Ethanol | 2 h | 10 | 87 | Reflux |
| 5 | Solvent free | 1 h | 10 | 92 | Reflux |
| 6 | Solvent free | 1 h | 2 | 96 | Reflux |
| 7 | Solvent free | 5 minutes | 2 | 70 | Microwave at 50 °C |
| 8 | Solvent free | 30 minutes | 2 | 74 | Sonication at 35 °C |
| 9 | Ethanol | 18 h | 2 | 40 | rt (25) |
| 10 | Solvent free | 14 h | 2 | 52 | rt |
 | ||
| Scheme 1 The model for preparation of DHPMs. | ||
| Entry | Product | R | X | Yield (%) | Mp (°C) | |
|---|---|---|---|---|---|---|
| Found | Reported | |||||
| 1 | 4a | C6H5 | O | 97 | 201–203 | 202–204 |
| 2 | 4b | C6H5 | S | 95 | 206–208 | 205–207 |
| 3 | 4c | 4-(Cl)–C6H4 | O | 93 | 209–211 | 210–212 |
| 4 | 4d | 2-(Cl)–C6H4 | O | 89 | 213–215 | 214 |
| 5 | 4e | 4-(Br)–C6H4 | O | 93 | 211–214 | 213–215 |
| 6 | 4f | 4-(MeO)–C6H4 | O | 96 | 201–204 | 201–203 |
| 7 | 4g | 2,3-(OMe)2–C6H3 | O | 94 | 176–177 | 178 |
| 8 | 4h | 2,4-(OMe)2–C6H3 | O | 95 | 157–160 | 158–160 |
| 9 | 4i | 3,4,5-(OMe)3–C6H2 | O | 97 | — | |
| 10 | 4j | 4-(NO2)–C6H4 | O | 79 | 207–208 | 209–211 |
| 11 | 4k | 4-(F)–C6H4 | O | 83 | — | |
| 12 | 4l | 2-(Br)-5-(OMe)–C6H3 | O | 87 | — | |
| 13 | 4m | Piperanal | O | 94 | — | |
| 14 | 4n | 2-(NO2)–C6H4 | O | 81 | 217–219 | 218–220 |
| 15 | 4o | 4-(OH)–C6H4 | O | 78 | 226–228 | 227–228 |
| 16 | 4p | 3-(OH)-4-(OMe)–C6H3 | O | 93 | 98–99 | 98–100 |
| 17 | 4q | 5-(Br)-2-(OMe)–C6H3 | O | 94 | — | |
The next part of the optimization study was to assess the recycled use of catalyst. It may be stated that due to the constantly rising environmental concerns in the field of chemistry, it is sensible to use easily recovered and recycled catalysts, especially expensive or toxic metallic ones for the next use,54 as only few of them meet the criterion of green chemistry. For example, the recovery of ytterbium triflate from water seems cumbersome since water must be removed through heating and then drying under vacuum at 100 °C for 2 hours,55 and in the case of polymer-supported Yb(III) resin, the activity of recycled resin is much lower than that of the original one thus limiting the recyclability.56 Therefore, there is still room for further search for recyclable catalysts to be used in the Biginelli reaction that can convert a variety of aldehydes to pyrimidinones in high yields under mild reaction conditions. In order to show the merit of the present work in comparison with some reported protocols, we compared the results of the synthesis of 5-ethoxycarbonyl-4-phenyl-6-methyl-3,4-dihydropyrimidin-2(1H)-one in the presence of montmorillonite KSF, sulfuric acid, zeolite, silica-sulfuric acid, BF3–OEt2/CuCl, H3PMo12O40 with HPA-Montmorillonite-KSF with respect to the reaction times (Table 3). The yield of product in presence HPA-Montmorillonite-KSF is comparable with these catalysts. However, the reaction in presence of these catalysts required longer reaction times than HPA-Montmorillonite-KSF. The catalyst could be reused several times (six times) and did not show any significant negative influence on the overall yields of the reaction (Fig. 1).
