
A first new porous d–p HMOF material with multiple active sites for excellent CO2 capture and catalysis†
Jiao
Liu
a,
Guo-Ping
Yang
 *a,
Jing
Jin
a,
Dan
Wu
a,
Lu-Fang
Ma
ab and
Yao-Yu
Wang
*a
*a,
Jing
Jin
a,
Dan
Wu
a,
Lu-Fang
Ma
ab and
Yao-Yu
Wang
*a
aKey Laboratory of Synthetic and Natural Functional Molecule Chemistry of the Ministry of Education, Shaanxi Key Laboratory of Physico-Inorganic Chemistry, College of Chemistry & Materials Science, Northwest University, Xi’an 710127, P. R. China. E-mail: ygp@nwu.edu.cn; wyaoyu@nwu.edu.cn
bCollege of Chemistry and Chemical Engineering, Henan Key Laboratory of Function-Oriented Porous Materials, Luoyang Normal University, Luoyang 471934, P. R. China
First published on 3rd February 2020
Abstract
In this work, a new monometallic metal–organic framework (MOF), namely {[Pb2(L)2(H2O)]·H2O}n (1), was prepared via a N,O-mixed 2-(imidazol-1-yl)terephthalic acid (H2L) ligand and Pb(II) ions. Then, a first new porous d–p heterometallic MOF (HMOF), namely {[PbZn(L)2]·DMA·H2O}n (2), was yielded via Zn(II) ions and MOF 1 as the precursor because of the different coordination affinity of oxygen and nitrogen atoms with various metal ions. MOF 1 is a condensed packing motif, whereas HMOF 2 is a porous three-dimensional (3D) framework with unsaturated Pb(II) and Zn(II) active sites, uncoordinated carboxylate oxygen atoms and two kinds of porous channels due to the introduction of Zn(II) ions into the framework of MOF 1, which thus endowed HMOF 2 with excellent sorption and selectivity for CO2 over CH4. Moreover, HMOF 2 was explored to be an efficient heterogeneous catalyst for the fixation of CO2 with various epoxides because of the existence of the abundant unsaturated bimetallic sites as the Lewis acid. It is hoped that this work may supply an effective strategy to build plenty of d–p HMOF materials with excellent gas adsorption and catalytic properties.
Metal–organic frameworks (MOFs) are spatial extension porous materials, which are supported by metal ions as nodes and different organic ligands as linkers via coordination bonds.1–4 They are of wide interest not only due to their structural diversities, but also for various applications. In particular, some strategies,5–8 such as forming more open metal centers as the active sites, introducing polar functional groups in the pores of MOFs or generating different composite materials with MOFs, have been explored to strengthen the CO2 adsorption affinity and catalysis.
To date, there are some works about d–f or d–d heterometallic MOFs (HMOFs) for different gas sorption or catalysis.9–13 However, there are still some demerits in the syntheses of d–f or d–d HMOFs. For example, for d–f HMOFs, the metal ions of the f block can be easily used to build high-dimensional condensed packing structure because of their high coordination numbers, which leads to a decrease in pore size and even formation of MOFs without pores. For the preparation of d–d HMOFs, the two different metal ions of the d block tend to produce monometallic MOFs rather than HMOFs when coordinated with the same ligand in the reaction. As a p block element, the Pb(II) ion has a stereochemically active electron lone-pair and large ionic radius; its coordination number can change from two to ten to form different coordination types, for instance, the holodirected and hemidirected geometries, which are of great significance for the construction of Pb(II)-based MOFs.14 Therefore, we attempt to use metal ions of d and p blocks to build new HMOFs with novel topological and wide applications in this work, as we know, which are not yet reported in the literature until now.
