
Gold nanoparticle amplification strategies for multiplex SPRi-based immunosensing of human pancreatic islet hormones†
F. Rafael
Castiello
a and
Maryam
Tabrizian
 *ab
*ab
aBiomedical and Biological Engineering Department, McGill University, Montreal, QC, Canada. E-mail: maryam.tabrizian@mcgill.ca
bFaculty of Dentistry, McGill University, Montreal, QC, Canada
First published on 13th March 2019
Abstract
In this work, we demonstrate the potential use of SPRi for secretion-monitoring of pancreatic islets, small micro-organs that regulate glucose homeostasis in the body. In the islets, somatostatin works as a paracrine inhibitor of insulin and glucagon secretion. However, this inhibitory effect is lost in diabetic individuals and little is known about its contribution to the pathology of diabetes. Thus, the simultaneous detection of insulin, glucagon and somatostatin could help understand such communications. Previously, the authors introduced an SPRi biosensor to simultaneously monitor insulin, glucagon and somatostatin using an indirect competitive immunoassay. However, our sensor achieved a relatively high LOD for somatostatin detection (246 nM), the smallest of the three hormones. For this reason, the present work aimed to improve the performance of our SPRi biosensor using gold nanoparticles (GNPs) as a means of ensuring somatostatin detection from a small group of islets. Although GNP amplification is frequently reported in the literature for individual detection of analytes using SPR, studies regarding the optimal strategy in a multiplexed SPR setup are missing. Therefore, with the aim of finding the optimal GNP amplification strategies for multiplex sensing we compared three architectures: (1) GNPs immobilized on the sensor surface, (2) GNPs conjugated with primary antibodies (GNP-Ab1) and (3) GNPs conjugated with a secondary antibody (GNP-Ab2). Among these strategies an immunoassay using GNP-Ab2 conjugates was able to achieve multiplex detection of the three hormones without cross-reactivity and with 9-fold LOD improvement for insulin, 10-fold for glucagon and 200-fold for somatostatin when compared to the SPRi biosensor without GNPs. The present work denotes the first report of the simultaneous detection of such hormones with a sensitivity level comparable to ELISA assays, particularly for somatostatin.
Introduction
Pancreatic islets are small micro-organs that regulate glucose homeostasis in the body.1 Deficiencies in the islet's secretory pathways give rise to diabetes mellitus. However, little is understood about the paracrine communications occurring during glucose regulation.1 For instance, it has been shown that somatostatin, secreted by the third most abundant cell type in the islets, is a potent paracrine inhibitor of the secretion of both insulin and glucagon.2,3 In a healthy adult, somatostatin secretion is normally stimulated by glucose; however, this triggering effect is lost in type 1 and type 2 diabetes. This has been proposed to contribute to the impaired regulation of glucagon secretion in diabetes.4,5 Thus, monitoring a secretory fingerprint (SF) of pancreatic islets including the three most abundant secreting cells in the islets could help to better understand such paracrine communications.Pancreatic islet research to date has involved mostly the use of traditional bioassays for hormone quantification such as ELISA,6–8 patch clamp,9–14 and capillary electrophoresis (CE).15–17 However, these techniques have a low throughput, are time-consuming, are labor intensive and can detect only one hormone at a time. Moreover, they face several challenges when attempting their implementation for multiplex analysis.
Most of these shortcomings could be addressed using optical, electrical, mechanical or magnetic biosensors.18 Among these biosensing technologies, sensors based on the optical excitation of surface plasmons have increased in popularity in the last decade due to their simple, easy to use, non-invasive and label-free nature.19 Surface plasmon resonance (SPR) biosensors present an additional advantage for the multiplex screening of biomolecular interactions when combined with imaging capabilities (SPRi).20 For instance, H. J. Lee et al.21 demonstrated the simultaneous detection of 3 low molecular weight protein biomarkers using SPRi, by creating a high-density antibody microarray achieving multiplex detection of the three protein markers down to 1 nM concentrations. This makes SPRi a very desirable technique for secretion-monitoring of pancreatic islets.
In a recent work reported by the authors, a multiplex SPRi-based biosensor was introduced as a viable tool for simultaneous quantification of insulin, glucagon, and somatostatin by performing three simultaneous competitive assays with monoclonal antibodies.22 In this work an innovative surface chemistry was introduced and optimized for the detection of the three targeted peptides in a competitive immunoassay format with high antifouling properties, obtaining limits of detection (LODs) of 1 nM for insulin, 4 nM for glucagon, and 246 nM for somatostatin in multiplexed mode with a total analysis time of 21 min per point. These LODs are satisfactory for the detection of insulin and glucagon as demonstrated by previous reports where these hormones were detected individually from secretions of a small group of islets within a microfluidic device.15,16 However, there are no available reports regarding the required LOD for somatostatin within a similar islet population. Moreover, it is known that the number of somatostatin secreting cells within pancreatic islets is usually lower than that of insulin or glucagon secreting cells.23
Considering the important role of somatostatin in regulating insulin and glucagon secretion,2,3 and before moving to precious and scarce human islet studies, the aim of the present work was to improve the performance of the previously developed SPRi biosensor to ensure that somatostatin secreted from a small group of islets can effectively, accurately and simultaneously be detected with other towed hormones.
Since their introduction by L. Lyon et al.,24 gold nanoparticles (GNPs) have been the most commonly used method for improving the performance of SPR immunosensors. Two configurations are typically considered for the use of GNPs in SPR signal amplification: (1) the sensor surface modification with GNPs and (2) the labeling of a recognition element with GNPs. These strategies rely either on the coupling of the local plasmon resonance of the GNP with the surface plasmon resonance of the system or on the increased mass attached to the recognition element for signal enhancement.25,26 Both GNP amplification strategies have been frequently reported in the literature for individual detection of hormones such as insulin,27 progesterone,28 and testosterone29 and other small analytes;26,30,31 however, studies regarding the application of these strategies for multiplex hormone detection are scarce. Moreover, few reports exist regarding the use of GNP amplification in a multiplex setting and they focus on the detection of DNA sequences32 or cancer biomarkers by means of localized SPR using microscopy.33 Although these reports demonstrated the detection of target analytes in femtomolar levels in multiplexed mode, there is a lack of formal studies regarding the optimal GNP amplification strategy for SPRi systems.
Thus, to establish the optimal signal amplification configuration for the multiplexed sensing of a SF of pancreatic islets with SPRi, three GNP amplification strategies were investigated including (1) GNPs immobilized on the sensor surface, (2) GNPs conjugated with primary antibodies (GNP-Ab1) and (3) GNPs conjugated with a secondary antibody (GNP-Ab2). For this study, somatostatin was used as the ‘reference’ hormone, as it is the smallest among the three SFs of pancreatic islets, to first test the performance of the aforementioned SPR signal amplification strategies in an indirect competitive assay. Then, the biosensor performance was assessed in multiplexed mode to determine the LOD and dynamic range for the three targeted hormones simultaneously.