| Entry | Catalyst | Time (h) | Yield (%) |
|---|---|---|---|
| 1 | Montmorillonite KSF | 48 | 82 |
| 2 | Sulphuric acid | 18 | 71 |
| 3 | Zeolite | 12 | 80 |
| 4 | Silica sulphur acid | 16 | 91 |
| 5 | BF3·OEt2/CuCl | 18 | 71 |
| 6 | H3PMo12O40 | 5 | 80 |
| 7 | HPA-Montmorilonite-KSF (present work) | 1 | 96 |
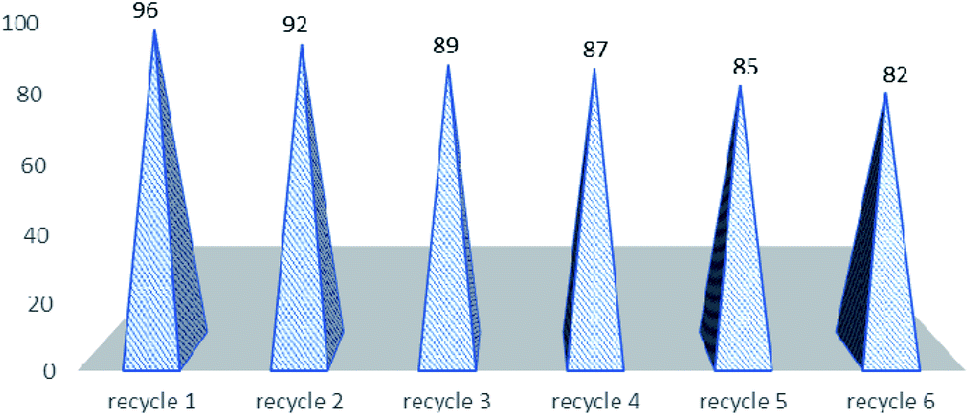 | ||
| Fig. 1 Recyclability of the catalyst. | ||
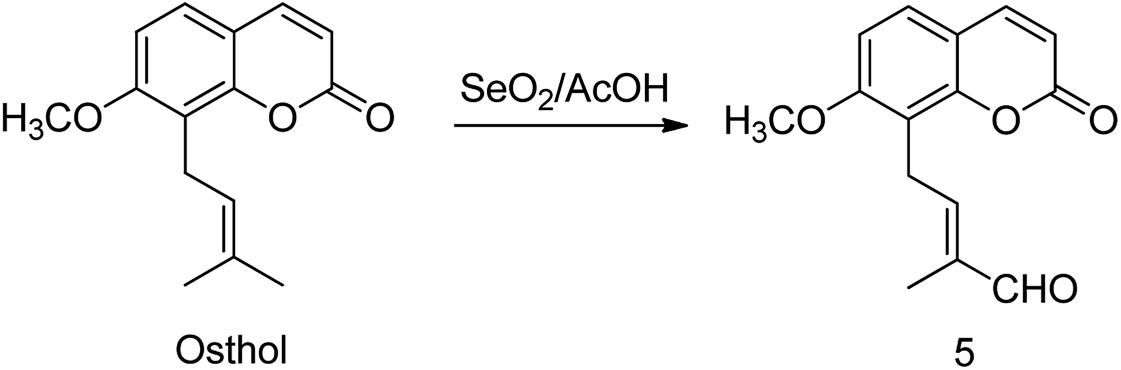 | ||
| Scheme 2 Oxidation of osthol to aldehyde 5. | ||
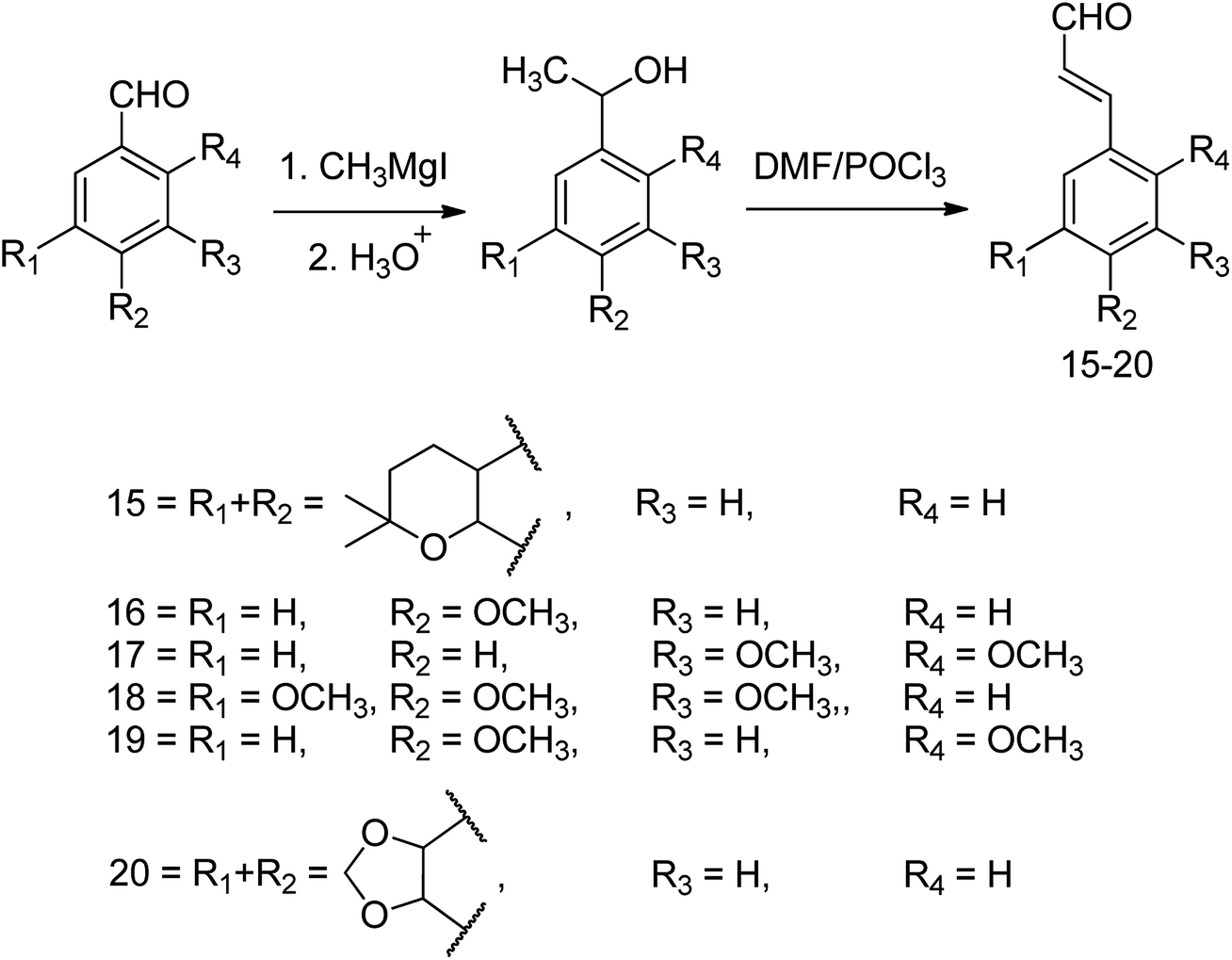 | ||
| Scheme 3 Preparation of aldehydes using Grignard and Vilsmeier reactions. | ||
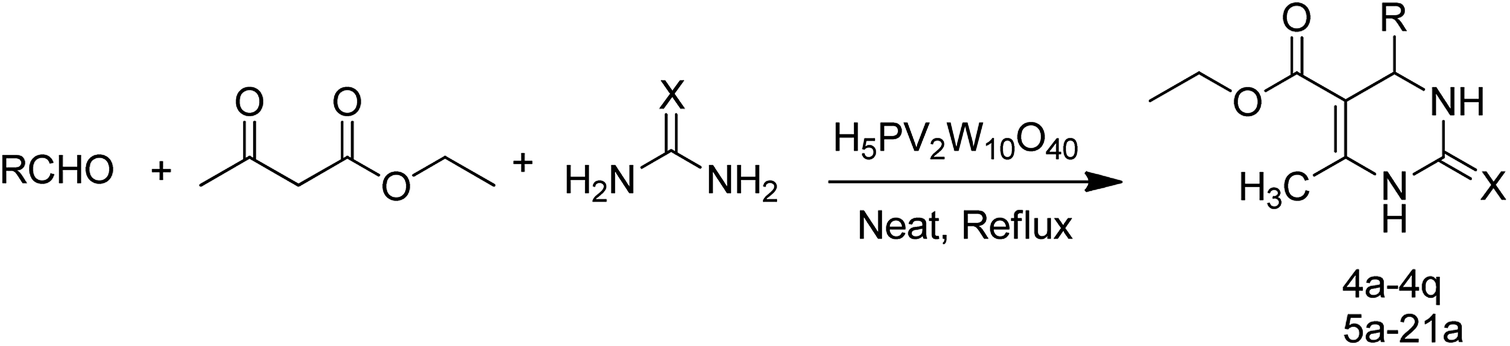 | ||
| Scheme 4 General procedure for preparation of novel DHPM analogs. | ||
| Entry | Product | R | X | Yield (%) |
|---|---|---|---|---|
| 1 | 5a | 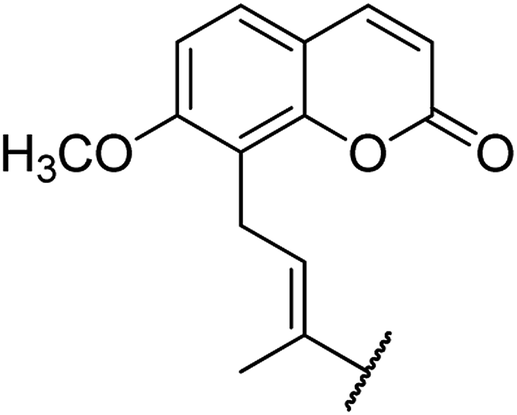 |
O | 96 |
| 2 | 6a | 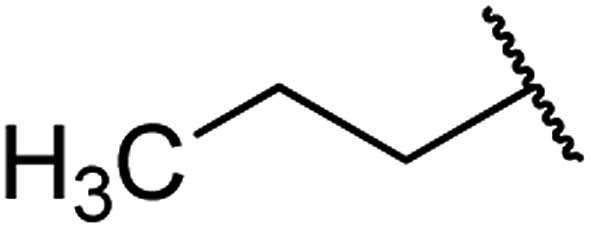 |
O | 93 |
| 3 | 7a | 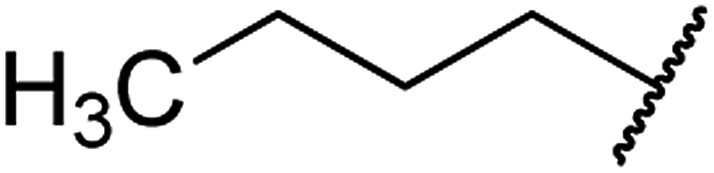 |
O | 95 |
| 4 | 8a | 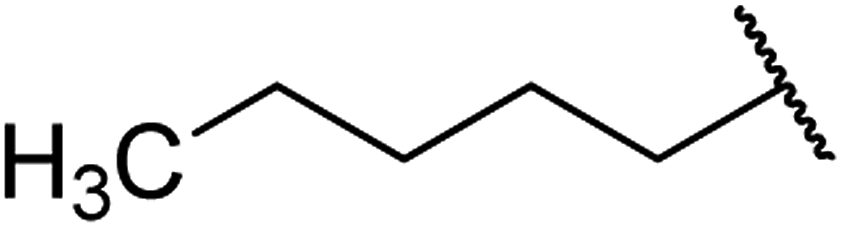 |
O | 93 |
| 5 | 9a | 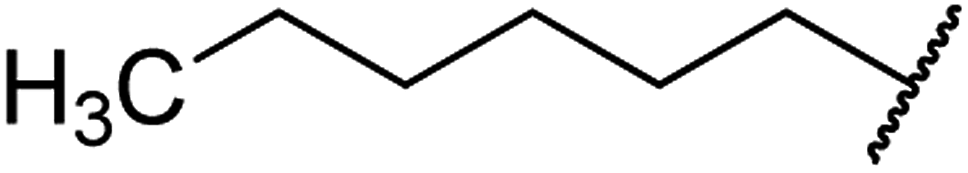 |
O | 89 |
| 6 | 10a | 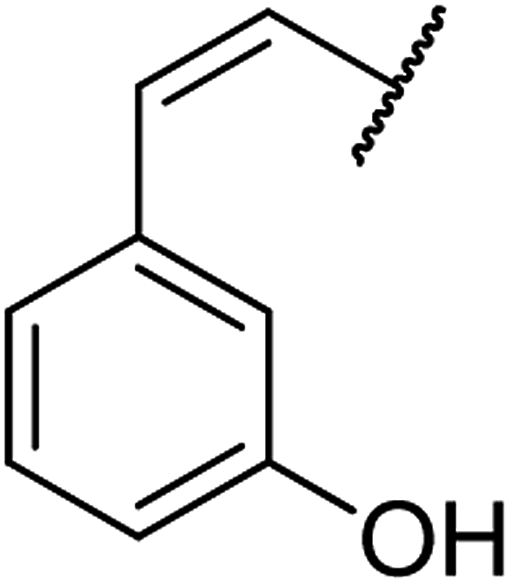 |
O | 93 |
| 7 | 11a | 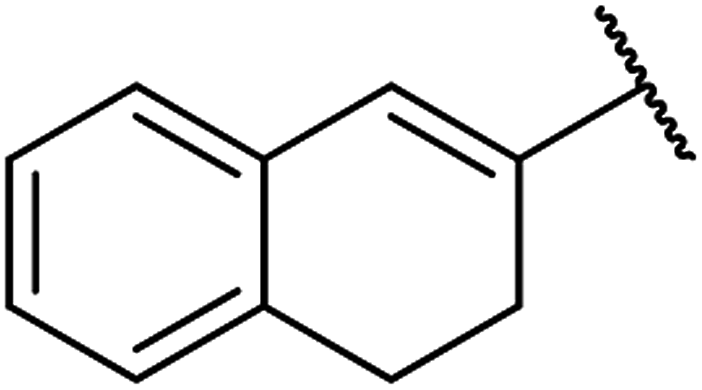 |
O | 96 |
| 8 | 12a | 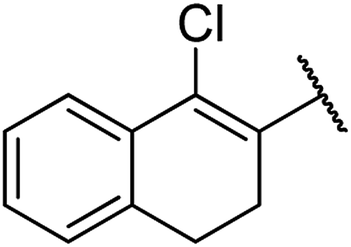 |
O | 94 |
| 9 | 13a | 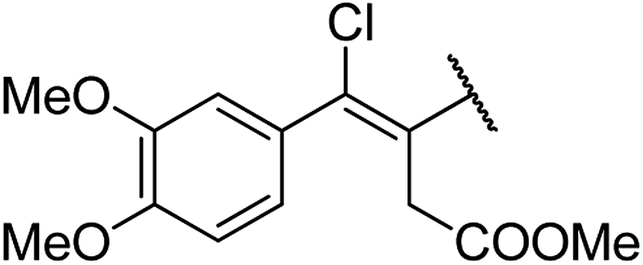 |
O | 95 |
| 10 | 14a | 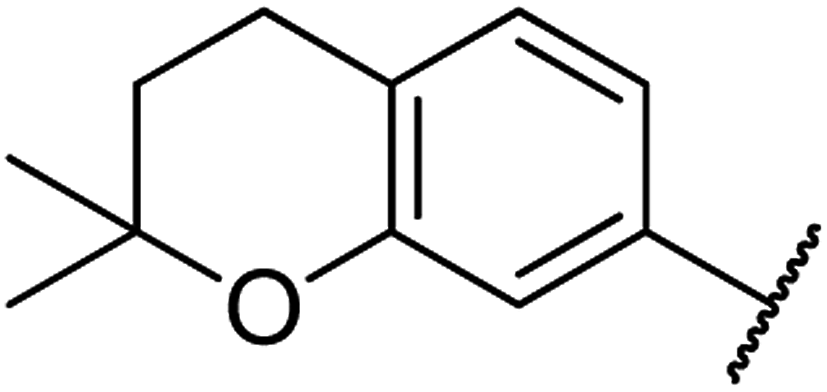 |
O | 92 |
| 11 | 15a | 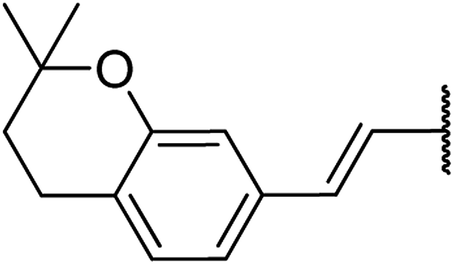 |
O | 92 |
| 12 | 16a | 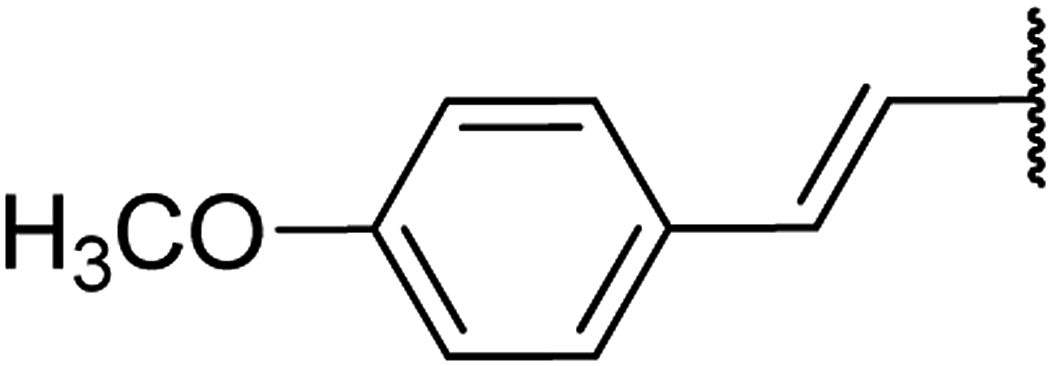 |
O | 92 |
| 13 | 17a | 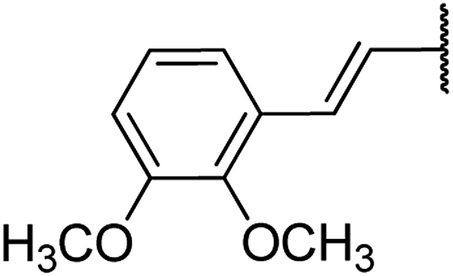 |
O | 87 |
| 14 | 18a | 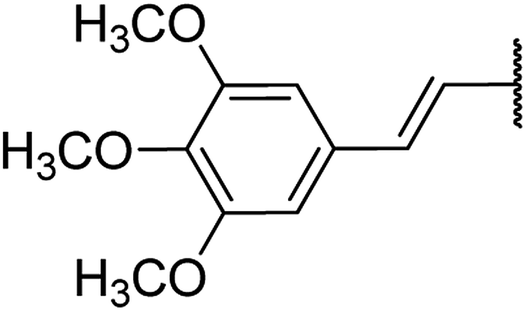 |
O | 89 |
| 15 | 19a | 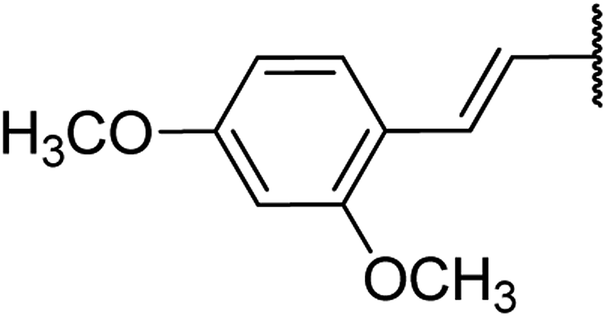 |
O | 88 |
| 16 | 20a | 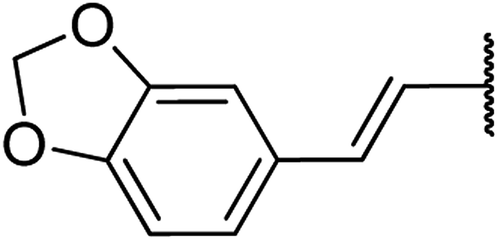 |
O | 90 |
| 17 | 21a | 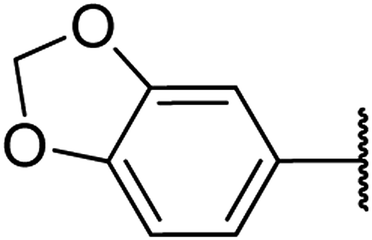 |
O | 93 |
2.2. Biology
| Compound | Colon (Colo-205) IC50 | Prostate (PC-3) IC50 | Leukemia (THP-1) IC50 | Lung (A549) IC50 | Normal (fR-2) IC50 |
|---|---|---|---|---|---|
| a IC50 values are expressed in μM concentration. 5-Fluorouracil (Colo-205, fR-2), paclitaxel (PC-3, A549) and doxorubicin (THP-1) were used as positive controls. | |||||
| 5a | 37.6 ± 1.1 | 40.8 ± 1.7 | 65.8 ± 1.4 | 67.8 ± 1.7 | 68.8 ± 1.1 |
| 6a | >70.0 | >70.0 | >70.0 | >70.0 | >70.0 |
| 7a | >70.0 | >70.0 | >70.0 | >70.0 | >70.0 |
