
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Waste not, want not: CO2 (re)cycling into block polymers†
Sumesh K.
Raman
a,
Robert
Raja
 b,
Polly L.
Arnold
c,
Matthew G.
Davidson
d and
Charlotte K.
Williams
*a
b,
Polly L.
Arnold
c,
Matthew G.
Davidson
d and
Charlotte K.
Williams
*a
aDepartment of Chemistry, University of Oxford, 12 Mansfield Road, OX1 3TA, Oxford, UK. E-mail: charlotte.williams@chem.ox.ac.uk
bCentre for Sustainable Chemical Technologies, Department of Chemistry, University of Bath, Bath, BA2 7AY, UK
cSchool of Chemistry, University of Southampton, University Road, Highfield, Southampton SO17 1BJ, UK
dEaStCHEM School of Chemistry, The University of Edinburgh, Joseph Black Building, David Brewster Road, Edinburgh, EH9 3FJ, UK
First published on 7th June 2019
Abstract
A new way to combine two different polymerisation reactions, using a single catalyst, results in efficient block polymer synthesis. The selective polymerisation of mixtures of L-lactide-O-carboxyanhydride and cyclohexene oxide, using a di-zinc catalyst in a one-pot procedure, allows the preparation of poly(L-lactide-b-cyclohexene carbonate). The catalysis near quantitatively recycles the carbon dioxide released during polyester formation into the subsequent polycarbonate block, with an atom economy of up to of 91%.
Aliphatic polycarbonates (PC) and polyesters (PE) are attracting attention as sustainable alternatives to petrochemicals.1–3 Many are biodegradable, recyclable and biocompatible and some are sourced from renewable resources.4–6 Here, block polymers comprising polylactide (PLA) and poly(cyclohexene carbonate) (PCHC) are targeted as they allow combination of two of the most widely studied materials in each class.2,6,7 PLA is a commercially produced bio-derived plastic, which is sourced from biomass and at end of life can be recycled or biodegraded.4 It serves as a replacement for petroleum derived plastics in sectors including packaging, house-hold goods, automotive and biomedicine.4,5 One limitation is its poor temperature stability, for example it has a moderate glass transition temperature (Tg) (∼60 °C), which prevents applications at higher temperatures.8 PCHC is a widely investigated amorphous polymer produced by CO2/cyclohexene oxide copolymerisation and, because of its rigid carbonate and ring structures, shows a higher Tg value (110–120 °C).6 Here, a new catalysis concept is explored as a means to deliver block polymers which in future may result in improved polymer thermal and mechanical properties. In this vein, several groups have already demonstrated the enhanced performances for PLA-b-PCHC but almost all these block polymers were prepared by sequential addition methods or by tandem catalysis.9–14 These prior studies have nicely demonstrated the enhanced properties