
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
New insights into the dissociation dynamics of methylated anilines†
Neil C.
Cole-Filipiak‡
 and
Vasilios G.
Stavros
*
and
Vasilios G.
Stavros
*
Department of Chemistry, University of Warwick, Coventry, CV4 7AL, UK. E-mail: v.stavros@warwick.ac.uk; Tel: +44 (0)2476 150172
First published on 12th March 2019
Abstract
Aniline, an important model system for biological chromophores, undergoes ultrafast H-atom loss upon absorption of an ultraviolet photon. By varying the number and position of methyl substituents on both the aromatic ring and amine functional group, we explore the ultrafast production of photofragments as a function of molecular structure. Both N-methyl aniline and 3,5-dimethyl aniline show altered H-atom loss behaviour compared to aniline, while no evidence for CH3 loss was found from either N-methyl aniline or N,N-dimethyl aniline. With the addition of time-resolved photoelectron spectroscopy, the photofragment appearance times are matched to excited state relaxation pathways. Evidence for a sequential excited state relaxation mechanism, potentially involving a valence-to-Rydberg decay mechanism, will be presented. Such a global, bottom-up approach to molecular photochemistry is crucial to understanding the dissociative pathways and excited state decay mechanisms of biomolecule photoprotection in nature.
1. Introduction
By investigating the basic building blocks of aromatic biomolecules, chemists and biologists may develop a fundamental understanding of the structure–dynamics–function relationship governing molecular biology. Towards that goal, the ultraviolet (UV) photochemistry of phenols, pyrroles, etc. have been studied for some time as both simple biomolecule model and as the basic chromophore of many biopolymers.1–3 Of particular importance was the revelation that these molecules have a weakly bound, πσ* electronic excited state which becomes dissociative at extended O–X or N–X distances (X = H, CH3, etc.), eventually intersecting the ground electronic state; a schematic potential energy cut is shown in Fig. 1.4 Evolution of excited state population along this dissociative coordinate affords an opportunity for rapid, non-radiative transfer to the ground state, a key clue to photoprotection in nature.4,5 As such, these heteroaromatic systems have attracted considerable interest from spectroscopists (see ref. 1, 2 and references therein). Modifications to these fundamental chromophores alters their photochemistry and, in addition to increasing complexity, is a powerful tool to explore and explain fundamental behaviour.6,7 Through such investigations, we ultimately seek to develop a complete, bottom-up understanding of biomolecule photochemistry.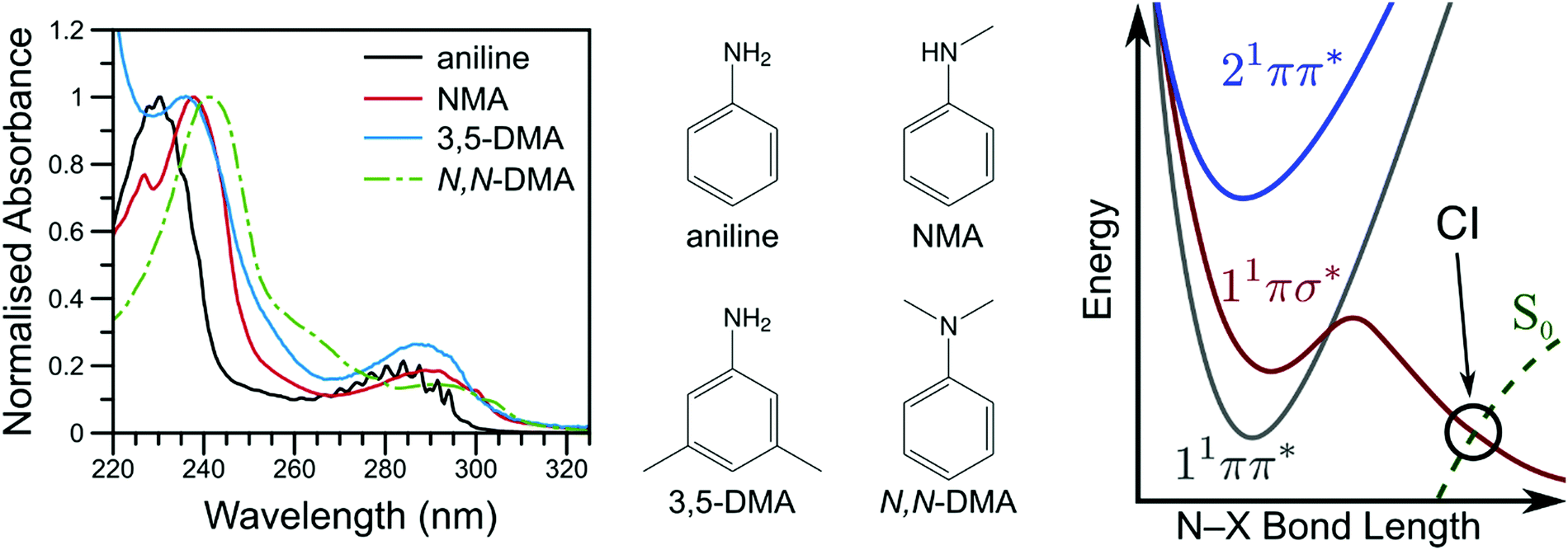 | ||
| Fig. 1 The vapour phase spectra of aniline (black line), NMA (red line), 3,5-DMA (blue line), and N,N-DMA (green, dot-dashed line) are shown on the left with the corresponding structures shown in the middle panel. A schematic potential energy cut along the aniline N–X reaction coordinate is shown on the right (adapted from ref. 16). | ||
The photochemistry of aniline, the simplest aromatic amine and a model subunit of many biomolecules, has been extensively studied both experimentally and theoretically. The first excited state of aniline is a bound, valence 1ππ* state8,9 (labelled 11ππ*, see Fig. 1), photoexcitation to which results in an extended excited state lifetime9–14 and high fluorescence quantum yield;15 photoexcitation to this state does not result in N–H bond dissociation.16,17 In the Franck–Condon (FC) region, the 11πσ* state is largely Rydberg in character (N 3s) and weakly bound18 while, at elongated N–H bond lengths, the state takes on increasing σ* character.1,8,9 From the 11πσ* state, photoexcited population may relax via two, competing pathways: dissociation or internal conversion (IC). H-atom loss from the 11πσ* state, due to N–H bond fission, results in high kinetic energy (KE) H-atoms16 dissociating on a sub-ps timescale.17 Alternatively, IC to the 11ππ* state results in ‘trapped’ population which again persists for an extended excited state lifetime.10,12,14,19
While several computational investigations have found several other low-lying Rydberg excited states in aniline,8,9,19 these states have received little experimental attention. Indeed, most of the recent experimental work on aniline photodynamics has focused on excitation to a second, higher lying 1ππ* state at ca. 240 nm. Relaxation from this state, henceforth referred to as 21ππ*, has been observed to occur in ≤50 fs14 and may decay through the aforementioned Rydberg states.9,20 Though a consensus on the exact decay pathways remains elusive (namely the degree to which 21ππ* state population decays to the 11πσ* and 11ππ* states, cf.ref. 11, 12, 14 and 17), photoexcitation at 240 nm results in high KE H-atoms16 with an appearance lifetime of 155 fs.17 Theoretical explorations have found several conical intersections (CIs) between the 21ππ*, 11πσ*, 11ππ*, and S0 states,17,21 including an apparent 21ππ*/11πσ*/11ππ* three-state CI which lies along the minimum energy 21ππ* → S0 relaxation pathway.21 Recent time-resolved photoelectron spectroscopy (TR-PES) by Fielding and co-workers14 has yielded considerable insight into the 21ππ* decay mechanisms, concluding that photoexcitation at 238 nm populates the 21ππ* which decays almost exclusively to S0 with some IC to both the 11πσ* and 11ππ* states via the aforementioned multi-state CI.21 The latter process is clearly revealed through a delayed rise in photoelectron signal due to the sequential population of the 11ππ* state.11,12
To help elucidate and explore the relaxation mechanisms of photoexcited aniline, recent TR-PES work by Townsend and co-workers19,22 has explored the excited state dynamics of N,N-dimethylaniline (N,N-DMA) and 3,5-dimethylaniline (3,5-DMA) at 250 and 240 nm. The 11πσ* state dynamics (as prepared by a 250 nm photon) of both species show remarkably similar behaviour to that of bare aniline,19 though N–H dissociation appears to be more favourable than direct 11πσ* → 11ππ* IC in 3,5-DMA than in aniline (as inferred by TR-PES results alone). At 240 nm,22 the ultrafast decay of PES features attributed to the 21ππ* and 3s/πσ* states (the latter nomenclature explicitly denoting the mixed N 3s and 1πσ* nature of the first dissociative excited state), was followed by a rapid rise and slow decay of a low eKE feature attributed to the 11ππ* state. Fits to these results suggested population transfer from the mixed 3s/πσ* state to the 11ππ* state in competition with rapid N–X bond fission, as supported by theoretical potential energy cuts (PECs) along the N–X stretch coordinate. Contrary to aniline,14 no direct evidence for population transfer from the 21ππ* to the lower lying excited states was found; the authors22 instead concluded that the observed TR-PES behaviour must be due to population of the 3s/πσ* state in the initial photoexcitation which subsequently decays to the lower lying 11ππ* state. Photoelectron angular distributions (PAD) in N,N-DMA also revealed evidence for the initial population of a high-lying, N 3p Rydberg state. Of particular interest from the work on 3,5-DMA and N,N-DMA at 240 nm is the insensitivity of the TR-PES lifetimes to methylation of either the amine or aromatic ring,22 a surprising find given the large amine geometry changes required to access previously reported CIs.17,21 To explain these results, Townsend and co-workers22 echoed previous speculation on the dynamics of aniline9,20 and suggested that one or more N centred Rydberg states may play a role in the 21ππ* state decay dynamics.
In the present work, we seek to measure the H-atom or CH3 appearance lifetime from N-methylaniline (NMA), N,N-DMA, and 3,5-DMA (structures shown in Fig. 1) to explore how simple alkyl substituents affect the dissociation dynamics of aniline. By correlating photofragment appearance lifetimes to excited state decays, as observed via TR-PES, we unveil both Rydberg-to-valence and valence-to-Rydberg dynamics in our attempt to develop a more complete picture of the excited state dynamics following photoexcitation to the 21ππ* state. We note that while previous investigations have referred to the mixed 3s/πσ* state using a variety of labels, in the present work we use 11πσ* to refer to the entire state and N 3s to refer to the bound, Rydberg-like local minimum near the Franck–Condon region.
