
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotoswitchable catalysis based on the isomerisation of double bonds
Ruth
Dorel
 and
Ben L.
Feringa
*
and
Ben L.
Feringa
*
Stratingh Institute for Chemistry, Zernike Institute for Advanced Materials, University of Groningen, Nijenborgh 4, 9747 AG Groningen, The Netherlands. E-mail: b.l.feringa@rug.nl
First published on 3rd May 2019
Abstract
Photoswitchable catalysis is a young but rapidly evolving field that offers great potential for non-invasive dynamic control of both activity and selectivity in catalysis. Within this context, the E/Z photoisomerisation of double bonds in molecular switches and motors is one of the most popular tools to control the catalytic activity essentially due to its reversible nature, the large concomitant geometrical changes, and the high tunability of such photochromic entities. This Feature Article summarises the key developments accomplished over the past years through the incorporation of photoswitchable double bonds into the structure of catalytically competent molecules and shows some perspectives on the remaining challenges and possibilities arising from this, yet still somehow immature, exciting area of research.
Introduction
The field of molecular catalysis has experienced during the past century a striking growth to the point that most industrial processes are nowadays inconceivable in its absence.1 Several generations of scientists have contributed to the design and improvement of a myriad of catalysts, which have enabled the development of new sophisticated chemical transformations that proceed with exquisite levels of efficiency and selectivity.2 Typically, the structural features of a catalyst are iteratively altered in order to achieve the desired outcome for a given chemical transformation, yet the function of such optimised system is often fixed once the reaction conditions are set. As a consequence, neither the regulation of its activity nor its direct transfer to a different chemical context are straightforward.The extraordinary reaction rates and selectivities achieved by enzymes in Nature have often served as a source of inspiration for the design of novel synthetic catalysts.3 However, while the flexibility of the active site of enzymes allows for effective regulation of their activity through allosteric effects and feedback loops,4 man-made catalysts are usually very sensitive to structural modifications as well as alterations of the reaction conditions.
The idea of modulating the activity of a catalyst merely as a response to an external stimulus and without the need of tedious and often expensive synthetic work constitutes an appealing concept and certainly holds great potential to substantially impact the way catalysis is regarded in the future. In order to achieve stimuli-responsive regulation of the catalytic activity of artificial molecular catalysts, it appears crucial to exploit the features of conformational flexibility like enzymes do. To this purpose, artificial molecular machines5 appear to be ideal scaffolds due to their ability to access geometrically very distinct states.6 The remarkable progress experienced in this field over the last two decades has prompted the development of highly creative designs by incorporating responsive units into complex functional molecules with the ultimate aim of obtaining on demand dynamic control over both activity and selectivity in catalysis. Hence, the incorporation of a responsive unit into the structure of a functional catalyst will, under the action of the proper stimulus, lead to an alteration of its intrinsic activity, which may translate into a change in the reaction rate, selectivity, or reaction type. In this context, efforts have been devoted to the development of artificial switchable catalysts responsive to external stimuli such as pH,7 redox processes,8 solvent change,9 or light.10
Light is perhaps the most advantageous stimulus to gate chemical transformations due to its non-invasive nature and its high spatiotemporal resolution. Photoswitchable catalysis relies on the embedment of a photochromic unit into a catalytically competent molecule so that the reversible changes that result from irradiation at the appropriate wavelength translate into a difference in catalytic function. In order to have a photoswitchable catalyst, the photogenerated isomer should revert back to the original state either thermally or by the action of light of a different wavelength. Furthermore, the photoinduced transformation should ideally occur (nearly) quantitatively, i.e. with high photostationary state (PSS) ratios, and repeatedly over time.
In this Feature Article, we present an overview of the recent advances in the growing field of photoswitchable catalysis with a specific focus on the E/Z isomerisation of double bonds11 that are integrated in the structure of the catalyst as the light-triggered event. The discussion has been primarily organised according to the nature of the photoisomerisable double bond. Photoswitchable catalysts based on photochromic units susceptible of photocyclization such as diarylethylenes12 or spiropyranes13 will therefore not be discussed.
Azobenzene-based photoswitchable catalysts
Azobenzenes are among the most studied and well-established photochromic entities.14 Upon irradiation, the N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond undergoes a photoisomerisation from the E to the Z isomer, which reverts back to the thermodynamically more stable E isomer in the dark (Scheme 1). This reverse isomerisation can also be triggered by irradiation with visible light.
N bond undergoes a photoisomerisation from the E to the Z isomer, which reverts back to the thermodynamically more stable E isomer in the dark (Scheme 1). This reverse isomerisation can also be triggered by irradiation with visible light.
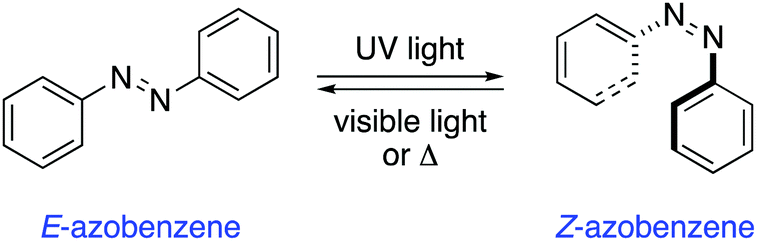 | ||
| Scheme 1 Photoisomerisation of azobenzene. | ||
Photoswitchable bifunctional catalysts
The photoisomerisation of azobenzenes is accompanied by a large geometrical change in the molecule, which in the case of catalytically active species can be exploited for the control of their activity. A pioneer example of a photoswitchable template 1 for cooperative bifunctional catalysis was described by Würthner and Rebek in 1995 utilising two carbazole-based adenine receptors linked by an azobenzene scaffold, which exerted control over the rate of the amide bond formation to give 4 (Scheme 2).15 After recognition of the adenine-containing substrates 2 and 3, the E-form of 1 places them apart from each other, which translates into a low reaction rate for the amide bond formation. On the other hand, Z-1 brings the two reacting partners into close proximity, which facilitates the coupling reaction as inferred from a significant increase in the reaction rate after irradiation with UV light. Thus, the use of the 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 mixture of E-1 and Z-1 that resulted from irradiation with 366 nm light caused a nearly nine-fold enhancement of the coupling rate. Nonetheless, the product 4 remains bound to 1 after amide bond formation, which precludes its turnover and therefore its task as a photoswitchable catalyst.
:50 mixture of E-1 and Z-1 that resulted from irradiation with 366 nm light caused a nearly nine-fold enhancement of the coupling rate. Nonetheless, the product 4 remains bound to 1 after amide bond formation, which precludes its turnover and therefore its task as a photoswitchable catalyst.
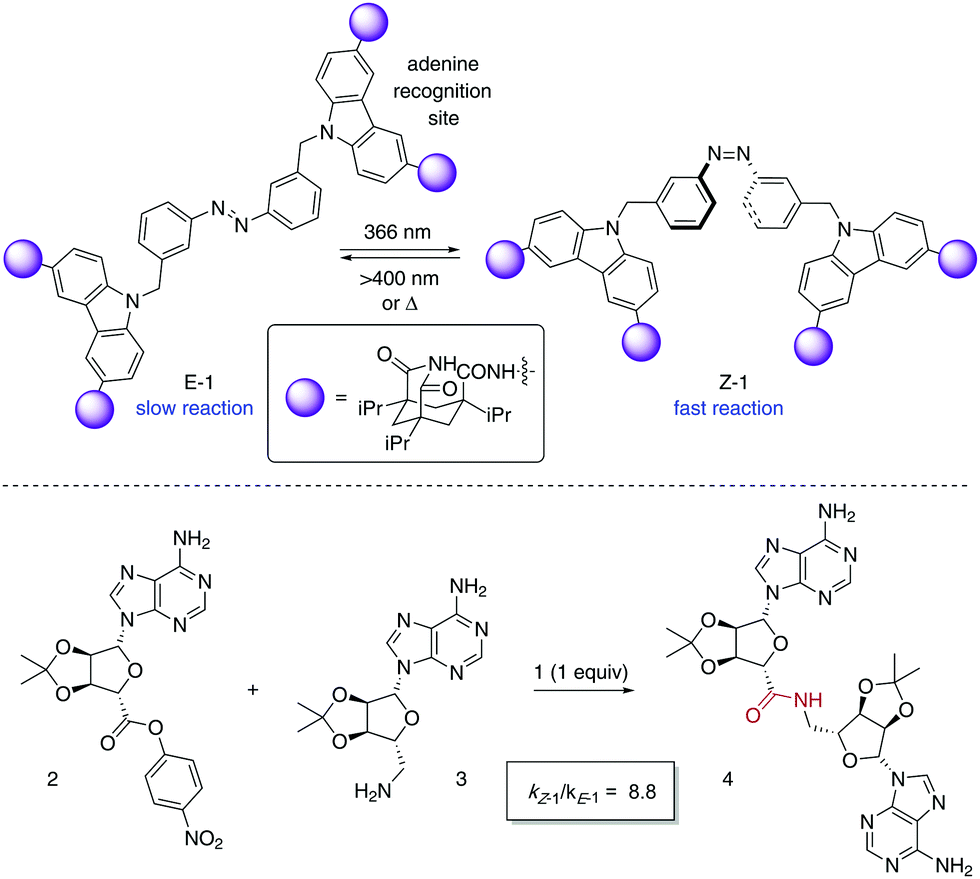 | ||
| Scheme 2 Photocontrol over amide bond formation between 2 and 3. | ||
The first successful example of a photoswitchable bifunctional catalyst was reported in 2003 by Cacciapaglia and co-workers who, inspired by the pioneer work by Shinkai on photoresponsive crown ethers,16 developed phototunable cooperative catalyst 5 for the ethanolysis of tertiary anilides 6 (Scheme 3).17 Dinuclear barium complexes of bis-crown ether hosts are known to catalyse the basic ethanolysis of tertiary acetanilides through a cooperative process in which one of the barium centres binds the carboxylate moiety of the substrate while the other barium centre activates the nucleophilic ethanoate and delivers it to the carbonyl of the acetanilide.18 The bis-barium complex of azobenzene-containing bis(benzo-18-crown-6) 5 exhibits low activity towards the ethanolysis of anilides 6 when in its E-form. In contrast, photoswitching of the azobenzene moiety to its Z-form (PSS ratio Z-5:E-5 = 95:5) turns 5 into a more active catalyst due to the proximity between two barium centres. However, the structural similarities between the reaction product and 6 still result in a notable product inhibition due to competitive coordination.