| 8a | 18.6 ± 1.0 | 22.2 ± 1.6 | 43.8 ± 1.4 | 63.4 ± 1.1 | 67.2 ± 1.5 |
| 9a | >70.0 | >70.0 | >70.0 | >70.0 | >70.0 |
| 10a | >70.0 | 29.4 ± 1.2 | 42.1 ± 1.7 | >70.0 | >70.0 |
| 11a | >70.0 | 29.3 ± 1.3 | >70.0 | >70.0 | >70.0 |
| 12a | >70.0 | >70.0 | >70.0 | >70.0 | >70.0 |
| 13a | 59.4 ± 1.6 | 35.4 ± 2.2 | 23.2 ± 1.4 | 30.3 ± 2.9 | 67.6 ± 2.5 |
| 14a | >70.0 | >70.0 | >70.0 | >70.0 | >70.0 |
| 15a | 27.3 ± 1.8 | >70.0 | >70.0 | >70.0 | >70.0 |
| 16a | 14.7 ± 1.1 | 13.8 ± 0.9 | 13.1 ± 1.4 | 7.1 ± 0.8 | >70.0 |
| 17a | 38.2 ± 1.4 | 47.9 ± 2.1 | >70.0 | 26.4 ± 1.1 | >70.0 |
| 18a | >70.0 | >70.0 | >70.0 | >70.0 | >70.0 |
| 19a | 33.7 ± 1.8 | >70.0 | >70.0 | 47.8 ± 2.4 | >70.0 |
| 20a | 23.4 ± 1.4 | >70.0 | >70.0 | >70.0 | 68.7 ± 1.1 |
| 21a | 48.1 ± 2.1 | >70.0 | >70.0 | >70.0 | >70.0 |
| 5-Fluorouracil | 33.9 ± 1.9 | — | — | — | 334.7 ± 3.8 |
| Paclitaxel | — | 0.048 ± 0.008 | — | 5.1 ± 0.4 | — |
| Doxorubicin | — | — | 0.018 ± 0.004 | — | — |
DHPM derivatives with varying carbon chain length of R group showed interesting results wherein compound 8a bearing n-pentyl chain showed improved cytotoxic effect against colon (Colo-205), prostate (PC-3) leukemia (THP-1) and lung (A549) cancer cell lines with IC50 of 18.6 ± 1.0, 22.2 ± 1.6, 43.8 ± 1.4 and 63.4 ± 1.1 μM, respectively; however, still toxic against normal cell line (fR-2) being with IC50 of 67.2 ± 1.5 μM. On the other hand compounds 6a, 7a and 9a bearing n-propyl, n-butyl and n-hexyl R groups lost their activity with IC50 values not in the range of ≤70 μM, thereby demonstrating the optimum chain length responsible for cytotoxicity. Compound 10a having hydroxyl-styryl R group showed activity against PC-3 and THP-1 with IC50 of 29.4 ± 1.2, and 42.1 ± 1.7 μM, respectively. Compound 11a demonstrated selective cytotoxicity against prostate (PC-3) cancer cell line with IC50 of 29.3 ± 1.3 μM while as its corresponding chloro-derivative (12a) totally lost its activity with IC50 values not in the range of ≤70 μM. Compound 13a showed broad spectrum cytotoxic effect against colon (Colo-205), prostate (PC-3), leukemia (THP-1) and lung (A549) cancer cell line with IC50 of 59.4 ± 1.6, 35.4 ± 2.2, 23.2 ± 1.4 and 30.3 ± 2.9 μM, respectively while toxicity against normal cell line (fR-2) determined with IC50 of 67.6 ± 2.5 μM. The role of an extra ethylene moiety in 15a as compared 14a is highlighted in terms of its selective cytotoxic activity against colon (Colo-205) cancer cell line with IC50 of 27.3 ± 1.8 μM, thereby making provision for synthesis of similar DHPM analogs having more activity and less toxicity. This observation is further supported in 20a and 21a wherein an extra ethylene group in 20a enhances the activity (23.4 ± 1.4 μM) by two fold as compared to 21a (48.1 ± 2.1 μM).
