
Recent advances of plasmonic nanoparticle-based optical analysis in homogeneous solution and at the single-nanoparticle level
Mingce
Tian
a,
Zhiqin
Yuan
 *a,
Ying
Liu
a,
Chao
Lu
a,
Zhongju
Ye
b and
Lehui
Xiao
*b
*a,
Ying
Liu
a,
Chao
Lu
a,
Zhongju
Ye
b and
Lehui
Xiao
*b
aState Key Laboratory of Chemical Resource Engineering, College of Chemistry, Beijing University of Chemical Technology, Beijing, 100029, China. E-mail: yuanzq@mail.buct.edu.cn; Tel: +86-10-64411957
bCollege of Chemistry, Nankai University, Tianjin, 300071, China. E-mail: lehuixiao@nankai.edu.cn; Fax: +86-022-23500201; Tel: +86-022-23500201
First published on 11th May 2020
Abstract
Plasmonic nanoparticles with special localized surface plasmon resonance (LSPR) characters have been widely applied for optical sensing of various targets. With the combination of single nanoparticle imaging techniques, dynamic information of reactions and biological processes is obtained, facilitating the deep understanding of their principle and design of outstanding nanomaterials. In this review, we summarize the recently adopted optical analysis of diverse analytes based on plasmonic nanoparticles both in homogeneous solution and at the single-nanoparticle level. A brief introduction of LSPR is first discussed. Colorimetric and fluorimetric homogeneous detection examples by using different sensing mechanisms and strategies are provided. Single plasmonic nanoparticle-based analysis is concluded in two aspects: visualization of chemical reactions and understanding of biological processes. The basic sensing mechanisms and performances of these systems are introduced. Finally, this review highlights the challenges and future trend of plasmonic nanoparticle-based optical analysis systems.
 Mingce Tian | Mingce Tian is currently a Master’s student at Beijing University of Chemical Technology, Beijing, China. He is engaged in the synthesis and characterization of metal nanoclusters and organic luminescent molecules. His study also covers the chemical and biological applications of metal nanoclusters and organic luminescent molecules. |
 Zhiqin Yuan | Zhiqin Yuan is currently an Associate Professor of State Key Laboratory of Chemical Resource Engineering, College of Chemistry, Beijing University of Chemical Technology. He obtained his PhD from the College of Chemistry and Chemical Engineering, Hunan University, in 2013 with Dr Yan He and Dr Edward S. Yeung. His research interests are focused on the synthesis and analytical application of metal nanoparticles and nanoclusters, carbon quantum dots, and layered nanomaterials. |
 Ying Liu | Ying Liu is currently a Master’s student at Beijing University of Chemical Technology, Beijing, China. She is engaged in the synthesis and characterization of metal nanoparticles and nanoclusters. Her study also covers the chemical and biological applications of metal nanoparticles and nanoclusters. |
 Chao Lu | Chao Lu received his Ph.D. degree in Materials Science from Chinese Academy of Sciences in 2004. He is currently a full professor at State Key Laboratory of Chemical Resource Engineering, Beijing University of Chemical Technology. In 2011, he was selected to participate in the ‘New Century Outstanding Talent’ scheme of the Ministry of Education. His research interests are focused on the synthesis and characterization of nanostructured materials, nanosensors, and chemiluminescence. He is responsible for national and international research projects and has published more than 100 papers. |
 Zhongju Ye | Zhongju Ye is currently pursuing her Ph.D. studies in the College of Chemistry at Nankai University. She obtained her Master’s degree in Analytical Chemistry from Hunan Normal University in 2017. Her research topic is focused on single-particle tracking, single-particle catalysis, and super-resolution imaging. |
 Lehui Xiao | Lehui Xiao is a professor in the College of Chemistry at Nankai University. He obtained his B.S., Master’s and Ph.D. degrees in Analytical Chemistry from Hunan University in 2006, 2008 and 2011, respectively. His research focuses on single-molecule imaging and detection, single-particle tracking, single-particle catalysis and super-resolution imaging. Website: http://www.xiaolhlab.cn. |
Introduction
There has been a consistent increase in quantitative and dynamic analysis of chemical and/or biological targets, which reveals their status and behaviors and explains their important roles in diverse fields, including environment, materials, food and life sciences.1 In the past few decades, a number of electronic detection methods and related-instruments have been developed for this purpose. In comparison with electronic sensing, optical analysis by measuring photons rather than electrons attracts increasing research interest because of the simplification of analytical instruments and low cost. In addition, optical analysis avoids the use of strong electronic or electromagnetic fields, and has no special demands on the properties of materials and provides high safety to the operators. Despite the exploration of many organic probe-based optical sensing systems, the complicated preparation/fabrication, as well as the photon instability of organics, partially limits their practical applications, i.e., long-term tracking. Therefore, it is appealing to construct stable optical systems with new materials.With the ever-increasing nanoscience, nanomaterials with a large specific surface area and unique optical/electronic properties are widely used for catalysis, adsorption, and detection.2–4 In particular, plasmonic nanoparticle-based optical analysis has been extensively applied for the investigation of various targets, such as chemicals, biomolecules, physical characteristics (e.g., temperature and viscosity), and so on.5–7 The localized surface plasmon resonance (LSPR) of noble metal nanoparticles associated with the collective oscillation of surface free electrons not only affects the color of the nanoparticles, but also interacts with nearby photons/electrons and results in the change of fluorescence. In this case, plasmonic nanoparticle-based optical analysis can be performed with absorption and fluorescence. Because of the high stability of LSPR, it enables long-term tracking of single plasmonic nanoparticles, which reveals the dynamic variations and detailed processes of chemical/biological reactions.8,9
The growing applications of plasmonic nanoparticles in various fields have been realized in the last decade. Although many review papers introducing plasmonic nanoparticle-based applications in individual research areas have been reported, a comprehensive summary of optical analysis both in homogeneous solution and at the single-nanoparticle level will facilitate the researchers to choose an appropriate system in the future work. In this review, we essentially focus on recent advances in optical analysis using plasmonic nanoparticles (e.g., gold and silver) that have been reported since 2015. The LSPR property of homogeneous solution and single nanoparticles is discussed briefly. Optical analysis in homogeneous solution based on colorimetry and fluorimetry follows with discussion of the detection of diverse chemical and physical characteristics. Single plasmonic nanoparticle analysis with enhanced detection sensitivity is introduced. Beyond detection, single nanoparticle-based dynamic analysis for tracking chemical reactions and biological processes is also delineated. Finally, the challenges and future outlooks of plasmonic nanoparticle-based optical analysis for multiplex target detection in complex surroundings are discussed based on our findings and experience.
LSPR of plasmonic nanoparticles
LSPR is an interesting optical property of plasmonic nanoparticles, which originates from the resonant oscillation of conduction electrons at the plasmonic nanoparticle surface with electromagnetic field.10 Taking spherical gold nanoparticles (Au NPs) as an example, the resonant oscillation of conduction electrons is related to the electromagnetic field (Fig. 1). The oscillation behaviors are largely dependent on the relationship between the light frequency and plasma frequency. It is reported that if the light frequency is higher than the plasma frequency, the dielectric constant of plasmonic nanoparticles is positive and light is transmitted.12 However, the dielectric constant becomes negative when the light frequency is smaller than the plasma frequency, leading to the reflection of most of the light. As a consequence, the resonant oscillation of conduction electrons not only enhances the ability of photon absorption, but also increases the scattering efficiency. Both photon absorption and scattering weaken the strength of incident light. Therefore, it is more accurate to represent the plasmonic nanoparticle-induced loss of light with extinction rather than absorbance only. Because of the collective oscillation of surface free electrons, it dramatically increases the photon absorption and scattering efficiency. As a result, in comparison with traditional organic dyes, the plasmonic nanoparticles usually show a high extinction coefficient, which is usually several orders of magnitude larger.29 Beyond the increment of extinction, the LSPR can also boost the electric fields near the plasmonic nanoparticle's surface, and the enhancement is reversely proportional to the distance to the nanoparticle surface. That is, the strongest enhancement is on the surface of plasmonic nanoparticles. Plasmonic nanoparticle-based optical analysis usually relies on these two effects.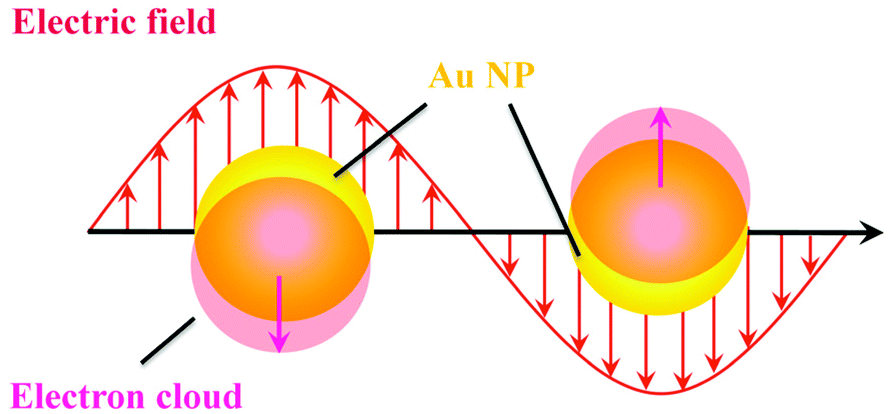 | ||
| Fig. 1 Schematic illustration of the origin of LSPR. | ||
Generally speaking, the LSPR peak of most plasmonic nanoparticles is in the visible range, resulting in bright color in solution or at the single-particle level. Interestingly, the LSPR characters of plasmonic nanoparticles are tunable by changing the size, morphology, interparticle distance, surrounding environment, etc.14 For example, regarding Au NPs, the maximum absorption wavelength is simply proportional to the size.30 The increased size of Au NPs increases the number of surface free electrons and decreases the discrete energy gap. Then, the maximum LSPR absorption of Au NPs shows a red-shift with the increase in size. However, for gold nanorods (Au NRs), they have two distinct LSPR bands, originating from longitudinal and transversal LSPR, respectively. The transversal one is proportional to the diameter of Au NRs, which is similar to that of Au NPs, while the longitudinal LSPR is only dependent on the aspect ratio (AR, length/diameter) of Au NRs. Despite the different LSPR properties of Au NPs and Au NRs, the increased size can increase the number of oscillation electrons, extinction coefficient and scattering intensity. In addition, resonant oscillation of conduction electrons can be affected by the interparticle distance. In other words, the interparticle plasmon coupling has effects on the LSPR of plasmonic nanoparticles. Note that the dramatic red-shift of LSPR absorption is observed during the aggregation of plasmonic nanoparticles, and the accompanying color change can be utilized as a signal report of the dispersity of plasmonic nanoparticles.