Since different metal ions have distinct coordination affinities for electron donor groups, a N,O-mixed 2-(imidazol-1-yl)terephthalic acid (H2L) ligand was chosen to construct the HMOFs in this work. Herein, a new monometallic MOF, namely {[Pb2(L)2(H2O)]·H2O}n (1), was first prepared via the H2L ligand and Pb(II) ions. Then, a porous d–p HMOF, namely {[PbZn(L)2]·DMA·H2O}n (2), was yielded for the first time via the introduction of Zn(II) ions into the framework of MOF 1. Structural analyses show that MOF 1 is a condense packing motif, while HMOF 2 is a 3D porous framework with two different 1D pores based on 1D metal rod-shaped chains. Furthermore, HMOF 2 has high CO2 sorption and catalytic capacity, and can thus be used as the functional material for CO2 storage and separation as well as a heterogeneous catalyst.
The solvothermal reaction of Pb(II) ions with the H2L ligand affords the crystalline monometallic MOF 1. Then HMOF 2 is progressively yielded via Zn(II) ions and MOF 1 as the precursor. In addition, HMOF 2 can also be obtained by one pot reaction with an equal mole ratio of the salts and ligand. The two complexes both crystallize in the triclinic P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group. There are two kinds of Pb(II) ions, two L2− ligands and one bonded H2O molecule in the asymmetric unit of 1. Pb1 displays a six-coordinated geometry formed by five oxygen atoms from three deprotonated L2− ligands and one nitrogen atom to form the distorted pentagonal pyramid configuration. On the contrary, Pb2 is penta-coordinated formed by four oxygen atoms and one nitrogen atom in a distorted tetragonal pyramid-shaped geometry (Fig. 1a). In 1, the carboxylate groups of the fully deprotonated L2− adopt η2μ2χ2 and η2μ1χ2 modes to link with Pb(II) ions (Fig. S1, ESI†), and the adjacent Pb(II) ions are connected by carboxylate groups from two deprotonated L2− ligands (Fig. S2a, ESI†), and are further extended via L2− spacers to generate a 3D porous framework with 1D pores of 5.9 × 7.4 Å2 (Fig. S2b, ESI†). After adopting topological analysis, MOF 1 can be simplified as a new tetranodal (4,5,6,7)-connected net with the (414·67)(44·62)(47·63)(49·66) point symbol (Fig. S2c, ESI†).
space group. There are two kinds of Pb(II) ions, two L2− ligands and one bonded H2O molecule in the asymmetric unit of 1. Pb1 displays a six-coordinated geometry formed by five oxygen atoms from three deprotonated L2− ligands and one nitrogen atom to form the distorted pentagonal pyramid configuration. On the contrary, Pb2 is penta-coordinated formed by four oxygen atoms and one nitrogen atom in a distorted tetragonal pyramid-shaped geometry (Fig. 1a). In 1, the carboxylate groups of the fully deprotonated L2− adopt η2μ2χ2 and η2μ1χ2 modes to link with Pb(II) ions (Fig. S1, ESI†), and the adjacent Pb(II) ions are connected by carboxylate groups from two deprotonated L2− ligands (Fig. S2a, ESI†), and are further extended via L2− spacers to generate a 3D porous framework with 1D pores of 5.9 × 7.4 Å2 (Fig. S2b, ESI†). After adopting topological analysis, MOF 1 can be simplified as a new tetranodal (4,5,6,7)-connected net with the (414·67)(44·62)(47·63)(49·66) point symbol (Fig. S2c, ESI†).