Among all the tested DHPM analogs, 16a bearing a p-methoxy phenyl moiety displayed the most potent cytotoxic effect with IC50 of 14.7 ± 1.1, 13.8 ± 0.9, 13.1 ± 1.4 and 7.1 ± 0.8 μM against colon (Colo-205), prostate (PC-3), leukemia (THP-1) and lung (A549) cancer lines respectively. 17a and 19a with dimethoxy phenyl moieties showed moderate cytotoxicity against lung (A549), prostate (PC-3) and colon (Colo-205) cancer lines, while as, 18a with trimethoxy phenyl moiety lost its activity with IC50 values not in the range of ≤70 μM. These observations demonstrate that the activity of such DHPM analogs increases as the number of methoxy groups decreases in the phenyl moiety of R group. All these results suggest that the structural features have profound influence on the biological profile of a compound. It was concluded that the compound 16a is the most potent derivative with minimum IC50 of 7.1 ± 0.8 μM against lung cancer line (A549). Additionally the derivative had least toxicity against normal epithelial cell line (fR-2) with IC50 value in the range of >70 μM.
One of the hallmark traits of cancer cells is their ability to undergo isolated clonal growth. Clonogenic assay was performed to test the colony formation property of single cell to grow into a colony. This assay confirmed that most potent compound 16a inhibits clonogenic property of A549 cells significantly at 7 and 15 μM when compared with vehicle DMSO; staurosporine is already known to have effect on mobility and invasiveness of A549 cells,58 hence was used as positive control (Fig. 2). To check the anti-metastatic activity, 16a was further evaluated by scratch motility (wound healing) assay against A549 cell line. After A549 cells got attached overnight incubation, the cell monolayers were wounded and PBS washed. This was followed by treatment with 16a/DMSO in culture media. The vehicle DMSO treated cells had almost completely filled in the cleared area, whereas treatment with 7 and 15 μM of compound 16a significantly abrogates motility and invasion potential of A549 cells (Fig. 3). Again staurosporine was used as positive control.62
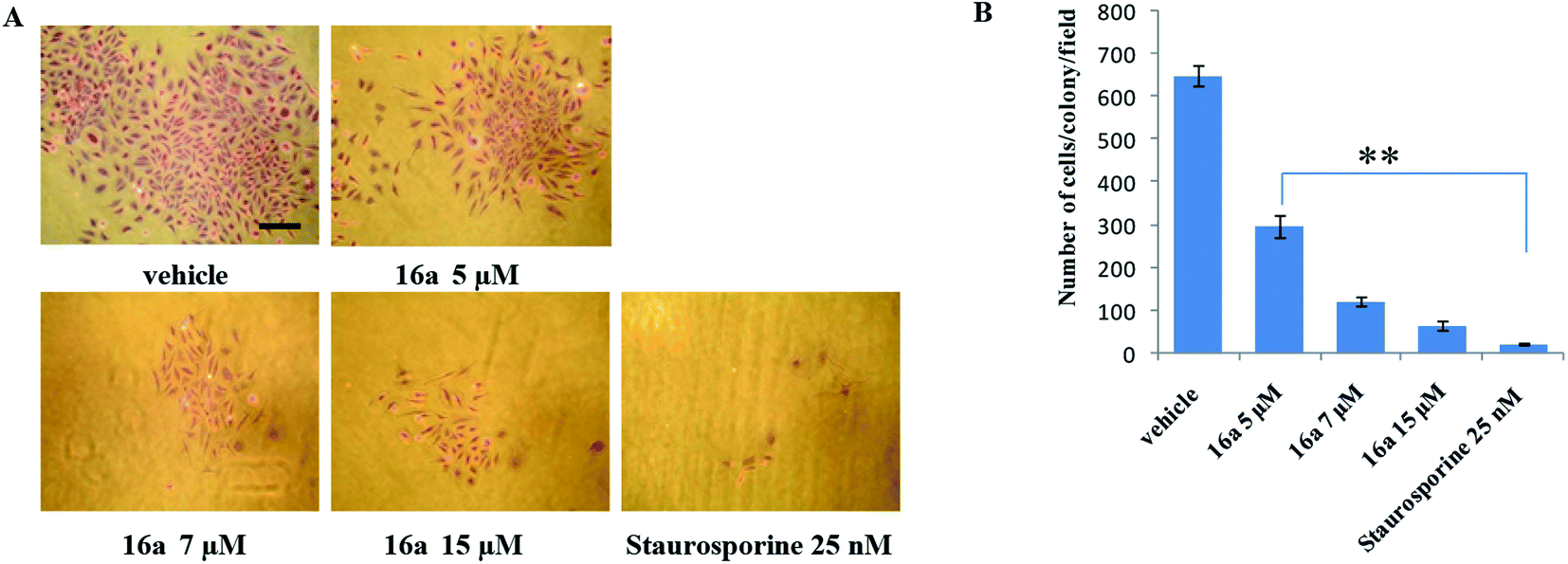 | ||
| Fig. 2 (A) A549 cells (1 × 103 cells per well) were cultured and treated with various concentration of compound 16a for 24 h at 37 °C and then stained with crystal violet (for details see Result section), number of stained colonies were counted, photographed (magnification, 100×; scale bar, 200 μm) and (B) data was calculated from three independent experiments. Columns mean; bars SD of three independent experiments. **P < 0.01 compared with untreated control. | ||
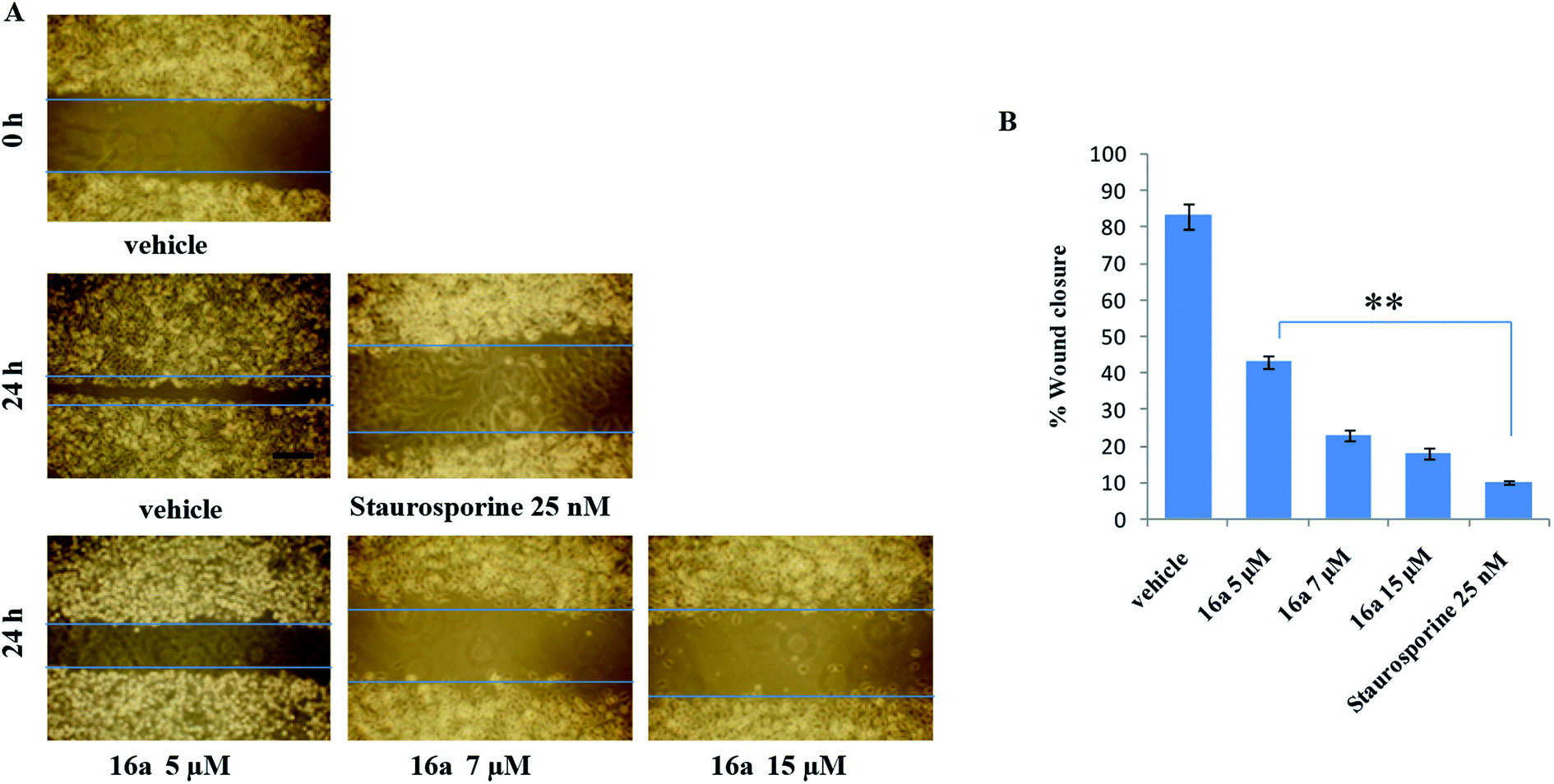 | ||
| Fig. 3 (A) A549 cells (0.5 × 105 cells per well) were grown to confluence in six well tissue culture plate and scratched with sterile tip, compound 16a was added to cultures as indicated. Scratched areas were photographed (magnification, 100×; scale bar, 200 μm) at zero hour and then subsequently again 24 h later to assess the degree of wound healing. (B) The scratched areas were quantified in three random fields in each treatment, and data were calculated from three independent experiments. Data were calculated from three independent experiments. Columns mean; bars SD of three independent experiments. **P < 0.01 compared with untreated control. | ||
2.3. Molecular docking studies
In order to rationalize the obtained biological data and understand the probable mode of binding, the most potent compound (16a) were docked into the crystal structure. On analysis, 16a was found to bind with better affinity on CDK2 and PI3Kγ which supports the in vitro cytotoxic activity data. The interaction figure analysis of compound 16a with CDK2 reveals that the dihydropyrimidinone moiety of 16a is involved in bidentate H-bonding with Leu83 (hydrophobic) amino acid residue and methoxy-benzene moiety is forming a pi–pi stacking with Phe80 (hydrophobic) which favors more affinity of the molecules for CDK2. Additionally, the “O” of methoxy-benzene moiety is for forming third H-bond with Asp-145 of CDK-2. Further the interaction figure analysis of compound 16a with PI3Kγ shows that the dihydropyrimidinone moiety of 16a is involved in bidentate H-bonding with Tyr867 (hydrophobic) and Asp964 (acidic) amino acid residue and esteric carbonyl oxygen is forming H-bonding with the hinge residue Val882 (hydrophobic) (Fig. 4). The three H-bondings enhance the affinity of the 16a molecule for both CDK-2 and PI3Kγ.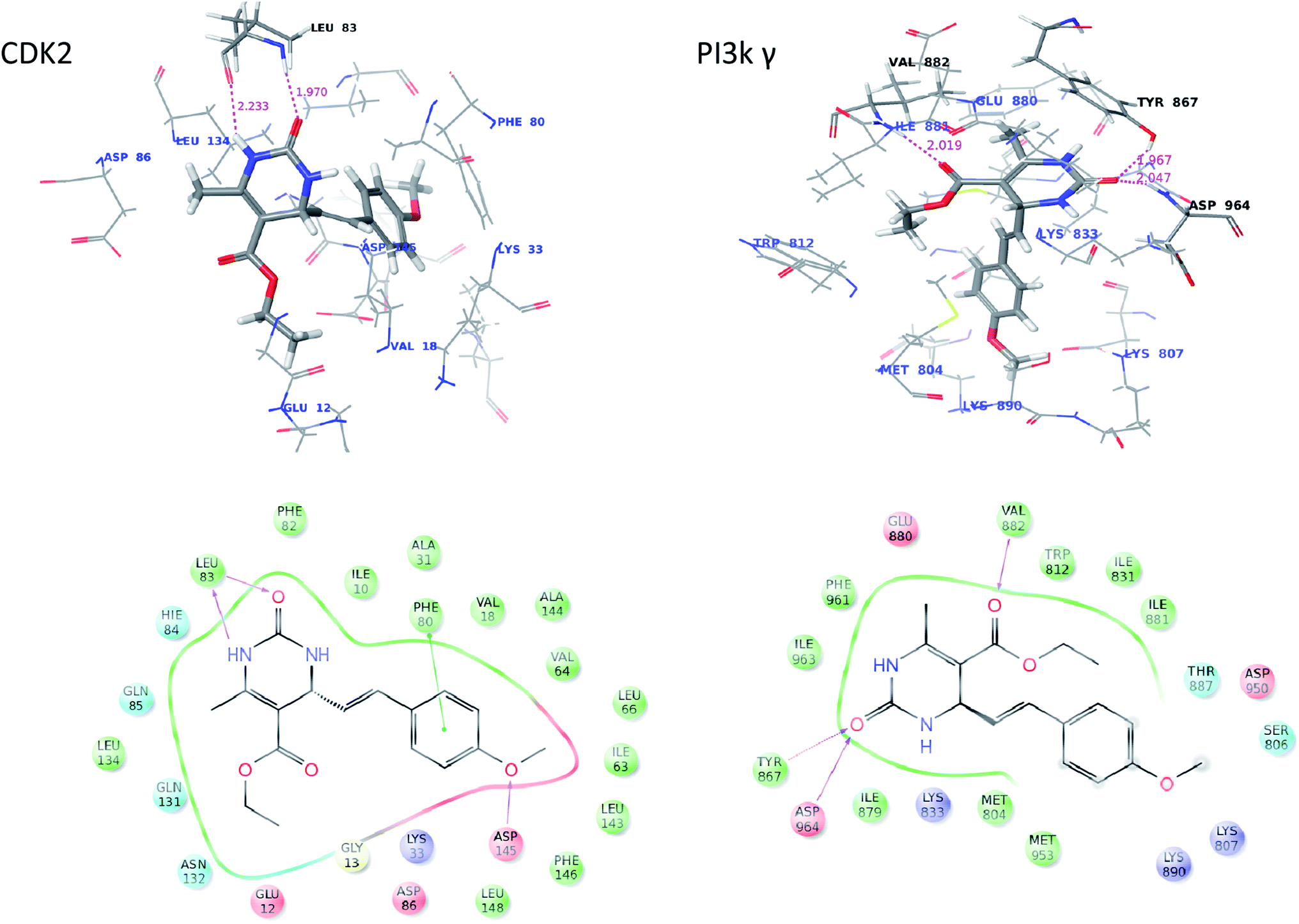 | ||