2. Experimental
Aniline (Acros Organics, 99.8%), NMA (Aldrich, 98%), N,N-DMA (Acros Organics, 99%), and 3,5-DMA (Acros Organics, 98%) were used without further purification. Room temperature vapour phase UV spectra (Fig. 1) were recorded on a Cary 60 spectrophotometer by placing a drop of sample into an empty quartz cuvette.The experimental apparatus used in the present work has been previously described in detail23–25 with a summary provided herein. The molecule of interest is gently heated (35 °C for aniline, NMA, and N,N-DMA; 55 °C for 3,5-DMA) and seeded in a molecular beam of ∼3 bar He. This gas mixture is then expanded into vacuum by an Even–Lavie pulsed solenoid valve26 operating at 125 Hz. The molecular beam then passes through a skimmer which separates the source and interaction chambers. Two, temporally delayed, ultraviolet laser beams—the ‘pump’ and ‘probe’—intersect the molecular beam: the former initiates photochemistry while the latter ionises either the photoexcited molecules or the resulting photoproducts. A set of ion optics designed for velocity map ion imaging27 (VMI) accelerate and focus the photoions or photoelectrons onto a position sensitive detector at the end of a time-of-flight tube such that scattered particles with the same initial velocity are projected onto the same point on the detector. The detector itself consists of two microchannel plates (MCP) coupled to a phosphor screen (Photek VID-240) and imaged by a CCD camera (Basler A-312f) interfaced with a computer. Images are recorded as a function of pump–probe time delay (Δt), thus producing time-resolved ion image (TR-VMI) or photoelectron spectroscopy (TR-PES) transients. Similarly, the total ion yield (TR-IY) transient of a particular m/z value may be measured using a recently installed MCP/metal anode detector that can rotate into the ion flight path, as shown in Section S1 of the ESI.† Ion signal, gated in flight time, is recorded as a function of time delay on an oscilloscope (LeCroy LT372 Waverunner) connected to a computer.
To generate the pump and probe laser beams, the pulsed (∼40 fs duration) 800 nm output of a commercial oscillator and regenerative amplifier (Spectra Physics Tsunami and Spitfire XP) operating at 1 kHz is split into three 1 W beams. The first two beams each pass through a tuneable UV optical parametric amplifier (Light Conversion TOPAS-C) to generate the pump and probe pulses of approximately 500 cm−1 full width at half maximum. The pulses are temporally delayed by a hollow corner gold retroreflector mounted on a motorised delay stage in the pump beam path prior to the TOPAS for a maximum Δt = 1.2 ns. The pump pulses (λpu) were, unless noted otherwise, centred at 238 nm. For TR-VMI, the probe wavelengths were centred at λpr = 333 or 243 nm to provide 2 + 1 resonance enhanced multiphoton ionisation (REMPI) of the CH3 or H-atom photofragments, respectively. TR-VMI experiments were also performed with the probe pulse tuned away from the REMPI wavelength while maintaining a similar photon energy (i.e. λpr = 245 nm while monitoring m/z = 1 and λpr = 307 nm while monitoring m/z = 15); these conditions will henceforth be described as an off-resonant or non-resonant probe. For TR-PES, the probe pulse wavelength was centred around 305 nm to minimise probe-initiated photodynamics (see Fig. 1), observe dynamics from multiple excited states, and for consistency with previous investigations.11,12,14,22 For TR-VMI the pump electric field vector was parallel to the plane of the detector (the probe polarisation is irrelevant); for TR-PES the probe pulse electric field vector was also held parallel to the plane of the detector. The laser beams are then focused to intersect the molecular beam within the ion optics. The instrument response function (IRF) was characterized by the non-resonant TR-IY of Xe+ at the temporal overlap of the pump and probe pulses (Δt = 0). The Xe+ transient was fit to a Gaussian distribution with a typical full width at half maximum (FWHM) of ∼100 fs; see Section S2 of the ESI.† Laser power studies were also performed to ensure single photon photochemistry.
Quantum chemical calculations were performed using the GAMESS computational package.28,29 The geometry of NMA was optimised on the ground electronic states of the neutral (S0) and cationic (D0+) surfaces at the CAM-B3LYP30/aug-cc-pVDZ31 level of theory,32–34 using the restricted Hartree–Fock35 or restricted open-shell Hartree–Fock36 methods respectively. Excitation energies and transition dipole moments were calculated from the S0 equilibrium geometry using the time-dependent version of the above level of theory; excited state character was determined by the component atomic orbital amplitudes and visual inspection of the three dimensional orbitals using the wxMacMolPlt program.37 Normal mode analysis was used to calculate the zero-point energies of the D0+ state at the equilibrium and Franck–Condon (i.e. S0 equilibrium) geometries. All computational results are presented in Section S3 of the ESI.†
3. Results and analysis
The velocity map ion images were deconvoluted to produce the one-dimensional velocity distribution using the polar onion peeling (POP) method.38 The velocity distributions were converted to total kinetic energy release (TKER) distributions by calibrating to the known dissociation energies of HBr39 and an appropriate mass weighting factor:| TKER = (mAB/mB)KEA | (1) |
| hc/λpu + E0 − BDE(A–B) = TKER + Eint | (2) |
POP deconvolution also produces the one-dimensional angular distributions of the photofragments. These are then fit to the known functional form40 to yield the anisotropy parameter β which, for the rapid fragmentation of polyatomic molecules, provides a measure of the alignment of the laser electric field vector and the dissociating bond. In the limiting cases, β may take a value of 2 or −1 corresponding to a parallel or perpendicular dissociation event, respectively; an isotropic distribution will result in β = 0.
Similar to the photofragment analysis, the photoelectron images were processed via POP deconvolution. The resulting photoelectron velocity distributions are converted to electron kinetic energy (eKE) by calibrating to the 2 + 1 REMPI of Xe.41 Similar to eqn (2), the photoelectron spectra unveil the excited state dynamics by:
| hc/λpu + hc/λpr + E0 − IE = eKE + Eint+ | (3) |
The two-dimensional TKER transients are then analysed by following the behaviour of a particular feature over Δt and fitting with a one-dimensional, parallel dynamics model consisting of a sum of exponential decay and rise functions convoluted with a Gaussian IRF; see Section S2 of the ESI† for further details of the kinetic model. Non-resonant production of H+ (i.e. dissociative ionisation) was similarly modelled using a sum of exponential decays convoluted with a Gaussian IRF. Conversely, the two-dimensional TR-PES results are analysed using global lifetime analysis42,43 to monitor changes to the entire spectrum as a function of time delay using the Glotaran software package.44 All TR-PES results were fit with two exponential decay models: one assuming sequential dynamics and, for comparison with previously reported results,11,12,14,22 a model assuming parallel (branched) dynamics. The evolution associated spectra (EAS) that are obtained from the sequential dynamics fit to the experimental data highlight the spectral feature changes associated with each step in the sequential model while, for the parallel model, the extracted decay associated spectra (DAS) are the wavelength dependent amplitudes for each exponential decay lifetime.42 Further details pertaining to how EAS and DAS are extracted from the experimental data using the aforementioned kinetic models, as well as details regarding the interpretation of these spectra, may be found in ref. 42–44. TR-PES lifetimes will be reported as τn, where increasing n denotes increasing lifetime. Similarly, H-atom appearance lifetimes will be reported as τHn. Photoelectron anisotropy parameters will be reported as β2 and β4 while a photofragment anisotropy parameter will be given as βA where A indicates the dissociated fragment (e.g. βH for N–H bond fission).