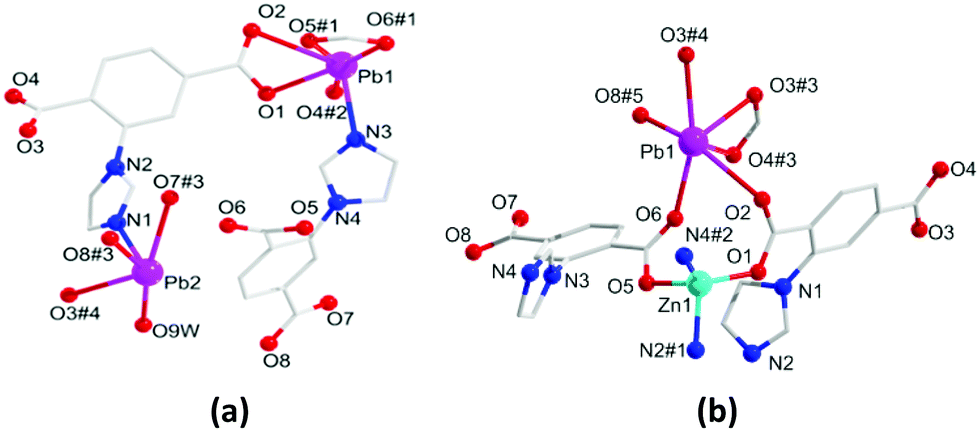 | ||
| Fig. 1 Coordination environment of Pb(II) and Zn(II) ions in 1 (a) and 2 (b). Symmetry codes: for 1: #1, 2 − x, −y, −z; #2, 2 − x, 1 − y, −z; #3, 1 + x, y, z; #4, 3 − x, 1 − y, 3 − z; for 2: #1, 1 − x, 1 − y, 1 − z; #2, −x, 2 − y, 1 − z; #3, 1 − x, 1 − y, −z; #4, −1 + x, −y, −z; #5, −x, 1 − y, 1 − z. | ||
HMOF 2 shows a 3D porous framework, and its asymmetric unit is composed of one Pb(II) ion and one Zn(II) ion, two L2− ligands, one dissociated DMA and one lattice water molecule. The Pb1 ion is six-coordinated with five carboxylate oxygen atoms to give rise to hemidirected geometry (Fig. 1b), which indicates that the electron lone-pairs of Pb(II) centers are stereochemically active. The Zn1 ion is four coordinated with two carboxylate oxygen atoms and two imidazole nitrogen atoms. The carboxylate groups of the L2− ligands adopt three kinds of bridging connection fashions in 2: η1μ1χ1, η3μ2χ3 and η2μ2χ2 to link Pb(II) and Zn(II) ions (Fig. S1, ESI†), giving a 1D metal rod-shaped chain (Fig. S3a, ESI†). Along the [722] axis, these 1D metal rod-shaped chains are further enlarged by deprotonated L2− to engender a 3D porous framework with 1D channel pores of 9.6 × 7.49 Å2 (Fig. S3b, ESI†). Interestingly, there exist uncoordinated carboxylate oxygen atoms on the surface of the 1D rectangular column channel of 5.8 × 6.3 Å2 along the [177] axis (Fig. S3c, ESI†). When all the dissociative molecules are removed, the effective free volume of 2 is ∼36%. On the principle of simplification, the whole framework of 2 can be simplified as a trinodal (5,7,11)-connected net with the point symbol of (311·410)(317·420·512·66)(32·43·54·6) (Fig. S3d, ESI†).
The thermogravimetric analysis curve of 2 shows that there is a 13.8% weight loss at ∼32–162 °C, which corresponds to the escape of dissociative solvent molecules (calcd 14.1%) (Fig. S4, ESI†). The main skeleton of the structure remains stable in the temperature range of ∼162–373 °C, and then collapses quickly. A powder X-ray diffraction data (PXRD) test indicated that the synthesized solid samples have high purity (Fig. S5, ESI†). In order to remove the guest molecules, the synthesized 2 was soaked in CH2Cl2 and heated to 105 °C for 4 h under high vacuum to obtain the desolvated 2a, the PXRD pattern of which proves that the main framework is intact and stable. Moreover, the infrared spectra demonstrate that there is no C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O vibration of DMA solvents in 2 (Fig. S6, ESI†).
O vibration of DMA solvents in 2 (Fig. S6, ESI†).