| Fig. 4 Molecular docking studies showing probable binding mode of 16a. | ||
We concluded earlier from SAR analysis that phenyl moiety as side chain of dihydropyrimidone increased the specificity and cytotoxicity of 16a. So, we did docking studies with 6a too which has one of the simplest aliphatic (propyl) as side chain for comparison. The docking data revealed that only two H-bonding were being formed between 6a and the enzymes, with Leu83 in case of CDK-2 and with Val882 and Glu880 in case of PI3Kγ compared to three in case of 16a. Further the absence of methoxy-benzene moiety in 6a leads to loss of pi–pi stacking. So, this docking comparison explains partially why phenyl moiety containing 16a molecule has increased cytotoxicity.
3. Conclusions
We have reported a catalytic method for the synthesis of dihydropyrimidinones using HPA-Montmorillonite-KSF as an efficient and eco-friendly heterogeneous inorganic catalyst. It is noteworthy to mention that the catalyst is reusable and even after five runs; the catalytic activity of H5PV2W10O40·10H2O was retained as that of the freshly used catalyst. The short reaction times, simple work-up and isolation of the products in high yields with high purity, and mild reaction conditions, are some of the highlights of current procedure. A diverse library of DHPM analogs have been synthesized by using this catalyst and most of the derivatives displayed impressive anti-proliferative effect at 50 μM, with best cytotoxic effect observed for 16a exhibiting the inhibition against all the tested cancer cell lines with most potency against lung (A549) cancer cells as 7.1 ± 0.8 μM while the toxicity against the normal epithelial cells was apparently more than 10 fold higher (>70 μM). However, in vivo studies are warranted to investigate the mechanisms of action responsible for the cytotoxic activity of the most active derivatives.4. Experimental
4.1. Chemistry
All reagents for chemical synthesis were obtained from Sigma Aldrich and the solvents used in reactions were distilled and dried prior to use. All the chemical reactions were monitored by TLC on 0.25 mm silica gel 60 F254 plates (E. Merck) using 2% ceric ammonium sulphate solution for detection of the spots. Purification of compounds was carried out by column chromatography using silica gel 60–120 mesh stationary phase. 1H NMR and 13C NMR spectra (with chemical shifts expressed in δ and coupling constants in hertz) were recorded on Bruker DPX 200, 400 and DPX 500 instruments using CDCl3 or CD3OD as the solvents with TMS as internal standard. High resolution mass spectra (HRMS) were recorded on Agilent Technologies 6540 instrument and IR recorded on an FT-IR Bruker (270-30) spectrophotometer. Melting points of compounds were recorded on Buchi melting point apparatus B-542.The HPA so formed was dissolved in 150 mL water and added dropwise to 500 mL aqueous suspension of 10 g Montmorillonite KSF. This mixture was stirred for 5 h and the water was evaporated over water bath to get a dry powder, which was kept overnight in hot air oven at 110 °C. A portion of this solid was then calcined at 425 °C for 3 h to get a heteropoly acid clay nano hybrid and was named as HCNH. The catalyst was characterized using X-ray powder diffraction (XRD) and scanning electron microscopy as discussed in ESI.†
4.1.4.1. Ethyl 4-(4-(6-methoxy-2-oxo-2H-chromen-7-yl)but-2-en-2-yl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate 5a. Yellowish solid, (mp: 144–146 °C). 1H NMR (400 MHz, CDCl3): δ 7.6 (1H, d, J = 9.5 Hz), 7.3 (1H, d, J = 8.6 Hz), 7.2 (1H, s), 6.8 (1H, d, J = 8.6 Hz), 6.5 (1H, t, J = 7.3 Hz), 6.2 (1H, d, J = 9.4 Hz), 4.9 (1H, s), 4.1 (2H, q, J = 8 Hz), 3.9 (3H, s), 3.8 (2H, d, J = 7.6 Hz), 2.3 (3H, s), 1.9 (3H, s), 1.2 (3H, t, J = 8 Hz). 13C NMR (100 MHz, CDCl3): δ 169.4, 161.8, 155.7, 154.6, 151.5, 149.3, 142.8, 134.5, 133.2, 126.1, 119.8, 119.3, 114.7, 106.8, 92.8, 61.5, 56.7, 48.9, 30.3, 18.9, 16.2, 14.6. IR (KBr) νmax cm−1: 3340, 3012, 1728, 1681, 1072, 832. HR-ESIMS m/z: 413.4424 (calculated for C22H24N2O6, 412.4358).
4.1.4.2. Ethyl 1,2,3,4-tetrahydro-6-methyl-2-oxo-4-propylpyrimidine-5-carboxylate 6a. White solid, (mp: 156–158 °C). 1H NMR (400 MHz, CDCl3): δ 4.2 (1H, m), 4.2 (2H, q, J = 6.7 Hz), 2.2 (3H, s), 1.4 (2H, m), 1.28 (4H, m), 0.9 (3H, t, J = 6.9 Hz). 13C NMR (100 MHz, CDCl3): δ 167.8, 156, 149.2, 102.2, 61.1, 51.9, 40.3, 18.4, 18.1, 14.6, 14.2. IR (KBr) νmax cm−1: 1744, 1717, 1637, 1440, 1215. HR-ESIMS m/z: 227.1393 (calculated for C11H18N2O3, 226.1317).