(a) N-Methylaniline
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cm−1, largely attributable to ionisation from the 21ππ* state, is initially populated. Next, the sharper feature at ∼8000 cm−1 is indicative of ionisation out of a Rydberg state and is consistent with ionisation out of the N 3s state observed in other aniline derivatives.11,12,14,22 Finally, a broad, low eKE feature is rapidly populated before narrowing and decaying (see Fig. S4 in the ESI†). This low eKE feature has previously been assigned to ionisation from the 11ππ* state, though ionisation of any long-lived 21ππ* state population to the first electronic excited state of the cation (D1+) may contribute to the TR-PES signal around 4000 cm−1.45 Similar to aniline,11,12 a clear temporal delay of ca. 50 fs is evident between the initial rise of the high eKE feature (10000–12000 cm−1) and all lower eKE features. Such an offset suggests sequential decay behaviour in NMA and, as such, results from the TR-PES fits using a sequential kinetic model will be presented here in the manuscript while the results of a parallel dynamics model fit are presented in the ESI.† Overall, a total of five lifetimes are needed to satisfactorily fit these data; time constants are given in Table 1, EAS for each time constant are shown in Fig. 3, and DAS and all fit residuals may be found in Fig. S4 of the ESI.†
000 cm−1, largely attributable to ionisation from the 21ππ* state, is initially populated. Next, the sharper feature at ∼8000 cm−1 is indicative of ionisation out of a Rydberg state and is consistent with ionisation out of the N 3s state observed in other aniline derivatives.11,12,14,22 Finally, a broad, low eKE feature is rapidly populated before narrowing and decaying (see Fig. S4 in the ESI†). This low eKE feature has previously been assigned to ionisation from the 11ππ* state, though ionisation of any long-lived 21ππ* state population to the first electronic excited state of the cation (D1+) may contribute to the TR-PES signal around 4000 cm−1.45 Similar to aniline,11,12 a clear temporal delay of ca. 50 fs is evident between the initial rise of the high eKE feature (10000–12000 cm−1) and all lower eKE features. Such an offset suggests sequential decay behaviour in NMA and, as such, results from the TR-PES fits using a sequential kinetic model will be presented here in the manuscript while the results of a parallel dynamics model fit are presented in the ESI.† Overall, a total of five lifetimes are needed to satisfactorily fit these data; time constants are given in Table 1, EAS for each time constant are shown in Fig. 3, and DAS and all fit residuals may be found in Fig. S4 of the ESI.†
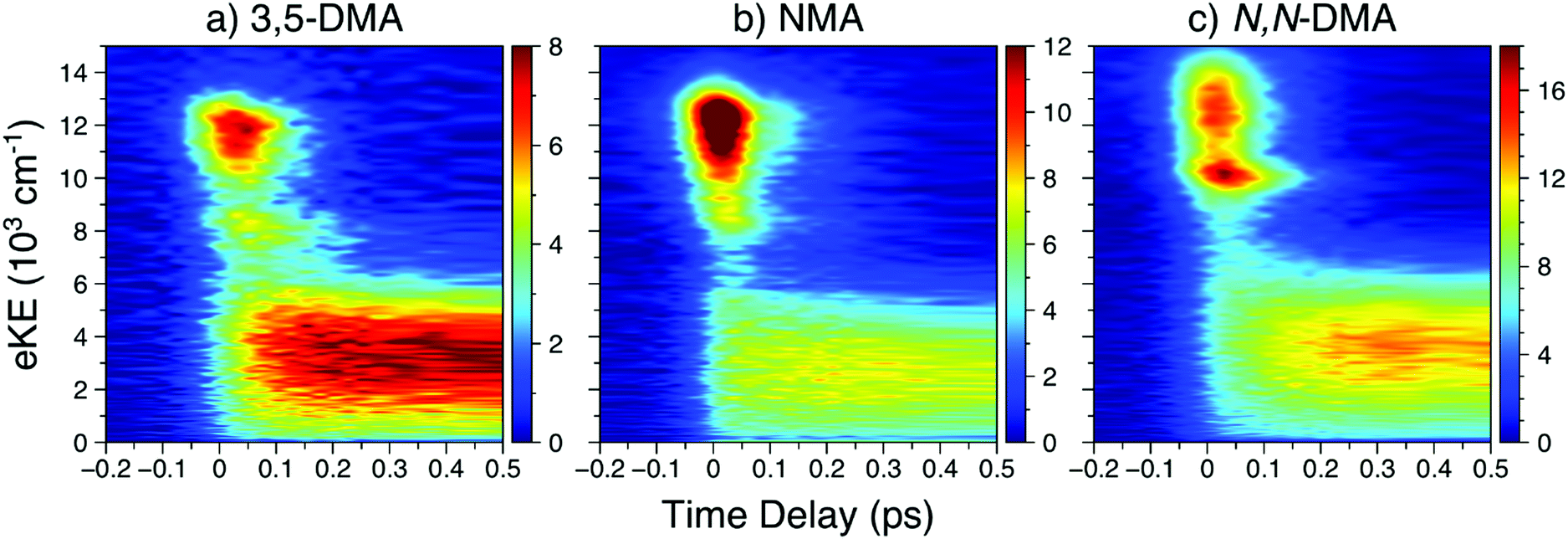 | ||
| Fig. 2 In order of increasing amine substitution, the short time delay TR-PES results at λpu = 238 nm and λpr = 305 nm from (a) 3,5-DMA, (b) NMA, and (c) N,N-DMA. | ||
| Molecule, λpu (nm) | TR-PES | H-atom | |||||
|---|---|---|---|---|---|---|---|
| τ 1 (fs) | τ 2 (fs) | τ 3 (ps) | τ 4 (ps) | τ 5 (ns) | τ H1 (fs) | τ H2 (ps) | |
| a Ref. 14. b Ref. 17. | |||||||
| Aniline, ∼240 | 50 ± 10a |
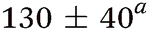
|
0.45a | 90 ± 5a | — |
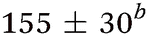
|
— |
| NMA, 238 | <50 |
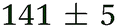
|
1.3 ± 0.1 |

|
>0.5 |
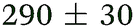
|

|
| NMA, 245 | <50 |
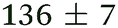
|
0.80 ± 0.05 |
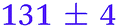
|
>1.2 |
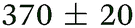
|

|
| 3,5-DMA, 238 | <50 |

|
1.05 ± 0.07 | 127 ± 2 | >1.2 |

|
— |
| N,N-DMA, 238 | <50 | 85 ± 2 | 1.03 ± 0.06 | 139 ± 2 | 0.88 ± 0.09 | — | — |
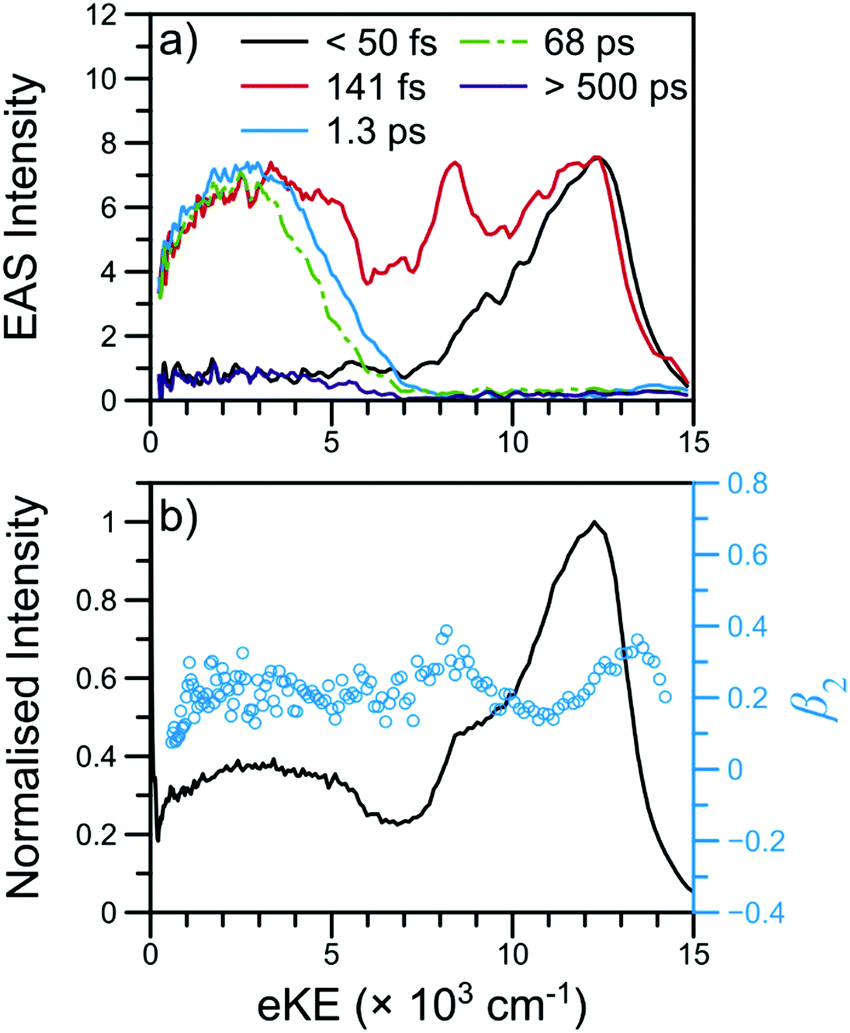 | ||
| Fig. 3 TR-PES results for NMA at 238 nm with (a) EAS and (b) the photoelectron signal (black line) and β2 values (blue circles) near Δt = 0. The short-time TR-PES transient for NMA may be found in Fig. 2; a full transient is shown in Fig. S4 of the ESI.† The EAS have been smoothed using a 3 point moving average; the τ1 EAS intensity has been scaled for visual clarity. | ||
The β2 values as a function of eKE near Δt = 0 are also shown in Fig. 3. Of particular interest are the two modest rises in β2 at ∼8000 and ∼13000 cm−1. As has been previously outlined,15 such β2 features are typically due to ionisation from Rydberg orbitals with well-defined energies and orbital angular momenta; the β2 feature at 8000 cm−1 is consistent with the assignment of the corresponding TR-PES feature to the N 3s state. The previous TR-PES investigation of N,N-DMA22 found a sharp rise in both β2 and β4 at the highest eKE, which the authors attributed to ionisation from a high lying N 3p state. In NMA, the β2 feature at ∼13000 cm−1 may also be due to ionisation from a N 3p state which yields only minimal TR-PES intensity. Unfortunately, large oscillations in the β4 distribution at these high eKE, shown in Fig. S5 of the ESI,† limit our confidence in this anisotropy parameter at the highest eKE and we cannot firmly assign the ∼13000 cm−1β2 feature to a N 3p state. Furthermore, the excited state calculations on NMA, presented in Section S3.1 of the ESI† and discussed below, reveal two candidate Rydberg states lying near the 21ππ* state energy. Thus, while the highest eKE edge of the 21ππ* TR-PES feature contains information about the dynamical behaviour of a Rydberg state, it is at present unclear from which Rydberg state these photoelectrons originate, whether the Rydberg state is populated in the initial photoexcitation, and how population of a Rydberg state affects the photodynamics of NMA.
Using the quantum defect value of δ = 0.912 from the aniline N 3s state18 as a qualitative estimate for NMA (as methylation of the amine will shift the value of δ), we can estimate the energy of the 3s state in NMA using
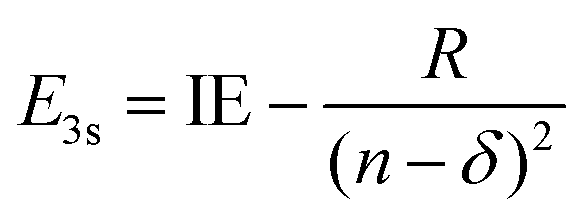 | (4) |
100 cm−1 (∼293 nm).45 Although the absorption spectrum of NMA in Fig. 1 does show additional intensity near 290 nm, there is not an obvious, additional feature when compared to aniline. Furthermore, similar spectral shifts are observed for the other substituted anilines suggesting that this behaviour is due to methyl group stabilisation of the 11ππ* state. Examining eqn (3) and (4) we may also estimate the eKE at which the N 3s state will appear using eKE = E3s + hc/λpr − IE. The resulting estimated eKE value of 7600 cm−1 is in remarkably good agreement with the experimental photoelectron spectra, further supporting the assignment of the ∼8000 cm−1 TR-PES feature to the N 3s state in NMA.
376 cm−1 is in relatively good agreement with the high TKER edge of the spectra, though the same publication reports an aniline BDE(N–H) approximately 1400 cm−1 smaller than Ashfold and co-workers.16 As also shown in Fig. 4, non-resonant production of H+ (independently detected using a non-resonant probe) contributes a small amount to the TKER signal at all kinetic energies and Δt. Fig. S6 of the ESI† reveals a nearly constant βH ≈ −0.3 across the entire main feature of the TKER spectrum; this value of βH is also conserved at all values of Δt. This value of βH is consistent with a perpendicular dissociation occurring before significant rotational randomisation of the photoexcited NMA. Due to rotational randomisation, we would expect the H+ appearing at later time delays to have an isotropic (βH = 0) distribution. The preservation of βH ≈ −0.3 at all time delays is likely due to the dominance of the initial, ultrafast H-atom loss mechanism to the total TKER spectral intensity. Thus, the absence of any significant change with increasing time delay is unsurprising given the small TKER contribution from H-atoms dissociating with lifetime τH2 (after rotational randomisation). The similarity of the TKER spectra to those taken at λpu = 252 nm (see Fig. S7 of the ESI†) where dissociation occurs above the 11πσ* barrier in aniline,7 suggests that the N–H bond fission is occurring from the same excited state (namely the 11πσ* state) for both pump wavelengths.