HMOF 2 has higher porosity than that of monometallic MOF 1 and possesses unsaturated bimetallic nodes and uncoordinated carboxylate oxygen atoms at the pore surface. Thus, we studied the gas sorption and separation properties of 2a for CO2 and CH4 systematically under different temperatures. At 195 K, the amount of CO2 reaches 128.2 cm3 g−1 (25.2 wt%) and the isotherm shows an obvious distributional sorption behavior (Fig. 2a). First of all, 2a adsorbs 35.5 cm3 g−1 (27.3 wt%) at P/P0 = 0.1, and then the tendency increases rapidly until saturation at 1 atm. The desorption isotherm does not trace the sorption process, and it is observed that there is a broad hysteresis phenomenon, which indicates that the sorption–desorption process is reversible and the gas molecules will be locked in the skeleton in the low pressure region. The reason for this phenomenon may be related to the contractive pores of the host skeleton which may be opened under the opening pressure, and 2 may thus be a case of a “breathing” MOF.16 The fitted Brunauer–Emmett–Teller (BET) and Langmuir surface areas are 221 and 611 m2 g−1 derived from the CO2 sorption curves at 195 K, respectively. Meanwhile, the pore size distribution was found in the range of 8.2–13.5 Å (Fig. S7, ESI†); in addition, the total micropore volume evaluated by using the Horvath–Kawazoe (H–K) method is 0.23 cm3 g−1, which is consistent with the structure analysis (0.21 cm3 g−1) of 2.
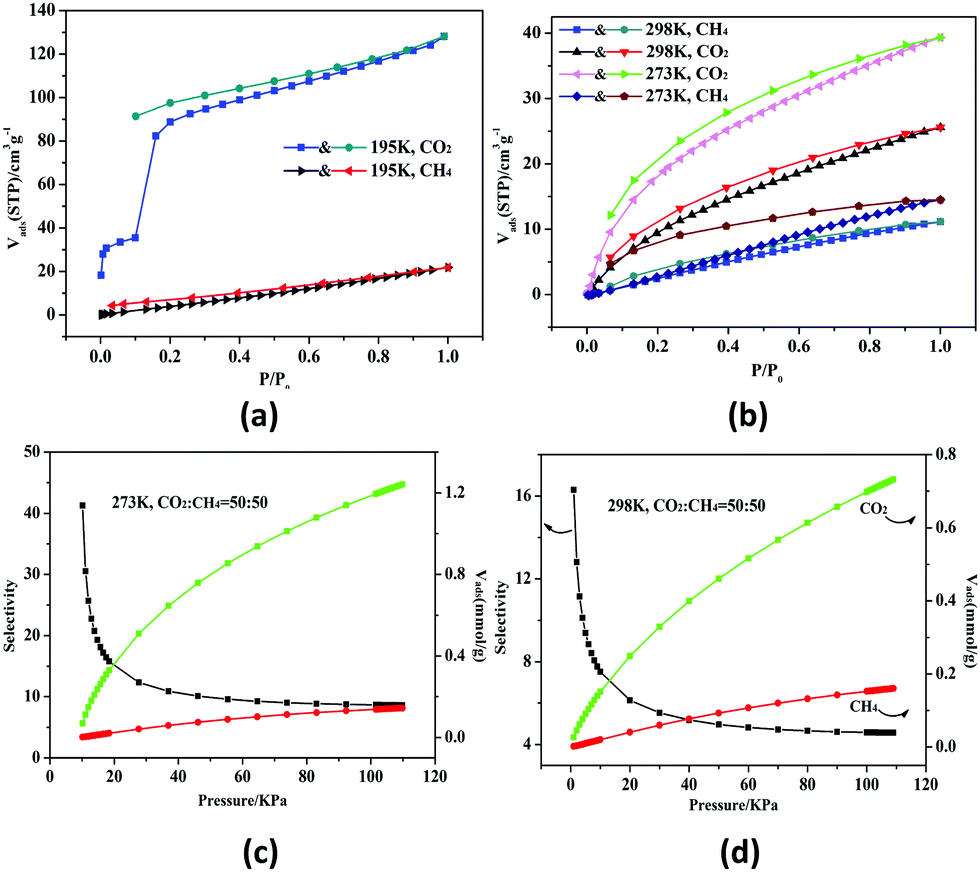 | ||
Fig. 2 CO2 and CH4 sorption and desorption isotherms at 195 K (a), and 273 and 298 K (b). The selectivity of 2a for CO2 and CH4 (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) at 273 K (c) and 298 K (d), respectively. :1) at 273 K (c) and 298 K (d), respectively. | ||
The existence of potential multi-active sites in 2a prompted us to continue to explore the separation performance. As shown in Fig. 2b, 2a has a higher sorption capacity of 39.3 cm3 g−1 (7.8 wt%) at 273 K and 25.5 cm3 g−1 (5.0 wt%) at 298 K, respectively. The CO2 uptake of 2a is superior to those of monometallic Zn(II) or Pb(II)-based MOFs with similar void volume at 298 K.15 On the contrary, the CH4 uptake is low at about 14.5 cm3 g−1 (1.03 wt%) at 273 K and 11.1 cm3 g−1 (0.79 wt%) at 298 K. Thus, the CO2/CH4 selectivity is calculated by the ideal adsorbed solution theory (Fig. S8, ESI†).17 The selectivity ranges from 4.5 to 16.3 at 298 K for the equimolar gas mixtures; remarkably, the initial value is 8.6 and then arrives to 