 | ||
| Scheme 3 Photoswitchable catalyst 5 for the ethanolysis of 6. | ||
A photoresponsive bifunctional organocatalyst was later developed based on the cooperative effects of two azobenzene-tethered trityl alcohol functionalities.19 The enhanced acidity of the Z form, which results from an intramolecular hydrogen bond between the two hydroxyl groups, translated into an acceleration of the reaction rate in a phosphine-catalysed Morita–Baylis–Hillman reaction. In a conceptually related approach, the photomodulation of pKa in an azobenzene-tethered biscarboxylic acid 7 was the key to design a photoresponsive glycosidase mimic (Scheme 4).20 The deprotonation of both carboxylic acid groups in E-7 occurs at the same pH, whereas in Z-7, which is obtained by irradiation with 366 nm light (PSS ratio Z-7:E-7 = 70:30), this takes place stepwise. Therefore, Z-7 exists between pH 4.7 and 6.5 as the monoanionic species, which parallels the active form of a glycosidase in terms of functionality. Hence, an enhancement in the reaction rate of six orders of magnitude was observed at pH 5.8 for the hydrolysis of 4-nitrophenyl-β-D-glycopyranoside 8 with respect to that of the background reaction.
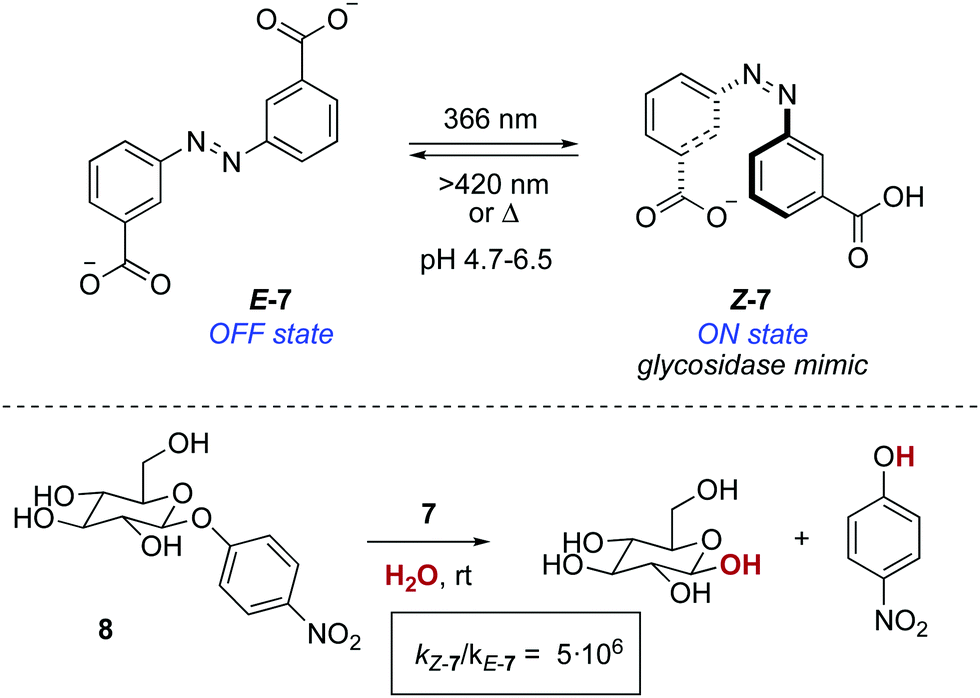 | ||
| Scheme 4 Photoresponsive glycosidase mimic 7. | ||
In a more recent example, the catalytic activity of bimetallic gold(I) complex 9 was modulated by photoisomerisation of the azobenzene linking group (Scheme 5).21 Both isomers of 9 could be isolated and tested independently as catalysts. The reaction rate of the intramolecular hydroamination of 10 turned out to be dependent on the configuration of the azobenzene moiety. Thus, Z-9, which results from the irradiation of E-9 with 320 nm light, exhibits a greater activity and this acceleration is tentatively attributed to a cooperative effect between the two metal centres.
 | ||
| Scheme 5 Photoswitchable bimetallic gold complex 9 and control over the rate of intramolecular hydroamination of 10. | ||
A complementary approach to control the catalytic activity consists of the switchable deactivation of the catalyst by means of intramolecular interactions when two groups are brought into close proximity. In this context, organocatalyst 12, in which a thiourea (hydrogen donor) and a nitro (hydrogen acceptor) groups are linked through an azobenzene bridge, was tested in a thiourea-catalysed nitro-Michael addition (Scheme 6).22 The thiourea reacting site in E-12 is available to efficiently catalyse the addition of acetylacetone to nitrostyrene 13. On the other hand, the nitro group in Z-12, which is obtained by irradiation of E-12 with 365 nm light (PSS ratio Z-12:E-12 = 56:44), is placed close enough to the thiourea unit to interact with it thereby blocking the active site. As a result, the activity upon irradiation is notably diminished, albeit not completely surppressed due to the low PSS ratio. When the nitro group was replaced by a methyl group in 12, the reactivity of E and Z isomers was not significantly different, which proves that the inhibition derives not only from a steric blocking of the active site but rather from a competitive intramolecular hydrogen bonding event. In a similar vein, 12 was later used as a photoswitchable catalyst in the ring-opening polymerization of lactide.23
 | ||
| Scheme 6 Photocontrol over the deactivation of organocatalyst 12. | ||
Photoswitchable catalysis by regulation of steric effects
An alternative strategy to regulate the activity of a catalyst relies on the alteration of the steric bulk around its reaction site. A pioneering example was reported in 1981 for the photoswitchable hydrolysis of p-nitrophenylbenzoate catalysed by β-cyclodextrin 15 to which a 4,4′-bis-(carboxy)azobenzene was attached as a capping unit (Scheme 7).24 The change in geometry of the azobenzene moiety upon irradiation translates into an alteration of the depth of the hydrophobic pocket of the β-cyclodextrin where the substrate needs to bind prior to hydrolysis. Thus, E-15 effectively prevents the binding of the ester substrate in the cavity, which impedes its hydrolysis, whereas the formation of Z-15 upon irradiation with 365 nm light resulted in up to a 5-fold increase in the hydrolysis reaction rate when 38% of the Z isomer was present. It was subsequently demonstrated that the use of a histidine linker for the attachment of the diazobenzene scaffold enabled photocontrol over a related β-cyclodextrin-catalysed hydrolysis, which was effectively switched by irradiation from an inactive state in the E-form to a catalytically active state in the Z-form.25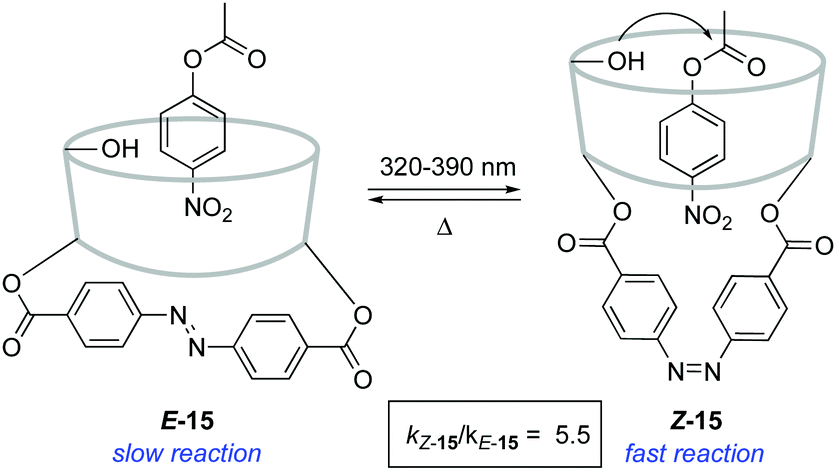 | ||
| Scheme 7 Photocontrol over ester hydrolysis catalysed by azobenzene-capped β-cyclodextrin 15. | ||
The light-gated modulation of the Brønsted basicity of a conformationally restricted N-alkylated piperidine was elegantly realised through the attachment of a bulky azobenzene fragment.26 The basic centre in 16 can be reversibly shielded by photoisomerisation of the azobenzene moiety, which was used to control the reaction rate in a base-catalysed Henry reaction (Scheme 8). The lone pair of the piperidine nitrogen is surrounded by the bulky aromatic fragment in the thermodynamically more stable E-form, which hinders its reactivity. Irradiation with 365 nm light generates Z-16 (PSS ratio Z-16:E-16 > 95:5), in which the active site becomes accessible. Azobenzene 16 proved to be a suitable photoswitchable catalyst for the reaction between p-nitrobenzaldehyde and nitroethane, where the isomerisation from E-16 to Z-16 resulted in a more than 35-fold increase in the rate of formation of 17. In a more recent study, the hydrogen bond formation ability of 16 was monitored in the infrared spectral range using MeOH as the hydrogen donor, which was used as a probe of the accessibility to the active site of the catalyst.27 The force generated by the azobenzene in the isomerisation process is enough to extrude MeOH from the active site within just a few picoseconds, thereby preventing access to the binding site. A related switchable piperidine base was also immobilised on silica gel,28 although its use as a catalyst has not yet been described.