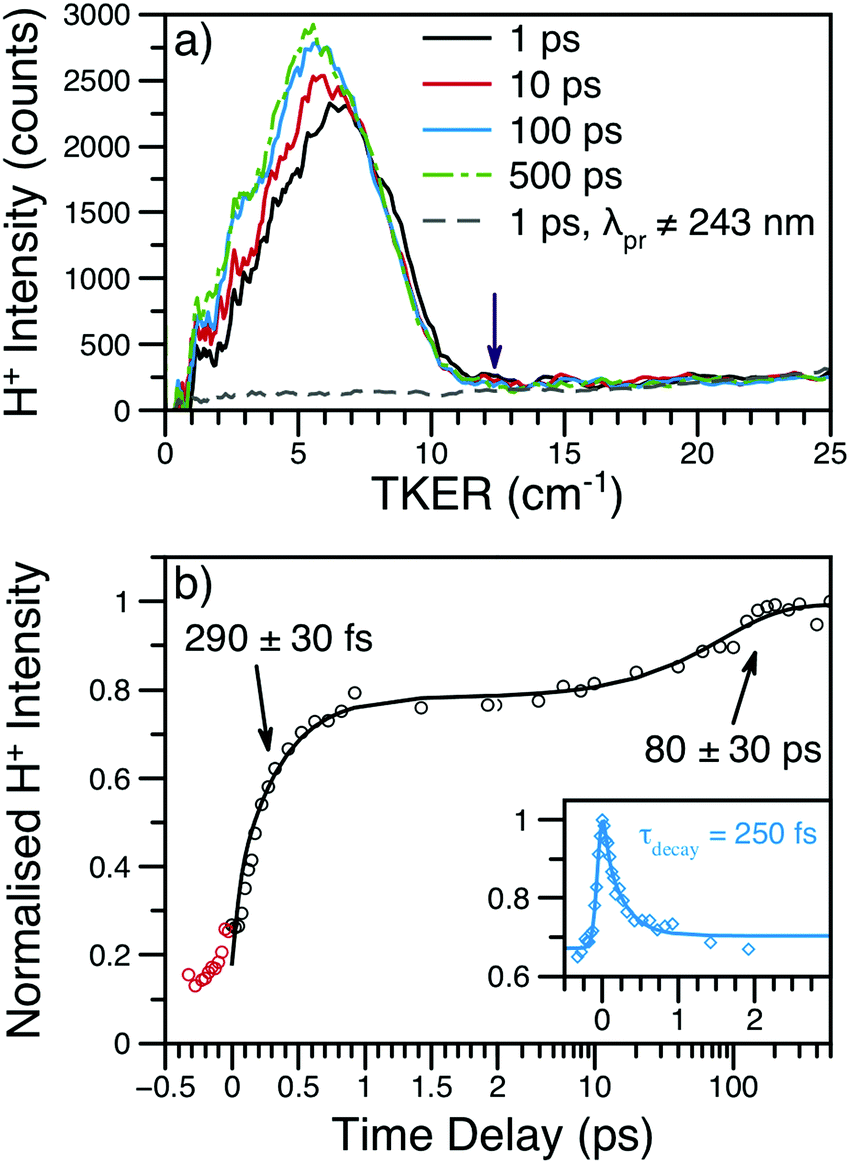 | ||
| Fig. 4 (a) H+ TKER spectra at several time delays and λpu = 238 nm; spectra have been smoothed using a 10 point moving average. The expected TKERmax at this wavelength is shown as a purple arrow.46 The dashed, grey line shows an example H+ TKER acquired with an off-resonant probe. (b) H+ transient for the main TKER feature at 238 nm (black circles) and the corresponding fit (black line). The two rises are due to the production of neutral H-atoms with appearance lifetimes assigned to each rise. The red circles are probe or multiphoton initiated H-atom loss dynamics and, for simplicity, have been excluded from the overall fit. Inset: The time-dependent, dissociative ionisation production of H+ is also shown (blue diamonds) along with the accompanying fit (blue line). The exponential decay lifetime from the inset transient (τdecay) was included in the fit to the main transient; see eqn (S1) (ESI†) for further details. | ||
Integration of the main TKER feature (ca. 2500–8000 cm−1) yields the transient also shown in Fig. 4. These transients show a clear, ultrafast rise in H+ intensity followed by an additional rise of ca. 100 ps. To account for the time-dependent dissociative ionisation captured in the integration of the main TKER feature, a separate H+ transient was produced by integrating TKER > 11000 cm−1 (Fig. 4b inset) and was fit using the exponential decay model described in Section S2.3 of the ESI.† The resulting H+ decay lifetime was included in the fit to the transient at lower TKER (i.e.Fig. 4b), yielding the two neutral H-atom appearance lifetimes given in Table 1. Further details on fitting and kinetic models may be found in Section S2 of the ESI.† It is interesting to note that the H-atom appearance lifetimes are similar to the TR-PES τ2 and τ4.
CH3 loss was also investigated at λpu = 238 nm; example transients and TKER distributions at m/z = 15 may be found in Fig. S7 of the ESI.† TKER distributions at several time delays show no features other than a sharp rise in intensity near zero kinetic energy, consistent with dissociative ionisation.47 The transients largely show a decay in signal, again due to dissociative ionisation of photoexcited NMA to m/z = 15. The transient appears to plateau at large Δt, in agreement with the behaviour observed in the TR-PES results at low eKE. The m/z = 15 transient also reveals an apparent exponential rise (with an estimated lifetime of 2 ps) superimposed on the overall decay that is not observed with an off-resonant probe, suggesting that this rise reports on the production of neutral CH3. However, a pump laser power study revealed that these neutral CH3 photofragments appear to be due to multiphoton pump photoexcitation, likely to a region of a cationic potential energy surface which, with a few picosecond lifetime, undergoes dissociation along the N–CH3 coordinate. Attempts to investigate single pump photon production of CH3 (i.e. by decreasing the pump laser power) were unsuccessful, as no time-dependent signal could be observed. Thus, no evidence was found for single pump photon initiated photodissociation to produce neutral CH3 photoproducts from NMA.
000 cm−1) is consistent with population of a Rydberg state lying above the 21ππ* state at λpu = 238 nm. Apart from the change in β2 behaviour, the TR-PES and H-atom results are very similar to those recorded at 238 nm, though with a noticeable increase in both τ4 and τH2; all time constants are given in Table 1. We note that while τH1 also increases with increasing pump wavelength, the time-dependent dissociative ionisation at both wavelengths reduces our confidence in the first H-atom appearance lifetime.
(b) Aniline, 3,5-dimethylaniline, and N,N-dimethylaniline
The short time TR-PES results from 3,5-DMA and N,N-DMA are shown in Fig. 2; full results are in Section S4.2 of the ESI† and lifetimes are given in Table 1. Similar to NMA, TR-PES results from 3,5-DMA and N,N-DMA reveal five decay lifetimes. While the previous study by Townsend and co-workers22 used four decay time constants, the new time constant is only apparent at Δt > 100 ps (larger time delays than the previous investigation). The additional time constant results in a slight contraction of the τ2 and τ3 lifetimes relative to their published values.22 Close examination of the N,N-DMA results shown in Fig. 2 show slightly different behaviour of the N 3s feature (the sharp rise in TR-PES intensity near 10000 cm−1 and Δt = 0) from the other two anilines studied herein: the feature has shifted to a higher eKE and now appears concurrently with the 21ππ* state rather than at a later time-delay.
The H-atom TKER spectrum from aniline at λpu = 238 nm was re-investigated to explicitly verify only one, sub-ns N–H bond dissociation process.17 As shown in Fig. S12 of the ESI,† TKER spectra at Δt = 1 ps and 1.2 ns are identical; the H+ transient was not re-investigated. Similar to aniline, H+ TKER spectra from 3,5-DMA (shown in Fig. 5) taken at Δt = 1 ps and 1.2 ns show no appreciable differences; both feature a clear peak at ∼6000 cm−1 which sits upon a structureless dissociative ionisation ‘baseline.’ Integration of the main TKER feature results in a H+ transient which reveals rapid H-atom loss with an appearance lifetime of ∼70 fs (also shown in Fig. 5).
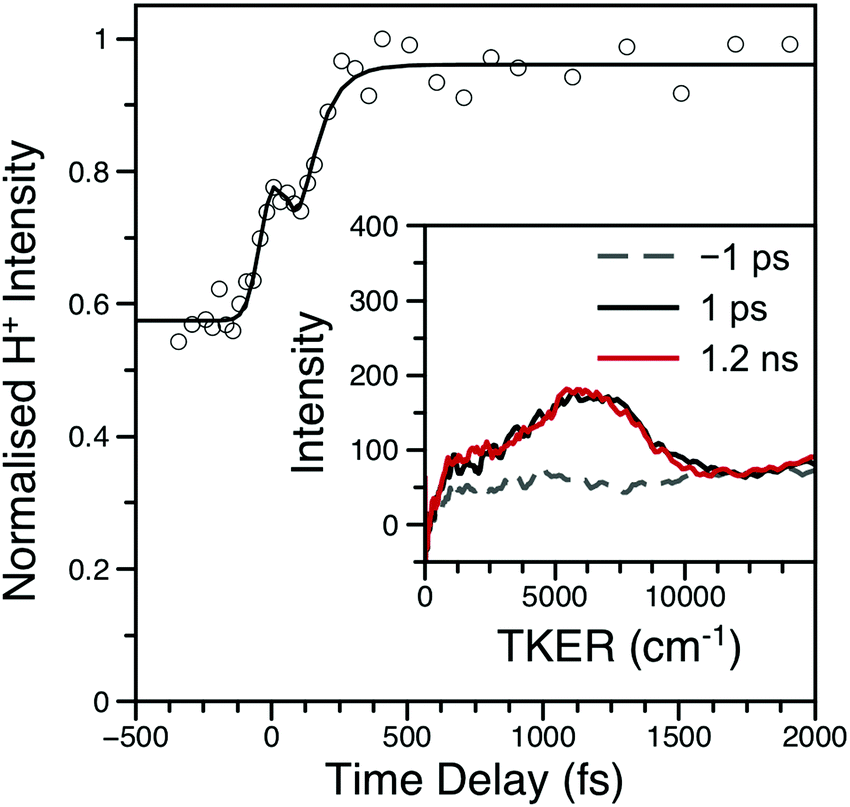 | ||
| Fig. 5 H+ transient from 3,5-DMA at λpu = 238 nm; data are shown as open circles while the black line is the fit. Inset: Example TKER spectra at λpu = 238 nm; the spectra have been smoothed using a 30 point moving average. Both the transient and TKER reveal that 3,5-DMA has only one ultrafast (i.e. within 1.2 ns) H-atom loss mechanism. The peak at Δt = 0 is due to multiphoton dissociative ionisation within the cross-correlation of the two laser pulses. | ||
To complement the previous TR-PES investigation of N,N-DMA,22 which speculated that rapid N–CH3 bond fission was occurring, CH3 loss from N,N-DMA was investigated in the present work; the results may be found in Fig. S15 of the ESI.† However, the TKER distributions and m/z = 15 transient are strikingly similar to the results from NMA, suggesting that the production of m/z = 15 photoions is dominated by dissociative ionisation of the parent cation. We note however that, unlike NMA, a pump laser power study suggests that the rise in the N,N-DMA m/z = 15 transient is from single pump photon photoexcitation. Given the absence of a TKER feature obviously attributable to neutral N–CH3 bond fission, we speculate that the rise in CH3+ intensity is due to neutral excited state evolution resulting in enhanced Franck–Condon factors for dissociative ionisation. Thus, while we cannot rule out CH3 loss from N,N-DMA, there is no clear evidence for single photon initiated N–CH3 bond dissociation.