41.3 at 273 K (Fig. 2c and d). Under the same conditions, the mole ratios for CO2/CH4 are set as 20/80 and 80/20 (Fig. S9, ESI†). The selectivity of 2a is better than lots of reported MOFs with polar pores or open metal sites (OMSs) (Tables S3 and S4, ESI†). The high selectivity may be caused by the synergistic effect of the OMSs and uncoordinated carboxylate oxygen atoms located within the wall of the porous surface, which gives rise to a highly polar framework to strengthen the interactions between the framework and CO2. In addition, the kinetic diameter of CO2 (3.30 Å) is smaller than that of CH4 (3.80 Å), promoting the sorption capacity of CO2.18,19
According to the Virial equation, the sorption enthalpies (Qst) of gas molecules were calculated for exploring the affinity of 2a (Fig. S10, ESI†). The Qst curve indicates that 2a has higher affinity to CO2, the main reason of which is that CO2 has a larger polarizability and quadrupole moment value (CO2, polarizability: 29.1 × 10−25 cm−3; quadrupole moment: 1.43 × 10−39 C m2; CH4, polarizability: 25.9 × 10−25 cm−3; quadrupole moment: 0 C m2).20,21 Overall, HMOF 2 can be used as a potential candidate for industrial gas purification.
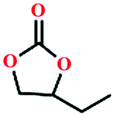
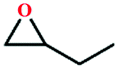
Due to the good thermal stability and CO2 sorption performance of HMOF 2, the cycloaddition of CO2 was investigated by exploring 2 as a catalyst. The OMSs adhere to the pore to serve as potential catalytic centers and CO2 binding sites in 2. In the first place, the relatively mild conditions were selected (0.1 MPa and 25 °C, Scheme 1), and butylene oxide (BO) reacts with CO2 at room temperature with a yield of 58% (Table 1), which indicates that 2 has certain activity in the presence of tetra-n-butyl ammonium bromide (TBAB) as a co-catalyst. Next, it is found that the reaction efficiency increases at a higher temperature (Fig. S11, ESI†). When the reaction temperature reaches 80 °C, the conversion is 97%. This conversion is comparable with that of the reported MOFs with Lewis acid catalytic centers.22 The high conversion should boil down to the existence of high density of Lewis acid sites and the rising of the reaction temperature, which accelerates the ring-opening of the substrates. Then, the epoxide substrates with different sizes of substituted groups (–CH3Br, –O–C4H9, –Ph and –CH2–OPh) were reacted under the same conditions. The experimental results show that the corresponding yields decreased with