of such block polymers and we were motivated to investigate an alternative preparation which couples together two different polymerisation cycles (Scheme 1). Our one-pot synthetic strategy targets PLA synthesis using the controlled ring opening polymerisation (ROP) of O-carboxyanhydride (LLA-OCA). The ROP is driven by reducing the ring-strain and results in the release of a molecule of CO2 per polymer repeat unit.15,16 We reasoned that it might be possible to recycle the carbon dioxide using controlled epoxide/CO2 ring opening copolymerisation (ROCOP) to produce PCHC. ROCOP is thermodynamically driven by epoxide ring-strain and several catalysts are reported showing rapid CO2 insertion rates.6,7,17 Indeed, some of us have already reported high activity dinuclear ROCOP catalysts which are effective at low pressures (1 bar) of CO2.18–21 Here, a di-zinc catalyst is targeted that is straightforward to prepare and which shows a zeroth order in CO2 pressure over the range 1–40 bar.22,23
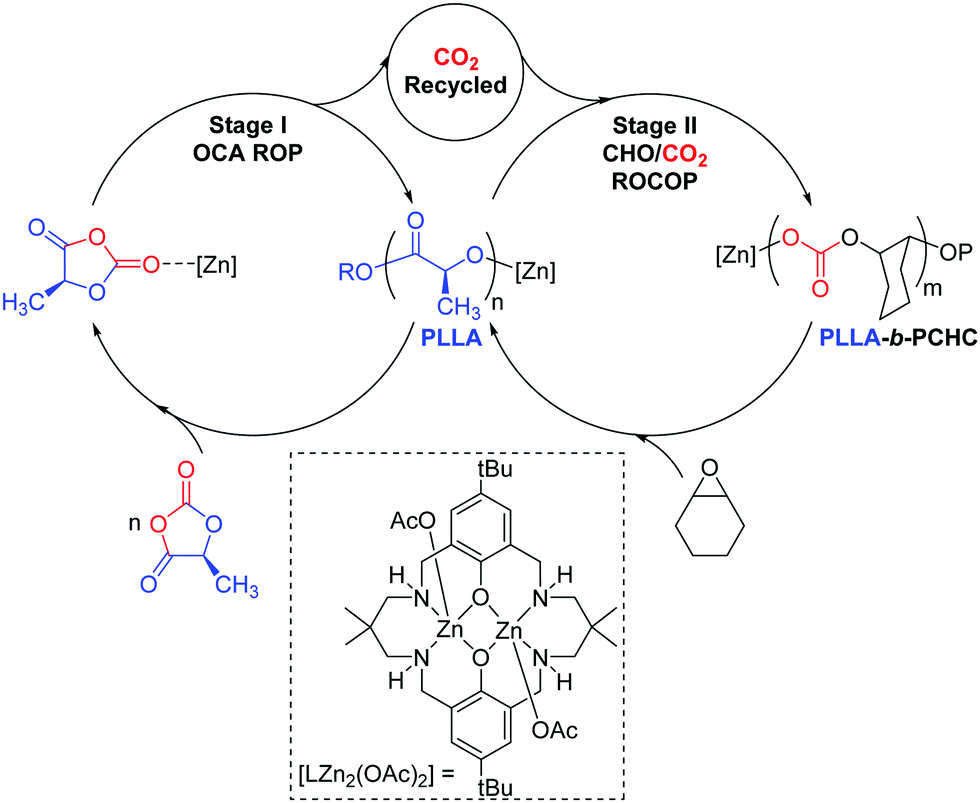 | ||
| Scheme 1 Proposed polymerisation pathway whereby the di-zinc catalyst bridges two catalytic cycles: LLAOCA ROP and CO2/CHO ROCOP. Where [Zn] represents [LZn2(OAc)2] during catalysis; R = initiating group and P = growing polymer chain. | ||
In pioneering earlier work, Thomas and co-workers demonstrated that N-carboxyanhydride ROP could be used to produce polypeptides and the released CO2 was subsequently used to make cyclic carbonates via tandem catalysis.24 Inspired by this intriguing recycling opportunity and with polymerisation catalysts in hand known to operate under low CO2 pressures,22,23 we envisaged a single catalyst could be used in a switchable process to deliver block polymer product (Scheme 1).
Firstly, [LZn2(OAc)2] was assessed for the ROP of L-lactide-O-carboxyanhydride (OCA) in THF, as a model solvent for subsequent processes in neat epoxide (Table 1). The OCA ROP was successful resulting in the formation of PLLA polymer over 16 h (Fig. S1 and S2, ESI†). The 1H NMR spectrum, of the crude product, showed ∼97% conversion to PLLA (Fig. S1 and S2, ESI†). The isolated PLLA was analysed by 1H{1H} and 13C{1H} NMR spectroscopy, the former showed a single resonance at 5.16 ppm corresponding to PLLA and confirming there was no significant epimerisation (Fig. S3 and S4, ESI†). The isolated polymer showed a molar mass of 4300 g mol−1 and narrow dispersity (Đ = 1.15) (Fig. S5, ESI†). It should be noted that the experimental molar mass values for the PLA samples are reproducibly lower than the values predicted on the basis of the reaction conversion (see Table S1, ESI†). Having established the success of the di-zinc catalyst for OCA ROP, its potential to recycle the liberated CO2 was tested using an equimolar mixture of THF and CHO (Table 1).