4.1.4.3. Ethyl 4-butyl-1,2,3,4-tetrahydro-6-methyl-2-oxopyrimidine-5-carboxylate 7a. White solid, (mp: 146–149 °C). 1H NMR (400 MHz, CDCl3): δ 4.2 (1H, m), 4.1 (2H, q, J = 7.1 Hz), 2.2 (3H, s), 1.4 (2H, m), 1.2 (7H, m), 0.9 (3H, t, J = 7.4 Hz). 13C NMR (100 MHz, CDCl3): δ 167.4, 154.8, 146.8, 101.5, 59.9, 51.4, 36.8, 31.4, 24, 22.5, 14.3, 14. IR (KBr) νmax cm−1: 1744, 1717, 1705, 1637, 1440, 1107. HR-ESIMS m/z: 241.1546 (calculated for C12H20N2O3, 240.1474).
4.1.4.4. Ethyl-methyl-2-oxo-4-pentyl-1,2,3,4-tetrahydropyrimidine-5-carboxylate 8a. White solid, (mp: 150–152 °C). 1H NMR (400 MHz, CDCl3): δ 4.8 (1H, s), 4.1 (1H, m), 4 (2H, q, J = 7.3 Hz), 3.2 (1H, s), 2.1 (3H, s), 1.4 (2H, m), 1.9 (6H, m), 0.8 (3H, t, J = 7.6 Hz). 13C NMR (100 MHz, CDCl3): δ 167.4, 154.8, 146.8, 101.5, 59.9, 51.4, 31.4, 37.4, 24, 22.5, 18.5, 14.3, 14. IR (KBr) νmax cm−1: 1744, 1705, 1637, 1617, 1440. HR-ESIMS m/z: 255.1703 (calculated for C13H22N2O3, 254.1630).
4.1.4.5. Ethyl-4-hexyl-1,2,3,4-tetrahydro-6-methyl-2-oxo-pyrimidine-5-carboxylate 9a. White solid, (mp: 153–156 °C). 1H NMR (400 MHz, CDCl3): δ 4.6 (1H, s), 4.1 (1H, m), 4 (2H, q, J = 7.2 Hz), 3 (1H, s), 2.1 (3H, s), 1.4 (2H, m), 1.2 (8H, m), 0.8 (3H, t, J = 7.3 Hz). 13C NMR (100 MHz, CDCl3): δ 166.7, 153.2, 145.8, 100.3, 59.5, 50.4, 32.2, 24.3, 23.1, 19.29, 15.3, 14.2, 13.9. IR (KBr) νmax cm−1: 1744, 1705, 1637, 1617, 1440. HR-ESIMS m/z: 269.1853 (calculated for C14H24N2O3, 268.1787).
4.1.4.6. Ethyl-4-(3-hydroxystyryl)-1,2,3,4-tetrahydro-6-methyl-2-oxopyrimidine-5-carboxylate 10a. Yellowish powder solid, (mp: 135–137 °C). 1H NMR (400 MHz, CDCl3): δ 7.3 (1H, m), 7.3 (1H, m), 7.2 (1H, m), 7.2 (1H, m), 6.5 (1H, d, J = 16 Hz), 6.2 (1H, dd, J = 3.9, 16 Hz), 4.2 (1H, m), 4.1 (2H, q, J = 7.4 Hz), 2.3 (3H, s), 1.3 (3H, t, J = 7.3 Hz). 13C NMR (100 MHz, CDCl3): δ 167.6, 158.4, 154.5, 144.6, 130.6, 130.1, 129.7, 128.8, 127.6, 118.3, 109.7, 101.8, 59.7, 53.7, 18.4, 14.9. IR (KBr) νmax cm−1: 3240, 3117, 2960, 1693, 1514, 1462. HR-ESIMS m/z: 303.1339 (calculated for C16H18N2O4, 302.1267).
4.1.4.7. Ethyl-4-(3,4-dihydronaphthalen-2-yl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate 11a. Pale yellow solid, (mp: 181–183 °C). 1H NMR (400 MHz, CDCl3): δ 7.1–7.2 (4H, m), 6.3 (1H, s), 5.3 (1H, s), 5 (1H, s), 4.1 (2H, q, J = 7.6 Hz), 2.8 (2H, t, J = 7.6 Hz), 2.3 (3H, s), 2.2 (2H, m), 1.2 (3H, t, J = 7.5 Hz). 13C NMR (100 MHz, CDCl3): δ 167.7, 155.7, 149.5, 142.3, 136.1, 135.2, 128.2, 128.4, 127.5, 125, 99.8, 61.1, 29.2, 23.9, 18.3, 14.8. IR (KBr) νmax cm−1: 3240, 3117, 2960, 1693, 1728, 1681, 1608, 1514. HR-ESIMS m/z: 313.1546 (calculated for C18H20N2O3, 312.1474).
4.1.4.8. Ethyl-4-(1-chloro-3,4-dihydronaphthalen-2-yl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate 12a. White solid, (mp: 162–164 °C). 1H NMR (400 MHz, CDCl3): δ 7.6 (1H, m), 7.2 (2H, m), 7.1 (1H, m), 5.8 (1H, s), 5.3 (1H, s), 5.2 (1H, s), 4.1 (2H, q, J = 6.7 Hz), 2.8 (2H, m), 2.3 (3H, s), 2.4 (2H, m), 1.1 (3H, t, J = 7.3 Hz). 13C NMR (100 MHz, CDCl3): δ 167.5, 154.8, 150.3, 137.7, 137.4, 134.14, 129.5, 128.3, 127.9, 127.4, 126.2, 98.6, 61.4, 29.2, 25.1, 18.9, 15.2. IR (KBr) νmax cm−1: 3235, 2936, 1693, 1700, 1681, 1596. HR-ESIMS m/z: 347.1156 (calculated for C18H19ClN2O3, 346.1084).
4.1.4.9. Ethyl-4-((Z)-3-(methoxycarbonyl)-1-chloro-1-(3,4-dimethoxyphenyl)prop-1-en-2-yl)-1,2,3,4-tetrahydro-6-methyl-2-oxopyrimidine-5-carboxylate 13a. Brown powder solid, (mp: 126–128 °C). 1H NMR (400 MHz, CDCl3): δ 7.1 (1H, d, J = 2.7 Hz), 7 (1H, dd, J = 2.3 & 8.3 Hz), 6.9 (1H, d, J = 8.3 Hz), 4.5 (1H, s), 4.2 (2H, q, J = 7.2 Hz), 3.6 (6H, s), 3.6 (3H, s), 2.7 (2H, s), 2.2 (3H, s), 1.3 (3H, t, J = 6.9 Hz). 13C NMR (100 MHz, CDCl3): δ 174, 169.9, 155.7, 151.3, 150.1, 148.5, 143.4, 134.3, 123.1, 121.6, 113.8, 113, 95.1, 61.5, 57.6, 56.7, 51.8, 36.3, 16.3, 14.7. IR (KBr) νmax cm−1: 3195, 2860, 1693, 1514, 1462. HR-ESIMS m/z: 453.1423 (calculated for C21H25ClN2O7, 452.1350).
4.1.4.10. Ethyl-1,2,3,4-tetrahydro-6-methyl-4-(2,2-dimethylchroman-6-yl)-2-oxo-pyrimidine-5-carboxylate 14a. Yellowish solid, (mp: 134–137 °C). 1H NMR (400 MHz, CDCl3): δ 7.5 (1H, s), 7 (2H, m), 6.6 (1H, d, J = 2 Hz), 5.4 (1H, m), 5.3 (1H, s), 4.1 (2H, q, J = 7.6 Hz), 2.7 (2H, t, J = 7.5 Hz), 2.3 (3H, s), 1.7 (2H, t, J = 7.5 Hz), 1.3 (6H, s), 1.2 (3H, t, J = 7.6 Hz). 13C NMR (100 MHz, CDCl3): δ 165.8, 153.7, 145.9, 135, 127.7, 125.7, 120.9, 117.2, 101.6, 74.3, 59.9, 55.2, 54, 32.6, 30.2, 26.8, 22.5, 18.6, 14.1. IR (KBr) νmax cm−1: 3335, 3156, 1693, 1700, 1608, 1576, 1488. HR-ESIMS m/z: 345.1822 (calculated for C19H24N2O4, 344.1736).
4.1.4.11. Ethyl-1,2,3,4-tetrahydro-6-methyl-4-((Z)-2-(2,2-dimethylchroman-6-yl)vinyl)-2-oxo-pyrimidine-5-carboxylate 15a. Yellowish solid, (mp: 120–123 °C) 1H NMR (400 MHz, CDCl3): δ 7.3 (1H, s), 7.2 (2H, m), 6.7 (1H, dd, J = 8.5 & 2.4 Hz), 6.2 (1H, d, J = 16 Hz), 5.9 (1H, dd, J = 3.2 & 16.0 Hz), 4.5 (1H, d, J = 4.0 Hz), 4.1 (2H, q, J = 7.3 Hz), 2.4 (3H, s), 2.7 (2H, t, J = 7.0 Hz) 1.7 (2H, t, J = 7.0 Hz), 1.2 (3H, t, J = 7.1 Hz), 1.2 (6H, s). 13C NMR (100 MHz, CDCl3): δ 168.6, 155.9, 154.6, 152.4, 130.5, 129, 128, 125.8, 122.8, 115.1, 100.7, 78.3, 61.5, 34.4, 27.4, 25, 19.5, 14.4. IR (KBr) νmax cm−1: 3240, 3117, 2960, 1693, 1514, 1097. HR-ESIMS m/z: 371.11979 (calculated for C21H26N2O4, 370.1893).