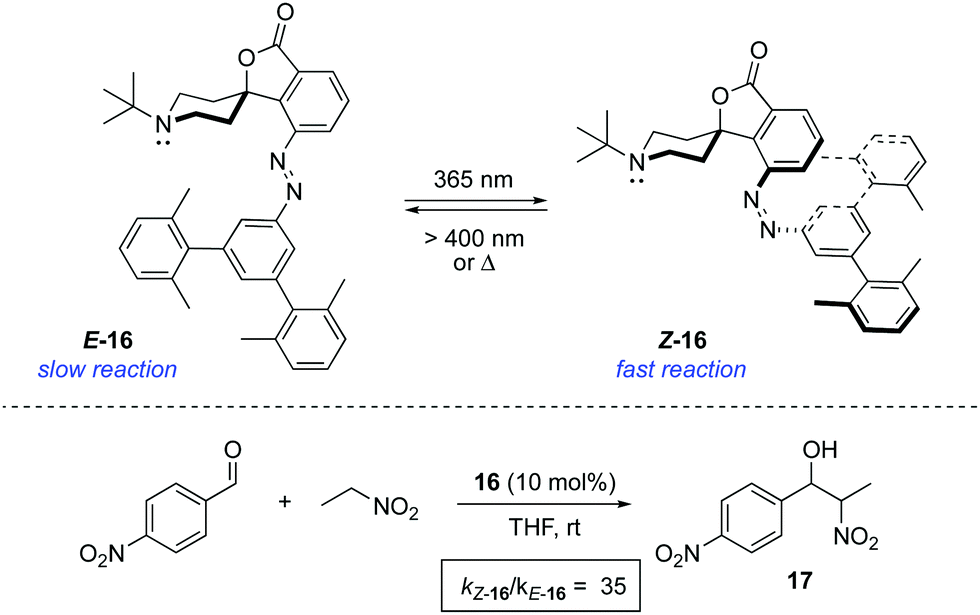 | ||
| Scheme 8 Photoswitchable base 16 and its activity in a Henry reaction. | ||
In a conceptually related study, the immobilization of a proline catalyst onto the surface of a gold nanoparticle through an azobenzene linker has been recently utilised to reversibly photocontrol the rate of the aldol reaction between cyclohexanone and p-nitrobenzaldehyde.29 The photoregulation was based on the active site being buried or exposed to the reaction mixture depending on the configuration of the azobenzene functionality.
Azobenzenes have also been incorporated in phosphine ligands to control the catalytic activity of the corresponding metal complexes. Thus, the differences in geometry derived from the photoinduced isomerisation of an azobenzene-linked Rh(I)-bisphosphine complex were used to gain photocontrol over the rate of hydrogenation of CO2, which is attributed to the change in the bite angle of the bisphosphine ligand.30 In addition, the photocontrolled generation of hydrogen by hydrolytic decomposition of ammonia-borane was achieved using a ruthenium(II) half-sandwich complex bearing and azobenzene-containing phosphine ligand.31
Photoswitchable aggregation state
An alternative approach to control the catalytic activity relies on the regulation of the aggregation/dispersion of catalytically active species. This concept was first realised for the hydrosilylation of p-anisaldehyde with diphenylsilane catalysed by Au nanoparticles 18 decorated with a mixed self-assembled monolayer of dodecylamine and azobenzene-terminated alkane thiols (Scheme 9).32 The nanoparticles remain dispersed in the absence of light (E-18) and aggregate upon UV light irradiation (Z-18). Thus, the exposed surface area gets significantly reduced in the presence of UV light and, as a consequence, the reaction rate for the formation of 19 gets dramatically lower. Irradiation with visible light regenerates the dispersed state and therefore restores the activity of the Au nanoparticles. This modulation of activity could be repeated up to three times.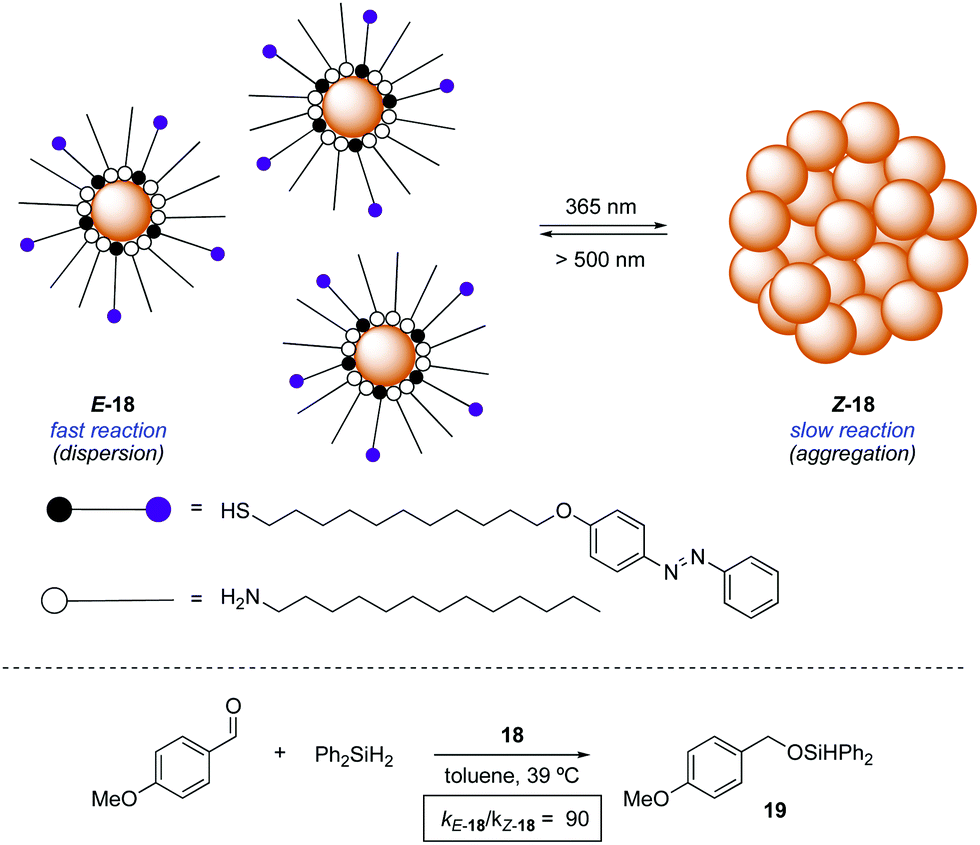 | ||
| Scheme 9 Photoswitchable aggregation of Au nanoparticles 18. | ||
In an different design, the modulation of the activity of a pyridyl-containing bisamide catalyst 20 was achieved through the light-triggered isomerisation of the attached azobenzene functionality, which had a direct influence on its aggregation state (Scheme 10).33 The catalyst in its E-form formed aggregates in a mixture of hexane/propionitrile 4:1, which were dissociated upon irradiation with UV light leading to a homogeneous solution due to the disruption of intermolecular hydrogen bonds (PSS ratio Z-20:E-20 = 96:4). The influence of the aggregation state of 20 was examined in the context of two transformations that are known to be catalysed by a nucleophilic base. The reaction of 1-naphthol with Boc2O barely gave rise to 11% of the protected naphthol 21 in the presence of 15 mol% of E-20 at room temperature after 3 h, whereas under the same reaction conditions using Z-20 the reaction proceeded smoothly to form the product in 69% yield. Likewise, the rearrangement of 2-acyloxybenzofuran 22 afforded 3-acyl-2-benzofuranone 23 in 80% yield in the presence of Z-20, while only 14% of this product was obtained in the presence of aggregated catalyst E-20. Nonetheless, this difference in activity is restricted to reactions carried out in solvents in which the difference in solubility between the Z and E forms is large, otherwise similar results are obtained irrespective of the catalyst state.