In addition to the size, morphology, and interparticle distance, the surrounding environment also plays an important role in the LSPR of plasmonic nanoparticles. As referred above, the resonant oscillation is related to both the light frequency and plasma frequency. With a certain plasmonic nanoparticle, the plasma frequency is fixed due to the definite dielectric function of the nanoparticle. Nevertheless, the frequency of light can be affected by the surrounding environments with different refractive indexes. Thus, surface ligands, media, temperature, and ionic strength can influence the LSPR of plasmonic nanoparticles. Such a phenomenon has been proved through theoretical calculations by Mie et al. using Au NPs as the model.31 The refractive index can change the incident electromagnetic field on the Au NP surface through the calculation, and subsequently alters the LSPR of Au NPs.
Optical assays in homogeneous solution
The extremely high extinction coefficient, as well as the tunable LSPR frequency, makes plasmonic nanoparticles a popular probe for the design of optical analysis systems.11,12 A parameter that changes or utilizes the LSPR of plasmonic nanoparticles can be explored for the development of versatile systems. Since the extinction of plasmonic nanoparticles is dependent on the refractive index, the change of the particle morphology, size, interparticle distance, and local environment caused by targets has been advanced for the exploitation of sensitive and selective optical systems. The LSPR of plasmonic nanoparticles not only induces the color change of colloid solution, but also interacts with organic dyes by modulating their fluorescence signals. Based on these facts, plasmonic nanoparticle-based optical analysis systems are usually designed through the change of absorption and fluorescence. Additionally, the LSPR of plasmonic nanoparticles greatly enhances their scattering intensity, making them adaptable for intensity analysis based on dynamic light scattering techniques.13Colorimetric assays
Among these plasmonic nanoparticles, Au NPs with easy preparation and surface functionalization have attracted great attention. As mentioned above, the interparticle distance affects the collective oscillation of free electrons, which results in the color change of colloid Au NPs from red to blue. Once the aggregated Au NPs become redispersive, color recovery from blue to red is observed. Such color variation suggests the feasibility for designing a general system toward different targets through colorimetric response. Taking this into consideration, a number of works have been reported for colorimetric sensing of diverse analytes including DNA, protein, amino acid, metal ions, anions, and even cells.3,12,14,15 For target-induced colorimetric changes, there are essentially two interaction pathways: target–ligand interaction and target–surface interaction.For instance, the classical DNA detection is based on target–ligand interaction induced aggregation with a sandwich structure, in which two different single-stranded DNAs (ssDNAs) are separately modified onto the Au NP surface. Upon the introduction of target DNA, both ssDNAs form stable double stranded DNA (dsDNA) through intermolecular hydrogen bonding and aggregate Au NPs subsequently. With this mechanism, scientists developed many systems for oncogene detection.12 It is well known that ssDNA shows high stability once attached onto the Au NP surface, which is partially due to the decreased accessibility toward DNAase. However, the hybridization efficiency of ssDNA on the Au NP surface is surprisingly higher than that of free ssDNA in solution. According to isothermal titration colorimetry, Fong et al. found that such an uncommon enhancement is attributed to enthalpy, about ∼20 kcal mol−1.16 Molecular dynamics simulations also proved that structural confinement of ssDNA on the Au NP surface can lead to the increment of enthalpy. In contrast, free ssDNA has the trend to form stable dsDNA without lower enthalpy. Thus, to some extent, ssDNA–Au NPs is better for DNA sensing in comparison with ssDNA only. Because of the similar molecular structure of DNA and RNA, such a mechanism is also applicable for the detection of RNA. By using RNA functionalized Au NPs as the optical reporter, Ding et al. realized the colorimetric analysis of microRNA-148a with a limit of ∼1.9 nM.17
DNA is one of the most important biomaterials with supernatural features. It can bind to DNA, RNA, protein, and even small molecules. Utilizing DNA–protein binding affinity, Mao et al. reported a fast and convenient gene detection protocol by modulating the interaction between dsDNA–protein and Au NPs.18 Using human papillomavirus (HPV) as the model, Sso7d diagnostic protein from hyperthermophilic archaebacteria Sulfolobus solfataricus was investigated. Sso7d protein with cysteine residues can bind to Au NPs and readily causes aggregation (Fig. 2A). Sso7d protein can also strongly bind to dsDNA with a specific sequence. However, Sso7d protein shows negligible affinity to ssDNA. In this case, the introduction of target gene (HPV types 16 and 18) induces the formation of dsDNA, which binds to Sso7d protein and suppresses the aggregation of Au NPs. With the assistance of polymerase chain reaction, they achieved HPV 16 and HPV 18 fragment detection in Pap smear clinical specimens (n = 52), with a sensitivity and specificity of 85.7%/100.0% and 85.7%/91.7%, respectively. Beyond DNA and proteins, Au NPs also show great potential in sensing of small molecules and ions. Using calix[4]arene derivative capped Au NPs as the probe, Yin et al. achieved colorimetric detection of aliphatic diamines at the submillimole level based on host–guest interaction.19 With this system, other monoamines, e.g., amino acids, show negligible influence. With the use of chelation, colorimetric Cu2+ detection has been reported using cysteamine modified Au NPs.20
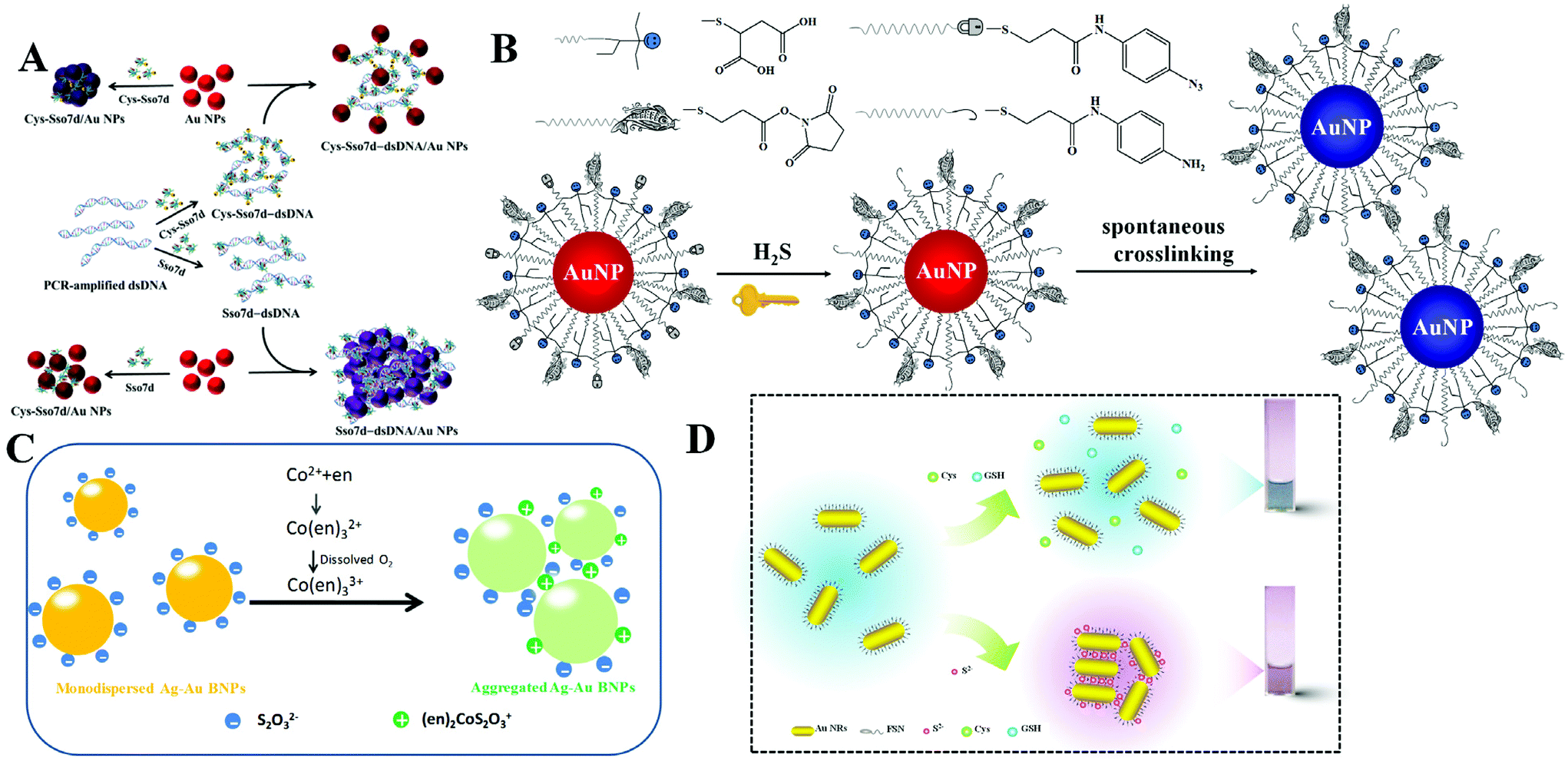 | ||
| Fig. 2 (A) Schematic representation of DNA-dependent Sso7d- or Cys-Sso7d-induced aggregation of Au NPs. Reprinted with permission from ref. 18. Copyright 2017, American Chemical Society. (B) Schematic representation of the sensing principle of AE-Au NP probes for the detection of H2S. Reprinted with permission from ref. 21. Copyright 2015, American Chemical Society. (C) Schematic illustration of colorimetric Co2+ detection using Ag–Au BNPs. Reprinted with permission from ref. 22. Copyright 2017, Royal Society of Chemistry. (D) Schematic illustration of H2S induced aggregation of the FSN-Au NRs. Reprinted with permission from ref. 29. Copyright 2015, Royal Society of Chemistry. | ||
To realize high selectivity, hydrogen-bonding, as well as chelation, has been explored for the design of specific systems. However, these interactions are sensitive to surrounding environments, and sometimes lead to false results. To overcome this issue, Yuan et al. reported the construction of sensing systems with satisfactory selectivity and rapidity by introducing chemical reactions.21 Chemical reactions are generally unique and respond to a specific target. Utilizing this tactic, they proposed a selective and rapid colorimetric detection platform for hydrogen sulfide (H2S) with the combination of azide-sulfide chemistry and an active ester-primary amine cross-linking reaction. The addition of H2S first reduces the azido derivative to primary amine, and the generated amine then reacts with active ester spontaneously to cross-link adjacent Au NPs, as shown in Fig. 2B. The rapid reaction rate makes the colorimetric response appear within 5 minutes. Since the azide-sulfide chemistry is reactant-specific, other anions including F−, Br−, I−, N3−, NO2−, NO3−, SO32−, SO42−, and glutathione (GSH), or other thiolates (i.e., tiopronin and thioglycolic acid) do not interfere in H2S sensing. By exploring the selective chelation reaction, they developed a colorimetric Co2+ detection method using S2O32− coated silver–gold bimetallic nanoparticles (Ag–Au BNPs).22 The sensing mechanism is that simultaneous addition of Co2+ and ethylenediamine (en) leads to the formation of a positively charged (en)2CoS2O3+ complex on the surface of Ag–Au BNPs (Fig. 2C). The formed (en)2CoS2O3+ decreases the electrostatic repulsion between adjacent Ag–Au BNPs and causes the aggregation. Such a chelation reaction is Co2+ selective, and other metal ions, such as Cu2+, Pb2+, Ag+, Zn2+, Ba2+, Mn2+, Fe2+, Fe3+, Ni2+ and Hg2+, cannot result in a conspicuous colorimetric response.