4.1.4.12. Ethyl-4-(4-methoxystyryl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxyl-ate 16a. Colourless liquid. 1H NMR (400 MHz, CDCl3): δ 7.2 (1H, s), 7 (2H, d, J = 7.9 Hz), 6.8 (2H, d, J = 8 Hz), 6.2 (1H, d, J = 16 Hz), 5.9 (1H, dd, J = 3.2, 16 Hz), 5.5 (1H, s), 4.5 (1H, d, J = 4 Hz), 4.1 (2H, q, J = 7.3 Hz), 3.7 (3H, s), 2.4 (3H, s), 1.2 (3H, t, J = 7.1 Hz). 13C NMR (100 MHz, CDCl3): δ 167.6, 154.5, 144.6, 132.9, 129.7, 129.3, 127.4, 127.3, 118.3, 109.7, 101.8, 85.3, 59.7, 55.2, 36.4, 19.5, 14.4. IR (KBr) νmax cm−1: 3240, 2960, 1693, 1514, 1462, 1097. HR-ESIMS m/z: 317.1495 (calculated for C17H20N2O4, 316.1423).
4.1.4.13. Ethyl 4-(2,3-dimethoxystyryl)-1,2,3,4-tetrahydro-6-methyl-2-oxopyrimidine-5-carboxylate 17a. Yellowish liquid, 1H NMR (400 MHz, CDCl3): δ 7.2 (1H, s), 6.8 (1H, d, J = 16 Hz), 6.5 (3H, m), 5.9 (1H, dd, J = 3.2, 16 Hz), 5.5 (1H, s), 4.5 (1H, d, J = 4 Hz), 4.1 (2H, q, J = 7.4 Hz), 3.7 (6H, s), 2.4 (3H, s), 1.2 (3H, t, J = 7.1 Hz). 13C NMR (100 MHz, CDCl3): δ 167.7, 156.8, 155.7, 151.2, 144.6, 130, 127.1, 122.5, 119.9, 104.5, 101.9, 93.8, 59.7, (2C, 55.3), 36.4, 19.5, 14.3. IR (KBr) νmax cm−1: 3240, 2960, 1693, 1514. HR-ESIMS m/z: 347.1601 (calculated for C18H22N2O5, 346.1529).
4.1.4.14. Ethyl 4-(3,4,5-trimethoxystyryl)-1,2,3,4-tetrahydro-6-methyl-2-oxopyrimidine-5-carboxylate 18a. Greyish liquid. 1H NMR (400 MHz, CDCl3): δ 7.2 (1H, s), 6.5 (2H, d, J = 16 Hz), 6.2 (2H, s), 5.9 (1H, dd, J = 3.2, 16 Hz), 5.5 (1H, s), 4.5 (1H, d, J = 3.4 Hz), 4.1 (2H, q, J = 7.6 Hz), 3.7 (9H, s), 2.4 (3H, s), 1.2 (3H, t, J = 7.1 Hz). 13C NMR (100 MHz, CDCl3): δ 167.8, 156.8, (2C, 155.7), 150.2, 144.6, 130, 127.1, 122.5, (2C, 104.5), 101.9, 59.7, 56.3, (2C, 55.3), 36.4, 19.5, 14.34. IR (KBr) νmax cm−1: 3240, 2960, 1693, 1514, 1462. HR-ESIMS m/z: 377.1822 (calculated for C19H24N2O6, 376.1634).
4.1.4.15. Ethyl-4-(2,4-dimethoxystyryl)-1,2,3,4-tetrahydro-6-methyl-2-oxopyrimidine-5-carboxylate 19a. Yellowish liquid. 1H NMR (400 MHz, CDCl3): δ 7.2 (1H, s), 7 (1H, m), 6.8 (1H, d, J = 16 Hz), 6.2 (2H, m), 5.96 (1H, dd, J = 3.2, 16 Hz), 5.5 (1H, s), 4.5 (1H, d, J = 4 Hz), 4.1 (2H, q, J = 7.3 Hz), 3.7 (6H, s), 2.4 (3H, s), 1.2 (3H, t, J = 7.1 Hz). 13C NMR (100 MHz, CDCl3): δ 167.7, 159.8, 156.7, 154.2, 144.6, 130, 127.1, 122.5, 119.9, 104.5, 101.9, 93.8, 59.7, (2C, 55.3), 36.4, 19.5, 14.3. IR (KBr) νmax cm−1: 3240, 2960, 1693, 1514, 1097. HR-ESIMS m/z: 347.1611 (calculated for C18H22N2O5, 346.1529).
4.1.4.16. Ethyl-4-((E)-2-(benzo[d][1,3]dioxol-6-yl)vinyl)-1,2,3,4-tetrahydro-6-methyl-2-oxo pyrimidine-5-carboxylate 20a. White solid, (mp: 181–184 °C). 1H NMR (200 MHz, CDCl3): δ 7.8 (1H, s), 6.9 (2H, m), 6.7 (1H, m), 6.6 (1H, d, J = 2.2 Hz), 6.3 (1H, d, J = 16.0 Hz), 6.1 (2H, s), 5.9 (1H, dd, J = 3.4 & 16.0 Hz), 5.6 (1H, s), 4.61 (1H, d, J = 3.4 Hz), 4.1 (2H, q, J = 7.2 Hz), 2.3 (3H, s), 1.2 (3H, t, J = 7.2 Hz). 13C NMR (100 MHz, CDCl3): δ 165.7, 154.2, 148.8, 146.9, 145.3, 136.8, 129.2, 124.3, 120.8, 109.1, 107.2, 102, 101.8, 60.2, 55.6, 19.4, 14.5. IR (KBr) νmax cm−1: 3230, 2930, 1710, 1655, 1520, 1107. HR-ESIMS m/z: 331.1309 (calculated for C18H22N2O5, 330.1216).
4.1.4.17. Ethyl-4-(benzo[d][1,3]dioxol-6-yl)-1,2,3,4-tetrahydro-6-methyl-2-oxopyrimidine-5-carboxylate 21a. White solid, (mp: 176–178 °C). 1H NMR (200 MHz, CDCl3): δ 7.9 (1H, s), 6.8 (1H, d, J = 8.7 Hz), 6.7 (1H, m), 6.7 (1H, d, J = 1.6 Hz), 5.9 (2H, s), 5.6 (1H, s), 5.3 (1H, s), 4 (2H, q, J = 7 Hz), 2.3 (3H, s), 1.18 (3H, t, J = 7 Hz). 13C NMR (100 MHz, CDCl3): δ 165.6, 153.6, (2C, 147.2), 146.3, 137.9, 120, 108.1, 107, (2C, 101.1), 60, 55.4, 18.5, 14.2. IR (KBr) νmax cm−1: 3225, 2928, 1710, 1651, 1514, 1097. HR-ESIMS m/z: 305.1131 (calculated for C18H22N2O5, 304.1059).
4.2. Biology
RPMI-1640 medium, Dulbecco's modified eagle's medium, penicillin, streptomycin, fetal calf serum, sodium bicarbonate, phosphate buffer saline, trypsin, gentamycin sulphate, tryphan blue, ethanol, DMSO, paraformaldehyde were purchased from Sigma Chemicals Co. Glacial acetic acid from Fischer scientific, PBS and trichloroacetic acid (TCA) from Merck specialties private limited. All the human cancer cell lines (Colo-205, PC-3, THP-1, and A549) as well as normal epithelial cell line (fR-2) were obtained from National Center for Cell Science, Ganeshkhind, Pune-4111007 (India) and National Cancer Institute, Biological Testing Branch DTP/DCTD/NCI, Frederick Cancer Research and Development Center, Fairview Center, Suite 205, 1003 West 7th Street, Frederick, MD 21701-8527 (USA).Wounded areas were progressively photographed with Olympus C-7070 with 700M camera (100× magnification). The percentage of wound closure was estimated by the following equation:
| Wound closure% = [1 − (wound area at t1/wound area at t0) × 100] |
4.3. Molecular docking studies
All the computational studies were carried out in the Schrodinger suite 2012 molecular modeling software. The 2D structure of the ligands was built in the maestro window. The structures were then converted to their respective 3D structures, with various conformers, tautomers and ionization states using the Ligprep and Confgen modules.64 The three dimensional structure of CDK2 and PI3Kγ i.e., PDB ID 2DUV and PDB ID 1E7U respectively were downloaded from Protein Data Bank.65 The receptor was prepared for docking using the Protein Preparation wizard. Extra precision (XP) scoring function of Glide was used for carrying out the docking studies.66Conflicts of interest
There are no conflicts to declare.Acknowledgements
The author (SF) is grateful to DST SERB, India for providing research funding bearing the Project No: TAR/2018/000088. The authors (FAA, AA) are also grateful to King Saud University, Riyadh, Saudi Arabia for providing research funding bearing the Researchers Supporting Project Number RSP-2020/160.References
- M. Gordaliza, Clin. Transl. Oncol., 2007, 9, 767–780 CrossRef CAS.
- A. J. V. Wangelin, H. Neumann, D. Grdes, S. Klaus, D. Strübing and M. Beller, Chem. – Eur. J., 2003, 9, 4286–4294 CrossRef.
- P. A. Wender, S. T. Handy and D. L. Wright, Chem. Ind., 1997, 6, 765 Search PubMed.
- J. Zhu and H. Bienaymee, Multicomponent Reactions, Willey-VCH, Weinheim, Germany, 2005 Search PubMed.
- A. Domling, Chem. Rev., 2006, 106, 17 CrossRef.
- D. J. Ramon and M. Yus, Angew. Chem., 2005, 44, 1602 CrossRef CAS.
- C. Simon, T. Constantieux and J. Rodriguez, Eur. J. Org. Chem., 2004, 24, 4957–4980 CrossRef.
- J. Zhu, Eur. J. Org. Chem., 2003, 7, 1133 CrossRef.
- R. V. A. Orru and M. de. Greef, Synthesis, 2003, 10, 1471 CrossRef.
- L. F. Tietze, F. Haunert, M. Shibasaki, J. F. Stoddart and F. Vgtle, Stimulating Concepts in Chemistry, Wiley-VCH, Weinhei, 2000, vol. 39, pp. 3168–3210 Search PubMed.
- H. Bienayme, C. Hulme, G. Oddon and P. Schmitt, Chem. – Eur. J., 2000, 6, 3321 CrossRef CAS.