Experimental section
Materials and methods
20 nm gold nanoparticles (GNPs) in citrate buffer, ethanolamine hydrochloride, N-hydroxysuccinimide (NHS), N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC), hexa(ethylene glycol) dithiol (HEGD), bovine serum albumin (BSA), hydrochloric acid (HCl), glycerol, sodium hydroxide (NaOH), human glucagon and human somatostatin were purchased from Sigma-Aldrich (St Louis, MO, USA). Absolute ethanol was purchased from Fisher Scientific (Fair Lawn, NJ, USA), and phosphate-buffered saline (PBS) tablets, Tween 20 and glycine were purchased from BioShop Canada Inc. (Burlington, Ontario, Canada). 3,3′-Dithiobis(sulfosuccinimidyl propionate) (DTSSP) was purchased from ThermoFisher Scientific. Tris-buffered saline (TBS) with 1% casein and goat anti-mouse IgG1 and goat anti-rat IgG2a secondary polyclonal antibodies were purchased from Bio-Rad. The anti-insulin antibody (6.2 mg mL−1) and human insulin were purchased from PROSPECT (Ness, Ziona, Israel). Anti-somatostatin monoclonal antibodies (200 μg mL−1 each) were purchased from Santa Cruz Biotechnology, Inc. (Mississauga, ON, Canada). Anti-glucagon monoclonal antibodies were purchased from Abcam (Cambridge, MA, USA). CH3O-PEG-SH (MW 1200 Da) was purchased from Rapp Polymers GmbH (Tübingen, Germany). 16-Mercaptohexadecanoic acid (MHDA) was purchased from ProChimia Surfaces Sp. (Zacisze, Sopot, Poland). Borate buffer (0.5 M, pH 8.5) was purchased from Alfa-Aesar (USA).Substrate preparation
Cleaned microscope glass slide (12 mm × 25 mm × 1 mm, n = 1.518) substrates were coated with 2 nm Cr as an adhesion layer, followed by the deposition of a thin Au layer of 48 nm using E-beam vapor deposition under high vacuum. The slides were then coupled to an SF11 equilateral triangular prism (nSF-11 = 1.765) using a refractive index matching liquid. Gold-coated prisms (n = 1.765) were purchased from Horiba Scientific, France, and used as received.SPRi measurements
SPRi detection was performed using a scanning-angle SPRi instrument (model SPRi Lab+, Horiba, France). The SPRi apparatus is equipped with an 800 nm LED source, a CCD camera, and a microfluidic flow cell. All experiments were performed at 25 °C by keeping the instrument inside an incubator (Memmert Peltier, Rose Scientific, Canada).To select the working angle for kinetic analysis, the slope of the plasmon curves was computed automatically using the instrument's software. The selected angle corresponds to the point of the plasmon curve at which the slope was maximum. Reflectivity shift (ΔR (%)) was measured upon stabilization of the baseline. After each analyte injection, the substrate was rinsed with running buffer PBS-T (PBS with 0.002% Tween 20), and ΔR was calculated by the difference between the buffer signals before and after the analyte injection. The signal was recorded at least on three spots for each analyte and controls to determine the average ΔR values. All experiments were performed using an injection loop with a fixed volume of 200 μL and a constant flow rate of 20 μL min−1, with the exception of functionalization steps where the flow rate was adjusted depending on the required contact time.
All SPR plots are presented as a function of reflectivity shift (ΔR (%)) vs. time. A Savitzky–Golay smoothing polynomial function of second order was applied to all plots using OriginLab 2018 (b.9.5.5.409).
Surface immobilization of hormones
Following a protocol previously developed by the authors,22 an ethanolic solution of 0.5 mM CH3O-PEG-SH and 0.5 mM MHDA was prepared and mixed at a molar ratio of 40% MHDA and 60% PEG (60-PEG/40-MHDA). Gold-coated prisms and slides were immersed in the ethanolic solution overnight to allow self-assembled monolayer (SAM) formation. Finally, they were thoroughly rinsed with absolute ethanol, DI water and dried under a stream of N2.For individual somatostatin sensing experiments, SAM-functionalized slides were placed on the SPRi system for subsequent functionalization. An initial conditioning step was performed by four serial injections of a 1 M glycine (pH 2.5) (1 M-Gly) solution (contact time of 2 min each). Then, PBS-T was allowed to run until a stable baseline was obtained. Next, covalent immobilization of insulin, glucagon or somatostatin via NHS/EDC chemistry was performed following the protocol reported by Gobi et al..34 Briefly, an aqueous solution containing 2 mg mL−1 NHS, 2 mg mL−1 EDC and 50 μg mL−1 of the desired hormone was injected into the system with a contact time of 1 h. Next, an injection of 1 M ethanolamine hydrochloride (pH 8.5) (contact time of 10 min) was performed to inactivate any unfunctionalized –COOH groups. Then, two serial injections of 1 M-Gly solution (contact time of 1 min each) were performed to remove weakly bound hormones. Finally, a blocking solution containing 5% BSA and 1% casein in TBS buffer was injected (contact time of 30 min) and subsequently, at least 3 injections of the 1 M-Gly solution were made to remove weakly bound proteins. Fig. 1A shows a schematic representation of a typical surface hormone functionalization.
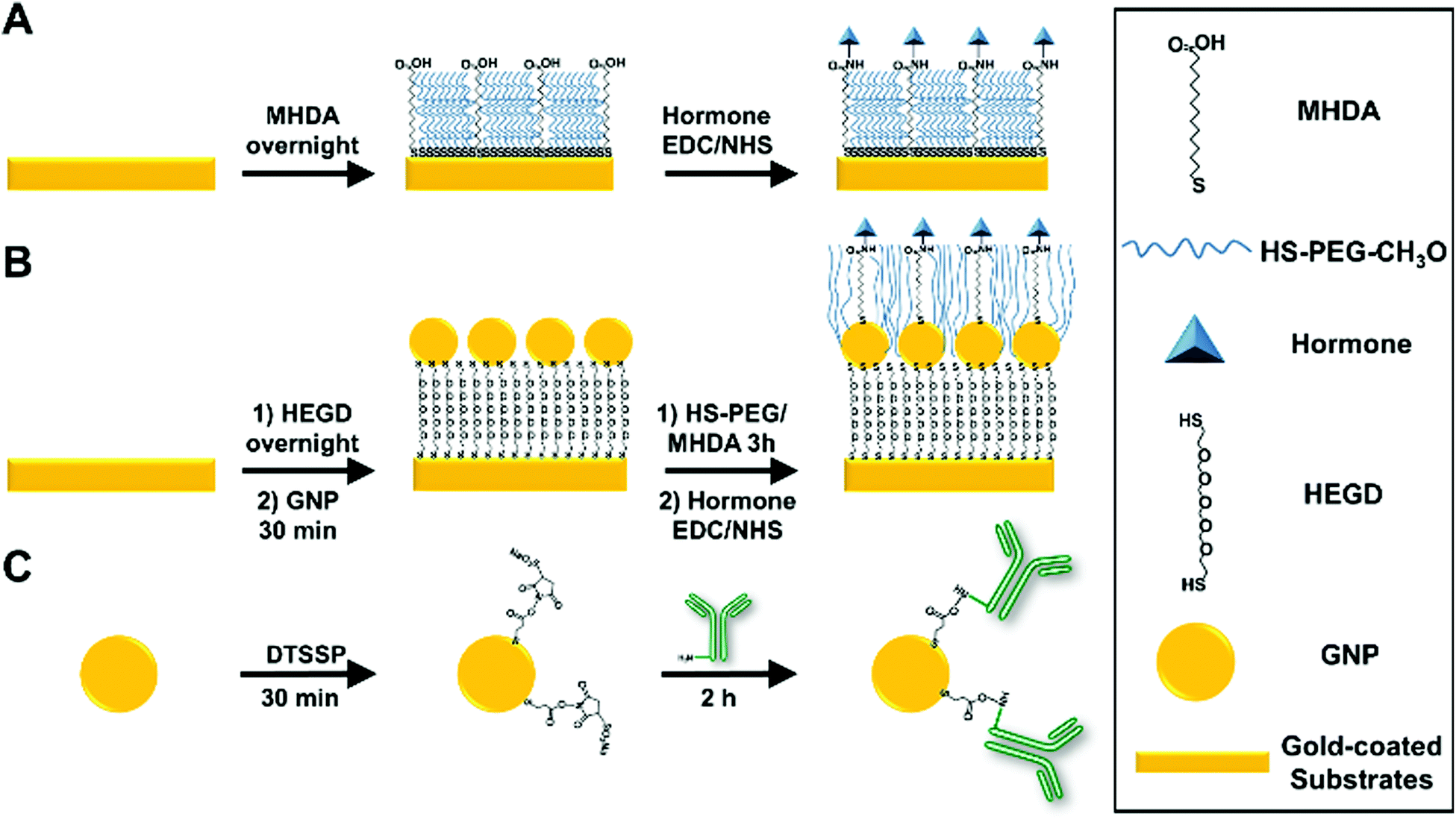 | ||
| Fig. 1 Schematic representation of the typical surface functionalization of (A) gold-coated substrates functionalized with a self-assembled monolayer (SAM) of 16-mercaptohexadecanoic acid (MHDA)/CH3O-PEG-SH (PEG) and subsequently with the targeted hormone, (B) gold-coated substrate surface functionalized with a SAM of hexa(ethylene glycol) dithiol (HEGD), GNPs and subsequently with the targeted hormone and (C) covalent antibody functionalization for the formation of GNP-conjugates using (3,3′-dithiobis(sulfosuccinimidyl propionate)) (DTSSP). | ||
For multiplex measurements, gold-coated prisms were functionalized using the procedure described for glass slides outside of the SPR system. After conditioning, four individual solutions containing NHS/EDC and each targeted hormone (insulin, glucagon, and somatostatin) or a control (BSA) were spotted (150 nL) in triplicate on the prisms and incubated in a humidity chamber for 1 h. After incubation, the prisms were rinsed with DI water and exposed to 1 M ethanolamine hydrochloride (pH 8.5) for 10 min. Next, the prisms were exposed to the blocking solution for 30 min and subsequently rinsed with PBS-T. Finally, the prisms were placed in the SPR system and a 1 M-Gly solution was injected at least three times to obtain a stable baseline before starting the experiments.