In addition to direct target-ligand interaction induced aggregation, the introduction of a special target (competitor) can inhibit the aggregation. For example, Zhang et al. reported a rapid and colorimetric DNA detection based on the competition strategy.23 SYBR Green I, a typical DNA dye with multi-charges, was utilized to induce the aggregation of citrate-capped Au NPs within 1 min. Target DNA drives the formation of dsDNA, which adsorbs SYBR Green I and inhibits the aggregation of Au NPs. This method allows the visual detection of target DNA at 0.1 nM. Using a DNA aptamer modified Au NP dimer as the optical reporter, Wang et al. explored a novel approach for the design of colorimetric assays.24 The Au NP dimer was formed by a contrivable DNA conformation with a head-to-hand structure. The specific aptamer-target binding provides good selectivity toward an expected object (microcystin-LR) over other interferents. Different from Au NP cluster formation, this work utilizes a Au NP dimer, providing high sensitivity with a LOD of 0.05 nM.
Targets with reducing ability can change metal ions to metal atoms, and then induce the growth of metal nanoparticles.25 With the silver mirror reaction-assisted formation of silver nanoparticles (Ag NPs), colorimetric detection of reducing sugars (glucose, galactose, fructose, mannose, maltose, and lactose) has been performed.26 The LOD toward glucose was determined to be 40 nM, which is much lower than that in other reported works. Beyond the redox reaction, the amalgamation reaction also affects the morphology of Au NPs. Using gold nanostars (Au NSs) as the probe, Xu et al. reported the colorimetric Hg2+ detection by amalgamation-induced morphology transition.27 The shape transition of tips on Au NSs dramatically affects the LSPR properties, resulting in sensitive color change. Because of the selective and rapid amalgamation reaction, this method displays high selectivity over other transition metal ions and exhibits excellent sensitivity toward Hg2+ with a LOD of 0.24 nM.
In the presence of an oxidant, Au NRs can be gradually oxidized into small Au NPs, accompanying colorimetric change. On the basis of this phenomenon, Mao et al. developed a portable method for the visual detection of different targets by integrating the DNA hydrogel and Au NRs together.28 In this work, a hydroxyl radical generated from H2O2 and Fe2+ acts as the oxidant. With such a system, targets that can produce H2O2 with appropriate treatment are theoretically detectable. In addition, targets that change the conformation of the DNA hydrogel are also detectable. By regulating the backbone of the DNA hydrogel, the colorimetric detection of Pb2+ and cocaine was proved.
Note that the most important role of surface ligands is to prevent Au NPs from aggregation. Once the target-gold surface binding affinity is stronger than ligand-surface interaction, the ligand would be replaced by the added target, leading to the aggregation of Au NPs. H2S with strong binding affinity to metal (gold and silver, comparable to the covalent bond) attracts growing research interest. Based on fluorosurfactant functionalized Au NRs (FSN-AuNRs), Yuan and Lu et al. proposed a colorimetric H2S detection platform with satisfactory selectivity (Fig. 2D).29 The condensed FSN ligand bilayer gives Au NRs high stability against salt, pH and temperature. The introduced H2S can cross the FSN bilayer interstices due to the small size and remove FSN because of the strong Au–S affinity. The decreased surface protection induced the formation of Au NR aggregates and color change of solution. With this designed nanoprobe, the colorimetric response induced by 0.2 μM H2S was detected. The combined Au–S binding and small size of H2S ruled out the interference from other sulfur containing anions (i.e., S2O32−, SO32− and SO42−) and thiolates (e.g., GSH, cysteine, and methionine). Moreover, this method was successfully applied for the evaluation of H2S synthetase activity with high accuracy. H2S has also forceful binding affinity to silver, and yields Ag2S on the Ag NP surface. Such a reaction suggests the possibility of colorimetric H2S sensing with Ag NPs. Employing DOPA capped Ag NPs as the reporter, a colorimetric H2S detection approach has been established by Sanskriti et al.30 The proposed system works in the pH value range from 5 to 11 and realizes sensitive H2S sensing with a LOD of 65 nM.
Fluorimetric assays
As is known, the LSPR originates from the collective oscillation of conduction electrons from a plasmonic nanoparticle surface. Those excited electrons can interact with the dipole of nearby fluorophores and change their fluorescence properties. In general, this interaction may cause two different results: fluorescence quenching and fluorescence enhancement. Specifically, quenching or enhancement induced by LSPR can be affected by many factors, including the size and the morphology of plasmonic nanoparticles, fluorescence characters of fluorophores, and the distance between fluorophores and plasmonic nanoparticles. The synergy of all factors decides the final influence on fluorescence. Among these factors, distance is the most important one as summarized in Aroca's report.31 When the distance is less than 10 nm, the fluorescence of fluorophores undergoes a distinct decrease due to the energy damping. A reasonable explanation is that the collective oscillation of conduction electrons generates intense electromagnetic local field on the surface and it decays exponentially on the nanometer scale outside the surface. Thus, LSPR induced fluorescence quenching usually occurs within 10 nm of the plasmonic nanoparticle surface. Interestingly, within such a short distance, the electromagnetic local field interacts with all kinds of dipoles with no dramatic difference. As a result, fluorescence of all dyes with different emission maxima can be efficiently quenched. In other words, spectral overlap between LSPR and fluorescence is not necessary. Once the distance becomes larger than 10 nm, the influence from local electromagnetic field may change, leading to different fluorescence regulation effects (i.e., fluorescence enhancement).32 In comparison with LSPR-induced fluorescence quenching, the latter requires suitable LSPR and fluorescence pairs. And maximum fluorescence enhancement was found when the excitation wavelength of the fluorophore matches the LSPR spectra.33,34 Interestingly, when the size of Au NPs becomes very small, the LSPR disappeared due to the decreased conduction electrons, which enlarges the discrete energy gap. As a consequence, Au NPs turn fluorescent.35The DNA–AuNP platforms have attracted growing interest during the last two decades.36,37 Different from colorimetric assays, dye-labelled DNA is linked onto the surface of Au NPs. The introduction of the target weakens the adhesion of dye-labelled DNA and results in the desorption of the probe from the surface. Then, the quenched fluorescence recovers. With a fluorescence “turn-on” strategy, this approach usually exhibits high sensitivity toward targets. Through the DNA–RNA hybridization, this strategy is also applied for fluorimetric detection of RNA using dye-labelled DNA-modified Au NPs.38 Through the amplification of nuclease, Pompa and Fiammengo et al. reported a universal route for the fluorimetric analysis of microRNA (Fig. 3A).39 In the proposed system, nuclease only acts on a dsDNA-like structure when the target microRNA combines the DNA probe. Such a design permits the sensitive target microRNA perception at 0.2 fmol. As the DNA-modified Au NPs show inherent high stability, this platform is also capable of microRNA sensing in live Hydra vulgaris (Fig. 3B).40 The large surface area of Au NPs allows high DNA density; thus, it is possible to detect multiple targets with diverse DNA functionalization on the same Au NPs. This assumption is demonstrated by many researchers. Li et al. realized simultaneous detection of Cu2+ and Zn2+ in HepG2 cancer cells using DNAzyme-modified Au NPs (Fig. 3C).41 Note that the surface functionalization of thiolated-DNA is usually time-consuming and not very efficient. To facilitate the attachment of dye-labelled DNA, Wu et al. proposed an electrostatic attraction approach.42 By this method, dye-labelled DNA is assembled onto a positively charged Au NP surface rather than Au–S covalent bonding, largely shortening the time cost of probe construction. By cooperating with the hybridization chain reaction, the proposed system provided sensitive fluorescence imaging of YM155 microRNA in HeLa cells.