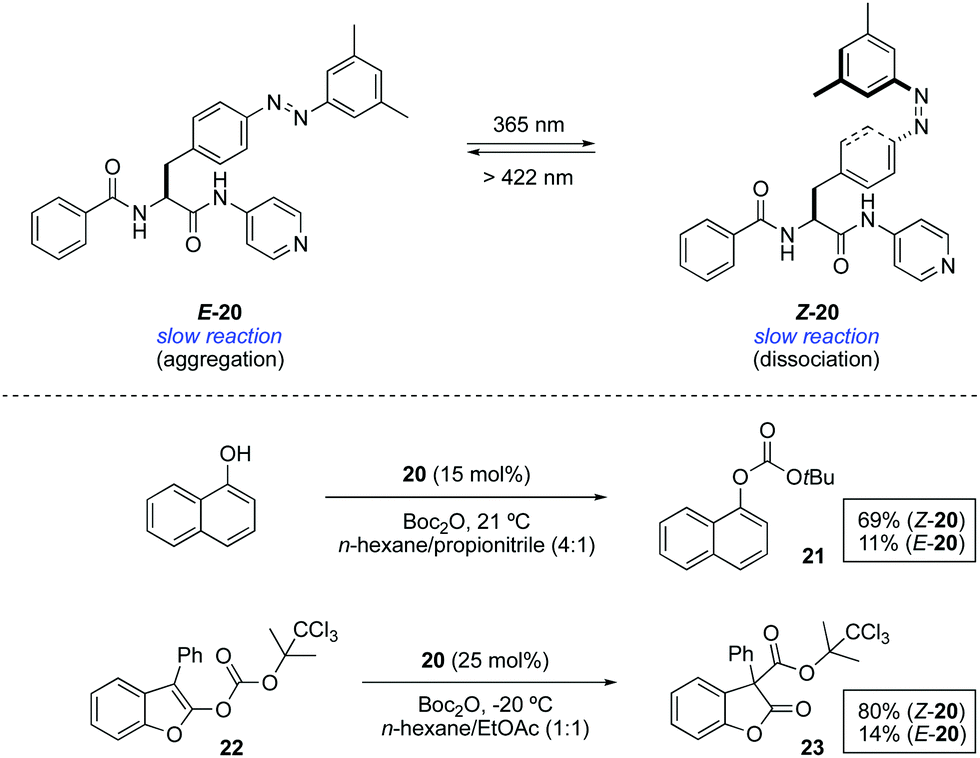 | ||
| Scheme 10 Photoswitchable aggregation of 20. | ||
The catalytic activity of a palladium complex of a water-soluble azobenzene-containing phosphane ligand could also be regulated by means of UV irradiation as a result of the different types of aggregates that are formed in the aqueous phase as a function of the azo-benzene geometry.34 This was applied to regulate the rate of the palladium-catalysed cleavage of allyl-undecyl-carbonate in a heptane/water biphasic reaction medium.
Hydrazone-based photoswitchable catalysts
Despite the growing interest in photoswitchable systems based on the isomerisation of CN bonds experienced over the last decade, both using imines35 and hydrazones36 as the photochromic entities, their applications in photoswitchable catalysis still remain scarce.
In 2017, a pair of enantioselective switchable organocatalysts 24 and 25 were developed based on a pyridyl-acyl hydrazone scaffold as the photoisomerisable moiety (Scheme 11).37 The active site in both catalysts is a bifunctional cinchona alkaloid-squaramide motif, which is able to catalyse Michael additions in an enantioselective fashion. On the other hand, a nitrobenzene hydrogen bond acceptor was linked through the photoswitchable hydrazone. In the case of 24, irradiation with UV light brings the nitro group into close proximity to the squaramide motif, which deactivates the catalyst as a result of intramolecular hydrogen bond interactions. In contrast, a change in the regiochemistry of the pyridine ring in 25 makes the same stimulus trigger the formation of the active species by placing the nitro group far apart from the squaramide centre. The PSS E:Z ratio reached after irradiation of E-24 and E-25 was in both cases 21:79. Nevertheless, in the case of 25 this ratio could be further improved up to 5:95 by treatment with substoichiometric amounts of trifluoroacetic acid, which was on the other hand not effective to switch 24 off. Catalysts E-24 and Z-25 efficiently generate opposite enantiomers in several conjugate additions such as the addition of malonitrile, masked thiol, or 1,3-dicarbonyl compounds to chalcone derivatives and β-nitrostyrene with good conversions and levels of stereoselectivity (up to 90% ee using E-24 and 86% ee with Z-25). Unfortunately, the pair of complementary catalysts cannot operate simultaneously in the same reaction vessel in order to access one or the other enantiomer on demand. It appears that the ON state of one catalyst binds to the OFF state of the other therefore preventing any catalysis.
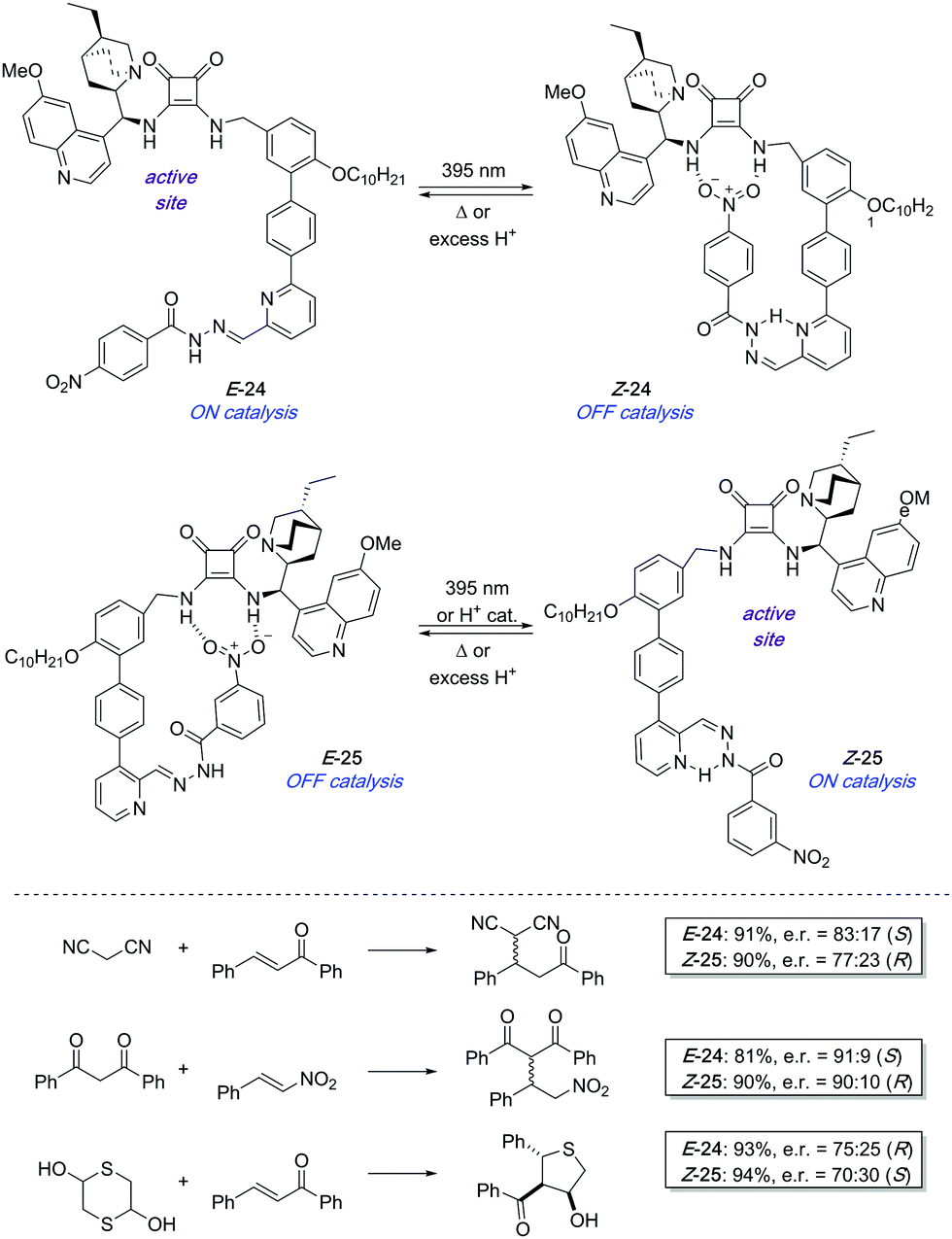 | ||
| Scheme 11 A complementary pair of hydrazone-based photoswitchable organocatalysts 24 and 25. | ||
Stilbene-based photoswitchable catalysts
Stilbenes, which undergo E–Z isomerisation around the CC bond upon irradiation, are a well-established and historically relevant class of photoswitches.38 Unlike Z-azobenzenes, Z-stilbenes can undergo irreversible oxidative electrocyclization to generate phenanthrene-based systems, which is one of the key factors to be taken into account in the design of functional light-driven stilbene-based switchable systems.