| Entry | Time (h) | LLAOCAb conv. (%) | PCHCc (%) | Polymer | M n,exp (g mol−1) | Đ |
|---|---|---|---|---|---|---|
| a All the polymerisations were conducted in a 15 mL Schlenk tube in THF, THF–CHO mixture or in neat CHO, [LLAOCA] = 1.25 M, [LZn2(OAc)2] = 0.2 mol%, at 80 °C. b Determined by 1H NMR spectroscopy from the normalized integrals for resonances from LLAOCA (1.76 ppm) and PLLA (1.56 ppm). c Determined by 1H NMR spectroscopy of the crude polymer from the normalized integrals for resonance from PLLA (1.56 ppm) and PCHC (4.92–4.18 ppm). d Determined by SEC in THF, calibrated with narrow polydisperse polystyrene standards. e Polymerisation conducted in THF, [LZn2(OAc)2]/[LLAOCA] = 500, [LLAOCA] = 1.25 M. f Polymerisation conducted in THF–CHO mixture with [LZn2(OAc)2]/[LLAOCA]/[CHO] = 1/500/500, [LLAOCA] = 1.25 M in THF–CHO mixture. g Molar mass values are corrected by multiplying by 0.58.25 | ||||||
| 1e | 16 | 97 | — | PLLA | 4300g | 1.15 |
| 2f | 16 | >99 | 0 | PLLA | 4100g | 1.98 |
| 3 | 0.16 | 23 | 0 | PLLA | 2800 | 1.66 |
| 4 | 1 | >99 | 8 | PLLA-b-PCHC | 4600 | 1.33 |
| 5 | 8 | >99 | 17 | PLLA-b-PCHC | 7600 | 1.30 |
| 6 | 16 | >99 | 53 | PLLA-b-PCHC | 8400 | 1.24 |
| 7 | 24 | >99 | 68 | PLLA-b-PCHC | 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
1.54 |
| 8 | 48 | >99 | 91 | PLLA-b-PCHC | 13500 |
1.45 |
A reaction at relative molar loadings of 1:500:500 ([Zn]:CHO:LLAOCA) was stopped after 16 h and showed complete OCA consumption. Both the LLAOCA conversion and the molar mass of the PLLA were similar to the reaction conducted in neat THF, confirming that the epoxide did not interfere with the OCA ring-opening polymerisation. Nonetheless, there was no evidence for any polycarbonate formation and we reasoned this was likely due to the low overall epoxide concentration. For this di-zinc catalyst, the epoxide/CO2 rate law is first-order in epoxide concentration and thus rates should increase at higher epoxide concentrations.22,23 All subsequent experiments were conducted using the same catalyst:monomer loadings but in neat epoxide. In order to systematically study the reaction, a series of experiments were conducted at different reaction times (Table 1). Initially, the reaction proceeds only with OCA ROP as is clear from the reaction stopped after ∼10 min which showed 23% conversion to PLLA but no resonances for PCHC or any other by-product (e.g. cyclic carbonate). After an hour, the complete OCA conversion to PLLA had occurred and there was ∼8% conversion to PCHC as apparent from a growing resonance at 4.65 ppm in the 1H NMR spectrum. Reactions were quenched from 1–48 h and showed progressively increased conversion to PCHC, with the final reaction delivering ∼91% CO2 conversion (Table 1). The reaction analysis indicated that there was a high catalytic selectivity, with OCA ROP occurring prior to CO2/epoxide ROCOP.