Immobilization of GNPs on the gold-coated sensor's surface
In this work, commercial 20 nm GNPs were selected since it has been reported in the literature as an optimal size for SPR amplification either when immobilized in the sensor's surface or for antibody modified GNP immunoassays of small molecules.28,35,36The immobilization of commercial 20 nm GNPs on the gold-coated sensor's surface was achieved according to a protocol developed by Taufik et al.37 with minor modifications. After cleaning, gold-coated slides were immersed in an ethanolic solution of 2 mM HEGD overnight to allow SAM formation. After rinsing with ethanol and DI water, the slides were exposed to an aqueous solution containing 20 nm GNPs (OD520 = 1.0) and incubated for one hour. Next, the slides were rinsed with DI water and placed in a 60-PEG/40-MHDA ethanolic solution for 3 hours. Finally, hormone functionalization was performed according to the procedure described in the previous section. Fig. 1B shows a schematic representation of the surface functionalization with GNPs and hormones.
Antibody–GNP functionalization
The functionalization protocol for commercial 20 nm GNPs (OD520 = 1) with primary and secondary antibodies was adapted from a previous report by J. D. Driskell et al.38 using DTSSP as a bifunctional crosslinker. Briefly, 134 μL of 50 mM borate buffer (pH 8.5) was added to a 1 mL suspension of 20 nm GNPs to adjust the pH. Next, 26 μL of 20 μM DTSSP was added to the GNPs and incubated for 30 min to form a thiolate monolayer through cleavage of the DTSSP disulfide bond. The suspension was then centrifuged at 17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500g for 30 min. Then, the supernatant containing excess DTSSP was removed and the GNPs were resuspended in 1 mL of 2 mM borate buffer. Immediately afterward, 20 μg of the desired antibody was added to the DTSSP-GNP suspension and incubated for 1.5 hours. The suspension was then centrifuged at 17500g for 30 min, the supernatant was removed, and the GNPs were resuspended in 1 mL of 2 mM borate buffer containing 1% (m/v) BSA and incubated for 30 min to allow the BSA to block any unreacted DTSSP and nonspecific binding sites. The centrifugation/resuspension cycle was repeated two additional times using 2 mM borate buffer for resuspension to remove excess antibody and BSA. The final volume of the solution after the centrifugation/resuspension cycles was fixed to approximately 200 μL (OD525 ≈ 4) and the solution was stored at 4 °C. The functionalized GNPs were diluted to the desired concentration in PBS-T prior to use in immunoassays. Fig. 1C shows a schematic representation of antibody–GNP functionalization. Successful GNP functionalization for all antibodies was confirmed by the shift in the absorbance maxima of the GNPs from 520 to 525 nm (Fig. S2 provided in the ESI†).
500g for 30 min. Then, the supernatant containing excess DTSSP was removed and the GNPs were resuspended in 1 mL of 2 mM borate buffer. Immediately afterward, 20 μg of the desired antibody was added to the DTSSP-GNP suspension and incubated for 1.5 hours. The suspension was then centrifuged at 17500g for 30 min, the supernatant was removed, and the GNPs were resuspended in 1 mL of 2 mM borate buffer containing 1% (m/v) BSA and incubated for 30 min to allow the BSA to block any unreacted DTSSP and nonspecific binding sites. The centrifugation/resuspension cycle was repeated two additional times using 2 mM borate buffer for resuspension to remove excess antibody and BSA. The final volume of the solution after the centrifugation/resuspension cycles was fixed to approximately 200 μL (OD525 ≈ 4) and the solution was stored at 4 °C. The functionalized GNPs were diluted to the desired concentration in PBS-T prior to use in immunoassays. Fig. 1C shows a schematic representation of antibody–GNP functionalization. Successful GNP functionalization for all antibodies was confirmed by the shift in the absorbance maxima of the GNPs from 520 to 525 nm (Fig. S2 provided in the ESI†).