Activity control
A seminal study in the area was reported in 1999 by Inoue and co-workers, who achieved the light-controlled fixation of CO2 mediated by aluminium porphyrin 26 (Scheme 12).39 The activity of this system is dependent on the coordination of a bulky 2-stilbazole to the aluminium centre, which can only occur in its Z configuration. Once the pyridine moiety is coordinated to the aluminium centre, the resulting complex accelerates the formation of propylene carbonate from propylene oxide and CO2.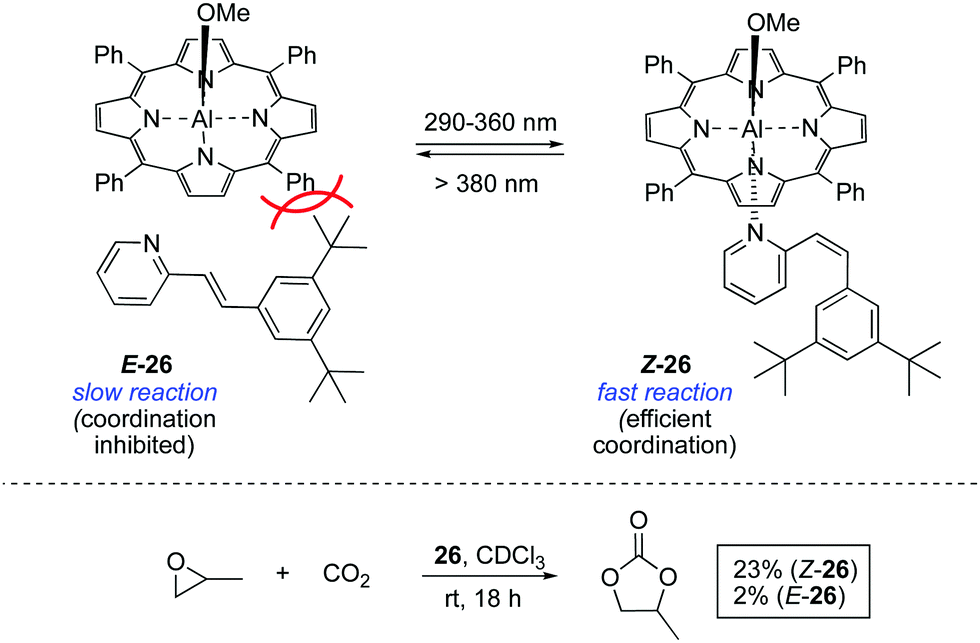 | ||
| Scheme 12 A photoswitchable coordination of stilbazole to a porphyrin to form catalyst 26. | ||
The bis-barium complex of a stibene-tethered bis-crown ether analogue to 5 was also developed and its activity triggered by irradiation of the E isomer with UV light.40 However, once activated, it was not possible to revert the complex to its E configuration and therefore this system did not fulfil the reversibility requirement for a photoswitchable catalyst.
Bifunctional molecular photoswitches based on the overcrowded alkene scaffold found in the so-called second generation molecular motors41 have been recently investigated as light-responsive catalysts.42 Photoresponsive thioureas 27 were synthesised in both E and Z forms, which showed efficient photoisomerisation upon irradiation with 312 nm light (Scheme 13). However, the photogenerated metastable states (M)-Z-27 and (M)-E-27 did not show reversible photoisomerisation upon irradiation with 365 nm light as happens in the unfunctionalised counterparts.38 Furthermore, they both turned out to be ineffective catalysts for the Morita–Baylis–Hillman reaction between 2-cyclohexen-1-one and 3-phenylpropionaldehyde regardless the configuration of the double bond, presumably due to the limited catalytic activity of the dimethylaniline moiety. Nonetheless, it was found that the catalytically active thiourea group was deactivated upon irradiation. In order to examine this phenomenon in detail, the addition of acetylacetone to 13 was chosen as a model reaction. In the absence of any catalyst, only 10% conversion was obtained after 18 h. In the presence of (P)-E-27, an increase in the reaction rate was observed, achieving 60% conversion under otherwise identical conditions. In contrast, when metastable form (M)-Z-27, obtained by irradiation with 312 nm light (PSS ratio (M)-Z-27:(P)-E-27 = 80:20), was used, the rate of the reaction was significantly decreased to afford 14 in only 19% after 18 h. Contrary to the expectations based on previously described azobenzene-based photoswitchable thiourea catalysts,22 the use of (P)-Z-27 under the same reaction conditions led to the formation of 14 in 40% yield. Thus, the lower activity of (M)-Z-27 compared to (P)-E-27 cannot be simply attributed to a deactivation by intramolecular hydrogen bonding between the thiourea and the dimethylamino groups as in the case of 12, since this would also be applicable to some extent in the case of (P)-Z-27. Irradiation with 312 nm light led, as in the previous case, to the formation of a less active species, (M)-E-27, which again points towards a deactivation mechanism different from simple shielding of the reactive site as in the case of 16.26 The rationale for the deactivation of 27 upon irradiation remains to this point unclear and further studies are still necessary in order to apply these systems as ON/OFF photoswitchable catalysts.
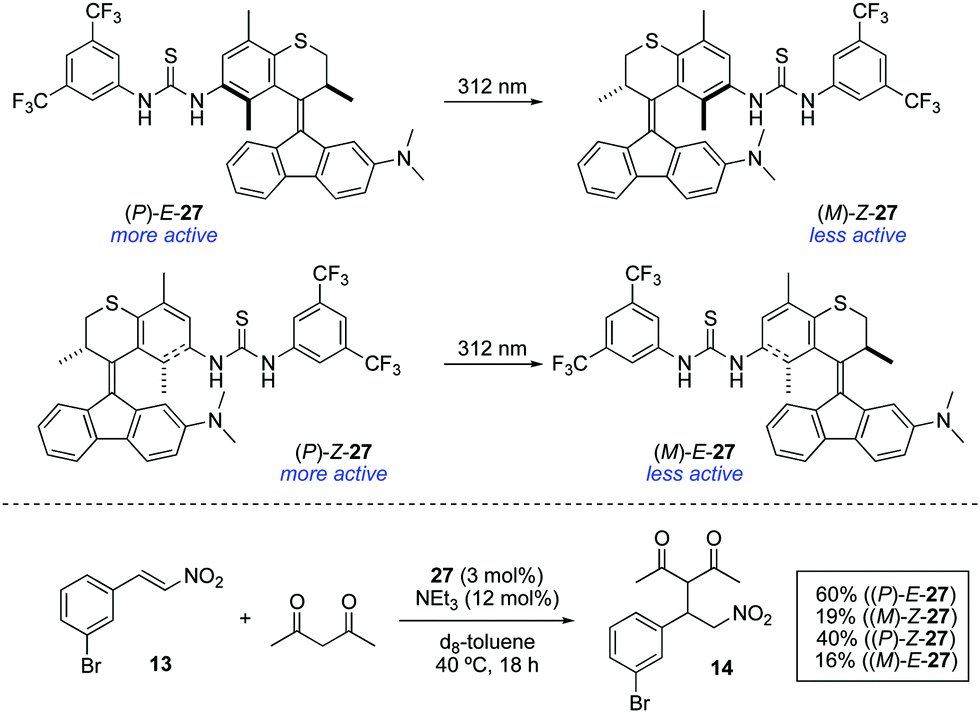 | ||
| Scheme 13 Light-triggered deactivation of thiourea catalysts 27. | ||
Stereoselectivity control
The attachment of a stiff-stilbene photoswitch to the backbone of a chiral biaryl bisphosphine ligand 28 enabled control over the stereochemical outcome of several palladium-catalysed transformations (Scheme 14).43 The irradiation of Z-28 with UV light (365 nm) induced the formation of two different E-28 diastereoisomers, which could be isolated by column chromatography and tested independently as ligands in palladium-catalysed asymmetric Heck reaction and Trost allylic alkylations. The enantioselectivities achieved in the presence of Z-28 were in all cases higher than with E-28, though the same enantiomer was always favoured. | ||
| Scheme 14 Photoresponsive biaryl bis(phosphine) ligand 28. | ||
The first example of stereodivergent photoswitchable catalysis was reported in 2011 by our group utilising a unidirectional first generation light-driven molecular motor44 as the photoresponsive core of the catalyst,45 which allowed for the sequential regulation of both the catalytic activity and the stereochemical outcome of a thia-Michael addition through application of the appropriate stimulus. The designed chiral motor-based organocatalyst 31 contains a thiourea functionality and a N,N-dimethyl-4-aminopyridine (DMAP) Brønsted base,46 which can cooperatively catalyse the conjugate addition of thiols to enones (Scheme 15). Unlike the majority of photoswitches, which are bistable, molecular motors based on overcrowded alkenes such as 31 can exist in four different isomeric forms, and this will dictate the relative orientation of the two catalytic moieties. Irradiation of (P,P)-E-31 (312 nm) induces the E–Z isomerisation to generate quantitatively the metastable (M,M)-Z-31 isomer, which upon heating at 70 °C can be converted into thermodynamically more stable (P,P)-Z-31. This isomer was purified by preparative HPLC prior to its use as a catalyst. Subsequent irradiation of (P,P)-Z-31 (312 nm) generates transient (M,M)-E-31, which readily isomerises back to (P,P)-E-31 even at low temperatures. Therefore, three isomeric states, namely (P,P)-E-31, (M,M)-Z-31, and (P,P)-Z-31, are in practice sufficiently stable to be employed as chiral catalysts. When the (P,P)-E-31 isomer was used, the thiourea and DMAP groups were far apart, which precluded any cooperative effect between them. As a result, 32 was formed slowly (7% in 15 h) and obtained as a racemate. On the other hand, the catalytic sites are close enough to act cooperatively in both Z configurations of 31, which resulted in high activity in the formation of 32. Moreover, (M,M)-Z-31 and (P,P)-Z-31 promoted the preferential formation of opposite enantiomers due to their opposite helicity. Hence, the coordination of the enone and thiol substrates to (R,R)–(M,M)-Z-31 favoured the addition to the Si-face of the prochiral enone, which resulted in the preferential formation of S-32 (e.r. = 75:25), while R-32 was the major enantiomer in the presence of (R,R)–(P,P)-Z-31 as a result of the addition to the Re-face of the enone (e.r. = 23:77). The choice of one or the other enantiomer of the motor catalyst 31 can also be used to control the sequential order of formation of the enantiomers of 32 ((R,S)-(R)-(S) versus (R,S)-(S)-(R)).
 | ||
| Scheme 15 Control of stereoselectivity in a thia-Michael reaction using molecular motor 31. | ||
A related light-responsive organocatalyst 33 lacking the phenyl spacer between the catalytic sites and the molecular motor core proved to be effective in exerting stereocontrol over the Henry reaction between nitromethane and α,α,α-trifluoromethylketones (Scheme 16).47 As in the case of 31, E-33 exhibited low activity, whereas (M,M)-Z-33 and (P,P)-Z-33 afforded preferentially opposite enantiomers of the products 35 (e.r. up to 86:14 for (R)-35versus 21:79 for (S)-35). It was subsequently demonstrated that it is not required to have the basic functional group attached to the motor.48 Thus, bisurea motor 34 was used for the stereodivergent Henry reaction using substoichiometric amounts of Hünig's base. The corresponding nitroalcohols 35 were obtained in up to 88:12 e.r. (S-35) in the presence of (M,M)-Z-34 and 80:20 (R-35) when (P,P)-Z-34 was used as the catalyst.