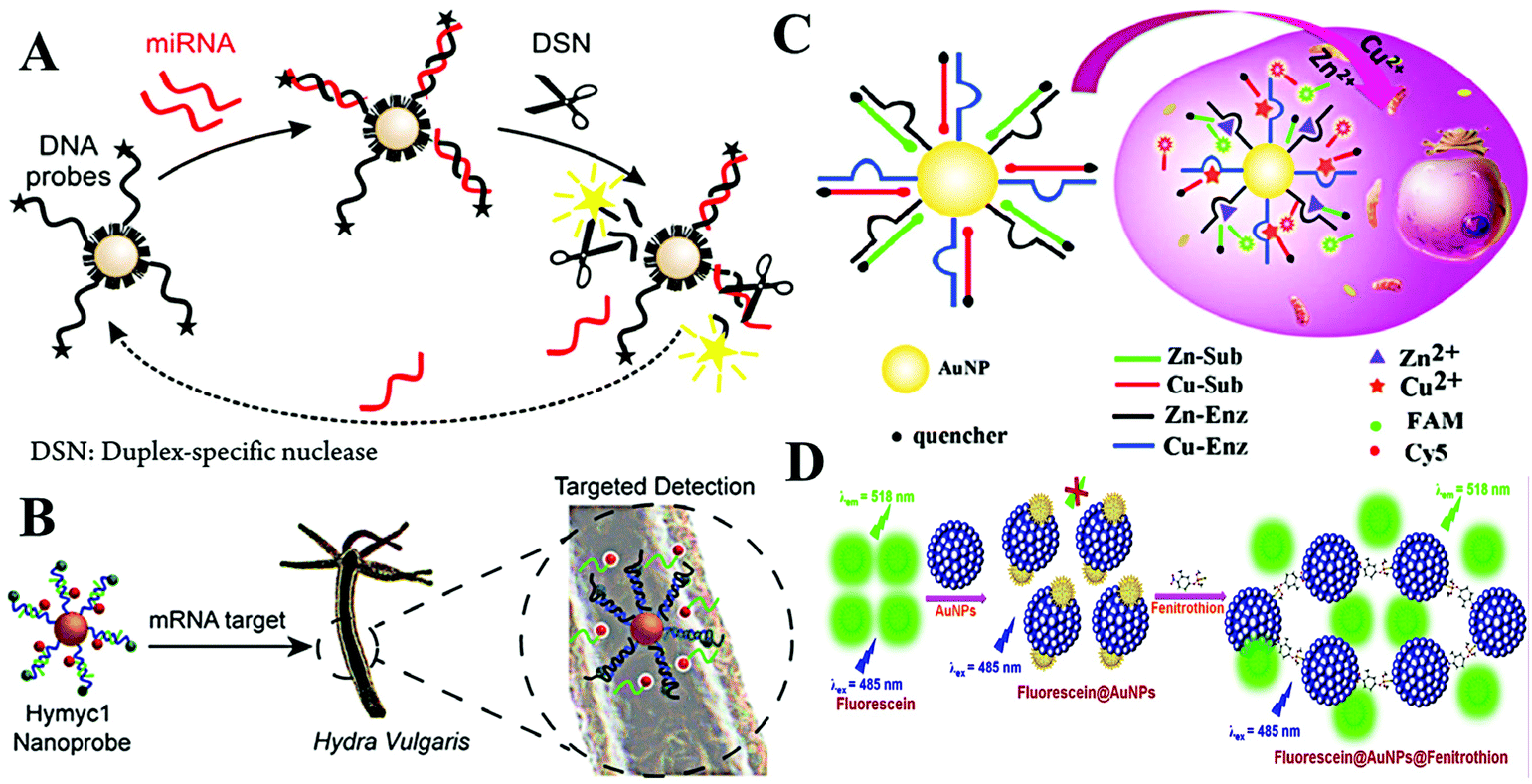 | ||
| Fig. 3 (A) Schematic of nuclease-amplified DNA detection. Reprinted with permission from ref. 39. Copyright 2014, American Chemical Society. (B) Scheme of the simultaneous imaging of Zn2+ and Cu2+ in living cells based on DNAzyme-modified AuNPs. Reprinted with permission from ref. 40. Copyright 2015, American Chemical Society. (C) Schematic illustration of the process of mRNA detection in Hydra. Reprinted with permission from ref. 41. Copyright 2019, American Chemical Society. (D) The schematic representation of the mechanism of quenching of fluorescein by Au NPs and turn-on response of Au NP quenched fluorescein in the presence of fenitrothion. Reprinted with permission from ref. 43. Copyright 2018, Elsevier. | ||
Beyond dye-labelled DNA, fluorescence of other fluorophores can also be efficiently quenched with direct interaction. Therefore, organic and inorganic fluorophores are used to construct fluorimetric sensing systems with Au NPs. Simple mixing of fluorescein and Au NPs leads to evident fluorescence quenching, after the addition of fenitrothion, a well-known pesticide containing phosphorus and sulfur atoms, causing visible fluorescence enhancement.43 The increased fluorescence is due to the fenitrothion-induced aggregation of Au NPs, which desorbs fluorescein and inhibits the fluorescence quenching from LSPR (Fig. 3D). Using this simple system, Nebu et al. successfully detected fenitrothion with a LOD of 6.05 nM. It is reported that perylene tends to form an excimer by intercalating the interparticle space of Ag NPs.44 The fluorescence of the excimer was boosted by the LSPR of Ag NPs. Thus, heparin-mediated aggregation of Ag NPs greatly enhances the fluorescence of perylene, making a perylene/Ag NP system a good candidate for fluorimetric detection of heparin and heparinise. The LODs toward heparin and heparinise were 5 nM and 0.005 U mL−1, respectively. In addition, inorganic fluorophores, such as semiconducting quantum dots, graphene quantum dots, upconversion nanoparticles, etc., have been explored to develop fluorimetric systems.45,46
The changed surface oxidation largely affects the metal centered electron transition. Lu et al. reported a fluorimetric hemoglobin detection method using f-Au NPs.49 The sensing mechanism is based on the hydroxyl radical-induced oxidation of the f-Au NP surface, which inhibits the metal centered electron transition and subsequently quenches the fluorescence. With this approach, a LOD of 5.0 nM toward hemoglobin is realized.
The ligand to metal/metal–metal charge transfer or metal/metal–metal to ligand charge transfer is very important to ligand (i.e., thiolates and polymers) stabilized f-Au NPs. Particularly, GSH stabilized f-Au NPs are brighter in aggregated state.50 Recently, Yuan and co-corkers found that 11-mercaptoundecanoic acid capped f-Au NPs have stronger emission in aqueous media (poor solvent) than in EtOH (good solvent).51 The enhanced emission is assigned to the rigidification of 11-mercaptoundecanoic acid (MUA), which restricts the rotation of MUA and enhances the ligand to metal/metal–metal charge transfer efficiency. This phenomenon has been utilized to detect H2S in an aqueous/EtOH mixture (Fig. 4A).
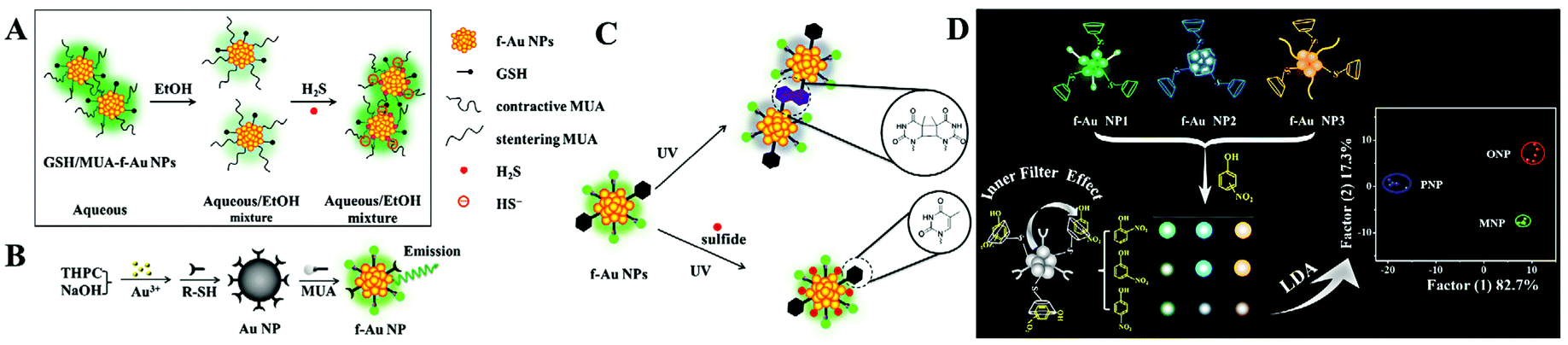 | ||
| Fig. 4 (A) Schematic illustration of the H2S detection principle using GSH/MUA-f-Au NP nanoprobes. Reprinted with permission from ref. 51. Copyright 2020, Springer. (B) Illustration of the synthetic route toward the preparation of dual-ligand protected f-Au NPs. Reprinted with permission from ref. 52. Copyright 2011, Royal Society of Chemistry. (C) Schematic principle of dual-ligand stabilized f-Au NPs for H2S sensing. Reprinted with permission from ref. 53. Copyright 2013, Royal Society of Chemistry. (D) Illustration of f-Au NP array-based discrimination of nitrophenol isomers by integrating host–guest interaction and inner filter effect. Reprinted with permission from ref. 55. Copyright 2018, American Chemical Society. | ||
To apply f-Au NPs into sensing application, the selectivity toward expected targets should be high. However, this demand is difficult to realize. A major problem is that the surface thiolate usually plays one role: maintaining the fluorescence or recognizing targets. To overcome this challenge, Yuan and co-workers developed a dual-ligand functionalization strategy. That is, one of the ligands (MUA) maintains the fluorescence of Au NPs, and the other (different thiolates) captures the designed targets (Fig. 4B). Using this approach, they reported the sensitive detection of Pb2+ with a LOD of 5 nM using glutathione and MUA co-functionalized f-Au NPs.52 The specific GSH-Pb2+ chelation provides high selectivity toward Pb2+ over other metal ions or anions. By exploring a fluorescence turn-on strategy, they also achieved selective H2S detection using 1-(10-mercaptodecyl)-5-methylpyrimidine-2,4-dione (TSH) and MUA co-functionalized f-Au NPs.53 The formation of the TSH dimer under UV light irradiation causes the aggregation of f-Au NPs and quenches the fluorescence. However, the added H2S adsorbs onto the f-Au NP surface and inhibits the aggregation through strong electrostatic repulsion (Fig. 4C). As a result, the fluorescence enhances with the increasing H2S concentration.
The above-mentioned systems detect only one target. To realize multiple target analysis, a sensor array-based technique is usually combined. Array-based detection strategies utilize the selective interaction between the sensing elements and target, which generates a unique response pattern for a specific target.54 Through the protein-f-Au NP interaction, Yuan et al. proposed a sensor array for the discrimination of eight proteins with different molecular weights and isoelectric points.35 They used dual-thiolates as the surface stabilizing ligands, one is to interact with proteins, and the other is to produce fluorescence. With low protein concentration (A280 = 0.005), they realized 100% discrimination of 48 unknown protein samples. In comparison with proteins, isomers of small molecules with tiny structural differences are more difficult to discriminate. To improve the differentiation ability, they introduced the host–guest interaction and inner filter effect.55 In this work, three f-Au NPs with diverse emission characters were modified with β-cyclodextrin. With the integration of the host–guest (β-cyclodextrin/nitrophenol) interaction and inner filter effect (nitrophenol/f-Au NPs), they achieved successful discrimination of three nitrophenol isomers (Fig. 4D). And interestingly, mixtures of two isomers have also been well distinguished under different compositions.