- L. F. Tietze and A. Modi, Med. Res. Rev., 2000, 20, 304–322 CrossRef CAS.
- M. Bell, K. Frisch and K. A. Jorgensen, J. Org. Chem., 2006, 71, 5407–5410 CrossRef CAS.
- R. W. Armstrong, A. P. Combs, P. A. Tempest, S. D. Brown and T. A. Keating, Acc. Chem. Res., 1996, 29, 123 CrossRef CAS.
- P. Eilbracht, L. Barfacker, C. Buss, C. Hollmann, B. E. Kitsos-Rzychon, C. L. Kranemann, T. Rische, R. Roggenbuck and A. Schimdt, Chem. Rev., 1999, 99, 3329 CrossRef CAS.
- U. Bora, A. Saikia and R. C. Boruah, Org. Lett., 2003, 5, 435–438 CrossRef CAS.
- C. O. Kappe, Acc. Chem. Res., 2000, 33, 879–888 CrossRef CAS.
- I. Ugi, Pure Appl. Chem., 2001, 73, 187–191 CAS.
- D. J. Ramon and M. Yus, Angew. Chem., Int. Ed., 2005, 44, 1602 CrossRef CAS.
- A. Fayol and J. Zhu, Org. Lett., 2005, 7, 239–242 CrossRef CAS.
- E. Rajanarendar, S. Raju, M. N. Reddy, S. R. Krishna, L. H. Kiran, A. R. N. Reddy and Y. N. Reddy, Eur. J. Med. Chem., 2012, 50, 274–279 CrossRef CAS.
- J. B. Sperry and D. L. Wright, Curr. Opin. Drug Discov. Dev., 2005, 8, 723–740 CAS.
- R. E. Ziegert, J. Toraeng, K. Knepper and S. Braese, J. Comb. Chem., 2005, 7, 147–169 CrossRef CAS.
- C. Hulme, in Multicomponent Reactions, ed. J. Zhu and H. Bienaymé, Wiley-VCH, Weinheim, 2005, p. 311 Search PubMed.
- L. Garuti, M. Roberti and D. Pizzirani, Med. Chem., 2007, 7, 481–490 CAS.
- C. Gil and S. J. Braese, Comb. Chem., 2009, 11, 175–197 CrossRef CAS.
- W. A. Loughlin, J. D. A. Tyndall, M. P. Glenn and D. P. Fairlie, Chem. Rev., 2004, 104, 6085–6117 CrossRef CAS.
- P. G. Baraldi, M. A. Tabrizi, S. Gessi and P. A. Borea, Chem. Rev., 2008, 108, 238–263 CrossRef CAS.
- E. Petricci, C. Mugnaini, M. Radi, A. Togninelli, C. Bernardini, F. Manetti, M. C. Parlato, M. L. Renzulli, M. Alongi, C. Falciani, F. Corelli and M. Botta, Arkivoc, 2006, 7, 452 Search PubMed.
- I. M. Lagoja, Chem. Biodiversity, 2005, 2, 1 CrossRef CAS.
- P. Biginelli, Gazz. Chim. Ital., 1893, 23, 360–461 Search PubMed.
- J. Lloyd, H. J. Finlay, K. Atwal, A. Kover, J. Prol, L. Yan, R. Bhandaru, W. Vaccaro, T. Huynh, C. S. Huang, M. Conder, T. Jenkins, H. Sun, D. Li and P. Levesque, Bioorg. Med. Chem. Lett., 2009, 19, 5469 CrossRef CAS.
- W. Vacarro, T. Huynh, J. Lloyd, K. S. Atwal, H. J. Finlay, P. C. Levesque, M. L. Conder, T. Jenkins-West, H. Shi and L. Sun, Bioorg. Med. Chem. Lett., 2008, 18, 6381 CrossRef.
- J. Lloyd, H. J. Finlay, W. Vacarro, T. Hyunh, A. Kover, R. Bhandaru, L. Yan, K. Atwal, M. L. Conder, T. Jenkins-West, H. Shi, C. Huang, D. Li, H. Sun and P. Levesque, Bioorg. Med. Chem. Lett., 2010, 20, 1436–1439 CrossRef CAS.
- C. O. Kappe, Eur. J. Med. Chem., 2000, 35, 1043–1052 CrossRef CAS.
- S. Chitra, D. Devanathan and K. Pandiarajan, Eur. J. Med. Chem., 2010, 45, 367–371 CrossRef CAS.
- M. B. Deshmukh, S. M. Salunkhe, D. R. Patil and P. V. Anbhule, Eur. J. Med. Chem., 2009, 44, 2651–2654 CrossRef CAS.
- T. U. Mayer, T. M. Kapoor, S. J. Haggarty, R. W. King, S. L. Schreiber and T. J. Mitchison, Science, 1999, 286, 971–974 CrossRef CAS.
- L. Ye, J. Liu, R. Zhang, Y. Guo, H. Wang, Q. Meng, Y. Sun and Z. Liu, Molecules, 2019, 24, 891 CrossRef.
- H. Y. K. Kaan, V. Ulaganathan, O. Rath, H. Prokopcov, D. Dallinger, C. O. Kappe and F. Kozielski, J. Med. Chem., 2010, 53, 5676–5683 CrossRef CAS.
- C. M. Wright, R. J. Chovatiya, N. E. Jameson, D. M. Turner, G. Zhu, S. Werner, D. Huryn, M. Pipas, J. M. Billy, W. Day, P. Wip and J. L. Brodskya, Bioorg. Med. Chem., 2008, 16, 3291 CrossRef CAS.
- O. C. Agbaje, O. O. Fadeyi, S. A. Fadeyi, L. E. Myles and C. O. Okoro, Bioorg. Med. Chem. Lett., 2011, 21, 989 CrossRef CAS.
- B. R. P. Kumar, G. Sankar, R. B. N. Baig and S. Chandrashekaran, Eur. J. Med. Chem., 2009, 44, 4192–4198 CrossRef.
- D. A. Ibrahim and A. M. El-Metwally, Eur. J. Med. Chem., 2010, 45, 1158–1166 CrossRef CAS.
- K. K. Pasunooti, H. Chai, C. N. Jensen, B. K. Gorityala, S. Wang and X. W. Liu, Tetrahedron Lett., 2011, 52, 80–82 CrossRef CAS.
- M. Wu, J. Yu, W. Zhao, J. Wu and S. Cao, J. Fluorine Chem., 2011, 132, 155–159 CrossRef CAS.
- J. Svetlik and V. Kettmann, Tetrahedron Lett., 2011, 52, 1062–1066 CrossRef CAS.
- P. M. Kumar, K. S. Kumar, S. R. Poreddy, P. K. Mohakhud, K. Mukkanti and M. Pal, Tetrahedron Lett., 2011, 52, 1187–1191 CrossRef CAS.
- T. Okuhara, N. Mizuno and M. Misono, Adv. Catal., 1996, 41, 113–252 CAS.
- M. M. Heravi and S. Sadjadi, J. Iran. Chem. Soc., 2009, 6, 1–54 CrossRef CAS.
- M. M. Heravi, K. Bakhtiari and F. F. Bamoharram, Catal. Commun., 2006, 7, 373–376 CrossRef CAS.
- I. V. Kozhevnikov, Chem. Rev., 1998, 98, 171–198 CrossRef CAS.
- A. Shaabani, A. Bazgir and F. Teimouri, Tetrahedron Lett., 2003, 44, 857–859 CrossRef CAS.
- P. T. Anastas and J. C. Warner, Green Chemistry, 1998 Search PubMed.
- Y. Ma, C. Qian, L. Wang and M. Yang, J. Org. Chem., 2000, 65, 3864–3868 CrossRef CAS.
- A. Dondoni and A. Massi, Tetrahedron Lett., 2001, 42, 7975–7978 CrossRef CAS.
- S. Koul, J. L. Koul, S. C. Taneja, K. L. Dhar, D. S. Jamwal, K. Singh, R. K. Reen and J. Singh, Bioorg. Med. Chem., 2000, 8, 251–268 CrossRef CAS.
- P. Y. Chen, J. D. Wu, K. Y. Tang, C. C. Yu, Y. H. Kuo, W. B. Zhong and C. K. Lee, Molecules, 2013, 18, 7600–7608 CrossRef CAS.
- R. Danesi, W. D. Figg, E. Reed and C. E. Myers, Mol. Pharmacol., 1995, 47, 1106–1111 CAS.
- J. E. Liebmann, J. A. Cook, C. Lipschultz, D. Teague, J. Fisher and J. B. Mitchell, Br. J. Cancer, 1993, 68, 1104–1109 CrossRef CAS.
- L. Zhang, Z. Wang, T. Khishignyam, T. Chen, C. Zhou, Z. Zhang, M. Jin, R. Wang, Y. Qiu and D. Kong, Biomed. Pharmacother., 2018, 103, 1069–1078 CrossRef CAS.
- Y. Wang, H. Yang, H. Liu, J. Huang and X. Song, BMC Cancer, 2009, 9(174), 1–12 Search PubMed.
- S. Farooq, S. U. Rehman, A. Hussain, A. Hamid, M. A. Qurishi and S. Koul, Eur. J. Med. Chem., 2014, 84, 545–554 CrossRef CAS.
- K. S. Watts, P. Dalal, R. B. Murphy, W. Sherman, R. A. Friesner and J. C. Shelley, J. Chem. Inf. Model., 2010, 50, 534–546 CrossRef CAS.
- J. Lee, T. Park, S. Jeong, K. H. Kim and C. Hong, Bioorg. Med. Chem. Lett., 2007, 17, 1284–1287 CrossRef CAS.
- T. A. Halgren, R. B. Murphy, R. A. Friesner, H. S. Beard, L. L. Frye, W. T. Pollard and J. L. Banks, J. Med. Chem., 2004, 47, 1750–1759 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra09072g |
| This journal is © The Royal Society of Chemistry 2020 |