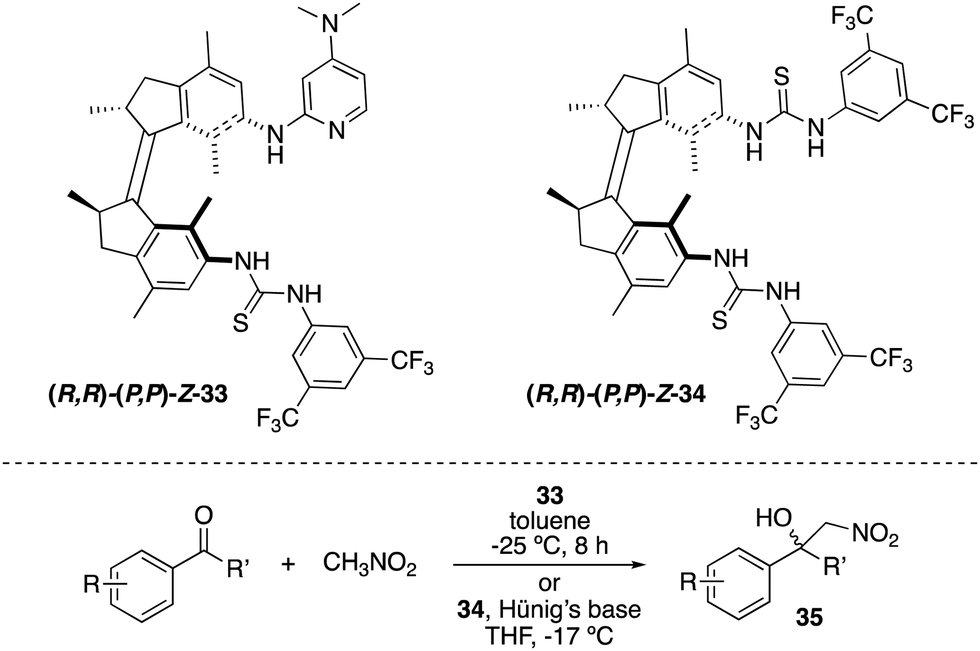 | ||
| Scheme 16 Light- and heat-responsive thiourea catalysts 33 and 34 for the stereocontrol over the Henry reaction. | ||
Not only switchable organocatalysts but also ligands for metal catalysis can be prepared based on first generation molecular motors. A bisphosphine ligand 36, reminiscent of Trost ligands,49 was obtained by linkage of the diphenylphosphine moieties to the motor core via an amide tether (Scheme 17).50 This ligand, which exhibited a rotational behaviour similar to that of 31, proved to be effective in the palladium catalysed intramolecular desymmetrisation of meso-biscarbamate 37. Thus, (3S,4R)-38 was obtained in the presence of (P,P)-Z-36 (e.r. = 94:6), whereas the use of (M,M)-Z-36 provided preferentially the opposite enantiomer (3R,4S)-38 (e.r. = 93:7) under identical reaction conditions.
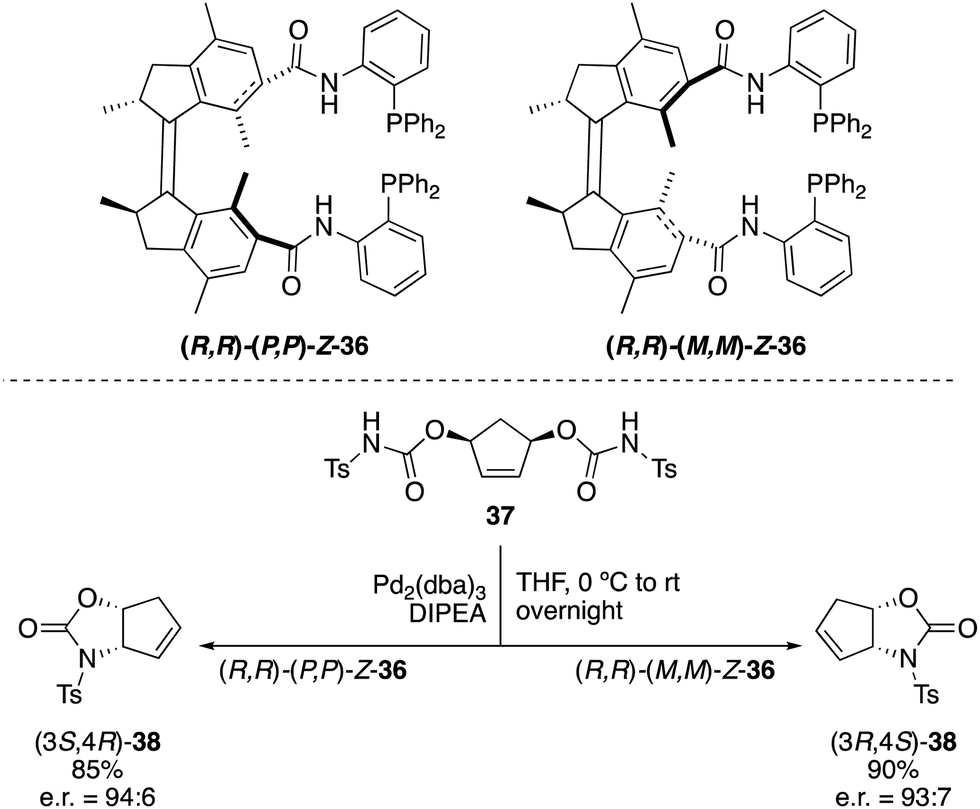 | ||
| Scheme 17 Palladium-catalysed desymmetrisation using molecular motor 36 as the ligand. | ||
Optically switchable dibenzosuberane-based helicene 39, which contains a 4-N-methylaminopyridine subunit, was later established as an efficient enantiodivergent catalyst for the Steglich rearrangement of O-carboxylazlactones 40 (Scheme 18).51 The two pseudoenantiomers (P)-39 and (M)-39 could be repeatedly interconverted by irradiation at two different wavelengths. Hence, irradiation with 290 nm converted (P)-39 into (M)-39 (PSS ratio (M)-39:(P)-39 > 99:1), while the latter could be reverted to (P)-39 with 340 nm light (PSS ratio (M)-39:(P)-39 = 9:91). The rearranged products 41 were obtained in up to 91% (R) and 94% (S) ee using (P)-39 and (M)-39, respectively.
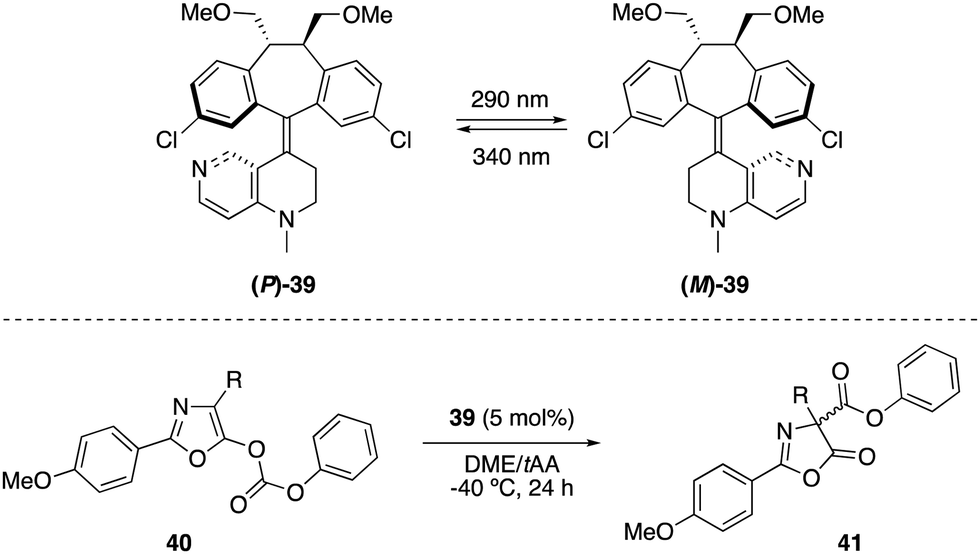 | ||
| Scheme 18 Enantiodivergent Steglich rearrangement using photoswitchable catalyst 39. | ||
Recently, a photoresponsive bis(2-phenol)-substituted photoswitch 42 was built based on the core of second generation molecular motors (Scheme 19).52 This design enables a dynamic central-helical-axial transfer of chirality in which the preferential chirality of the biaryl motif is coupled to the reversible photoswitching of the helicity of the motor core upon irradiation, which is dictated by the configuration of the stereogenic centre adjacent to the overcrowded alkene axis. Thus, (R,P,Sa)-42 and (R,M,Ra)-42 can be interconverted by irradiation with 365 nm (PSS ratio = 17:83) and 420 nm light (PSS ratio = 50:50), respectively, which was used to control the stereochemical outcome of the addition of diethylzinc to aromatic aldehydes. The enantioselectivity of this process was successfully reversed upon irradiation, giving rise preferentially to R-43 in the presence of (R,P,Sa)-42 (up to 68% ee) and S-43 after irradiation with 365 nm light (up to 55% ee).