Competitive immunoassays
The four configurations used for indirect competitive immunoassays consisted of a surface with (Fig. 2A) and without GNPs (Fig. 2B), primary antibody–GNP (GNP-Ab1) conjugates (Fig. 2C) and secondary antibody–GNP (GNP-Ab2) conjugates (Fig. 2D). Since somatostatin was the smallest of the targeted hormones, it was used as the “reference” hormone in all individual hormone assays. The optimal primary anti-somatostatin antibody, GNP-Ab1 and GNP-Ab2 concentrations were defined as the concentration that could generate a small but detectable SPR signal of ΔR ≈ 1, previously reported as a reliable ΔR for this type of assay.39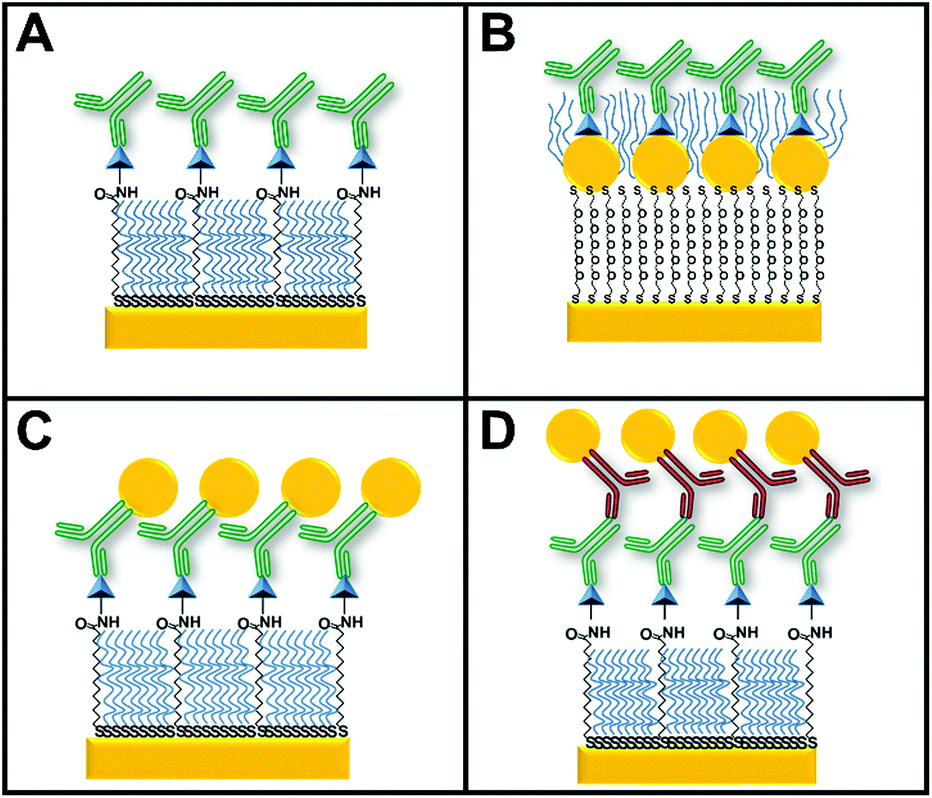 | ||
| Fig. 2 Schematic representation of the four configurations of competitive immunoassays used for this work: (A) an assay involving only primary antibodies and the hormone immobilized on a gold surface used as the control, (B) an assay where GNPs are present on the surface and the hormone is immobilized on top of the GNPs, (C) an assay where GNPs are conjugated with monoclonal primary antibodies (GNP-Ab1) and (D) an assay involving amplification using GNPs conjugated with a polyclonal secondary antibody (GNP-Ab2) after a primary competitive assay. | ||
Standard somatostatin solutions were prepared by serial dilution in PBS-T buffer with concentrations ranging between 0.01 and 4000 ng mL−1. These solutions were then mixed with 2 μg mL−1 of anti-somatostatin antibody for assays involving only primary antibodies, 0.6 μg mL−1 of antibody for the assay with GNP-Ab2 conjugates or a 1:50 dilution (from OD525 ≈ 4) of GNP-Ab1 conjugates. All mixtures were incubated for 2 min with gentle mixing by inverting upside down and then injected into the system from the highest to the lowest hormone concentration (contact time of 10 min) starting with a blank solution containing only anti-somatostatin antibodies or GNP-Ab1 conjugates. For secondary antibody conjugate assays, immediately after anti-somatostatin antibody injection, GNP-Ab2 conjugates were injected (1:50 dilution from OD525 ≈ 4). The optimal primary anti-somatostatin antibody, GNP-Ab1 and GNP-Ab2 concentrations were defined as the concentration that could generate a small but detectable SPR signal of ΔR ≈ 1, previously reported as a reliable ΔR for this type of assay.39
During calibration curve experiments, different sensor regeneration solutions were tested including 10–50 mM NaOH, 50 mM NaOH with 5–50% (v/v) glycerol, 0.1–1 M glycine (pH 1.5), 0.1–1 M glycine (pH 1.5) with 1% (v/v) DMSO, and 2 M MgCl2. From these solutions, 50 mM NaOH with 25% glycerol provided the most efficient conditions for regeneration and it was used throughout all the experiments.
For multiplex assays, standard solutions having a mixture of insulin, glucagon, and somatostatin were prepared in PBS-T buffer in a concentration range of 0.01–4000 ng mL−1 and mixed with a cocktail of primary antibodies containing anti-insulin (0.2 μg mL−1), anti-glucagon (0.05 μg mL−1) and anti-somatostatin (0.6 μg mL−1). Similar to individual somatostatin assays, the mixtures were incubated for 2 min with gentle mixing and serially injected over the spotted sensor chip from the highest to the lowest hormone concentration (contact time of 10 min) starting with a blank solution containing only the antibody cocktail. Subsequently, a GNP-Ab2 mixture containing GNPs conjugated to anti-mouse IgG1 (1:100 dilution from OD525 ≈ 4) and anti-rat Ig2a (1:50 dilution from OD525 ≈ 4) was injected into the system. Each sensing cycle comprised: hormone primary antibody mixing and incubation for 2 min, cocktail injection for 10 min, 3 min buffer washing, 10 min injection of a GNP-Ab2 mixture and 2 injections of a regeneration solution with a contact time of 30 s with 3 min washing with buffer in between.
Statistics
Relative binding (C/C0) was calculated for all competitive immunoassays, by dividing the response of the standard solutions containing hormones (C) by the response of the blank solution containing only a fixed concentration of antibodies (C0). To generate calibration curves C/C0 was plotted against the hormone concentration. The calibration curves were fitted using a non-linear 4 parameter logistic (4PL) model using OriginLab 2018 (b.9.5.5.409). The LOD for all immunoassays was calculated from the calibration curves as the blank signal (C0) minus three times its standard deviation. The dynamic range for the competitive immunoassay was established between 0.2C/C0 and 0.8C/C0. All data are expressed as the average of at least 3 independent experiments ± standard deviation (SD).Results and discussion
GNP amplification strategy comparison for competitive immunoassays
First, the formation of a chemically linked layer of GNPs was performed using a SAM of a dithiol alkane (HEGD). HEGD allowed anchoring of the GNPs to the gold-coated sensor's surface through the thiol group at each end of the molecule.37 AFM analysis indicated the successful immobilization of the GNPs by a significant change in surface morphology from a clean gold surface to a GNP-modified surface as clearly observed in Fig. S1 provided in the ESI.† This was further confirmed by a change in surface RMS roughness from 0.68 nm to 2.41 nm and later to 5.36 nm from a clean surface to a SAM-functionalized surface and to a GNP-functionalized surface. The signal amplification rationale here is that the activation of the GNP localized SPR due to the proximity of the immobilized GNPs to the sensor's surface can lead to different resonance properties of the overall SPR system with additional resonance shifts, resulting in an enhanced sensitivity of the biosensor.25For all GNP–antibody conjugates, functionalization was confirmed by a shift observed in the maximum absorption peak from 520 to 525 nm (Fig. S2 in the ESI†). In the case of GNP-Ab1 conjugates, the rationale behind this strategy is that the increased mass of the antibody due to the linked GNPs will result in a higher refractive index change on the SPR surface, thereby producing a larger SPR shift.26 Additionally, due to the close proximity of the GNPs to the SPR surface (<15 nm) signal enhancement is also linked to electromagnetic field coupling between surface SPR and the GNP localized SPR.26 Regarding the use of GNP-Ab2 conjugates, the signal amplification is only expected from the increased mass of the antibody due to the linked GNPs, as the GNPs are quite far from the surface.26
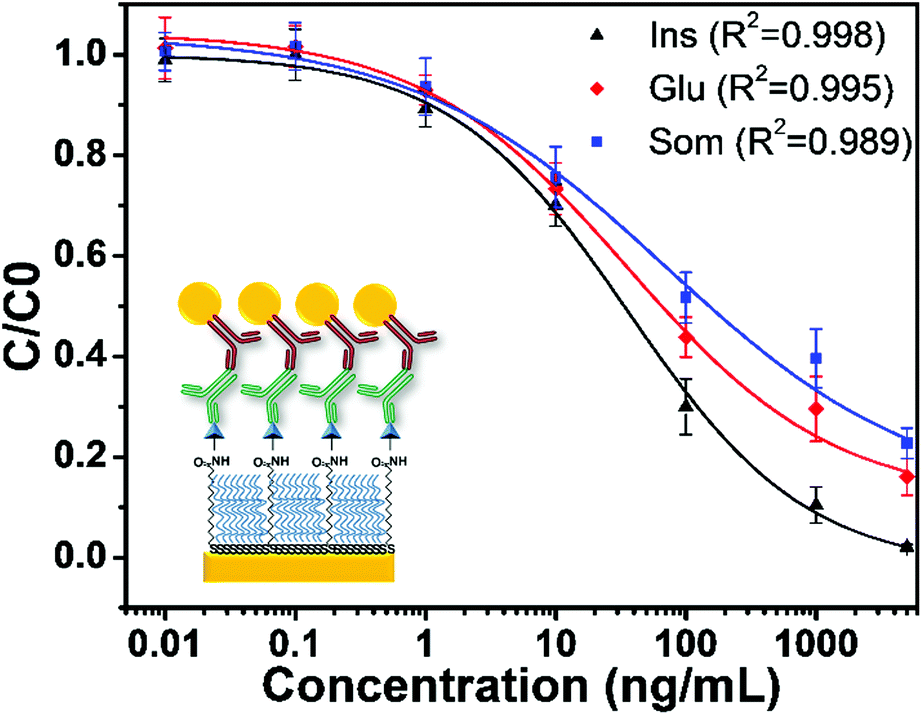
Fig. 3 shows the sensor calibration curves for somatostatin detection where the mean relative binding values (C/C0) are plotted as a function of hormone concentration for all sensing strategies. For these experiments, detection without GNPs was used as a control assay (Fig. 3A). Additionally, the entire sensor surface was functionalized with somatostatin, and the SPR shift for all sensing events was obtained as the mean of at least 10 spots from different regions of the chip. Then, an average of ΔR of three independently prepared chips was calculated representing their corresponding standard deviation (SD). The LOD and dynamic range for each sensing strategy are shown in Table 1.