To establish that block polymers rather than mixtures of polymers formed, the polymer products were analysed by GPC. The reactions showed progressively increasing molar mass with conversion to PCHC and of the final polymer sample showed a molar mass 13500 g mol−1 with the corresponding highest CO2 incorporation. The 1H NMR spectrum shows resonances at 5.10–4.95 ppm and 4.93–4.76 ppm which correspond to junction units between the ester and carbonate blocks (Fig. S6, ESI†). Low intensity resonances at 74.67 ppm and 71.81 ppm, in 13C{1H} NMR spectrum of isolated PLLA-b-PCHC, are also attributed to methine junctions (Fig. S7, ESI†). The block polymer structure was further indicated by HMBC NMR spectroscopy which confirmed junction unit assignments and was consistent with previous reports of PLLA-b-PCHC copolymer prepared by sequential addition routes (Fig. S8, ESI†).9,10 The DOSY NMR spectrum showed a single diffusion coefficient for all resonances, as expected for a block polymer, whereas a physical mixture of the two constituent polymers (PLA + PCHC) shows two different diffusion coefficients (Fig. S9 and S10, ESI†). Overall the analyses indicate sequential catalysis to deliver a block polymer.
To improve understanding and allow for preliminary kinetic analysis, reactions were repeated using both in situ1H NMR and in situ ATR-IR spectroscopies. For the 1H NMR experiments, known concentrations of monomers and catalyst, with mesitylene as an internal standard, were reacted in a J Young NMR tube, at 80 °C. 1H NMR spectra were acquired at regular intervals over 53 h. Monomer conversions were determined by analysing the normalized integrals of selected signals (Fig. S11 and S12, ESI†). The monomer conversion vs. time plot shows clearly that OCA ROP occurs fastest and is complete in around 3.5 h (Fig. 1). At this stage of the reaction there is no evidence for any formation of PCHC or cyclic carbonate. Furthermore, the linear fit to the experimental data indicates a zero order in OCA concentration (kobs = 1.08 × 10−4 s−1) (Fig. S11, ESI†). After the OCA has been completely consumed, there is a steady increase in the concentration of polycarbonate and low quantities (<3%) of trans-cyclohexene