4. Discussion
The above results raise several pertinent questions regarding the excited state dynamics of aniline-based systems. For the present discussion, we will focus on the nature of the initial photoexcitation, the excited state population leading to the second H-atom appearance in NMA, and the absence of CH3 loss from NMA and N,N-DMA in an attempt to synthesise a global relaxation mechanism and elucidate the structure–dynamics relationships underpinning these results. Before the discussion of an excited state decay mechanism, two important points must be made. First, the following discussion will focus on the relaxation of the 21ππ* state to the other electronic excited states. Previous investigations have either found no evidence for 21ππ* state population transfer to the 11πσ* or 11ππ* states22 or concluded that a barrierless 21ππ* → S0 excited state pathway was dominant.11,12,14 While we are unable to directly probe repopulation of S0, the former conclusion (exclusive 21ππ* → S0 population transfer) is in apparent conflict with our present results. As a quantitative determination of the excited state branching ratios is difficult,48 we instead accept the second conclusion that relaxation of the 21ππ* state to the lower lying singlet excited states may only be a minor decay pathway. However, the importance of each pathway remains a key question in the photodynamics of these aniline-based systems; excited state trajectory calculations to elucidate the initial, ultrafast branching ratios would be of considerable interest. The second point is a caveat regarding TR-PES assignments: the strong similarity of TR-PES results across molecules does not guarantee that the NMA assignments are transferrable to other anilines. However, as the H-atom results provide compelling evidence for overlapping TR-PES assignments in NMA and 3,5-DMA, we follow previous examples19,22 and will assume (unless otherwise noted) that the global dynamics of these anilines may be discussed across species. As a final aside, Fig. 6 shows a proposed decay mechanism along a schematic cut through the potential energy surfaces important to the dynamics. This mechanism is presented first as a reference point and will be explored throughout the following text.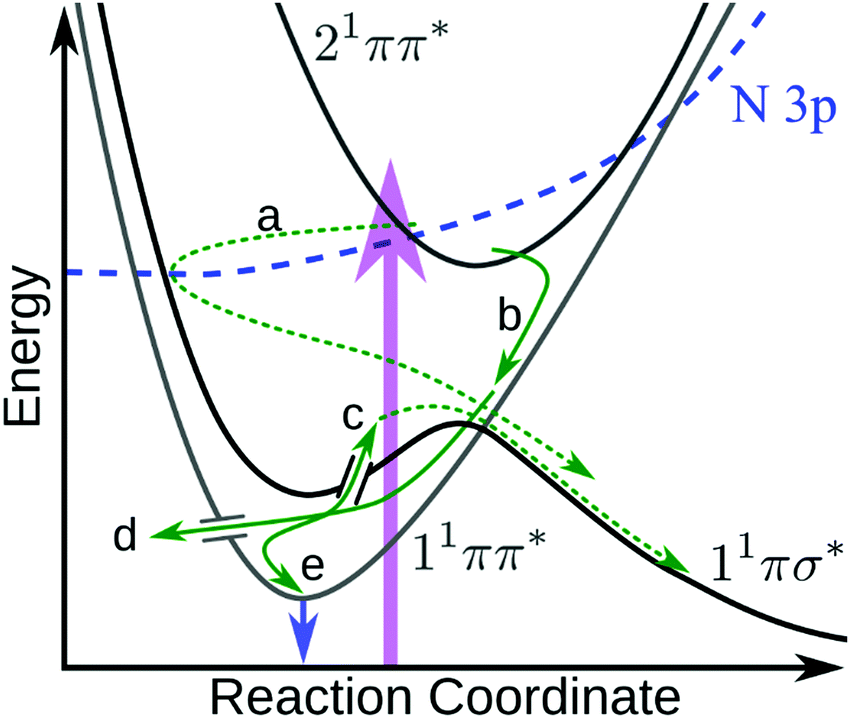 | ||
| Fig. 6 Schematic diagram showing the proposed 21ππ* de-excitation pathways. Photoexcitation (purple arrow) to the 21ππ* state undergoes IC to the N 3p state, which couples through the N 3s region of the 11πσ* state (a) where it immediately scatters to dissociate. Simultaneously, the 21ππ* state undergoes IC onto 11ππ* (b) and undergoes IVR. From the 11ππ* state, population can either sample the 11πσ* state via a CI17,21 and dissociate (c), undergo IC to the ground state (d), or fluoresce (e and blue arrow). Dashed green arrows indicate dissociative mechanisms while solid green arrows represent non-dissociative mechanisms. In the case of N,N-DMA, the initial photoexcitation may be to both the 21ππ* and N 3s states. Not shown are any possible direct 21ππ* → 11πσ* population transfer or intersystem crossing(s) to the triplet manifold. | ||
(a) Photoexcitation
The first, and perhaps most fundamental, task is to determine which excited states the pump photon populates. From Fig. 1, it is clear that photoexcitation at 238 nm will principally populate the strongly absorbing 21ππ* state. However, as previous work has concluded that other excited states must also be photoprepared in the dimethyl anilines,22 we explore that possibility herein. The DAS for each species, particularly 3,5-DMA, show clear evidence for delayed population of the lower-lying excited states; see Fig. S4, S9, S13, and S14 in the ESI.† For NMA and 3,5-DMA, the N 3s and 11ππ* TR-PES features show a clear time delay offset from the appearance of the 21ππ* feature (see further discussion below) and we thus conclude that the corresponding states are not populated by the pump laser pulse.Based on the PAD results from NMA at 238 nm, and supported by the previous investigation of N,N-DMA,22 a high-lying Rydberg orbital is populated at or near Δt = 0 and the initial photoexcitation may thus populate both the 21ππ* and a nearby Rydberg state (though not the N 3s state, see above). While direct excitation to a Rydberg state is generally less favourable than a valence-to-valence transition, as evidenced by the weak oscillator strengths calculated for other anilines,9,22 the oscillator strengths for the two Rydberg orbitals closest to the 21ππ* state in NMA are only a factor of two and four smaller than for excitation to the 21ππ* state (see Table S1 in the ESI†). At λpu = 245 nm, the absence of the highest eKE Rydberg-like β2 feature suggests that the 238 nm pump photon populates a Rydberg state lying above the 21ππ* state energy. The carbon-centred Rydberg state, described in Table S1 in the ESI,† is the most likely source of the highest eKE β2 feature as this state's energy – calculated to lie ∼400 cm−1 above the 21ππ* state – is within the pump laser bandwidth of the 21ππ* state. In contrast, the energy difference (from both electronic structure and quantum defect calculations45) between the 21ππ* and lower-lying N 3p states is greater than the pump laser bandwidth. Thus, the results from NMA at λpu = 238 nm support the photoexcitation of a high-lying, carbon-centred Rydberg orbital, though the otherwise strong similarity of the NMA TR-PES and H-atom temporal behaviour at both pump wavelengths suggests that this Rydberg state does not play a significant role in the observed photodynamics.
(b) Initial TR-PES and lifetime assignments
The TR-PES results from NMA reveal similar dynamics to the overall behaviour previously observed in aniline, N,N-DMA, and 3,5-DMA.11,12,14,22 The length of the newly observed ns lifetime (τ5) suggests that this process is the fluorescent decay of the 11ππ* state15 and supports the assignment of the low eKE TR-PES feature to this state; measurement of the fluorescence lifetimes and quantum yields following photoexcitation to the higher lying states of aniline would help confirm the assignment of τ5 and add insight to the other decay dynamics. As mentioned above, it is possible that 21ππ* state dynamics are appearing in the low eKE feature. Indeed, excited state dynamics calculations of aniline using a model Hamiltonian suggested that the 21ππ* state lives for >300 fs.9 However, given that we can neither support nor eliminate this possibility, we accept previous assignments11–14,19,22 that the low eKE TR-PES feature itself is principally due to ionisation from the 11ππ* state.While τ1 is still clearly dominated by decay of the 21ππ* TR-PES feature within the IRF, some of the 21ππ* feature persists to decay along with the N 3s state (τ2). The τ2 EAS also involves population of the lowest eKE feature, suggesting a sequential decay mechanism from the 21ππ* or N 3s states. Indeed, the τ2 DAS for all species (see Fig. S4, S9, S13, and S14 in the ESI†) show negative amplitude for the lowest TR-PES feature, indicative of population transfer from the higher lying states. Such a sequential mechanism is supported by the delayed rise of the 11ππ* TR-PES feature, as shown in Fig. 2. As discussed below, the time constants of τ2 and τ4 are in good agreement with the H-atom appearance times in NMA and (for τ2 only) 3,5-DMA. It thus seems reasonable to conclude that these time constants are related to population sampling the 11πσ* state.
The dynamics described by τ3 are not simple to assign, as altering the methyl substitution and pump wavelength both substantially change the best-fit value of this lifetime (see Table 1). For 3,5-DMA and N,N-DMA,19,22 the equivalents of τ3 and τ4 have been assigned to IVR on and decay of the 11ππ* state. The assignment of τ4 to 11ππ* relaxation is reasonable, further analysis of this lifetime may be found along with the second H-atom loss discussion below. While energy redistribution seems to be supported by the negative DAS amplitudes associated with τ3 (see Fig. S14 of the ESI† for a clear example), the assignment of τ3 to IVR is at odds with the increased lifetime observed upon methyl substitution (see Table 1). The possibility of a N 3s → 11ππ* population transfer was a proposed explanation for the low eKE PAD change on a similar timescale in N,N-DMA and 3,5-DMA,22 however we find no evidence for the N 3s TR-PES feature on this timescale. The equivalent aniline lifetime to τ3 has been assigned to population dynamics on the 11πσ* state located away from the Franck–Condon region.14 Such dynamics may contribute to the lowest eKE TR-PES signal, as previous work on alkyl substituted amines has revealed a broad, structureless σ* feature at low eKE (see, for example, ref. 22 and 49). Indeed, the second H-atom appearance from NMA suggests that the 11πσ* state must be populated or remain accessible for several 10s of ps, though we attribute that dissociative population to 11ππ* → 11πσ* IC with lifetime τ4 (see below). Diffusion of the excited state wavepacket, particularly along a relatively flat potential energy surface, has been observed to occur with a ∼1 ps lifetime in the related system 2-hydroxypyridine50 and may also contribute to the present TR-PES changes describe by τ3. Thus, while dynamics on both the 11ππ* and 11πσ* states are possible, and some form of population redistribution must be occurring, the excited state processes described by τ3 may be both highly mixed and strongly dependent upon molecular structure. As such, we are hesitant to assign one mechanism across all aniline species to the changes described by τ3.