Optical assays at the single-nanoparticle level
The signal from homogeneous solution represents the ensemble averaging results, that is, all events are recorded through averaging. If the signals from individual plasmonic nanoparticles can be collected, more information might be obtained. Toward this purpose, many works on measuring the optical signals (LSPR absorption spectrum and scattering intensity) have been explored. In comparison with conventional analysis methods, optical assays with single plasmonic nanoparticles generally supply high sensitivity due to the reduced number of probes and targets. On the other hand, ensemble averaging measurement cannot reveal the detailed feature of individual events; thus the events with low possibilities might be neglected. Optical analysis at the single plasmonic nanoparticle level can collect information from individual objects and show dynamic characters, providing a promising tool for the deep understanding of reaction dynamics and biological processes with the integration of time-resolved and/or spatially resolved strategies.Optical detection based on single plasmonic nanoparticles
The design of single plasmonic nanoparticle-based detection systems is similar to homogeneous assays, while the detected signal originates from individual single plasmonic nanoparticles rather than from the bulk solution.8 There are three categories of detection ways: aggregation of single plasmonic nanoparticles, surface/morphology variation of single plasmonic nanoparticles and single plasmonic nanoparticle-regulated fluorescence. | ||
| Fig. 5 (A) Schematic diagram of the single-molecule sandwich assay with plasmonic Au NPs. Reprinted with permission from ref. 56. Copyright 2010, American Chemical Society. (B) Schematic illustration of colorimetric antigen sensing at the single-particle level with Au NP-Ag NP pairs. Reprinted with permission from ref. 60. Copyright 2016, American Chemical Society. (C) Working principle of single-nanoparticle analysis for PPi assay with Au PNLs. Reprinted with permission from ref. 65. Copyright 2019, American Chemical Society. (D) Diagram of the light path for the optical microscopic imaging of single nanoparticles and the principle of the color-coded single-particle detection for PPi Assay. Reprinted with permission from ref. 68. Copyright 2019, American Chemical Society. | ||
The aggregation of plasmonic nanoparticles not only causes a red-shift in scattering color, but also enhances the scattering intensity. Through enhancing the scattering intensity of single Au NPs, Poon et al. proposed a general immunoassay for cancer biomarkers (Fig. 5B).60 Individual Au NPs and Ag NPs were linked by an antigen–antibody immunoreaction. The scattering intensity of the resulting Au NP–Ag NP pair was much higher than that of a sole Au NP or Ag NP. Using the scattering intensity as the signal transmitter, they realized quantification of three typical antigens, including carcinoembryonic antigen (CEA), prostate-specific antigen (PSA), and alpha fetoprotein (AFP). The LODs toward these antigens were found to be 1.7, 3.3, and 5.9 pM, respectively. By changing Au NPs with 100 nm gold nanospots, Kang and co-workers reported a very sensitive single-particle immunosensor based on 3D light sheet microscopy.61 With this approach, they detected norovirus at a LOD of 7.8 zM with a wide linear dynamic range of 7.8 zM–240 aM. This LOD is 102 to 106 times lower than that of other reported methods. The immunoassay allows the design of various probes for multiple targets with suitable antigens. Ma et al. reported a simple system to simultaneously detect CEA and AFP by target-induced fixing of antigen-capped Ag NPs and Au NPs.62 The digital counting of Ag NPs and Au NPs represented the concentration of CEA and AFP, respectively. With this method, CEA and AFP were simultaneously detected in the concentration range of 0.5–10 ng mL−1.
By using gold triangular nanoplates (Au PNLs) as the optical probe, Gu et al. observed the etching of Au NPLs in the presence of Cu2+ and I− (Fig. 5C).65 The blue-shifted LSPR and reduced scattering intensity of single Au PNLs upon adding Cu2+ and I− were dramatically inhibited with the introduction of pyrophosphate (PPi). Through linear expression of the scattering intensity ratio, PPi was indirectly detected in the concentration range from 7 to 100 nM. The LOD was calculated to be 1.9 nM. Meanwhile, colorimetric analysis based on single plasmonic nanoparticle etching has been applied for other chemicals. For example, Au NPs with surface coating by the MnO2 layer (Au NP@MnO2) showed a distinct red-shift in maximum scattering wavelength. Taking this feature, the Xiao group developed a colorimetric assay for alkaline phosphatase by using Au NP@MnO2 and 2-phosphol-L-ascorbic acid.66 The introduction of alkaline phosphatase led to the dephosphorylation of 2-phosphol-L-ascorbic acid and formation of L-ascorbic acid, which reduces MnO2 into Mn2+. As a result, a blue-shift in LSPR spectra of single Au NPs was observed. On the basis of this approach, alkaline phosphatase analysis with a linear dynamic range of 0.06 to 2.48 mU mL−1 was conducted with a LOD of 5.8 μU mL−1. Through the oxidation of silver by MnO4—, they developed a sensitive MnO4— detection method based on the LSPR red-shift of Au@Ag NPs.67
It is reported that Au NPs can catalyze the surface deposition of copper in the presence of reduced nicotinamide adenine dinucleotide (NADH) and Cu2+. The surface copper deposition causes a LSPR red-shift. However, the deposition of copper can be inhibited by PPi. Xiao and co-workers thus proposed a single Au NP-based colorimetric assay for the detection of PPi based on the above mechanism (Fig. 5D).68 With the calculation of the yellow/all particle ratio, PPi was detected with a LOD of 1.49 nM. Beyond the chemical reaction, surface adsorption of biomolecules (e.g., protein and DNA) also leads to the change of the refractive index.69,70 With the use of Au NRs, Lin et al. realized in situ protein corona detection by comparing rotational diffusivity.70
With the use of a color CCD, the RGB tricolor information of single plasmonic nanoparticles can be obtained. Based on the RGB analysis, Liu et al. revealed the binding process of thiols to single Au NPs.71 The mechanism is that binding of thiols results in the change of the surface refractive index and causes the variation of LSPR characters. However, spherical Au NPs respond to a refractive index with low sensitivity, leading to a slight RGB ratio change. In comparison with Au NPs, Ag NPs are more sensitive toward the refractive index. Using spherical Au@Ag core–shell NPs as the probes, Hao et al. realized H2S detection at 50 nM through R/G ratio analysis of single Au@Ag NPs.72
Single plasmonic nanoparticle tracking
The reaction difference between the homogeneous area and microscale is also an important factor for the discovery of new pathways and the design of new materials. Single plasmonic nanoparticle imaging reveals the details of individual events; thus spatial and long-term tracking of single plasmonic nanoparticles would benefit the deep understanding of chemical and biological processes. According to many reports, single plasmonic nanoparticle tracking has shown great potential in various fields, especially chemical reaction visualization and biological process understanding.78–80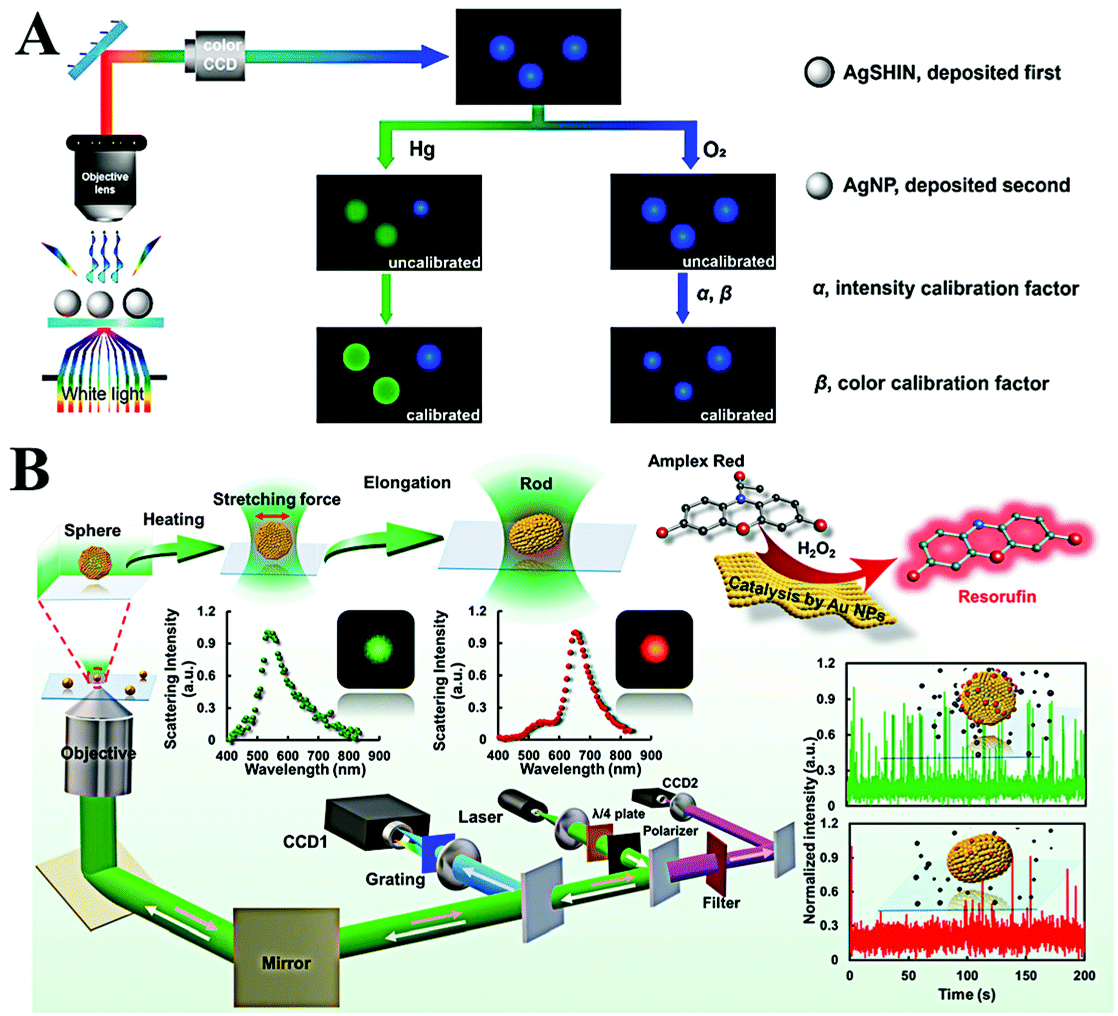 | ||
| Fig. 6 (A) Schematic of the enlarged color-coded dark-field image of amalgamation using Ag NPs as the reference. Reprinted with permission from ref. 81. Copyright 2019, American Chemical Society. (B) Schematic diagram of the procedures for laser-controlled shape modulation and single-molecule catalysis from gold nanostructures with different morphologies. Reprinted with permission from ref. 88. Copyright 2019, Royal Society of Chemistry. | ||
Direct visualization of the catalytic process is of significance to explore highly efficient catalysts. The peroxidase-like catalytic activity of Au NPs has been widely reported and applied for colorimetric sensing of glucose or H2O2 in homogeneous solution. However, the origin of catalytic activity and a detailed catalytic process are still unclear. A real-time peroxidase-like catalytic reaction was observed by Li et al. through constructing a halo-like Au nanostructure (nanohalo), which links 13 nm Au NPs onto the surface of 50 nm Au NPs with DNA hybridization.85 The interparticle distance was found to be ∼9 nm via controlling the number of DNA base pairs; such a distance favors strong plasmon coupling. According to their result, three distinct LSPR variations versus reaction time were observed. Upon adding glucose, the maximum LSPR showed an immediate red-shift of 2.52 nm, then a rapid blue-shift of 6.88 nm, and finally a red-shift of 3.53 nm. The first red-shift of LSPR was assigned to the adsorption of glucose, which changes the surface refractive index. The following blue-shift and red-shift were attributed to the charging and discharging of 13 nm Au NPs. Combining the electrochemical measurement, the intrinsic peroxidase-like catalytic activity of single Au NPs was also detected by Hafez et al.86 Their results demonstrated the important roles of a highly accessible surface area and particle collision in the high activity of single Au NPs. In view of the high resolution and low background of a single-molecule fluorescence imaging technique, Fang and co-workers used a fluorimetric method to evaluate the catalysis of SiO2 coated platinum nanoparticles.87 Through the single-molecule fluorescence measurement, they found that the molecular transport and reaction kinetics were distinctly affected by the confined metal reaction centres, which is essential to the rational design of effective nanocatalysts. In order to have a better understanding on morphology dependent catalytic kinetics from single nanoparticles, the Xiao group explored the morphology dependent catalytic property of single gold nanoparticles after shape transition (Fig. 6B).88 The morphology of spherical gold nanoparticles was modulated to a rod shape with a tightly focused Gaussian laser beam based on the photothermal effect. Then, the distinct catalytic property of single gold nanoparticles before and after symmetry breaking was disclosed at the single-particle level for the first time. According to the single-molecule fluorescence imaging results, they found that the catalytic efficiency decreased when the heterogeneous lattice structure was destroyed after the morphology adjustment. In situ kinetic evidence indicated that the lattice defect was one of the most import factors to control the catalytic efficiency of nanoparticle-based heterogeneous catalysts.