carbonate (CHC) by-product. The formation of trans-CHC occurs by back-biting of the growing polymer chain,26,27 and, in this case, becomes more prominent at the later reaction stages. This increasing by-product formation is attributed to progressive depletion of CO2 resulting in the catalytic cycle resting state changing to the metal-alkoxide intermediate (which is the precursor to trans-CHC) (Fig. S12, ESI†). It should be emphasised that overall the reaction is highly selective, yielding up to 91% conversion of the CO2 into the block polymer and only 9% into the cyclic carbonate.
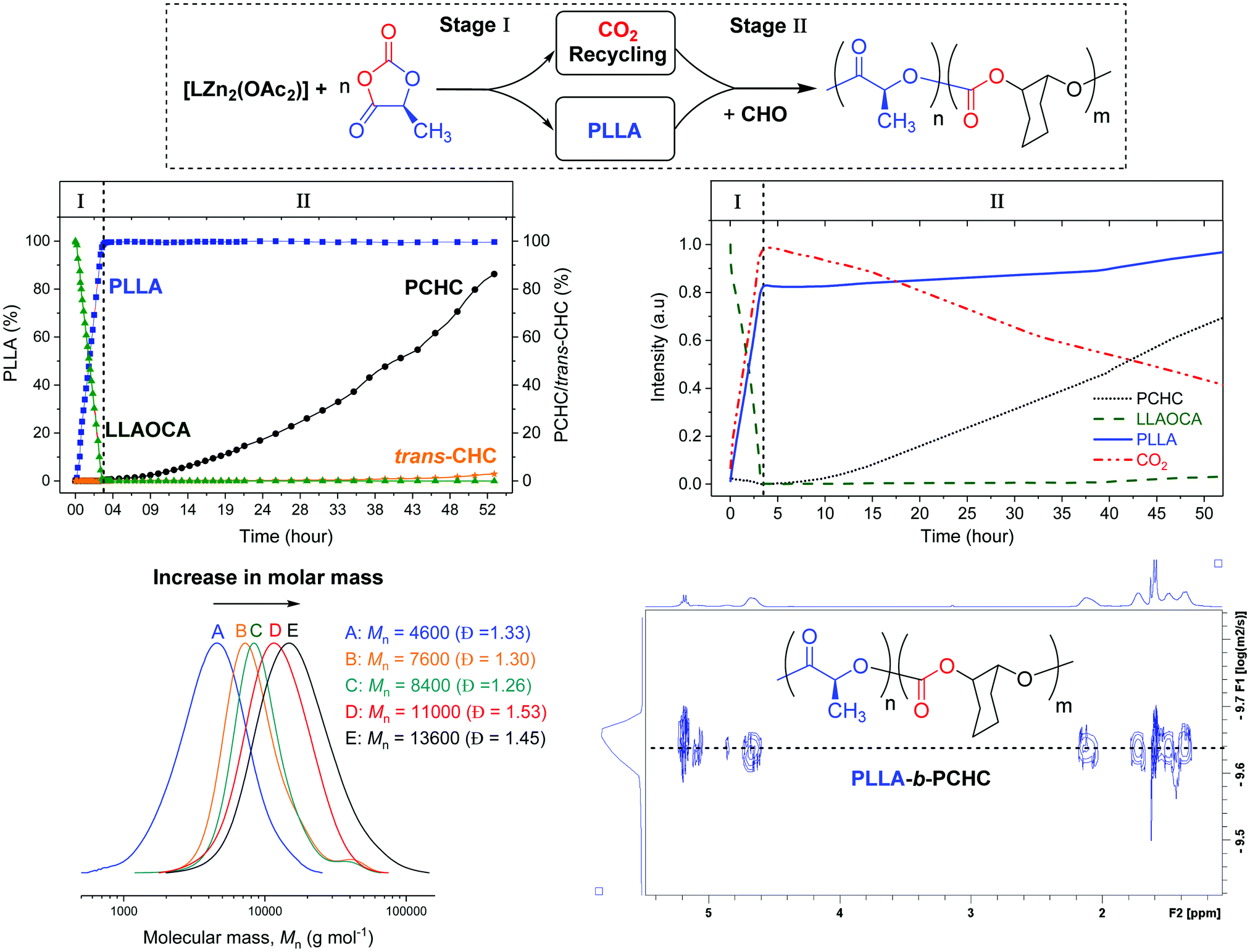 | ||
| Fig. 1 The conversion vs. time data, determined from in situ1H NMR spectroscopy, during formation of PLLA-b-PCHC (top left). The changes in absorption intensity vs. time for selected resonances, from in situ IR spectroscopy (top right). Overlaid GPC analyses for isolated PLLA-b-PCHC samples (bottom left). The DOSY NMR spectrum of PLLA-b-PCHC (bottom right). Note: the structure of trans-CHC is illustrated in Fig. S23 (ESI†). | ||
One limitation of NMR spectroscopy for monitoring polymerisation rates could be that the system is not stirred and this raises concerns that the block selectivity might arise from CO2 diffusion limitations. To test the relevance of this factor, the reaction was also monitored using in situ ATR-IR spectroscopy using a Schlenk tube equipped with an ATR-IR probe and with magnetic stirring. Changes in intensity of characteristic IR absorptions were monitored including for CO2 (2340 cm−1), LLAOCA (1184 cm−1), PLLA (1191 cm−1) and PCHC (1237 cm−1) (Fig. 2 and ESI† for details). Initially, the LLAOCA resonance showed a decrease in intensity and there were concomitant increases in the resonances assigned to PLLA and CO2. Over this time period, there was no intensity for the resonance attributed to PCHC, substantiating the selectivity observed by NMR spectroscopy. Only after the complete consumption of the LLAOCA resonance were signals observed for PCHC. Both findings are fully consistent with the in situ1H NMR studies and suggest that the selectivity is independent of CO2 diffusion since rates are broadly the same with or without stirring.