the increasing of the size of the substrate molecules as indicated by the 95% formation from 1-bromo-2,3-epoxypropane, 96% formation from butyl glycidyl ether, 78% formation from styrene oxide and 56% formation of 3-phenoxy-1,2-propylene carbonate from phenyl glycidyl ether. This phenomenon could be attributed to the limited diffusion of the sterically hindered substrate with large-size into the pore of 2.23,24
 | ||
| Scheme 1 Reaction scheme of CO2 and epoxide catalyzed by HMOFs. | ||
| Entry | Epoxide | Product | T (°C) | Yields (%) |
|---|---|---|---|---|
| Mole amounts: epoxides (20 mmol), 2 (0.2 mmol), and TBAB (2 mmol). | ||||
| 1 |
|
|
25 | 58 |
| 2 |
|
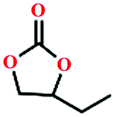
|
40 | 67 |
| 3 |
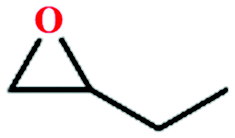
|
|
60 | 76 |
| 4 |
|
|
80 | 97 |
| 5 |
|
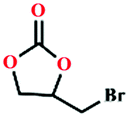
|
80 | 95 |
| 6 |
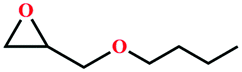
|
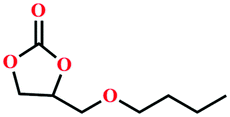
|
80 | 96 |
| 7 |
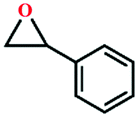
|
|
80 | 78 |
| 8 |
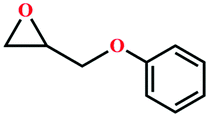
|
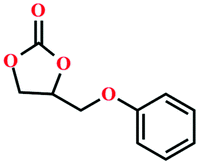
|
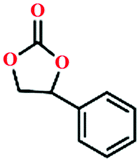
80 | 56 |
Meanwhile, the mechanism for the cycloaddition of the CO2 reaction by 2 and TBAB as a co-catalyst was proposed via the reported mechanism (Scheme 2).25,26 The epoxide substrates were first activated by linking the reactants to the unsaturated bimetal nodes (where Zn(II) and Pb(II) ions act as the Lewis acidic sites) in 2 through interactions of the coordination bonds. Then the Br− of TBAB attacked carbon atoms on the less sterically hindered carbons to activate the substrates, thereby causing the ring-opening of the substrates to form the active oxygen anions. Thirdly, the active oxygen anion was reacted with CO2 to engender alkylcarbonate anions. The corresponding cyclic carbonate and the catalyst for the next reaction were finally formed by a ring-closing step. And among these steps, the ring-opening is considered to be a decisive step for the rate of CO2 cycloaddition reaction. Furthermore, the cycling catalytic efficiency of HMOF 2 was also studied carefully, which was kept at the beginning. However, the PXRD of HMOF 2 was changed after more cycling experiments, and the catalysis was decreased gradually, a possibility of which is the longer distance of Pb–O in HMOF 2, and its coordination bonds of the Pb(II) ions are weaker than those of rare earth ions with oxygen atoms, causing the breakdown of the framework.