 | ||
| Scheme 19 Central-helical-axial transfer of chirality in 42 and its application in stereodivergent addition of ZnEt2 to aromatic aldehydes. | ||
Conclusions and outlook
The emergent field of photoswitchable catalysis has experienced over the past 20 years a remarkable progress through the advent of light-responsive switches and molecular machines with increased levels of sophistication. The judicious choice of a photochromic unit and its precise incorporation into the structure of a catalyst offers unparalleled opportunities to control in a non-invasive way its activity and/or selectivity with exquisite spatiotemporal precision. However, there are still several unresolved challenges that need to be addressed in order to turn the field from a proof of concept into a practical tool in synthesis.A number of photoswitchable homogeneous catalysts have been successfully developed by implementing azobenzene, hydrazone, or stilbene motifs into the catalyst structure. Furthermore, the immobilization of photoswitchable scaffolds onto the surface of catalytically active nanoparticles has also been realised. The majority of the reported examples target the ON/OFF control of the activity of the catalyst through a light-triggered geometrical change in its structure. More recently, the use of chiral stilbene-based overcrowded alkenes has enabled the development of photoswitchable stereodivergent catalysts able to exert dual stereocontrol on chemical transformations, which is particularly desirable from the perspective of pharmaceutical industry. This concept would allow for the on-demand preparation of the stereoisomer of interest starting from a single isomer of the catalyst, which could lead to more versatile and efficient processes. Nonetheless, these examples still remain scarce and further investigations will be required in order to demonstrate their generality. In general, most of the examples reported to date do not comprehensively analyse how the differences in catalytic activity are affected over several photoswitching cycles. Furthermore, the in situ regulation of the catalytic activity, which could be of particular interest to control multitasking catalysts or reactions in flow, often remains a challenge despite the high spatiotemporal resolution of light, and preirradiation of the catalyst is frequently required. Fully compatible photoswitchable catalysts that are able to on-demand catalyse two different orthogonal reactions, which has been recently accomplished with a pH-responsive rotaxane-based molecular machine,53 also remain a challenge in the field. In addition, the use of (photo)switchable catalysts for the regulation of tacticity, chain length and/or topology in polymers may lead to new classes of materials with unprecedented properties.54
Despite the field of photoswitchable catalysis is still at an early stage, the outstanding advances herein described set the basis for further investigations towards more robust, multifunctional, and broadly applicable light-responsive catalysts. The growth of the field of molecular machines combined with the progress achieved in metal catalysis, organocatalysis, and supramolecular chemistry holds great potential for the development of new generations of (photo)responsive catalysts able to perform complex sequences of tasks or show on command adaptive behaviour in the near future.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the Ministry of Education, Culture and Science (Gravitation Program 024.001.035), the Ramón Areces Foundation (Postdoctoral fellowship to R. D.), and the European Research Council (Advanced Investigator Grant No. 694345 to B. L. F.) are gratefully acknowledged.References
- (a) J. Hagen, Industrial Catalysis: A Practical Approach, Wiley-VCH, Weinheim, 2nd edn, 2006 Search PubMed; (b) H. U. Blaser and E. Schmidt, Asymmetric Catalysis on Industrial Scale, Wiley-VCH, Weinheim, 2004 Search PubMed.
- (a) Catalysis: From Principles to Applications, ed. M. Beller, A. Renken and R. A. van Santen, Wiley-VCH, Weinheim, 2012 Search PubMed; (b) J. F. Hartwig, Organotransition Metal Chemistry: From Bonding to Catalysis, University Science Books, New York, 2010 Search PubMed.
- (a) E. Kuah, S. Toh, J. Yee, Q. Ma and Z. Gao, Chem. – Eur. J., 2016, 22, 8404 CrossRef CAS PubMed; (b) J. Meeuwissen and J. N. H. Reek, Nat. Chem., 2010, 2, 615 CrossRef CAS PubMed; (c) M. D. Nothling, Z. Xiao, A. Bhaskaran, M. T. Blyth, C. W. Bennett, M. L. Coote and L. A. Connal, ACS Catal., 2019, 9, 168 CrossRef CAS; (d) M. T. Reetz, Adv. Catal., 2006, 49, 1 CAS; (e) F. H. Arnold, Angew. Chem., Int. Ed., 2018, 57, 4143 CrossRef CAS PubMed.
- T. W. Traut, Allosteric Regulatory Enzymes, Springer, New York, 2008 Search PubMed.
- (a) Molecular Devices and Machines: Concepts and Perspectives for the Nanoworld, ed. V. Balzani, A. Credi and M. Venturi, Wiley-VCH, Weinheim, 2008 Search PubMed; (b) W. R. Browne and B. L. Feringa, Nat. Nanotechnol., 2006, 1, 25 CrossRef CAS PubMed; (c) Molecular Switches, ed. W. R. Browne and B. L. Feringa, Wiley-VCH, Winheim, 2nd edn, 2011 Search PubMed; (d) The Nature of the Mechanical Bond: Form Molecules to Machines, ed. C. J. Bruns and J. F. Stoddart, Wiley-VCH, 2016 Search PubMed; (e) S. Erbas-Cakmak, D. A. Leigh, C. T. McTernan and A. L. Nussbaumer, Chem. Rev., 2015, 115, 10081 CrossRef CAS PubMed; (f) A. S. Lubbe, T. van Leeuwen, S. J. Wezenberg and B. L. Feringa, Tetrahedron, 2017, 73, 4837 CrossRef CAS; (g) T. van Leeuwen, A. S. Lubbe, P. Štacko, S. J. Wezenberg and B. L. Feringa, Nat. Rev. Chem., 2017, 1, 0096 CrossRef CAS.
- For recent reviews on switchable catalysis, see; (a) V. Blanco, D. A. Leigh and V. Marcos, Chem. Soc. Rev., 2015, 44, 5341 RSC; (b) M. Vlatković, B. S. L. Collins and B. L. Feringa, Chem. – Eur. J., 2016, 22, 17080 CrossRef PubMed; (c) J. Choudhury, Tetrahedron Lett., 2018, 59, 487 CrossRef CAS; (d) J. Choudhury and S. Semwal, Synlett, 2018, 141 CAS; (e) G. Romanazzi, L. Degennaro, P. Mastrorilli and R. Luisi, ACS Catal., 2017, 7, 4100 CrossRef CAS; (f) L. van Dijk, M. J. Tilby, R. Szpera, O. A. Smith, H. A. P. Bunce and S. P. Fletcher, Nat. Rev. Chem., 2018, 2, 0117 CrossRef CAS.
- For selected examples, see: (a) S. L. Balof, S. J. P'Pool, N. J. Berger, E. J. Valente, A. M. Shiller and H.-J. Schanz, Dalton Trans., 2008, 5791 RSC; (b) V. Blanco, A. Carlone, K. D. Hänni, D. A. Leigh and B. Lewandowski, Angew. Chem., Int. Ed., 2012, 51, 5166 CrossRef CAS PubMed; (c) S. Semwal and J. Choudhury, Angew. Chem., Int. Ed., 2017, 56, 5556 CrossRef CAS PubMed.
- For selected examples, see: (a) I. M. Lorkovic, R. R. Duff and M. S. Wrighton, J. Am. Chem. Soc., 1995, 117, 3617 CrossRef CAS; (b) C. S. Slone, C. A. Mirkin, G. P. A. Yap, I. A. Guzei and A. L. Rheingold, J. Am. Chem. Soc., 1997, 119, 10743 CrossRef CAS; (c) K. Arumugam, C. D. Varnado, S. Sproules, V. M. Lynch and C. W. Bielawski, Chem. – Eur. J., 2013, 19, 10866 CrossRef CAS PubMed; (d) J. Wei and P. L. Diaconescu, Acc. Chem. Res., 2019, 52, 415 CrossRef CAS PubMed.
- For selected examples, see: (a) Y. Sohtome, S. Tanaka, K. Takada, T. Yamaguchi and K. Nagasawa, Angew. Chem., Int. Ed., 2010, 49, 9254 CrossRef CAS PubMed; (b) R. J. Chew, X.-R. Li, Y. Li, S. A. Pullarkat and P.-H. Leung, Chem. – Eur. J., 2015, 21, 4800 CrossRef CAS PubMed.
- (a) R. S. Stoll and S. Hecht, Angew. Chem., Int. Ed., 2010, 49, 5054 CrossRef CAS PubMed; (b) B. M. Neilson and C. W. Bielawski, ACS Catal., 1874, 2013, 3 Search PubMed; (c) R. Göstl, A. Senf and S. Hecht, Chem. Soc. Rev., 2014, 43, 1982 RSC.