In terms of reaction pathway, the process is proposed to operate via two distinct catalytic cycles which are bridged by a common zinc-alkoxide intermediate (Scheme 1). MALDI-ToF mass spectrometry analysis of the PLLA homopolymer shows chains that are end-capped with a cyclohexyl acetate end group (Fig. S13 and S14, ESI†). This finding is consistent with initiation occurring via ring-opening of a cyclohexene oxide molecule to produce the key zinc-alkoxide intermediate. This intermediate can propagate LLAOCA ring opening polymerisation. Polymerisation of the LLAOCA is proposed to proceed by elimination of a CO2 molecule and re-generation of a zinc alkoxide with each monomer insertion (Fig. S15 and S16, ESI†). At this stage it is unclear precisely why the high selectivity for block polymer formation arises. It could be that a minimum pressure (concentration) of CO2 is required to allow the ROCOP process to occur, a notion which is consistent with previous studies that at pressures below 1 bar CO2 the ROCOP reaction shuts down.28,29 On the other hand, it could be that the L-LA zinc-alkoxide intermediate is stabilised towards CO2 insertion by formation of a five-membered chelate, a finding in line with other ROP catalysts.30 It is clear that the CHO/CO2 ROCOP does not occur until after the complete consumption of the LLAOCA. It is also apparent that nearly all the released CO2 can be recycled to yield the PLLA-b-PCHC block polymer.
Differential Scanning Calorimetry (DSC) was used to determine the thermal properties of PLLA-b-PCHC polymers (Table S3 and Fig. S17, ESI†). All materials showed higher glass transition temperatures (Tg) (61–78 °C) than PLLA and values which correlate with the wt% of PCHC in the polymer. Switchable catalysis using vinyl-CHO and LLAOCA yielded a related block polymer: PLA-b-PVCHC as did the reaction between L-phenyl lactic acid O-carboxyanhydride (LPheLAOCA) and CHO (Table S2 and Fig. S18–S30, ESI†). These polymerisations indicate that the selective catalysis may be more generally applicable and could deliver other block polyester-carbonates in future.
In summary, a new catalytic process starting from mixtures of OCA and epoxide selectively produced poly(L-lactide-b-cyclohexene carbonate). The CO2 liberated in one polymerisation reaction (OCA ROP) is efficiently recycled into the second polycarbonate block. The catalysis is highly selective and permits near quantitative consumption of carbon dioxide and yields block polymer with a maximum atom economy of 91%. Although at an early stage, the process appears promising both from the point of view of CO2 utilisation and as means to modify properties of oxygenated polymers. Further research to investigate the detailed polymerisation mechanism, to expand the range of catalysts and to apply it to other monomers is recommended.
The EPSRC and UK Catalysis Hub are acknowledged for financial support (EP/K014668/1).
Conflicts of interest
CKW is the Director of Econic Technologies.References
- X. Y. Zhang, M. Fevre, G. O. Jones and R. M. Waymouth, Chem. Rev., 2018, 118, 839–885 CrossRef CAS.
- S. J. Poland and D. J. Darensbourg, Green Chem., 2017, 19, 4990–5011 RSC.
- Y. Q. Zhu, C. Romain and C. K. Williams, Nature, 2016, 540, 354–362 CrossRef CAS PubMed.
- M. A. Hillmyer and W. B. Tolman, Acc. Chem. Res., 2014, 47, 2390–2396 CrossRef CAS PubMed.
- M. Rabnawaz, I. Wyman, R. Auras and S. Cheng, Green Chem., 2017, 19, 4737–4753 RSC.
- S. Paul, Y. Zhu, C. Romain, R. Brooks, P. K. Saini and C. K. Williams, Chem. Commun., 2015, 51, 6459–6479 RSC.
- G. Trott, P. K. Saini and C. K. Williams, Philos. Trans. R. Soc., A, 2016, 374, 20150085 CrossRef PubMed.
- H. T. H. Nguyen, P. X. Qi, M. Rostagno, A. Feteha and S. A. Miller, J. Mater. Chem. A, 2018, 6, 9298–9331 RSC.
- M. R. Kember, J. Copley, A. Buchard and C. K. Williams, Polym. Chem., 2012, 3, 1196–1201 RSC.