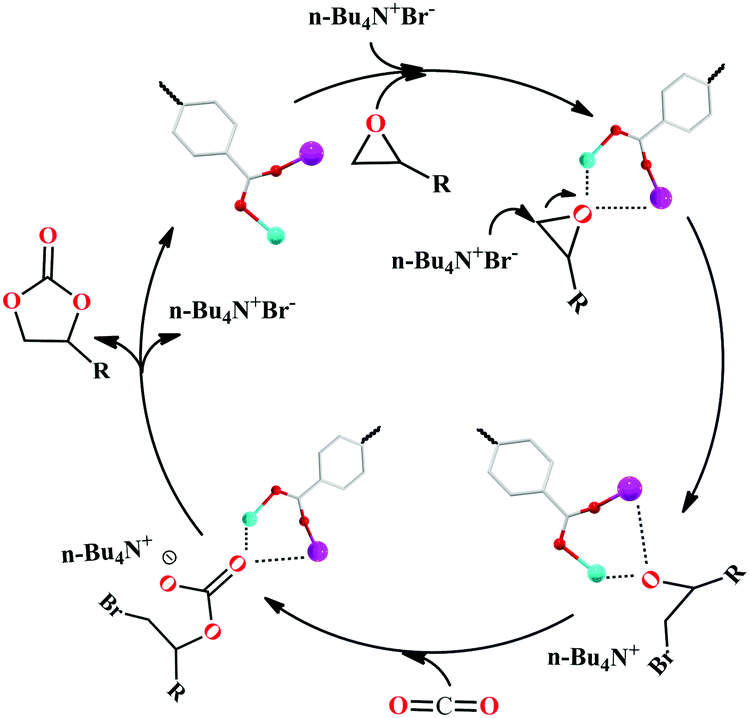 | ||
| Scheme 2 Proposed mechanism of CO2 cycloaddition via HMOF 2 and TBAB as a co-catalyst. | ||
In summary, a new porous d–p HMOF 2 has been successfully assembled by monometallic 1 as the precursor via a stepwise strategy. More interestingly, HMOF 2 displays permanent porosity with unsaturated bimetallic nodes and uncoordinated carboxylate oxygen atoms at the pore surface, affording highly selective CO2 capture and catalytic fixation. This work may supply an effective approach to isolate new d–p HMOF materials possessing CO2 sorption and catalytic performance.
We acknowledge the financial support from the NSFC (21531007 and 21971207), the NSF of Shaanxi Province (2019JM-013, and 2017KJXX-59), the Key Science and Technology Innovation Team of Shaanxi Province (2019TD-007), the China Postdoctoral Science Foundation (2016M600807 and 2017T100765), and the Technology Foundation for Selected Overseas Scholars of Shaanxi Province (2018041).
Conflicts of interest
There are no conflicts to declare.Notes and references
- V. Guillerm, D. Kim, J. F. Eubank, R. Luebke, X. Liu, K. Adil, M. Soo Lah and M. Eddaoudi, Chem. Soc. Rev., 2014, 43, 6141–6172 RSC.
- Y.-X. Tan, F. Wang and J. Zhang, Chem. Soc. Rev., 2018, 47, 2130–2144 RSC.
- B. Zhao, X.-Y. Chen, Z. Chen, W. Shi, P. Cheng, S.-P. Yan and D.-Z. Liao, Chem. Commun., 2009, 3113–3115 RSC.
- Y. P. Wu, J. W. Tian, S. Liu, B. Li, J. Zhao, L. F. Ma, D. S. Li, Y. Q. Lan and X. Bu, Angew. Chem., Int. Ed., 2019, 58, 12185–12189 CrossRef CAS PubMed.
- E. Fotopoulou, J. Martinez-Lillo, M. Siczek, T. Lis, V. Tangoulis, M. Evangelisti, E. K. Brechin and C. J. Milios, Chem. Commun., 2018, 54, 6153–6156 RSC.
- Z.-L. Wu, J. Dong, W.-Y. Ni, B.-W. Zhang, J.-Z. Cui and B. Zhao, Inorg. Chem., 2015, 54, 5266–5272 CrossRef CAS PubMed.
- J.-H. Liu, Y.-J. Qi, D. Zhao, H.-H. Li and S.-T. Zheng, Inorg. Chem., 2019, 58, 516–523 CrossRef CAS PubMed.
- G. Ayoub, B. Karadeniz, A. J. Howarth, O. K. Farha, I. Đilović, L. S. Germann, R. E. D. K. Užarević and T. Friščića, Chem. Mater., 2019, 31, 5494–5501 CrossRef CAS.