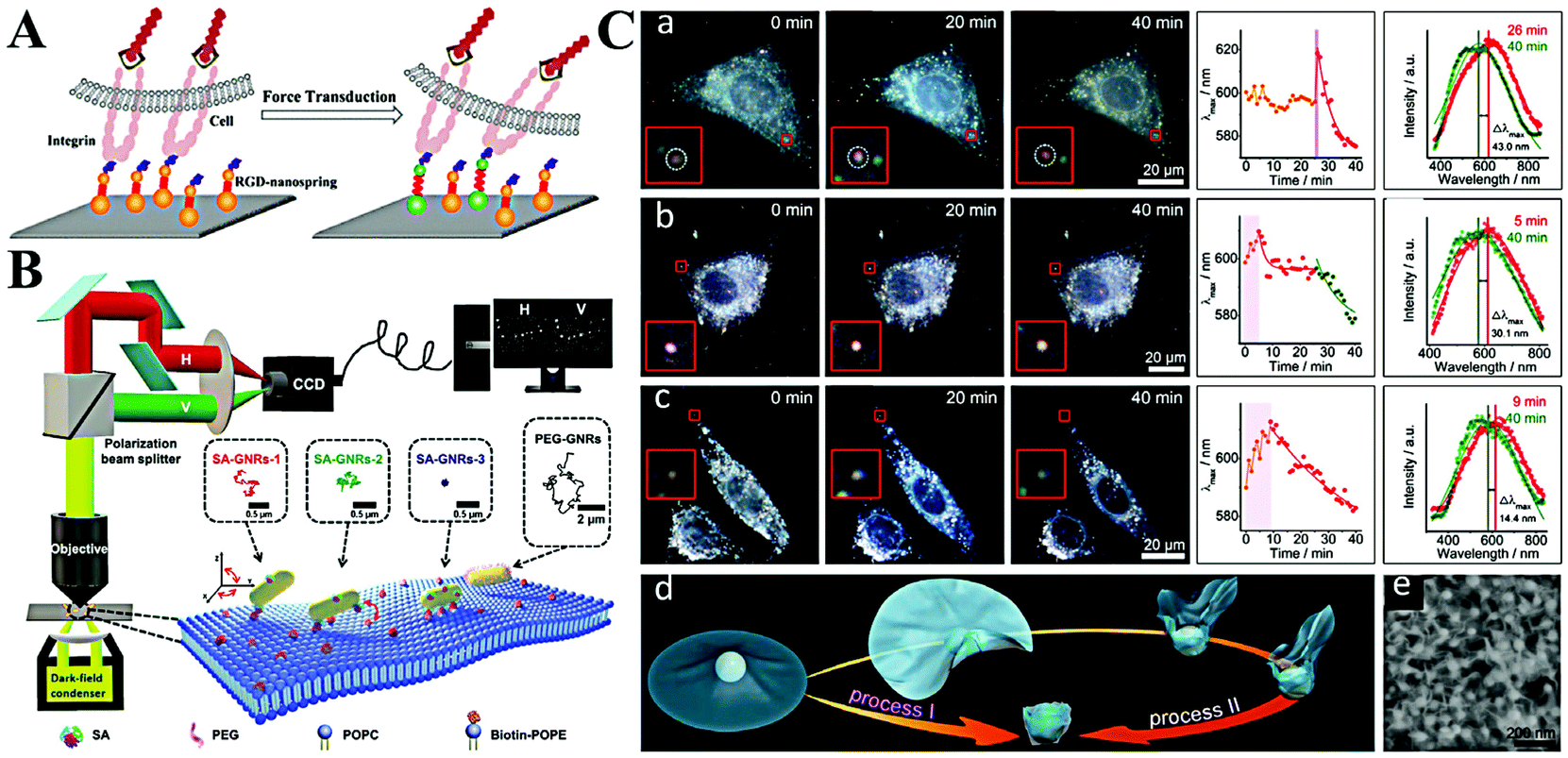 | ||
| Fig. 7 (A) Schematic illustration of imaging mechanical force transduction with plasmonic nanosprings in live cells. Reprinted with permission from ref. 94. Copyright 2017, American Chemical Society. (B) The light path for single-particle tracking of individual Au NRs (GNRs) on a lipid membrane and a schematic illustration of the binding-mediated translational and rotational diffusion from individual functionalized Au NRs on a lipid membrane. Reprinted with permission from ref. 99. Copyright 2019, Royal Society of Chemistry. (C) Representative dark-field images and scattering spectra changes of single Au NP@MnO2 from different living cells. (a) Represents the HepG2 cells, (b) represents 3 T3 cells, and (c) represents HepG2 cells. (d) Schematic illustration of two kinds of transmembrane processes for the Au NP@MnO2. (e) SEM image of the Au NP@MnO2 at different deformation states. Reprinted with permission from ref. 101. Copyright 2019, Wiley-VCH. | ||
The cellular uptake of nanomaterials attracts growing attention due to their high performances and large specific surface area. The most important part is the cytomembrane–nanomaterial interaction, which decides the entrance pathway and efficiency of nanomaterials.9 Toward this point, many researchers focus on understanding the membrane–nanomaterial interaction by using plasmonic nanoparticles as the optical probe.96 To simplify the physical structure and preparation of membranes, some of them study the lipid membrane instead of the cell membrane. As an example, the Xiao group reported lots of work on monitoring the diffusion of Au NRs on lipid membranes.97,98 They proved that Au NRs are not static on the membrane but experience hopping diffusion (Fig. 7B).99 That is, when Au NRs adsorb onto the membrane, both translational diffusion and rotational diffusion exist. Based on the single Au NR tracking, they found that the translational diffusion was primarily affected by the mobility of the ligands on the binding sites, while the translational diffusion could be facilitated by the rotational diffusion. A similar phenomenon was also observed by Xu et al.93
Despite having a similar structure, a lipid membrane still cannot stand for a real cytomembrane. Thus, the single Au NR tracking on a living cell membrane was conducted through a polarization-resolved dual-channel imaging system.100 The binding sites of Au NRs were modified by surface coating with viral capsid proteins. Interestingly, anomalous confined diffusion of Au NRs with randomly distributed large walking steps was observed. This does not appear in non-coated Au NRs. Accordingly, the non-Gaussian step distribution also demonstrates the heterogeneous binding and desorption processes on the cytomembrane. On the basis of Au NR-cytomembrane interaction and the single plasmonic nanoparticle tracking, Ling et al. monitored real-time cell membrane vesiculation by using hybrid 2D Au NP@MnO2 nanomaterials (Fig. 7C).101 The formation of the 2D Au NP@MnO2 structure first caused a red-shift of LSPR. After endocytosis, the intracellular molecules (e.g., GSH) etched the MnO2 and led to the blue-shift of the LSPR. The blue-shifted LSPR allowed the confirmation of completion of the engulfment process. In addition, this vesiculation-induced etching also provided promise to screen local environment redox states at the single-cell level.
Conclusion and perspectives
In this review, we have presented an overview of the recent progress of plasmonic nanoparticle-based optical analysis both in homogeneous solution and at the single-nanoparticle level. The LSPR of plasmonic nanoparticles enables the sensitive detection of various targets in homogeneous solution based on colorimetry and fluorimetry. With a reduced number of nanoparticles, optical analysis at the single-nanoparticle level provides higher sensitivity. Most sensing systems show a LOD at the pM level for the expected targets. With a rational design, ultra-high sensitivity with a LOD at fM and even zM is achieved with single-nanoparticle assay. Additionally, the time-resolved and spatially-resolved single-nanoparticle tracking allows the understanding of the actual process of chemical reactions and biological functions.Despite the successful analysis of various targets in homogeneous solution and at the single-nanoparticle level using plasmonic nanoparticle-based optical systems, there are still some challenges. For homogeneous assays, three major limitations need to be improved in future. First, the poor stability of plasmonic nanoparticles limits their applications in relatively pure media (in vitro) with low interference and ionic strength. However, biological assays are usually conducted in complicated media with high salt concentration and a strong matrix effect. The salt-induced electrostatic screening and matrix-mediated non-specific interaction change the dispersive state of plasmonic nanoparticles, and sometimes cause false results. As a result, quantitation of trace amounts of biomarkers (e.g., microRNA and protein) in biological media is difficult. Second, the plasmonic nanoparticle-based optical analysis generally focuses on a sole target. Howbeit, as indicated in many reports recently, most diseases (i.e., cancers and tumors) are associated with more than one biomarker, and some biomarkers are related to several tumors. Simultaneous detection of multiple targets is thus more reliable to diagnose or predict cancer diseases. For example, with the combination of electrochemiluminescence and LSPR-regulated luminescence, simultaneous detection of multiple targets has been widely explored through spatial-resolved, potential-resolved, and spectrum-resolved strategies.102 They however may suffer from irreversible oxidation and desorption of luminophores, plasmonic nanoparticles of special optical properties, and fussy electrode modification. To boost the analysis of multiple targets, microchip-based multichannel analysis with the combination of plasmonic nanoparticles is worth being tried.