(c) The N 3s feature
The energetic and temporal behaviour of the N 3s TR-PES feature is worth discussing in some greater detail. While previous TR-PES experiments have observed this Rydberg state,11–14,19,22 the feature has not received a close, comparative analysis as a function of chemical modification. First, as seen in Fig. 2 and previous investigations,11–14,19,22 the N 3s feature (in aniline, 3,5-DMA, and NMA) is separated from the low eKE feature by ∼5000–6000 cm−1. As the separation of the 11ππ* and 11πσ* states in aniline is only ∼3700 cm−1,18 the remaining eKE difference is likely due to the projection of the vibrationally excited 11ππ* state onto the vibrationally excited D0+ state while the N 3s state projects closer to the v = 0 level of the ground cationic state (see Section S3.3 of the ESI†). The 3s feature clearly shifts to higher eKE with increasing alkylation of the amine, possibly due to the Rydberg state becoming more ‘atom-like’ (less dependent upon molecular geometry) and able to project closer to the vibrationally cold, electronic ground state of the cation.There is also a clear shift in the temporal behaviour of the N 3s feature with increasing amine alkylation: the rise in 3,5-DMA is noticeably offset to later Δt from the 21ππ* onset, while in N,N-DMA the two features seem to appear simultaneously. The appearance of the N 3s peak in NMA is less clear due to overlap with the 21ππ* peak along the energy axis, though a slight shift to later time delays with decreasing eKE is evident. In the case of N,N-DMA, the 11πσ* absorption itself may have shifted to higher energies and may therefore be prepared by the initial photoexcitation, however this interpretation is inconsistent with both published electronic structure calculations22 and expectations using quantum defect arguments.45 As the N 3s feature appears most intense after the peak intensity of the 21ππ* feature, we will continue with our conclusion that for all of the anilines studied herein the N 3s state is not populated by the initial photoexcitation.
(d) Decay of N 3s and rapid H-atom loss
Due to the strong similarity in TKER distributions and H-atom appearance lifetimes, the first H-atom appearance in NMA and 3,5-DMA appears to be dissociation along a similar 11πσ* state as in aniline.17 While photoexcitation to the N 3p, 21ππ*, or carbon-centred Rydberg states could reproduce the observed H-atom angular distribution of βH ≈ −0.3 from NMA photodissociation (see Section S3.2 of the ESI†), as discussed above direct photoexcitation to the N 3p state seems unlikely. Furthermore, the similarity of the NMA H-atom results at λpu = 238 and 245 nm (i.e. with and without the carbon-centred Rydberg state) suggests that the observed H-atom signal is due to photoexcitation of the 21ππ* state. Thus the observed H-atom anisotropy, along with the delayed appearance of the N 3s TR-PES feature, reinforces the conclusion that H-atom loss from NMA and 3,5-DMA is due to rapid population transfer from the 21ππ* state onto the dissociative 11πσ* state.In NMA, the elongation of τH1 (relative to aniline) is likely due to the heavier CH3 group retarding any amine geometry change prior to or during the dissociation. One such motion was proposed21 as a three state 21ππ*/11πσ*/11ππ* CI where the H2N–C bond is approximately perpendicular to the plane of the ring. While motion through the three state CI would explain the increase in τH1 for NMA, such a motion should also retard the population of the lower lying states. However, the appearance Δt of the lower lying TR-PES features appears to remain constant upon methylation of the amine, e.g. comparing 3,5-DMA (Fig. 2a) to NMA (Fig. 2b). The increased τH1 in NMA is thus most likely due to the dissociation coordinate involving larger changes to the amine geometry (such as planarisation towards the anilino radical equilibrium geometry16) in addition to the N–H stretch.
Thus, either the decay of the 21ππ* state is due to the aforementioned CI21 with the expected changes to the 21ππ* and 11ππ* lifetimes obscured by the IRF or population transfers to the N 3s state by another mechanism, such as sequential transfer through the N 3p state (see ref. 49 and 51–53 for related examples). This latter mechanism would explain the absence of any evidence for direct 21ππ* → 11πσ* transfer from previous TR-PES work11,12,14,22 and is qualitatively supported by (i) the highly mixed nature of the 21ππ* transition (which includes contributions from nearby N- and C-centred Rydberg orbitals; see Table S1 in the ESI†) and (ii) visual inspection of the molecular orbitals shown in Fig. S3 of the ESI,† which suggest good overlap between the N 3s and lowest-lying N 3p Rydberg orbitals. Unfortunately, we are unable to further support or disprove either mechanism in NMA at this point: the temporal resolution of our experiment limits the information we can obtain near Δt = 0 and we would expect any N 3p state population to yield little or no discernible TR-PES structure.22,48 Regardless of the mechanism, population transferred into the N 3s state in 3,5-DMA and NMA rapidly escapes the ionisation window towards dissociation (shown schematically in Fig. 6 as the dashed green arrow). We note that the previously reported 3,5-DMA DAS,22 which do not show the same ‘sequential’ behaviour as is shown in Fig. S13 in the ESI,† are reproducible with the previously published kinetic model of four exponential decays.
(e) 11ππ*, τ4, and the second H-atom loss mechanism
With a small decrease in TKER, i.e. an increase in Eint, the second H-atom appearance lifetime in NMA is most likely due to dissociation following excited state IVR. Building on the possible mechanisms discussed above, a 21ππ* → 11ππ* transition (likely also described by τ2, see DAS in Fig. S4 of the ESI†) will result in a highly vibrationally excited 11ππ* state. The excellent agreement between τH2 and τ4 thus implies that some of the vibrationally excited 11ππ* population must sample a 11πσ*/11ππ* CI17,21 and transiently populate the 11πσ* state, ultimately accessing the repulsive portion of the potential (shown schematically in Fig. 6c). The absence of any obvious, long-lived 11πσ* TR-PES features and the modest growth in H+ signal with τH2 (see Fig. 4) suggest that a relatively small amount of 11ππ* population relaxes via dissociation on the 11πσ* state. Furthermore, the increase in both τH2 and τ4 with increasing pump wavelength suggest that non-dissociative mechanisms (e.g. IC) control the excited state relaxation time τ4 and thus the H-atom appearance lifetime τH2; similar behaviour has been observed in the excited state dissociation of 1,3-dihydroxybenzene.54 Thus, τ4 is likely describing the majority of the 11ππ* state population undergoing IC to the S0 state (via CIs such as the prefulvenic geometries previously reported17,21) or intersystem crossing to the triplet manifold,15,55 though some population appears to remain in the 11ππ* state and likely fluoresces15 with lifetime τ5 as discussed above.Assuming the 11ππ* → 11πσ* pathway outlined above ultimately leads to N–H fission with lifetime τH2, there are several possible explanations for the absence of this second H-atom loss mechanism in aniline and 3,5-DMA. First, IC back out of the 11πσ* state (to either the 11ππ* state or S0) may be more facile than dissociation for aniline and 3,5-DMA; methylation of the amine may shift a relevant CI. Alternatively, and not exclusively, dissociative tunnelling out of the 11πσ* state minimum is likely mediated by particular vibrational modes13,16 which are sensitive to methylation. In this case, amine methylation would apparently enhance the rate of tunnelling while methylation of the ring would disrupt a mode-specific mechanism (in contrast, alkylation of the pyrrole ring reduces the contribution of tunnelling mechanisms to dissociation25,56,57). The higher symmetry of aniline and 3,5-DMA versus NMA may also play a role in the disappearance of τH2, as both the number and positions of substituents have been shown to play a crucial role in H-atom loss dynamics (see, for examples, ref. 7, 58 and 59). Ultimately, further study of the second H-atom loss pathway – such as high-resolution photofragment translational spectroscopy60 – will be needed to fully elucidate the excited state dynamics at work.
(f) Absence of CH3 loss
The apparent absence of CH3 loss from both NMA and N,N-DMA may be understood by comparison to the related system N-methylpyrrole. In a previous study of N-methylpyrrole, Ashfold and co-workers61 only observed excited state N–CH3 bond fission over a narrow range of pump photon energies above the N–CH3 stretch barrier. This behaviour was attributed to the heavier and slower CH3 group helping facilitate efficient 11πσ*/S0 coupling at the CI at extended N–CH3 bond lengths (see the schematic potential energy cut in Fig. 1). In these substituted anilines, the 11πσ*/11ππ* CI is accessed faster in N,N-DMA than in 3,5-DMA,22 thus reducing the relative importance of the dissociative coordinate in the relaxation dynamics of N,N-DMA. However, as the same investigation22 concluded that N,N-DMA photoexcited at 240 nm (above the 11πσ* state barrier) must undergo some ultrafast N–CH3 bond cleavage, our present results would thus indicate that this dissociative population returns to the ground state via the 11πσ*/S0 CI. Thus, while our present results do not support ultrafast CH3 loss as a competitive dissociation product from photoexcited NMA or N,N-DMA, neither do our results disprove this relaxation pathway. Indeed, the qualitative agreement between our results and the absence of ‘fast’ CH3 loss from N-methylpyrrole at short pump wavelengths61 suggests that CH3 loss from these anilines may occur near the 11πσ* absorption onset; further study of these systems would substantially contribute to the understanding of non-hydride πσ* states.(g) Remaining questions
From the above discussion, it is evident that relaxation of these substituted anilines following photoexcitation at 238 nm involves competing population and decay of the lower lying 11πσ* and 11ππ* states. The correlation between excited state decay times and photofragment appearance times strongly supports the assignment of several PES features and dynamics to loss of population via dissociation on the 11πσ* excited state. While the mechanisms discussed provide a reasonable picture for the dynamics, some lingering questions remain. First, the apparent acceleration of τH1 upon methylation of the ring (aniline versus 3,5-DMA) seems at odds with preparation of a N 3s Rydberg orbital. However, the previously reported TR-PES results from aniline at 238 nm show the 21ππ* and 11πσ* features appearing simultaneously,11,12,14 suggesting that direct population of and dissociation from the 11πσ* state may overlap with and obscure dynamics from the 21ππ* state of aniline. TR-PES and H-atom appearance measurements from aniline following photoexcitation at ∼225 nm (the peak of the aniline 21ππ* state absorption shown in Fig. 1) could be of significant interest, though aniline photoexcitation at both 200 and 240 nm results in a similar H-atom appearance time.17 Similarly, a more systematic study of N–H bond dissociation time as a function of ring substitution would add considerable insight to the connections between ring substituents and ultrafast dissociation from a mixed valence/Rydberg state.Another dynamical question is the nature of the N 3s state preparation in these substituted anilines. Principally, is population of the N 3s Rydberg state due to direct ultrafast transfer from the 21ππ* state? Or does population flow through a more complicated pathway, such as through the N 3p state? As discussed above, the initial photoexcitation appears to be dominated by the 21ππ* state; the EAS/DAS for all of the substituted anilines studied at λpu = 238 nm show loss of the highest eKE signal (i.e. from the most internally excited 21ππ* state) with lifetime τ1 followed by a slightly longer decay of the remaining 21ππ* feature with lifetime τ2. Indeed, the 3,5-DMA τ1 DAS (shown in Fig. S13 of the ESI†) shows negative amplitude in the N 3s eKE region, indicative of sequential dynamics from the highest eKE feature into the N 3s state. While direct population from the 21ππ* state is the simplest explanation consistent with the observed dynamics, the previously reported 21ππ*/11ππ* and 21ππ*/11πσ* CIs show considerable out-of-plane motion of the amine,17,21 which neither previous22 nor the present work can find evidence to support. However the current results may also be interpreted using a valence-to-Rydberg decay mechanism, i.e. 21ππ* → N 3p, a pathway which has only been experimentally identified in a few small molecules (see, for examples, ref. 53 and 62–65) and was previously alluded to play a role in the dynamics of N,N-DMA.22 While such a 21ππ* → N 3p transition would also have to be within the IRF, and thus cannot be confirmed in the present work, the possibility further reinforces the conclusion that Rydberg states play a vital and complicated role in the relaxation dynamics of many systems.