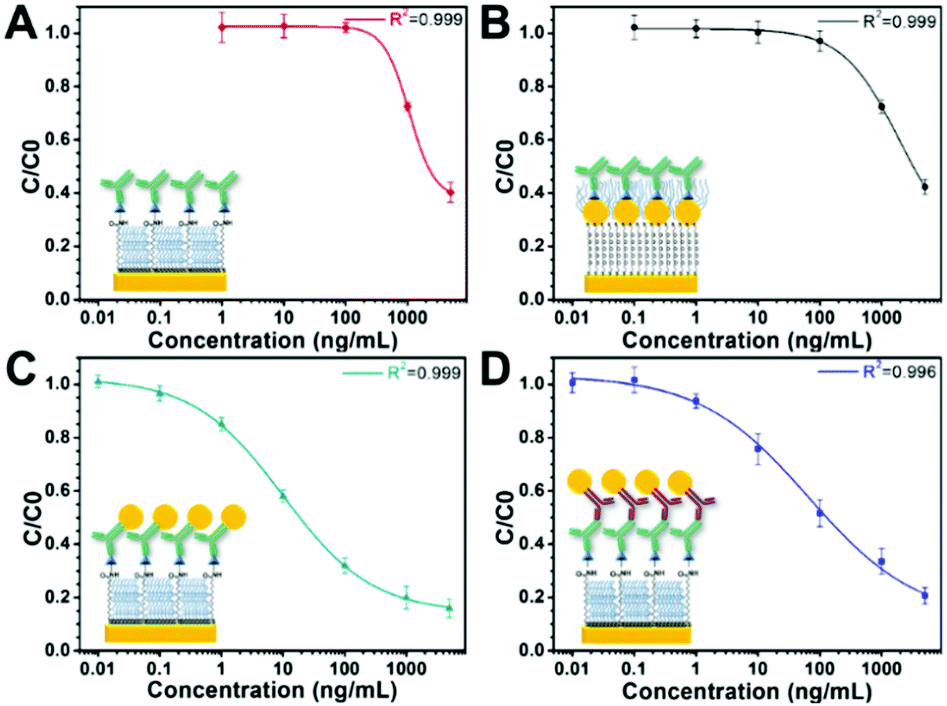 | ||
| Fig. 3 Somatostatin sensing calibration curves in PBS-T for competitive immunoassays involving (A) only primary antibodies, (B) GNPs on the sensor's surface, (C) GNPs conjugated with monoclonal primary antibodies (GNP-Ab1) and (D) an assay with GNPs conjugated with a polyclonal secondary antibody (GNP-Ab2). Solid lines correspond to the fitting of a nonlinear 4PL model. Error bars represent the standard deviation from 3 independent experiments (n = 3). | ||
It is noteworthy that the concentration of the control assay (Fig. 3A) and the strategy using a GNP-modified surface (Fig. 3B) was set to 2 μg mL−1 to facilitate comparison and to easily observe SPR signal enhancement. Interestingly, these two sensing strategies presented similar LODs. However, the GNP-modified surface showed a higher SPR signal (Fig. S3 in the ESI†) compared to the signal obtained for the surface without GNPs as clearly observable in the ΔR (%) (C0) values in Table 1. Indeed, this seems consistent with recent reports where the LOD of the calibration curve of an indirect competitive immunoassay using GNP-modified sensors did not change even when the SPR signal was enhanced.31,40 A possible explanation is that the performance of an indirect competitive immunoassay highly depends on the affinity constant of the immunoreaction.31 Therefore, only strategies affecting the affinity of the antigen–antibody system such as the GNP-Ab conjugation could improve the LOD of the SPR sensor.31
Finally, from all immunosensing strategies, the competitive immunoassay using GNP-Ab1 conjugates presented the best performance decreasing the LOD by three orders of magnitude compared to the control assay from 450 ng mL−1 to 240 pg mL−1. Hence, this immunoassay amplification strategy was selected for further development of the multiplex hormone-sensing assay.