Single plasmonic nanoparticle-based optical analysis provides high sensitivity and allows the dynamic understanding of reaction and biological processes. Imaging of single plasmonic nanoparticles with distinct LSPR and different surface modifications may facilitate multiple target analysis. In comparison with homogeneous solution assays, single-nanoparticle assays perform all experiments on individual nanoparticles and require the usage of a photon detector. To achieve effective optical analysis at the single-nanoparticle level, there are two problems that need to be fixed in the future work. One is the uniform morphology of plasmonic nanoparticles. It is well known that the size and shape largely affect the LSPR of plasmonic nanoparticles. Wide distribution in size and/or morphology leads to inconsistent results. For example, the dark-field images of both Au NRs and aggregated Au NPs are red color spots, making their differentiation difficult. Also, limited by the wavelength dependent-photo collection efficiency of optical detectors, single-nanoparticle imaging with a high signal-to-noise ratio can only be obtained when the LSPR matches the detector's optical response curve. Thus, the exploration of a simple approach for precisely and reproducibly tuning the LSPR of plasmonic nanoparticles is still a challenging job in future. The other problem is the urgent demand for a new imaging technique with super resolution. The interference of halos from adjacent plasmonic nanoparticles extremely restricts the accurate analysis. That is, the discrimination of two individual plasmonic nanoparticles with a distance less than the light diffraction limit (∼200 nm) is very hard. Although a few works have reported the sub-diffraction limit imaging of single plasmonic nanoparticles, the specially designed optical system may hinder their widespread dissemination and application.91,95
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Beijing Natural Science Foundation (2202038), the National Natural Foundation of China (21605003 and 21974073), and the Fundamental Research Funds for the Central Universities (buctrc201619).Notes and references
- K. P. Carter, A. M. Young and A. E. Palmer, Chem. Rev., 2014, 114, 4564–4601 CrossRef CAS PubMed.
- J. Beik, M. Khateri, Z. Khosravi, S. K. Kamrava, S. Kooranifar, H. Ghaznavi and A. Shakeri-Zadeh, Chem. Soc. Rev., 2019, 387, 299–324 CAS.
- Y. W. Lin, C. C. Huang and H. T. Chang, Analyst, 2011, 136, 863–871 RSC.
- X. Huang, R. O'Connor and E. A. Kwizera, Nanotheranostics, 2017, 1, 80–102 CrossRef PubMed.
- K. Saha, S. S. Agasti, C. Kim, X. Li and V. M. Rotello, Chem. Rev., 2012, 112, 2739–2779 CrossRef CAS PubMed.
- W. Zhou, X. Gao, D. Liu and X. Chen, Chem. Rev., 2015, 115, 10575–10636 CrossRef CAS PubMed.
- L. Qin, G. Zeng, C. Lai, D. Huang, P. Xu, C. Zhang, M. Cheng, X. Liu, S. Liu, B. Li and H. Yi, Chem. Soc. Rev., 2018, 359, 1–31 CAS.
- S. Lee, Y. Sun, Y. Cao and S. H. Kang, TrAC, Trends Anal. Chem., 2019, 117, 58–68 CrossRef CAS.
- A. S. Stender, K. Marchuk, C. Liu, S. Sander, M. W. Meyer, E. A. Smith, B. Neupane, G. Wang, J. Li, J.-X. Cheng, B. Huang and N. Fang, Chem. Rev., 2013, 113, 2469–2527 CrossRef CAS PubMed.
- K. M. Mayer and J. H. Hafner, Chem. Rev., 2011, 111, 3828–3857 CrossRef CAS PubMed.
- M. Li, S. K. Cushing and N. Wu, Analyst, 2015, 140, 386–406 RSC.
- Z. Yuan, C. C. Hu, H. T. Chang and C. Lu, Analyst, 2016, 141, 1611–1626 RSC.
- T. Zheng, N. Pierre-Pierre, X. Yan, Q. Huo, A. J. Almodovar, F. Valerio, I. Rivera-Ramirez, E. Griffith, D. D. Decker, S. Chen and N. Zhu, ACS Appl. Mater. Interfaces, 2015, 7, 6819–6827 CrossRef CAS PubMed.
- I. S. Che Sulaiman, B. W. Chieng, M. J. Osman, K. K. Ong, J. I. A. Rashid, W. M. Z. Wan Yunus, S. A. M. Noor, N. A. M. Kasim, N. A. Halim and A. Mohamad, Microchim. Acta, 2020, 187, 131 CrossRef CAS PubMed.
- H. Zhou, H. Yang, G. Wang, A. Gao and Z. Yuan, Curr. Pharm. Des., 2019, 25, 4861–4876 CrossRef CAS PubMed.
- L.-K. Fong, Z. Wang, G. C. Schatz, E. Luijten and C. A. Mirkin, J. Am. Chem. Soc., 2018, 140, 6226–6230 CrossRef CAS PubMed.
- J. Cai, L. Ding, P. Gong and J. Huang, Nanotechnology, 2019, 31, 095501 CrossRef PubMed.
- J.-Y. Mao, H.-W. Li, S.-C. Wei, S. G. Harroun, M.-Y. Lee, H.-Y. Lin, C.-Y. Chung, C.-H. Hsu, Y.-R. Chen and H.-J. Lin, ACS Appl. Mater. Interfaces, 2017, 9, 44307–44315 CrossRef CAS PubMed.
- Y. Chen, J. Zhang, Y. Gao, J. Lee, H. Chen and Y. Yin, Biosens. Bioelectron., 2015, 72, 306–312 CrossRef CAS PubMed.
- L. Li, Z. Yuan, X. Peng, L. Li, J. He and Y. Zhang, J. Chin. Chem. Soc., 2014, 61, 1371–1376 CrossRef CAS.
- Z. Yuan, F. Lu, M. Peng, C. W. Wang, Y. T. Tseng, Y. Du, N. Cai, C. W. Lien, H. T. Chang, Y. He and E. S. Yeung, Anal. Chem., 2015, 87, 7267–7273 CrossRef CAS PubMed.
- D. Xu, H. Chen, Q. Lin, Z. Li, T. Yang and Z. Yuan, RSC Adv., 2017, 7, 16295–16301 RSC.
- X. Zhang, X. Fan, Y. Wang, F. Lei, L. Li, J. Liu and P. Wu, Anal. Chem., 2020, 92, 1455–1462 CrossRef CAS PubMed.
- F. Wang, S. Liu, M. Lin, X. Chen, S. Lin, X. Du, H. Li, H. Ye, B. Qiu, Z. Lin, L. Guo and G. Chen, Biosens. Bioelectron., 2015, 68, 475–480 CrossRef CAS PubMed.
- J. H. Soh, Y. Lin, S. Rana, J. Y. Ying and M. M. Stevens, Anal. Chem., 2015, 87, 7644–7652 CrossRef CAS PubMed.
- S. Durmazel, A. Üzer, B. Erbil, B. Sayın and R. Apak, ACS Omega, 2019, 4, 7596–7604 CrossRef CAS PubMed.
- D. Xu, S. Yu, Y. Yin, S. Wang, Q. Lin and Z. Yuan, Front. Chem., 2018, 6, 566 CrossRef CAS PubMed.
- Y. Mao, J. Li, J. Yan, Y. Ma, Y. Song, T. Tian, X. Liu, Z. Zhu, L. Zhou and C. Yang, Chem. Commun., 2017, 53, 6375–6378 RSC.
- X. Zhang, W. Zhou, Z. Yuan and C. Lu, Analyst, 2015, 140, 7443–7450 RSC.
- I. Sanskriti and K. K. Upadhyay, ChemistrySelect, 2019, 4, 3803–3810 CrossRef CAS.
- A. Guerrero and R. Aroca, Angew. Chem., Int. Ed., 2011, 50, 665–668 CrossRef CAS PubMed.
- S. Abalde-Cela, S. Carregal-Romero, J. P. Coelho and A. Guerrero-Martínez, Adv. Colloid Interface Sci., 2016, 233, 255–270 CrossRef CAS PubMed.
- W. Hong, Y. Zhang, L. Gan, X. Chen and M. Zhang, J. Mater. Chem. C, 2015, 3, 6185–6191 RSC.
- K. Sugawa, T. Tamura, H. Tahara, D. Yamaguchi, T. Akiyama, J. Otsuki, Y. Kusaka, N. Fukuda and H. Ushijima, ACS Nano, 2013, 7, 9997–10010 CrossRef CAS PubMed.
- Z. Yuan, Y. Du, Y.-T. Tseng, M. Peng, N. Cai, Y. He, H.-T. Chang and E. S. Yeung, Anal. Chem., 2015, 87, 4253–4259 CrossRef CAS PubMed.
- A. Heuer-Jungemann, P. K. Harimech, T. Brown and A. G. Kanaras, Nanoscale, 2013, 5, 9503–9510 RSC.
- C. Sun, S. Zhao, F. Qu, W. Han and J. You, Microchim. Acta, 2019, 187, 34 CrossRef PubMed.
- Y. Gao, J. Tian, X. Zhang, B. Qiao, Y. Cao, X. Wang and Q. Wu, Analyst, 2020, 145, 1190–1194 RSC.
- F. Degliangeli, P. Kshirsagar, V. Brunetti, P. P. Pompa and R. Fiammengo, J. Am. Chem. Soc., 2014, 136, 2264–2267 CrossRef CAS PubMed.
- M. Moros, M. E. Kyriazi, A. H. El-Sagheer, T. Brown, C. Tortiglione and A. G. Kanaras, ACS Appl. Mater. Interfaces, 2019, 11, 13905–13911 CrossRef CAS PubMed.
- L. Li, J. Feng, Y. Fan and B. Tang, Anal. Chem., 2015, 87, 4829–4835 CrossRef CAS PubMed.
- Z. Wu, G.-Q. Liu, X.-L. Yang and J.-H. Jiang, J. Am. Chem. Soc., 2015, 137, 6829–6836 CrossRef CAS PubMed.
- J. Nebu, J. S. Anjali Devi, R. S. Aparna, B. Aswathy, G. M. Lekha and G. Sony, Sens. Actuators, B, 2018, 277, 271–280 CrossRef CAS.