- (a) D. Cameron and S. Eisler, J. Phys. Org. Chem., 2018, 31, e3858 CrossRef; (b) T. Niazov, B. Shlyahovsky and I. Willner, J. Am. Chem. Soc., 2007, 129, 6374 CrossRef CAS PubMed.
- (a) M. Irie, Chem. Rev., 2000, 100, 1685 CrossRef CAS PubMed; (b) D. Sud, T. B. Norsten and N. R. Branda, Angew. Chem., Int. Ed., 2005, 44, 2019 CrossRef CAS PubMed; (c) D. Wilson and N. R. Branda, Angew. Chem., Int. Ed., 2012, 51, 5431 CrossRef CAS PubMed; (d) B. M. Neilson and C. W. Bielawski, Organometallics, 2013, 32, 3121 CrossRef CAS.
- G. Berkovic, V. Krongauz and V. Weiss, Chem. Rev., 2000, 100, 1741 CrossRef CAS.
- H. M. D. Bandara and S. C. Burdette, Chem. Soc. Rev., 2012, 41, 1809 RSC.
- F. Würthner and J. Rebek, Angew. Chem., Int. Ed. Engl., 1995, 34, 446 CrossRef.
- (a) S. Shinkai, T. Nakaji, Y. Nishida, T. Ogawa and O. Manabe, J. Am. Chem. Soc., 1980, 102, 5860 CrossRef CAS; (b) S. Shinkai, T. Nakaji, T. Ogawa, K. Shigematsu and O. Manabe, J. Am. Chem. Soc., 1981, 103, 111 CrossRef CAS; (c) S. Shinkai, K. Shigematsu, Y. Kusano and O. Manabe, J. Chem. Soc., Perkin Trans. 1, 1981, 3279 RSC; (d) S. Shinkai, T. Ogawa, Y. Kusano, O. Manabe, K. Kikukawa, T. Goto and T. Matsuda, J. Am. Chem. Soc., 1982, 104, 1960 CrossRef CAS.
- R. Cacciapaglia, S. Di Stefano and L. Mandolini, J. Am. Chem. Soc., 2003, 125, 2224 CrossRef CAS PubMed.
- R. Cacciapaglia, S. Di Stefano, E. Kelderman and L. Mandolini, Angew. Chem., Int. Ed., 1999, 38, 348 CrossRef CAS PubMed.
- T. Imahori, R. Yamaguchi and S. Kurihara, Chem. – Eur. J., 2012, 18, 10802 CrossRef CAS PubMed.
- M. Samanta, V. S. R. Krishna and S. Bandyopadhyay, Chem. Commun., 2014, 50, 10577 RSC.
- T. Arif, C. Cazorla, N. Bogliotti, N. Saleh, F. Blanchard, V. Gandon, R. Métivier, J. Xie, A. Voituriez and A. Marinetti, Catal. Sci. Technol., 2018, 8, 710 RSC.
- L. Osorio-Planes, C. Rodríguez-Esrich and M. A. Pericàs, Org. Lett., 2014, 16, 1704 CrossRef CAS PubMed.
- Z. Dai, Y. Cui, C. Chen and J. Wu, Chem. Commun., 2016, 52, 8826 RSC.
- A. Ueno, K. Takahashi and T. Osa, J. Chem. Soc., Chem. Commun., 1981, 94 RSC.
- W.-S. Lee and A. Ueno, Macromol. Rapid Commun., 2001, 22, 448 CrossRef CAS.
- (a) M. V. Peters, R. S. Stoll, A. Kühn and S. Hecht, Angew. Chem., Int. Ed., 2008, 47, 5968 CrossRef CAS PubMed; (b) R. S. Stoll, M. V. Peters, A. Kühn, S. Heiles, R. Goddard, M. Bühl, C. M. Thiele and S. Hecht, J. Am. Chem. Soc., 2009, 131, 357 CrossRef CAS PubMed.
- M. Pescher, L. van Wilderen, S. Grützner, C. Slavov, J. Wachtveitl, S. Hecht and J. Bredenbeck, Angew. Chem., Int. Ed., 2017, 56, 12092 CrossRef CAS PubMed.
- R. S. Stoll and S. Hecht, Org. Lett., 2009, 11, 4790 CrossRef CAS PubMed.
- M. Szewczyk, G. Sobczak and V. Sashuk, ACS Catal., 2018, 8, 2810 CrossRef CAS.
- N. Priyadarshani, B. Ginovska, J. T. Bays, J. C. Linehan and W. J. Shaw, Dalton Trans., 2015, 44, 14854 RSC.
- A. Telleria, P. W. N. M. van Leeuwen and Z. Freixa, Dalton Trans., 2017, 46, 3569 RSC.
- Y. Wei, S. Han, J. Kim, S. Soh and B. A. Grzybowski, J. Am. Chem. Soc., 2010, 132, 11018 CrossRef CAS PubMed.
- A. Nojiri, N. Kumagai and M. Shibasaki, Chem. Commun., 2013, 49, 4628 RSC.
- H. Bricout, E. Banaszak, C. Len, F. Hapiot and E. Monflier, Chem. Commun., 2010, 46, 7813 RSC.
- (a) J.-M. Lehn, Chem. – Eur. J., 2006, 12, 5910 CrossRef CAS PubMed; (b) L. Greb and J.-M. Lehn, J. Am. Chem. Soc., 2014, 136, 13114 CrossRef CAS PubMed; (c) L. Greb, A. Eichhöfer and J.-M. Lehn, Angew. Chem., Int. Ed., 2015, 54, 14345 CrossRef CAS PubMed.
- I. Aprahamian, Chem. Commun., 2017, 53, 6674 RSC.
- G. de Bo, D. A. Leigh, C. R. McTernan and S. Wang, Chem. Sci., 2017, 8, 7077 RSC.
- (a) H. Suzuki, Bull. Chem. Soc. Jpn., 1952, 25, 145 CrossRef CAS; (b) D. H. Waldeck, Chem. Rev., 1991, 91, 415 CrossRef CAS.
- H. Sugimoto, T. Kimura and S. Inoue, J. Am. Chem. Soc., 1999, 121, 2325 CrossRef CAS.
- R. Cacciapaglia, S. Di Stefano and L. Mandolini, J. Org. Chem., 2002, 67, 521 CrossRef CAS PubMed.
- N. Koumura, E. M. Geertsema, M. B. van Gelder, A. Meetsma and B. L. Feringa, J. Am. Chem. Soc., 2002, 124, 5037 CrossRef CAS PubMed.
- S. F. Pizzolato, B. S. L. Collins, T. van Leeuwen and B. L. Feringa, Chem. – Eur. J., 2017, 23, 6174 CrossRef CAS PubMed.
- Z. S. Kean, S. Akbulatov, Y. Tian, R. A. Widenhoefer, R. Boulatov and S. L. Craig, Angew. Chem., Int. Ed., 2014, 53, 14508 CrossRef CAS PubMed.
- N. Koumura, R. W. Zijlstra, R. A. van Delden, N. Harada and B. L. Feringa, Nature, 1999, 401, 174 CrossRef PubMed.
- J. Wang and B. L. Feringa, Science, 2011, 331, 1429 CrossRef CAS PubMed.
- A. G. Doyle and E. N. Jacobsen, Chem. Rev., 2007, 107, 5713 CrossRef CAS PubMed.
- M. Vlatković, L. Bernardi, E. Otten and B. L. Feringa, Chem. Commun., 2014, 50, 7773 RSC.
- M. Vlatković, J. Volarić, B. S. L. Collins, L. Bernardi and B. L. Feringa, Org. Biomol. Chem., 2017, 15, 8285 RSC.
- B. M. Trost, D. L. van Vranken and C. A. Bingel, J. Am. Chem. Soc., 1992, 114, 9327 CrossRef CAS.
- D. Zhao, T. M. Neubauer and B. L. Feringa, Nat. Commun., 2015, 6, 6652 CrossRef CAS PubMed.
- C.-T. Chen, C.-C. Tsai, P.-K. Tsou, C.-T. Huang and C.-H. Yu, Chem. Sci., 2017, 8, 524 RSC.
- S. F. Pizzolato, P. Štacko, J. C. M. Kistemaker, T. van Leeuwen, E. Otten and B. L. Feringa, J. Am. Chem. Soc., 2018, 140, 17278 CrossRef CAS PubMed.
- K. Eichstaedt, J. Jaramillo-García, D. A. Leigh, V. Marcos, S. Pisano and T. A. Singleton, J. Am. Chem. Soc., 2017, 139, 9376 CrossRef CAS PubMed.
- (a) N. E. Kamber, W. Jeong, R. M. Waymouth, R. C. Pratt, B. G. G. Lohmeijer and J. L. Hedrick, Chem. Rev., 2007, 107, 5813 CrossRef CAS PubMed; (b) A. J. Teator, D. N. Lastovickova and C. W. Bielawski, Chem. Rev., 2016, 116, 1969 CrossRef CAS PubMed; (c) F. Eisenreich, M. Kathan, A. Dallmann, S. P. Ihrig, T. Schwaar, B. M. Schmidt and S. Hecht, Nat. Catal., 2018, 1, 516 CrossRef.
| This journal is © The Royal Society of Chemistry 2019 |