GNP-amplified multiplex hormone sensing
Fig. 4A–C show the assessment of cross-reactivity for primary antibodies. Individual injection of each antibody caused an increase in the SPR signal only on its relevant spot, demonstrating specificity and low cross-contamination between spots. Moreover, there was a negligible response on the control spots, indicating good antifouling properties.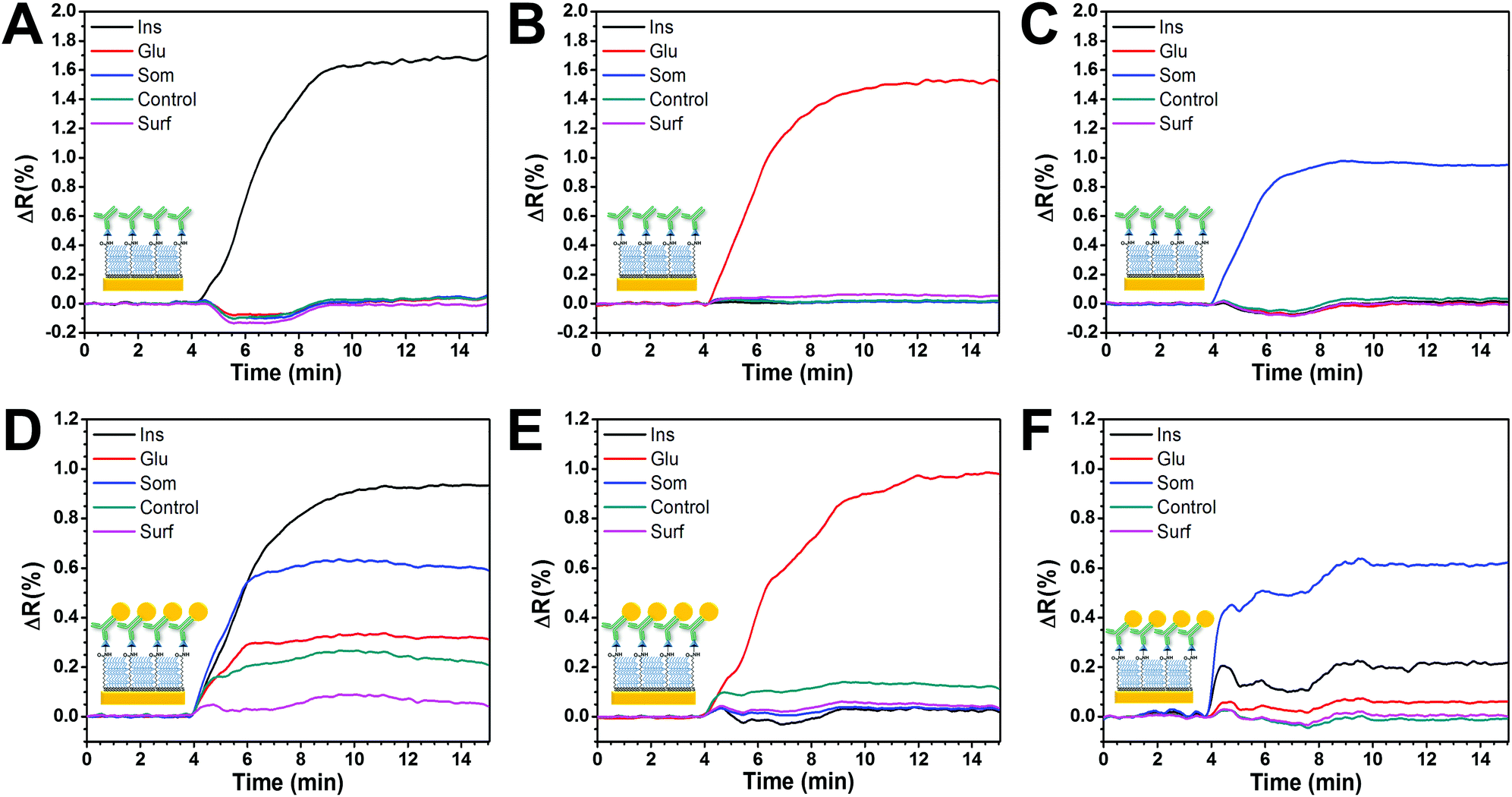 | ||
| Fig. 4 Comparison of the sensor specificity in multiplexed mode. The first row shows the specificity of the sensor without signal amplification for (A) anti-insulin, (B) anti-glucagon, and (C) anti-somatostatin. The second row shows the specificity of the sensor on the same surface with signal amplification for (D) GNP–anti-insulin, (E) GNP–anti-glucagon and (F) GNP–anti-somatostatin. Immobilized BSA (“Control”) and the bare SAM surface (“Surf”) were used as negative controls. | ||
However, when a similar experiment was performed using GNP-Ab1 conjugates (Fig. 4D–G), high cross-reactivity was observed. This effect was particularly large for GNP-anti-insulin conjugates which generated a non-specific signal increase in all the functionalized spots including the two negative controls (Fig. 4D). When GNP-anti-glucagon conjugates were injected, cross-reactivity with insulin and somatostatin spots was not observed; however some degree of non-specific interactions was detected for the negative control spots (Fig. 4F). For GNP-anti-somatostatin conjugates, cross-reactivity was also observed mostly with insulin spots (Fig. 4G). Additional antibodies from different species and companies were used for GNP-Ab1 conjugates for anti-insulin and anti-somatostatin. However cross-reactivity was always present (data not shown). This has been reported in the literature as a recurring problem for multiplex immunoassays41,42 particularly for GNP conjugates since it has been shown that such conjugations can modify the activity of the antibodies. In theory, a combination of GNP-Ab1 conjugates with little or null cross-reactivity for our system could be achieved; however testing a library of antibodies would be time consuming and cost-ineffective. Due to this dilemma, the second-best amplification strategy (GNP-Ab2) was selected for further development of our multiplex immunosensor. For this strategy, some degree of cross-reactivity could occur since the secondary antibodies are similarly conjugated to GNPs. In general, antibody conjugation is known to affect the antibody's affinity;31 however this did not seem to hinder the possibility for multiplex sensing as later demonstrated in this section.
The nonspecific binding effect of the GNP-Ab2 conjugates on the analyte spots was determined prior to the multiplex assay through the injection of a mixture of GNP-goat anti-mouse IgG1 and GNP-goat anti-rat IgG2a over the sensor surface. As seen in Fig. 5A, a minimum SPR angle shift was detected during the injection with the signal returning to similar baseline levels after a few minutes of PBS-T washing, indicating a negligible nonspecific binding effect of the GNP-Ab2 conjugates to the different surface spots.
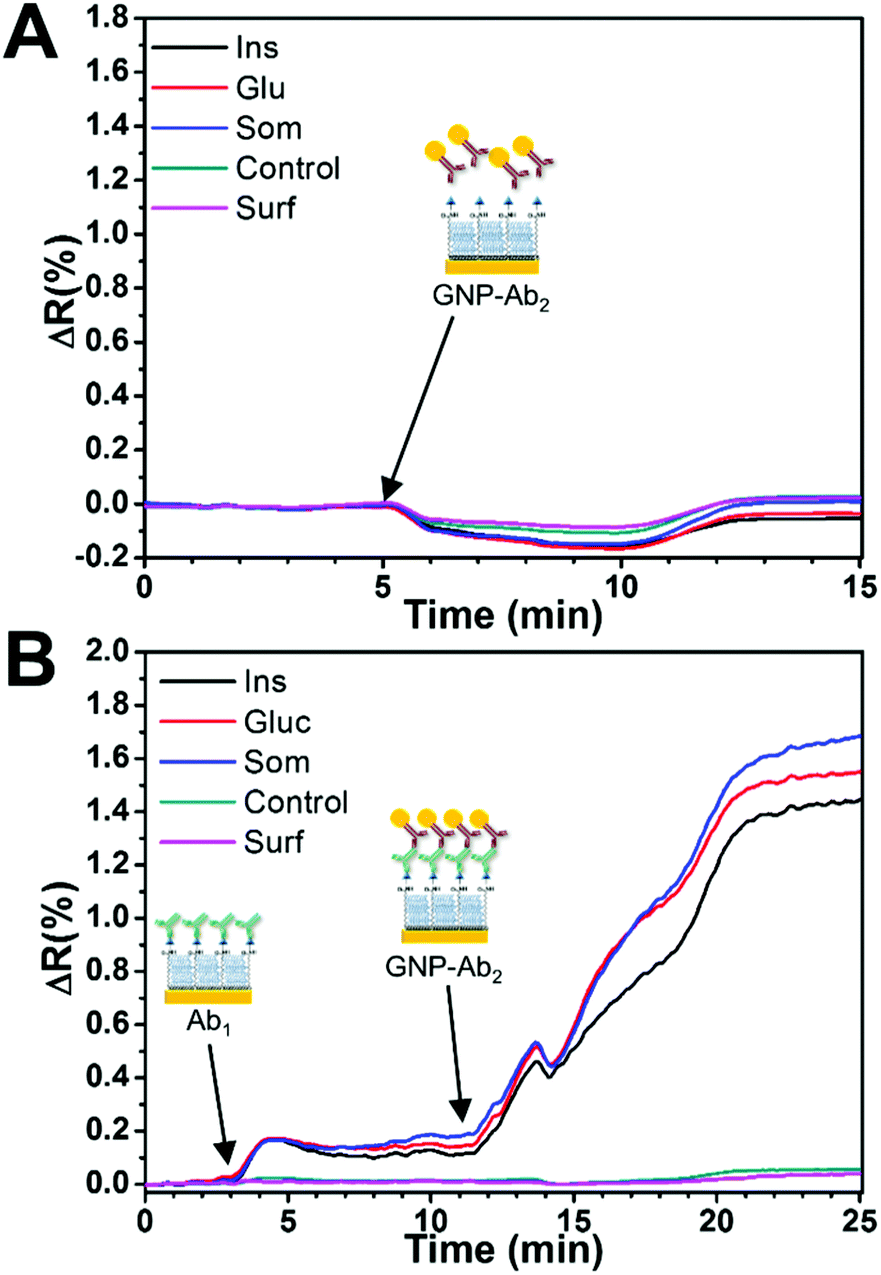 | ||
| Fig. 5 Real-time SPR angle shift sensorgrams of (A) the nonspecific binding effect of GNP-Ab2 conjugates and (B) the specific binding effect of GNP-Ab2 conjugates after primary antibody injection (blank injection C0). Immobilized BSA and the bare SAM surface identified as “Control” and “Surf” were used as negative controls. | ||
Fig. 5B shows a typical sensor response to a blank solution (mixture of all antibodies) and the subsequent amplification effect of the GNP-Ab2 conjugates. First, the injection caused a small increase in the SPR signal on the relevant hormone spots while an almost no response for the control spots, indicating specific binding. For these experiments, the initial concentration of primary antibodies (C0) was fixed to 0.2 μg mL−1 for anti-insulin, 0.05 μg mL−1 for anti-glucagon and 0.6 μg mL−1 for anti-somatostatin. This Ab1 concentration produced a small signal of ≈0.15ΔR for all hormone spots. The subsequent injection of GNP-Ab2 conjugates generated an SPR signal enhancement of ≈10 times, which was consistent with previous literature reports where 20 nm GNP-Ab2 conjugates have been used for signal amplification.28,43
It is noteworthy that the C0 signal obtained for somatostatin during individual GNP-Ab2 was smaller (ΔR = 0.93, Table 1) compared to that obtained during the multiplex assay (ΔR = 1.51, Table 2). This difference is likely due to some cross-reactivity between the different species of secondary antibodies in GNP-Ab2. However, due to the absence of non-specific interactions with the hormone spots and the controls, it was possible to use these conjugates for multiplex detection since the sensor response (ΔR) was always consistent and reproducible for all targeted hormones.