- J. Li, J. Xu, W. Guo, W. Zhong, Q. Li, L. Tan and L. Shang, Sens. Actuators, B, 2020, 305, 127422 CrossRef.
- X. Miao, Z. Cheng, H. Ma, Z. Li, N. Xue and P. Wang, Anal. Chem., 2018, 90, 1098–1103 CrossRef CAS PubMed.
- R. Lv, P. Yang, Y. Dai, S. Gai, F. He and J. Lin, ACS Appl. Mater. Interfaces, 2014, 6, 15550–15563 CrossRef CAS PubMed.
- Z. Yuan, N. Cai, Y. Du, Y. He and E. S. Yeung, Anal. Chem., 2014, 86, 419–426 CrossRef CAS PubMed.
- Z. Yuan, Y. Du and Y. He, Methods Appl. Fluoresc., 2017, 5, 014011 CrossRef PubMed.
- F. Lu, H. Yang, Z. Yuan, T. Nakanishi, C. Lu and Y. He, Sens. Actuators, B, 2019, 291, 170–176 CrossRef CAS.
- Z. Luo, X. Yuan, Y. Yu, Q. Zhang, D. T. Leong, J. Y. Lee and J. Xie, J. Am. Chem. Soc., 2012, 134, 16662–16670 CrossRef CAS PubMed.
- F. Lu, H. Yang, Y. Tang, C.-J. Yu, G. Wang, Z. Yuan and H. Quan, Microchim. Acta, 2020, 187, 200 CrossRef CAS PubMed.
- Z. Yuan, M. Peng, Y. He and E. S. Yeung, Chem. Commun., 2011, 47, 11981–11983 RSC.
- Z. Yuan, M. Peng, L. Shi, Y. Du, N. Cai, Y. He, H.-T. Chang and E. S. Yeung, Nanoscale, 2013, 5, 4683–4686 RSC.
- Y. Sun, F. Lu, H. Yang, C. Ding, Z. Yuan and C. Lu, Nanoscale, 2019, 11, 12889–12897 RSC.
- H. Yang, F. Lu, Y. Sun, Z. Yuan and C. Lu, Anal. Chem., 2018, 90, 12846–12853 CrossRef CAS PubMed.
- L. Xiao, L. Wei, Y. He and E. S. Yeung, Anal. Chem., 2010, 82, 6308–6314 CrossRef CAS PubMed.
- Z. Yuan, J. Cheng, X. Cheng, Y. He and E. S. Yeung, Analyst, 2012, 137, 2930–2932 RSC.
- L. Peng, X. Cao, B. Xiong, Y. He and E. S. Yeung, Chem. Commun., 2016, 52, 7616–7619 RSC.
- Y. F. Xie, Y. Y. Cheng, M. L. Liu, H. Y. Zou and C. Z. Huang, Analyst, 2019, 144, 2011–2016 RSC.
- C.-Y. Poon, L. Wei, Y. Xu, B. Chen, L. Xiao and H.-W. Li, Anal. Chem., 2016, 88, 8849–8856 CrossRef CAS PubMed.
- S. K. Chakkarapani, Y. Sun and S. H. Kang, Sens. Actuators, B, 2019, 284, 81–90 CrossRef CAS.
- J. Ma, L. Zhan, R. S. Li, P. F. Gao and C. Z. Huang, Anal. Chem., 2017, 89, 8484–8489 CrossRef CAS PubMed.
- F. Medeghini, M. Hettich, R. Rouxel, S. D. Silva Santos, S. Hermelin, E. Pertreux, A. Torres Dias, F. Legrand, P. Maioli, A. Crut, F. Vallée, A. San Miguel and N. Del Fatti, ACS Nano, 2018, 12, 10310–10316 CrossRef CAS PubMed.
- B. Xiong, R. Zhou, J. Hao, Y. Jia, Y. He and E. S. Yeung, Nat. Commun., 2013, 4, 1708 CrossRef PubMed.
- X. Y. Gu, J. J. Liu, P. F. Gao, Y. F. Li and C. Z. Huang, Anal. Chem., 2019, 91, 15798–15803 CrossRef CAS PubMed.
- F. Wang, Y. Li, Y. Han, Z. Ye, L. Wei, H.-B. Luo and L. Xiao, Anal. Chem., 2019, 91, 6329–6339 CrossRef CAS PubMed.
- Z. Ye, R. Weng, Y. Ma, F. Wang, H. Liu, L. Wei and L. Xiao, Anal. Chem., 2018, 90, 13044–13050 CrossRef CAS PubMed.
- F. Qi, Y. Han, Z. Ye, H. Liu, L. Wei and L. Xiao, Anal. Chem., 2018, 90, 11146–11153 CrossRef CAS PubMed.
- C. Qiao, J. Wu, Z. Huang, X. Cao, J. Liu, B. Xiong, Y. He and E. S. Yeung, Anal. Chem., 2017, 89, 5592–5597 CrossRef CAS PubMed.
- X. Lin, Q. Pan and Y. He, Nanoscale, 2019, 11, 18367–18374 RSC.
- Y. Liu, J. Ling and C. Z. Huang, Chem. Commun., 2011, 47, 8121–8123 RSC.
- J. Hao, B. Xiong, X. Cheng, Y. He and E. S. Yeung, Anal. Chem., 2014, 86, 4663–4667 CrossRef CAS PubMed.
- M. M. Azab, R. Cherif, A. L. Finnie, M. M. A. El-Alamin, M. A. Sultan and A. W. Wark, Analyst, 2018, 143, 1635–1643 RSC.
- X. Li, L. Wei, L. Pan, Z. Yi, X. Wang, Z. Ye, L. Xiao, H.-W. Li and J. Wang, Anal. Chem., 2018, 90, 4807–4814 CrossRef CAS PubMed.
- Y. Han, T. Chen, Y. Li, L. Chen, L. Wei and L. Xiao, Anal. Chem., 2019, 91, 11146–11153 CrossRef CAS PubMed.
- F. Wang, Y. Han, S. Wang, Z. Ye, L. Wei and L. Xiao, Anal. Chem., 2019, 91, 11856–11863 CrossRef CAS PubMed.
- S. K. Chakkarapani, S. Lee, B. Park, H.-Y. Seo and S. H. Kang, ACS Sens., 2019, 4, 953–960 CrossRef CAS PubMed.
- H. Liu, Z. Ye, X. Wang, L. Wei and L. Xiao, Analyst, 2019, 144, 859–871 RSC.
- Z. Ye, X. Wang and L. Xiao, Anal. Chem., 2019, 91, 15327–15334 CrossRef CAS PubMed.
- Q. Pan, H. Zhao, X. Lin and Y. He, Angew. Chem., Int. Ed., 2019, 58, 8389–8393 CrossRef CAS PubMed.
- W. Feng, W. He, J. Zhou, X. Y. Gu, Y. F. Li and C. Z. Huang, Anal. Chem., 2019, 91, 3002–3008 CrossRef CAS PubMed.
- J. Zhou, T. Yang, W. He, Z. Y. Pan and C. Z. Huang, Nanoscale, 2018, 10, 12805–12812 RSC.
- X. Cao, J. Feng, Q. Pan, B. Xiong, Y. He and E. S. Yeung, Anal. Chem., 2017, 89, 2692–2697 CrossRef CAS PubMed.
- J. Feng, X. Cao, Q. Pan and Y. He, Electrophoresis, 2019, 40, 2227–2234 CrossRef CAS PubMed.
- K. Li, K. Wang, W. Qin, S. Deng, D. Li, J. Shi, Q. Huang and C. Fan, J. Am. Chem. Soc., 2015, 137, 4292–4295 CrossRef CAS PubMed.
- M. E. Hafez, H. Ma, W. Ma and Y.-T. Long, Angew. Chem., Int. Ed., 2019, 58, 6327–6332 CrossRef CAS PubMed.
- B. Dong, Y. Pei, N. Mansour, X. Lu, K. Yang, W. Huang and N. Fang, Nat. Commun., 2019, 10, 4815 CrossRef PubMed.
- Z. Ye, L. Wei, L. Xiao and J. Wang, Chem. Sci., 2019, 10, 5793–5800 RSC.
- L. M. Browning, K. J. Lee, P. K. Cherukuri, T. Huang, P. Songkiatisak, S. Warren and X.-H. N. Xu, Analyst, 2018, 143, 1599–1608 RSC.
- S. K. Chakkarapani, Y. Sun, S. Lee, N. Fang and S. H. Kang, ACS Nano, 2018, 12, 4156–4163 CrossRef CAS PubMed.
- X. Cheng, X. Cao, B. Xiong, Y. He and E. S. Yeung, Nano Res., 2017, 10, 1423–1433 CrossRef CAS.
- H. Sun, Z. Wang and Y. He, ACS Nano, 2019, 13, 11334–11342 CrossRef CAS PubMed.
- D. Xu, Y. He and E. S. Yeung, Angew. Chem., Int. Ed., 2014, 53, 6951–6955 CrossRef CAS PubMed.
- B. Xiong, Z. Huang, H. Zou, C. Qiao, Y. He and E. S. Yeung, ACS Nano, 2017, 11, 541–548 CrossRef CAS PubMed.
- L. Wei, Y. Ma, X. Zhu, J. Xu, Y. Wang, H. Duan and L. Xiao, Nanoscale, 2017, 9, 8747–8755 RSC.
- D. Xu, Y. He and E. S. Yeung, Anal. Chem., 2014, 86, 3397–3404 CrossRef CAS PubMed.
- L. Wei, Z. Ye, Y. Xu, B. Chen, E. S. Yeung and L. Xiao, Anal. Chem., 2016, 88, 11973–11977 CrossRef CAS PubMed.
- L. He, Y. Li, L. Wei, Z. Ye, H. Liu and L. Xiao, Nanoscale, 2019, 11, 10080–10087 RSC.
- Z. Ye, H. Liu, F. Wang, X. Wang, L. Wei and L. Xiao, Chem. Sci., 2019, 10, 1351–1359 RSC.
- Z. Ye, L. Wei, X. Zeng, R. Weng, X. Shi, N. Wang, L. Chen and L. Xiao, Anal. Chem., 2018, 90, 1177–1185 CrossRef CAS PubMed.
- Y. Ling, D. Zhang, X. Cui, M. Wei, T. Zhang, J. Wang, L. Xiao and Y. Xia, Angew. Chem., Int. Ed., 2019, 58, 10542–10546 CrossRef CAS PubMed.
- W. Lv, H. Ye, Z. Yuan, X. Liu, X. Chen and W. Yang, TrAC, Trends Anal. Chem., 2020, 123, 115767 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2020 |