- A. K. Diallo, W. Guerin, M. Slawinski, J.-M. Brusson, J.-F. Carpentier and S. M. Guillaume, Macromolecules, 2015, 48, 3247–3256 CrossRef CAS.
- G.-P. Wu, D. J. Darensbourg and X.-B. Lu, J. Am. Chem. Soc., 2012, 134, 17739–17745 CrossRef CAS PubMed.
- D. J. Darensbourg and G.-P. Wu, Angew. Chem., Int. Ed., 2013, 52, 10602–10606 CrossRef CAS PubMed.
- Y.-Y. Zhang, G.-W. Yang, Y. Wang, X.-Y. Lu, G.-P. Wu, Z.-S. Zhang, K. Wang, R.-Y. Zhang, P. F. Nealey, D. J. Darensbourg and Z.-K. Xu, Macromolecules, 2018, 51, 791–800 CrossRef CAS.
- C. Y. Hu, R. L. Duan, S. C. Yang, X. Pang and X. S. Chen, Macromolecules, 2018, 51, 4699–4704 CrossRef CAS.
- B. M. Vaca and D. Bourissou, ACS Macro Lett., 2015, 4, 792–798 CrossRef.
- A. Buchard, D. R. Carbery, M. G. Davidson, P. K. Ivanova, B. J. Jeffery, G. I. Kociok-Köhn and J. P. Lowe, Angew. Chem., Int. Ed., 2014, 53, 13858–13861 CrossRef CAS PubMed.
- Y. Y. Wang and D. J. Darensbourg, Coord. Chem. Rev., 2018, 372, 85–100 CrossRef CAS.
- M. R. Kember, P. D. Knight, P. T. R. Reung and C. K. Williams, Angew. Chem., Int. Ed., 2009, 121, 949–951 CrossRef.
- C. Romain and C. K. Williams, Angew. Chem., Int. Ed., 2014, 126, 1633–1636 CrossRef.
- A. C. Deacy, C. B. Durr, J. A. Garden, A. J. P. White and C. K. Williams, Inorg. Chem., 2018, 57, 15575–15583 CrossRef CAS.
- G. Trott, J. A. Garden and C. K. Williams, Chem. Sci., 2019, 10, 4618–4627 RSC.
- A. Buchard, F. Jutz, M. R. Kember, A. J. P. White, H. S. Rzepa and C. K. Williams, Macromolecules, 2012, 45, 6781–6795 CrossRef CAS.
- F. Jutz, A. Buchard, M. R. Kember, S. B. Fredriksen and C. K. Williams, J. Am. Chem. Soc., 2011, 133, 17395–17405 CrossRef CAS PubMed.
- S. K. Raman, E. Brule, M. J. L. Tschan and C. M. Thomas, Chem. Commun., 2014, 50, 13773–13776 RSC.
- J. Baran, A. Duda, A. Kowalski, R. Szymanski and S. Penczek, Macromol. Rapid Commun., 1997, 18, 325–333 CrossRef CAS.
- D. J. Darensbourg, J. C. Yarbrough, C. Ortiz and C. C. Fang, J. Am. Chem. Soc., 2003, 125, 7586–7591 CrossRef CAS PubMed.
- D. J. Darensbourg and A. D. Yeung, Polym. Chem., 2015, 6, 1103–1117 RSC.
- The size of the reaction vessel, and in particular, the head-space volume was observed to influence the rate of switching from OCA ROP to CO2/epoxide ROCOP indicating a relationship between CO2 pressure and rate.
- A. M. Chapman, C. Keyworth, M. R. Kember, A. J. J. Lennox and C. K. Williams, ACS Catal., 2015, 5, 1581–1588 CrossRef CAS.
- V. Poirier, T. Roisnel, S. Sinbandhit, M. Bochmann, J.-F. Carpentier and Y. Sarazin, Chem. – Eur. J., 2012, 18, 2998–3013 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Materials, method, experimental procedures and other data. See DOI: 10.1039/c9cc02459j |
| This journal is © The Royal Society of Chemistry 2019 |