To further assess the sensor's resistance to non-specific adsorption of proteins, at the end of each calibration curve, two solutions containing either BSA or LYZ, both with a final concentration of 1 mg mL−1, were injected to the system following the same protocol as that for the hormone immunoassay. After an abrupt increase in the SPR signal during the injections due to the high bulk refractive index of the solution, the shift in reflectivity was measured after 10 minutes of contact time and 5 minutes of PBS-T wash. Since LYZ presented the largest ΔR among the two tested proteins, it was used as a reference to quantitatively evaluate the antifouling properties of the sensor. The mean (n = 3) ΔR response to BSA and LYZ from the hormone-functionalized spots and controls is shown in Table S1 in the ESI.† It was found that the adsorbed amount of LYZ was ≈100 pg mm−2 for all spots assuming that 1 RU = 1 μRIU = 1 pg mm−2 of surface mass shift for a fixed wavelength of 800 nm.44 This was very consistent with the definition of an antifouling surface.45 Moreover, a short (30 s) injection of a regeneration solution returned the baseline to its original value, indicating a weak interaction of LYZ and BSA on the sensor's surface.
Fig. 6 shows the average calibration curves for the multiplex sensing of serially diluted mixtures of insulin, glucagon, and somatostatin in PBS-T obtained from three independent sensor chips. The calculated LOD and dynamic range obtained (Table 2) for the three hormones in multiplexed mode were somehow comparable to those of ELISA kits (0.001–40 ng mL−1 depending on the hormone targeted) assessing individual hormone quantification. However, our sensing approach has the advantage of multiplexing, a larger working range and a relatively low analysis time of 32 min per point. Moreover, each multiplex SPRi immunosensing surface showed good stability on which over 21 binding/regeneration cycles were performed.
 | ||
| Fig. 6 Calibration curves for a multiplex immunoassay using GNP-Ab2 conjugates as the amplification strategy in PBS-T. Solid lines correspond to the fitting of a nonlinear 4PL model for different concentrations of insulin (blue), glucagon (red), and somatostatin (black). For each hormone, mean relative binding values (C/C0) were plotted as a function of hormone concentration (ng mL−1). The mean SPR shift was measured in sets of triplicate spots for each hormone and the controls. Then, the average SPR shift was calculated using 3 independent multiplex sensors (n = 3) corresponding to the reported SD. | ||
The use of GNPs for SPR signal amplification led to a remarkable LOD improvement for all tested hormones. An increase of 9-fold for insulin, 10-fold for glucagon and 200-fold for somatostatin detection was obtained as compared to the multiplex sensing approach without using GNP conjugate amplification.22 Noteworthy is the fact that somatostatin showed a dramatic improvement in the LOD. This could be explained by the fact that somatostatin is the smallest of the targeted hormones in this study (MW = 1637.88 Da). Thus, under the same immobilization conditions, the maximum amount of immobilization is expected to be lower than that of the higher MW hormones.46 This could lead to less steric hindrance for binding of the large GNP-Ab2 conjugates. This is corroborated by the fact that despite showing a similar response for primary antibody injection, somatostatin produced a slightly larger SPR shift for GNP-Ab2 conjugates (Fig. 5).
The LOD achieved in this study is in accordance with previous studies where detection of insulin15 and glucagon16 secreted from 10 islets was achieved at 15 mM glucose. Therefore, the somatostatin secreted from this small population of islets could be effectively and accurately detected by our proposed approach, opening the possibility of gaining better understanding of its paracrine communications associated with abnormal islets’ function in diabetes.
Conclusions
To address the challenges in the detection of low molecular weight hormones secreted in very low concentrations by human islets such as somatostatin, we investigated three GNP amplification strategies using an SPRi-based biosensing approach. Although the amplification method involving the conjugation of primary antibodies with GNPs showed the best performance for sensing of individual hormones, it presented large cross-reactivity during multiplex experiments. This cross-reactivity was successfully circumvented using an immunoassay with secondary antibodies conjugated to GNPs as amplification. We successfully achieved multiplex detection of three pancreatic islet related hormones with LODs of 0.15 nM, 0.39 nM and 1.22 nM for insulin, glucagon and somatostatin, respectively, with a total analysis time of 32 min per point. This performance is comparable with the previously reported detection sensitivity of insulin and glucagon secreted from 10 islets as well with the individual hormone sensing using conventional ELISA kits.The possibility of working with a small pancreatic islet population combined with the advantage of multiplexing, a wide working concentration window and a low analysis time makes our sensor very suitable for its future application in secretion-monitoring of pancreatic islets, particularly for understanding the paracrine effect of somatostatin on glucose-induced insulin and glucagon secretion. Furthermore, integrating a microfluidic perfusion platform with the proposed sensing approach could allow one to perform multiparametric analysis of a SF of pancreatic islets in the context of discovery of novel drugs for diabetes treatment.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the financial support from the Natural Science and Engineering Research Council of Canada (NSERC) and the NSERC-CREATE in ISS grants and the FRC scholarship from Consejo Nacional de Ciencia y Teconología de México (CONACyT) and Secretaría de Educación Pública de México (SEP). The authors are also thankful to Dr. F Melaine for her help during the initial exploration of GNPs as the amplification strategy.References
- F. R. Castiello, K. Heileman and M. Tabrizian, Lab Chip, 2016, 16, 409–431 RSC
.