- A. Szécsényi, G. Li, J. Gascon and E. A. Pidko, Chem. Sci., 2018, 9, 6765–6773 RSC.
- J. F. Kurisingal, R. Babu, S.-H. Kim, Y. X. Li, J.-S. Chang, S. J. Cho and D.-W. Park, Catal. Sci. Technol., 2018, 8, 591–600 RSC.
- Y.-J. Qi, D. Zhao, X.-X. Li, X. Ma, W.-X. Zheng and S.-T. Zheng, Cryst. Growth Des., 2017, 17, 1159–1165 CrossRef CAS.
- B. Iqbal, M. Saleem, S. N. Arshad, J. Rashid, N. Hussain and M. Zaheer, Chem. – Eur. J., 2019, 25, 1–10 CrossRef.
- T.-T. Liu, J. Liang, R. Xu, Y.-B. Huang and R. Cao, Chem. Commun., 2019, 55, 4063–4066 RSC.
- E.-C. Yang, J. Li, B. Ding, Q.-Q. Liang, X.-G. Wang and X.-J. Zhao, CrystEngComm, 2008, 10, 158–161 RSC.
- X.-Y. Li, L.-N. Ma, Y. Liu, L. Hou, Y.-Y. Wang and Z. Zhu, ACS Appl. Mater. Interfaces, 2018, 10, 10965–10973 CrossRef CAS PubMed.
- J. Liu, G.-P. Yang, Y. Wu, Y. Deng, Q. Tan, W.-Y. Zhang and Y.-Y. Wang, Growth Des., 2017, 17, 2059–2065 CrossRef CAS.
- M. Du, C.-P. Li, M. Chen, Z.-W. Ge, X. Wang, L. Wang and C.-S. Liu, J. Am. Chem. Soc., 2014, 136, 10906–10909 CrossRef CAS PubMed.
- L. Zou, X. Sun, J. Yuan, G. Li and Y. Liu, Inorg. Chem., 2018, 57, 10679–10684 CrossRef CAS PubMed.
- H. R. Fu, Y. Zhao, Z. Zhou, X. G. Yang and L. F. Ma, Dalton Trans., 2018, 47, 3725–3732 RSC.
- L.-T. Du, Z.-Y. Lu, K.-Y. Zheng, J.-Y. Wang, X. Zheng, Y. Pan, X.-Z. You and J.-F. Bai, J. Am. Chem. Soc., 2013, 135, 562–565 CrossRef CAS PubMed.
- M.-Q. Zhu and M. A. J. Carreon, Appl. Polym. Sci., 2014, 131, 39738–39750 Search PubMed.
- J. Dong, H. Xu, S. Hou, Z. Wu and B. Zhao, Inorg. Chem., 2017, 56, 6244–6250 CrossRef CAS PubMed.
- Y.-Z. Li, H.-H. Yang, H.-Y. Wang, L. Hou, Y.-Y. Wang and Z. Zhu, Chem. – Eur. J., 2018, 24, 865–871 CrossRef CAS PubMed.
- H. Xu, C.-K. Cao, H.-S. Hu, S.-B. Wang, J.-C. Liu, P. Cheng, N. Kaltsoyannis, J. Li and B. Zhao, Angew. Chem., Int. Ed., 2019, 58, 6022–6027 CrossRef CAS PubMed.
- B. Ugale, S. S. Dhankhar and C. M. Nagaraja, Inorg. Chem., 2016, 55, 9757–9766 CrossRef CAS PubMed.
- Y. Yuan, J. Li, X. Sun, G. Li, Y. Liu, G. Verma and S. Ma, Chem. Mater., 2019, 31, 1084–1091 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: The syntheses of the crystals, X-ray crystallographic data in CIF format, the additional structural figures, the 1H NMR spectra, the details on sorption, and bond length/angle tables. CCDC 1961111 and 1961112. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9cc09664g |
| This journal is © The Royal Society of Chemistry 2020 |