5. Conclusions
In the present work we have investigated the ultrafast dissociation dynamics of several substituted anilines. By correlating the photoproduct appearance times to TR-PES decay lifetimes, we are able to assign some of the observed lifetimes to specific excited states. Our results strongly support a sequential mechanism whereby population first moves from the photoprepared 21ππ* state to the 11πσ* state, where the wavepacket scatters to produce photoproducts. Concurrently, the 21ππ* state relaxes to the lower lying 11ππ* state where, for NMA, population samples the 11ππ*/11πσ* CI, resulting in a second H-atom loss pathway. While population of these states have been previously reported,10–14,19,22 our experiments are the first to clearly match specific TR-PES features and lifetimes to dissociative processes. Ultimately, the work presented herein further highlights the rapid depopulation of higher-lying, potentially reactive excited states in these model biomolecules. While ultrafast dissociation is observed along the N–H bond, the absence of a CH3 loss channel may indicate efficient 11πσ*/S0 coupling in these anilines, reinforcing the role of these states as central to photoprotection in molecular biology. Despite the substantial insight afforded by these two techniques, important dynamical questions remain including the nature of the 11πσ* excited state population (direct versus valence-to-Rydberg population transfer from the 21ππ* state) and the exact structure–dynamics relationship underlying the second H-atom loss channel. Furthermore, the decay of the 21ππ* state appears to be largely insensitive to molecular geometry; as this study is one of several that is unable to firmly identify the cause, ab initio quantum dynamics calculations would be of considerable interest.Data availability
Data underpinning the present work are available through the Zenodo data repository at doi: 10.5281/zenodo.1488380; a detailed description of the archive may be found in the ESI.†Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
The authors would like to thank N. d. N. Rodrigues, S. Trouton, G. Thornton, and K. Krokidi for laboratory assistance; Dr M. Staniforth for many helpful discussions; and the Warwick Centre for Ultrafast Spectroscopy for the use of the Cary 60 spectrophotometer. N. C. C. F. thanks the Leverhulme Trust for post-doctoral funding (RPG-2015-190) and V. G. S. thanks the EPSRC for an equipment grant (EP/J007153) and the Royal Society and Leverhulme Trust for a Royal Society Leverhulme Trust Senior Research Fellowship.References
- M. N. R. Ashfold, G. A. King, D. Murdock, M. G. D. Nix, T. A. A. Oliver and A. G. Sage, πσ* excited states in molecular photochemistry, Phys. Chem. Chem. Phys., 2010, 12, 1218–1238 RSC.
- G. M. Roberts and V. G. Stavros, The role of πσ* states in the photochemistry of heteroaromatic biomolecules and their subunits: insights from gas-phase femtosecond spectroscopy, Chem. Sci., 2014, 5, 1698–1722 RSC.
- V. G. Stavros and J. R. R. Verlet, Gas-Phase Femtosecond Particle Spectroscopy: A Bottom-Up Approach to Nucleotide Dynamics, Annu. Rev. Phys. Chem., 2016, 67, 211–232 CrossRef CAS PubMed.
- A. L. Sobolewski, W. Domcke, C. Dedonder-Lardeux and C. Jouvet, Excited-state hydrogen detachment and hydrogen transfer driven by repulsive 1πσ* states: a new paradigm for nonradiative decay in aromatic biomolecules, Phys. Chem. Chem. Phys., 2002, 4, 1093–1100 RSC.
- M. N. R. Ashfold, B. Cronin, A. L. Devine, R. N. Dixon and M. G. D. Nix, The Role of πσ* Excited States in the Photodissociation of Heteroaromatic Molecules, Science, 2006, 312, 1637 CrossRef CAS PubMed.
- T. N. V. Karsili, B. Marchetti, R. Moca and M. N. R. Ashfold, UV Photodissociation of Pyrroles: Symmetry and Substituent Effects, J. Phys. Chem. A, 2013, 117, 12067–12074 CrossRef CAS PubMed.
- T. N. V. Karsili, A. M. Wenge, B. Marchetti and M. N. R. Ashfold, Symmetry matters: photodissociation dynamics of symmetrically versus asymmetrically substituted phenols, Phys. Chem. Chem. Phys., 2014, 16, 588–598 RSC.
- Y. Honda, M. Hada, M. Ehara and H. Nakatsuji, Excited and ionized states of aniline: symmetry adapted cluster configuration interaction theoretical study, J. Chem. Phys., 2002, 117, 2045–2052 CrossRef CAS.
- F. Wang, S. P. Neville, R. Wang and G. A. Worth, Quantum Dynamics Study of Photoexcited Aniline, J. Phys. Chem. A, 2013, 117, 7298–7307 CrossRef CAS PubMed.
- R. Montero, Á. P. Conde, V. Ovejas, R. Martínez, F. Castaño and A. Longarte, Ultrafast dynamics of aniline in the 294–234 nm excitation range: the role of the πσ* state, J. Chem. Phys., 2011, 135, 54308 CrossRef PubMed.
- R. Spesyvtsev, O. M. Kirkby, M. Vacher and H. H. Fielding, Shedding new light on the role of the Rydberg state in the photochemistry of aniline, Phys. Chem. Chem. Phys., 2012, 14, 9942–9947 RSC.
- R. Spesyvtsev, O. M. Kirkby and H. H. Fielding, Ultrafast dynamics of aniline following 269–238 nm excitation and the role of the S2(π3s/πσ*) state, Faraday Discuss., 2012, 157, 165–179 RSC.
- J. O. F. Thompson, R. A. Livingstone and D. Townsend, Following the relaxation dynamics of photoexcited aniline in the 273–266 nm region using time-resolved photoelectron imaging, J. Chem. Phys., 2013, 139, 34316 CrossRef PubMed.
- O. M. Kirkby, M. Sala, G. Balerdi, R. de Nalda, L. Banares, S. Guerin and H. H. Fielding, Comparing the electronic relaxation dynamics of aniline and d7-aniline following excitation at 272–238 nm, Phys. Chem. Chem. Phys., 2015, 17, 16270–16276 RSC.
- F. Kraus, E. Gregorek and H. von Weyssenhoff, Absolute Fluorescence Lifetimes and Quantum Yields of Substituted Anilines in the Gas Phase, Z. Phys. Chem., 1972, 82, 139–146 CrossRef CAS.
- G. A. King, T. A. A. Oliver and M. N. R. Ashfold, Dynamical insights into 1πσ* state mediated photodissociation of aniline, J. Chem. Phys., 2010, 132, 214307 CrossRef PubMed.
- G. M. Roberts, C. A. Williams, J. D. Young, S. Ullrich, M. J. Paterson and V. G. Stavros, Unraveling Ultrafast Dynamics in Photoexcited Aniline, J. Am. Chem. Soc., 2012, 134, 12578–12589 CrossRef CAS PubMed.
- T. Ebata, C. Minejima and N. Mikami, A New Electronic State of Aniline Observed in the Transient IR Absorption Spectrum from S1 in a Supersonic Jet, J. Phys. Chem. A, 2002, 106, 11070–11074 CrossRef CAS.
- J. O. F. Thompson, L. Saalbach, S. W. Crane, M. J. Paterson and D. Townsend, Ultraviolet relaxation dynamics of aniline, N,N-dimethylaniline and 3,5-dimethylaniline at 250 nm, J. Chem. Phys., 2015, 142, 114309 CrossRef PubMed.
- B. N. Rajasekhar, A. Veeraiah, K. Sunanda and B. N. Jagatap, Excited states of aniline by photoabsorption spectroscopy in the 30 000–90 000 cm−1 region using synchrotron radiation, J. Chem. Phys., 2013, 139, 64303 CrossRef CAS PubMed.
- M. Sala, O. M. Kirkby, S. Guerin and H. H. Fielding, New insight into the potential energy landscape and relaxation pathways of photoexcited aniline from CASSCF and XMCQDPT2 electronic structure calculations, Phys. Chem. Chem. Phys., 2014, 16, 3122–3133 RSC.
- M. M. Zawadzki, M. Candelaresi, L. Saalbach, S. W. Crane, M. J. Paterson and D. Townsend, Observation of multi-channel non-adiabatic dynamics in aniline derivatives using time-resolved photoelectron imaging, Faraday Discuss., 2016, 194, 185–208 RSC.
- A. Iqbal, L.-J. Pegg and V. G. Stavros, Direct versus Indirect H Atom Elimination from Photoexcited Phenol Molecules, J. Phys. Chem. A, 2008, 112, 9531–9534 CrossRef CAS PubMed.
- K. L. Wells, G. Perriam and V. G. Stavros, Time-resolved velocity map ion imaging study of NH3 photodissociation, J. Chem. Phys., 2009, 130, 74308 CrossRef PubMed.
- M. Staniforth, J. D. Young, D. R. Cole, T. N. V. Karsili, M. N. R. Ashfold and V. G. Stavros, Ultrafast Excited-State Dynamics of 2,4-Dimethylpyrrole, J. Phys. Chem. A, 2014, 118, 10909–10918 CrossRef CAS PubMed.