- M. Braun, R. Ramracheya, S. Amisten, M. Bengtsson, Y. Moritoh, Q. Zhang, P. R. Johnson and P. Rorsman, Diabetologia, 2009, 52, 1566–1578 CrossRef CAS PubMed
- S. J. Le Marchand and D. W. Piston, J. Biol. Chem., 2010, 285, 14389–14398 CrossRef CAS PubMed
- V. Grill, M. Gutniak, A. Roovete and S. EfendiĆ, J. Clin. Endocrinol. Metab., 1984, 59, 293–297 CrossRef CAS PubMed
- O. Segers, M. Vroede, Y. Michotte and G. Somers, Diabetic Med., 1989, 6, 232–238 CrossRef CAS PubMed
- K. Ramachandran, X. Peng, K. Bokvist and L. Stehno-Bittel, Br. J. Pharmacol., 2014, 171, 3010–3022 CrossRef CAS PubMed
- D. T. Nguyen, D. van Noort, I. K. Jeong and S. Park, Biofabrication, 2017, 9, 015021 CrossRef PubMed
- Y. Xing, M. Nourmohammadzadeh, J. E. M. Elias, M. W. Chan, Z. Q. Chen, J. J. McGarrigle, J. Oberholzer and Y. Wang, Biomed. Microdevices, 2016, 18, 9 CrossRef PubMed
- M. Braun, R. Ramracheya, P. R. Johnson and P. Rorsman, Ann. N. Y. Acad. Sci., 2009, 1152, 187–193 CrossRef CAS PubMed
- S. T. Hanna, G. M. Pigeau, J. Galvanovskis, A. Clark, P. Rorsman and P. E. MacDonald, Pflug. Arch. Eur. J. Physiol., 2009, 457, 1343–1350 CrossRef CAS PubMed
- D. S. Luciani and J. D. Johnson, Mol. Cell. Endocrinol., 2005, 241, 88–98 CrossRef CAS PubMed
- M. Braun, R. Ramracheya, M. Bengtsson, A. Clark, J. N. Walker, P. R. Johnson and P. Rorsman, Diabetes, 2010, 59, 1694–1701 CrossRef CAS PubMed
- R. Ramracheya, C. Ward, M. Shigeto, J. N. Walker, S. Amisten, Q. Zhang, P. R. Johnson, P. Rorsman and M. Braun, Diabetes, 2010, 59, 2198–2208 CrossRef CAS PubMed
- K. Terao, M. Gel, A. Okonogi, A. Fuke, T. Okitsu, T. Tada, T. Suzuki, S. Nagamatsu, M. Washizu and H. Kotera, Sci. Rep., 2014, 4, 6 Search PubMed
- M. G. Roper, J. G. Shackman, G. M. Dahlgren and R. T. Kennedy, Anal. Chem., 2003, 75, 4711–4717 CrossRef CAS PubMed
- J. G. Shackman, K. R. Reid, C. E. Dugan and R. T. Kennedy, Anal. Bioanal. Chem., 2012, 402, 2797–2803 CrossRef CAS PubMed
- A. R. Lomasney, L. Yi and M. G. Roper, Anal. Chem., 2013, 85, 7919–7925 CrossRef CAS PubMed
- J. P. Lafleur, A. Jonsson, S. Senkbeil and J. P. Kutter, Biosens. Bioelectron., 2016, 76, 213–233 CrossRef CAS PubMed
- S. S. Hinman, K. S. McKeating and Q. Cheng, Anal. Chem., 2018, 90, 19–39 CrossRef CAS PubMed
- G. Spoto and M. Minunni, J. Phys. Chem. Lett., 2012, 3, 2682–2691 CrossRef CAS PubMed
- H. J. Lee, D. Nedelkov and R. M. Corn, Anal. Chem., 2006, 78, 6504–6510 CrossRef CAS PubMed
- F. R. Castiello and M. Tabrizian, Anal. Chem., 2018, 90, 3132–3139 CrossRef CAS PubMed
- O. Cabrera, D. M. Berman, N. S. Kenyon, C. Ricordi, P. O. Berggrern and A. Caicedo, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 2334–2339 CrossRef CAS PubMed
- L. A. Lyon, M. D. Musick and M. J. Natan, Anal. Chem., 1998, 70, 5177–5183 CrossRef CAS PubMed
- J. Jung, K. Na, J. Lee, K. W. Kim and J. Hyun, Anal. Chim. Acta, 2009, 651, 91–97 CrossRef CAS PubMed
- S. Szunerits, J. Spadavecchia and R. Boukherroub, Rev. Anal. Chem., 2014, 33, 153–164 CAS
- M. Frasconi, C. Tortolini, F. Botre and F. Mazzei, Anal. Chem., 2010, 82, 7335–7342 CrossRef CAS PubMed
- J. S. Mitchell, Y. Wu, C. J. Cook and L. Main, Anal. Biochem., 2005, 343, 125–135 CrossRef CAS PubMed
- J. S. Mitchell and T. E. Lowe, Biosens. Bioelectron., 2009, 24, 2177–2183 CrossRef CAS PubMed
- S. W. Zeng, D. Baillargeat, H. P. Ho and K. T. Yong, Chem. Soc. Rev., 2014, 43, 3426–3452 RSC
- D. C. Kabiraz, K. Morita, K. Sakamoto and T. Kawaguchi, Talanta, 2017, 172, 1–7 CrossRef CAS PubMed
- I. E. Sendroiu, L. K. Gifford, A. Lupták and R. M. Corn, J. Am. Chem. Soc., 2011, 133, 4271–4273 CrossRef CAS PubMed
- J. U. Lee, A. H. Nguyen and S. J. Sim, Biosens. Bioelectron., 2015, 74, 341–346 CrossRef CAS PubMed
- K. V. Gobi, H. Iwasaka and N. Miura, Biosens. Bioelectron., 2007, 22, 1382–1389 CrossRef CAS PubMed
- S. Takae, Y. Akiyama, Y. Yamasaki, Y. Nagasaki and K. Kataoka, Bioconjugate Chem., 2007, 18, 1241–1245 CrossRef CAS PubMed
- L. Huang, G. Reekmans, D. Saerens, J.-M. Friedt, F. Frederix, L. Francis, S. Muyldermans, A. Campitelli and C. V. Hoof, Biosens. Bioelectron., 2005, 21, 483–490 CrossRef CAS PubMed
- S. Taufik, A. Barfidokht, M. T. Alam, C. Jiang, S. G. Parker and J. J. Gooding, J. Electroanal. Chem., 2016, 779, 229–235 CrossRef CAS
- J. D. Driskell, C. A. Jones, S. M. Tompkins and R. A. Tripp, Analyst, 2011, 136, 3083–3090 RSC
- M. C. Estevez, J. Belenguer, S. Gomez-Montes, J. Miralles, A. M. Escuela, A. Montoya and L. M. Lechuga, Analyst, 2012, 137, 5659–5665 RSC
- T. Kawaguchi, D. R. Shankaran, S. J. Kim, K. Matsumoto, K. Toko and N. Miura, Sens. Actuators, B, 2008, 133, 467–472 CrossRef CAS
- D. Juncker, S. Bergeron, V. Laforte and H. Li, Curr. Opin. Chem. Biol., 2014, 18, 29–37 CrossRef CAS PubMed
- J. T. Dias, L. Lama, J. Gantelius and H. Andersson-Svahn, Nanoscale, 2016, 8, 8195–8201 RSC
- J. Martinez-Perdiguero, A. Retolaza, L. Bujanda and S. Merino, Talanta, 2014, 119, 492–497 CrossRef CAS PubMed
-
R. B. Schasfoort, Handbook of surface plasmon resonance, Royal Society of Chemistry, United Kingdom, 2nd edn, 2017 Search PubMed
- H. Vaisocherova, E. Brynda and J. Homola, Anal. Bioanal. Chem., 2015, 407, 3927–3953 CrossRef CAS PubMed
- J. Lahiri, L. Isaacs, J. Tien and G. M. Whitesides, Anal. Chem., 1999, 71, 777–790 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Gold nanoparticle functionalization characterization: including surfaces and GNP-Ab conjugates. See DOI: 10.1039/c9an00140a |
| This journal is © The Royal Society of Chemistry 2019 |