- U. Even, The Even–Lavie valve as a source for high intensity supersonic beam, EPJ Techn. Instrum., 2015, 2, 17 CrossRef.
- A. T. J. B. Eppink and D. H. Parker, Velocity map imaging of ions and electrons using electrostatic lenses: application in photoelectron and photofragment ion imaging of molecular oxygen, Rev. Sci. Instrum., 1997, 68, 3477–3484 CrossRef CAS.
- M. W. Schmidt, K. K. Baldridge, J. A. Boatz, S. T. Elbert, M. S. Gordon, J. H. Jensen, S. Koseki, N. Matsunaga, K. A. Nguyen, S. Su, T. L. Windus, M. Dupuis and J. A. Montgomery, General atomic and molecular electronic structure system, J. Comput. Chem., 1993, 14, 1347–1363 CrossRef CAS.
- M. S. Gordon and M. W. Schmidt, in Theory and Applications of Computational Chemistry, ed. C. E. Dykstra, G. Frenking, K. S. Kim and G. E. Scuseria, Elsevier, Amsterdam, 2005, pp. 1167–1189 Search PubMed.
- T. Yanai, D. P. Tew and N. C. Handy, A new hybrid exchange–correlation functional using the Coulomb-attenuating method (CAM-B3LYP), Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS.
- R. A. Kendall, T. H. Dunning and R. J. Harrison, Electron affinities of the first-row atoms revisited. Systematic basis sets and wave functions, J. Chem. Phys., 1992, 96, 6796–6806 CrossRef CAS.
- Y. Tawada, T. Tsuneda, S. Yanagisawa, T. Yanai and K. Hirao, A long-range-corrected time-dependent density functional theory, J. Chem. Phys., 2004, 120, 8425–8433 CrossRef CAS PubMed.
- M. Chiba, T. Tsuneda and K. Hirao, Excited state geometry optimizations by analytical energy gradient of long-range corrected time-dependent density functional theory, J. Chem. Phys., 2006, 124, 144106 CrossRef PubMed.
- K. A. Nguyen, P. N. Day and R. Pachter, Analytical energy gradients of Coulomb-attenuated time-dependent density functional methods for excited states, Int. J. Quantum Chem., 2010, 110, 2247–2255 CrossRef CAS.
- C. C. J. Roothaan, New Developments in Molecular Orbital Theory, Rev. Mod. Phys., 1951, 23, 69–89 CrossRef CAS.
- C. C. J. Roothaan, Self-Consistent Field Theory for Open Shells of Electronic Systems, Rev. Mod. Phys., 1960, 32, 179–185 CrossRef.
- B. M. Bode and M. S. Gordon, Macmolplt: a graphical user interface for GAMESS, J. Mol. Graphics Modell., 1998, 16, 133–138 CrossRef CAS PubMed.
- G. M. Roberts, J. L. Nixon, J. Lecointre, E. Wrede and J. R. R. Verlet, Toward real-time charged-particle image reconstruction using polar onion-peeling, Rev. Sci. Instrum., 2009, 80, 53104 CrossRef CAS PubMed.
- P. M. Regan, S. R. Langford, A. J. Orr-Ewing and M. N. R. Ashfold, The ultraviolet photodissociation dynamics of hydrogen bromide, J. Chem. Phys., 1998, 110, 281–288 CrossRef.
- R. N. Zare, Photoejection dynamics, Mol. Photochem., 1972, 4, 1–37 CAS.
- E. Altiere, E. R. Miller, T. Hayamizu, D. J. Jones, K. W. Madison and T. Momose, High-resolution two-photon spectroscopy of a 5p56p ← 5p6 transition of xenon, Phys. Rev. A, 2018, 97, 12507 CrossRef.
- J. R. Knutson, D. G. Walbridge and L. Brand, Decay-associated fluorescence spectra and the heterogeneous emission of alcohol dehydrogenase, Biochemistry, 1982, 21, 4671–4679 CrossRef CAS PubMed.
- I. H. M. van Stokkum, D. S. Larsen and R. van Grondelle, Global and target analysis of time-resolved spectra, Biochim. Biophys. Acta, Bioenerg., 2004, 1657, 82–104 CrossRef CAS PubMed.
- J. J. Snellenburg, S. Laptenok, R. Seger, K. M. Mullen and I. H. M. Van Stokkum, Glotaran: A Java-based graphical user interface for the R package TIMP, J. Stat. Softw., 2012, 49, 1–22 Search PubMed.
- J. P. Maier and D. W. Turner, Steric inhibition of resonance studied by molecular photoelectron spectroscopy. Part 3—Anilines, phenols and related compounds, J. Chem. Soc., Faraday Trans. 2, 1973, 69, 521–531 RSC.
- A. J. Colussi and S. W. Benson, Very-low-pressure pyrolysis of N-methyl aniline and N,N-dimethyl aniline. Enthalpy of formation of the anilino and N-methyl anilino radicals, Int. J. Chem. Kinet., 1978, 10, 1139–1149 CrossRef CAS.
- D. J. Hadden, C. A. Williams, G. M. Roberts and V. G. Stavros, Time-resolved velocity map imaging of methyl elimination from photoexcited anisole, Phys. Chem. Chem. Phys., 2011, 13, 4494–4499 RSC.
- N. Kotsina and D. Townsend, Relative detection sensitivity in ultrafast spectroscopy: state lifetime and laser pulse duration effects, Phys. Chem. Chem. Phys., 2017, 19, 29409–29417 RSC.
- L. B. Klein, J. O. F. Thompson, S. W. Crane, L. Saalbach, T. I. Sølling, M. J. Paterson and D. Townsend, Ultrafast relaxation dynamics of electronically excited piperidine: ionization signatures of Rydberg/valence evolution, Phys. Chem. Chem. Phys., 2016, 18, 25070–25079 RSC.
- L. Poisson, D. Nandi, B. Soep, M. Hochlaf, M. Boggio-Pasqua and J.-M. Mestdagh, A roaming wavepacket in the dynamics of electronically excited 2-hydroxypyridine, Phys. Chem. Chem. Phys., 2014, 16, 581–587 RSC.
- G. Wu, S. P. Neville, O. Schalk, T. Sekikawa, M. N. R. Ashfold, G. A. Worth and A. Stolow, Excited state non-adiabatic dynamics of N-methylpyrrole: a time-resolved photoelectron spectroscopy and quantum dynamics study, J. Chem. Phys., 2016, 144, 14309 CrossRef PubMed.
- D. Yang, Z. Chen, Z. He, H. Wang, Y. Min, K. Yuan, D. Dai, G. Wu and X. Yang, Ultrafast excited-state dynamics of 2,4-dimethylpyrrole, Phys. Chem. Chem. Phys., 2017, 19, 29146–29152 RSC.
- T. Geng, O. Schalk, S. P. Neville, T. Hansson and R. D. Thomas, Dynamics in higher lying excited states: valence to Rydberg transitions in the relaxation paths of pyrrole and methylated derivatives, J. Chem. Phys., 2017, 146, 144307 CrossRef PubMed.
- J. D. Young, M. Staniforth, A. S. Chatterley, M. J. Paterson, G. M. Roberts and V. G. Stavros, Relaxation dynamics of photoexcited resorcinol: internal conversion versus H atom tunnelling, Phys. Chem. Chem. Phys., 2014, 16, 550–562 RSC.
- B. Kim, C. P. Schick and P. M. Weber, Time-delayed two-color photoelectron spectra of aniline, 2-aminopyridine, and 3-aminopyridine: snapshots of the nonadiabatic curve crossings, J. Chem. Phys., 1995, 103, 6903–6913 CrossRef CAS.
- V. Ovejas, R. Montero, M. Fernández-Fernández and A. Longarte, Tracking the Relaxation of 2,5-Dimethylpyrrole by Femtosecond Time-Resolved Photoelectron and Photoion Detection, J. Phys. Chem. A, 2015, 119, 3355–3365 CrossRef CAS PubMed.
- N. C. Cole-Filipiak, M. Staniforth, N. D. N. Rodrigues, Y. Peperstraete and V. G. Stavros, Ultrafast Dissociation Dynamics of 2-Ethylpyrrole, J. Phys. Chem. A, 2017, 121, 969–976 CrossRef CAS PubMed.
- G. A. King, A. L. Devine, M. G. D. Nix, D. E. Kelly and M. N. R. Ashfold, Near-UV photolysis of substituted phenols Part II. 4-, 3- and 2-methylphenol, Phys. Chem. Chem. Phys., 2008, 10, 6417–6429 RSC.
- T. A. A. Oliver, G. A. King and M. N. R. Ashfold, Position matters: competing O–H and N–H photodissociation pathways in hydroxy- and methoxy-substituted indoles, Phys. Chem. Chem. Phys., 2011, 13, 14646–14662 RSC.
- M. N. R. Ashfold, G. A. King, M. G. D. Nix and T. A. A. Oliver, Handb. High-resolution Spectrosc., 2011 Search PubMed.
- A. G. Sage, M. G. D. Nix and M. N. R. Ashfold, UV photodissociation of N-methylpyrrole: the role of 1πσ* states in non-hydride heteroaromatic systems, Chem. Phys., 2008, 347, 300–308 CrossRef CAS.
- M. Ben-Nun and T. J. Martínez, Ab initio molecular dynamics study of cis–trans photoisomerization in ethylene, Chem. Phys. Lett., 1998, 298, 57–65 CrossRef CAS.
- H. Tao, T. K. Allison, T. W. Wright, A. M. Stooke, C. Khurmi, J. van Tilborg, Y. Liu, R. W. Falcone, A. Belkacem and T. J. Martinez, Ultrafast internal conversion in ethylene. I. The excited state lifetime, J. Chem. Phys., 2011, 134, 244306 CrossRef CAS PubMed.
- R. Spesyvtsev, T. Horio, Y. I. Suzuki and T. Suzuki, Excited-state dynamics of furan studied by sub-20-fs time-resolved photoelectron imaging using 159-nm pulses, J. Chem. Phys., 2015, 143, 14302 CrossRef CAS PubMed.
- E. G. Champenois, N. H. Shivaram, T. W. Wright, C.-S. Yang, A. Belkacem and J. P. Cryan, Involvement of a low-lying Rydberg state in the ultrafast relaxation dynamics of ethylene, J. Chem. Phys., 2016, 144, 14303 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Description of the new ion detector, example Xe+ transient, details of the instrument response function and kinetic models, computational results, and additional experimental results (including TR-PES fit residuals; additional H+ TKER; m/z = 15 TKER and transients; and the results from aniline and N,N-DMA) may be found. See DOI: 10.1039/c8cp07061j |
| ‡ Present address: Combustion Research Facility, Mail Stop 9055, Sandia National Laboratories, Livermore, CA 94551-0969, USA. |
| This journal is © the Owner Societies 2019 |