
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Matrix-assisted laser desorption/ionization tandem mass spectrometry for identification of organic tattoo pigments in inks and tissue samples†
Ines
Schreiver‡
 *,
Lisa-Marie
Eschner‡
and
Andreas
Luch
*,
Lisa-Marie
Eschner‡
and
Andreas
Luch
German Federal Institute for Risk Assessment (BfR), Department of Chemical and Product Safety, Max-Dohrn-Straße 8-10, 10589 Berlin, Germany. E-mail: ines.schreiver@bfr.bund.de; Tel: +49 30 18412 4181
First published on 27th July 2018
Abstract
With regard to the increasing number of tattooed people, legal regulations for tattoo inks were implemented across Europe—aiming for higher consumer safety. To control the laws’ abidance, analytical methods are needed to identify banned ingredients from the given negative lists. Since specific organic pigments are often associated with tattoo side effects, their identification in tattoo inks as well as in biological samples is of great importance. Particularly, poorly soluble organic pigments are challenging to detect. In the past, matrix-assisted laser desorption/ionization mass spectrometry (MALDI-MS) was reported as a promising tool for organic pigment identification. Here, we present a MALDI tandem mass spectrometry (MS/MS) approach to increase identification specificity and sensitivity in the process based on pigment fragment ions which is of special importance in tissue samples. For further verification of pigment identities, alkali metal cation attachment was used. Sample preparation was optimized and included mechanical disruption followed by the application as dried droplets with the matrices α-cyano-4-phenylcinnamic acid and sinapinic acid as well as ethanol. Pigments were identified by spectral comparison to reference libraries containing 40 pigments and following a decision tree. Additionally, successful pigment identification in biological samples was carried out. The implemented automated MALDI-MS and -MS/MS acquisitions make the hereby proposed pigment identification suitable for routine application.
Introduction
In response to the increasing popularity of tattooing in the past decades, the European Commission published the resolution ResAP(2008)1 to improve the safety of the used inks.1 However, only few European countries implemented laws based on this resolution.2 Currently, the European Chemical Agency is proposing a restriction for hazardous substances in tattoo inks and permanent make-up.3 Common to all, regulation is based on a negative list banning critical substances from their use in tattoo inks which also comprises specific pigments.2,4 To accomplish the compliance of products in the market, state laboratories have to identify banned pigments based upon reliable analytical methods. Moreover, the distinct identification of pigments in skin biopsies is also of great importance in terms of deciphering adverse tattoo reactions. Especially, tattoo allergies are often correlated with the use of certain pigments.5Nowadays, pigments used in tattoo colorants are mostly organic substances besides the frequently used carbon black and white titanium dioxide.6 Due to their poor solubility in common solvents, it is challenging to analyze organic pigments in both tattoo inks as well as skin samples.
In the past, Fourier-transform infrared spectroscopy (FT-IR)7 as well as Raman spectroscopy8 have been used for pigment identification. These methods are suitable to pinpoint the most abundant pigments. However, they are limited in the analysis of pigment mixtures due to missing separation techniques. Pyrolysis-gas chromatography mass spectrometry (py-GC/MS) is a further approach helpful in the analysis of tattoo inks.9,10 Yet, a major drawback of this is the requirement of characteristic cleavage patterns for pigment identification.
Other studies focused on the use of high performance liquid chromatography (HPLC) which offers the possibility of upstream separation prior to substance detection, but which is again limited by the poor solubility of the pigments.11,12
Laser desorption/ionization- (LDI) and matrix-assisted laser desorption/ionization (MALDI) time-of-flight (TOF)-MS were also described to be suitable for organic pigment analysis.12–14 Due to the pigment separation and high mass accuracy of TOF-MS, pigment identification should theoretically become feasible by the exact molecular weight, the isotope pattern and pigment fragments. Unfortunately, pure pigments might agglomerate causing a lowered mass accuracy due to the uneven distribution on the target plate. Identification of pigments without specific isotope or fragment patterns in MS acquisition will therefore suffer in specificity—especially when interfering substances are present. In a recent study, we investigated human tissue samples15 and found the identification of pigments via LDI-MS acquisition impeded by skin matrix molecules and low pigment concentrations. Characteristic molecular peak ions or fragment ions are sometimes of low abundance or missing when compared to reference spectra of pure pigments.
We here aim to improve the identification of tattoo pigments in complex matrices comprising inks and skin samples through MALDI-TOF-MS/MS. Especially upon equivocal results in MS acquisition, characteristic fragmentation patterns achieve a higher specificity. We also focused on a technical implementation of automated MALDI-MS and -MS/MS acquisition for an increased sensitivity in pigment identification in the presence of highly abundant interfering peaks. Additionally, tissue sample preparation was optimized. We established a decision tree for efficient and reproducible analysis of large sample numbers.
Experimental
Chemicals
Pigments used in this investigation were PV Fast Blue BG (pigment blue (P.B.) 15, C.I. 74160), PV Fast Green GNX (pigment green (P.G.) 7, C.I. 74260), Graphtol Orange GPS (pigment orange (P.O.) 13, C.I. 21110), Graphtol Red F3RK 70-CN09 (pigment red (P.R.) 170, C.I. 12475), PV Fast Red D3G (P.R.254, C.I. 56110) and PV Fast Red E3B (pigment violet (P.V.) 19, C.I. 73900) from Clariant (Frankfurt am Main, Germany). Heliogen Green D 9360 (P.G.36, C.I. 74265), Cinquasia Magenta K 4535 (P.R.202, C.I. 73907) and Paliotol Yellow D 0960 (pigment yellow (P.Y.) 138, C.I. 56300) were obtained from BASF (Ludwigshafen, Germany). MALDI matrix α-cyano-4-phenylcinnamic acid (CPA) was obtained from Sigma Aldrich (Munich, Germany). Sinapinic acid (SA) was purchased from Bruker Daltonik (Bremen, Germany). The enzyme collagenase from Clostridium histolyticum type IA was obtained from Sigma Aldrich (Munich, Germany), proteinase K from Tritirachium album from Quiagen (Hilden, Germany). 2-Propanol (≥99.5%), glycerol (reagent plus, ≥99.0%), lithium trifluoroacetate (95%), polyvinylpyrrolidone (PVP, K60, 45% in H2O) and propylene glycol (FCC, ≥99.5%) were purchased from Sigma Aldrich (Munich, Germany). Also, LC-MS grade acetonitrile (ACN), ethanol (EtOH) (absolute for analysis) and sodium carbonate (anhydrous, for analysis), all from Merck (Darmstadt, Germany), buffer ATL from Quiagen (Hilden, Germany), Dulbecco's phosphate buffered saline (DPBS) from PAN Biotech (Aidenbach, Germany), potassium dihydrogen phosphate (≥99%) from Carl Roth (Karlsruhe, Germany) as well as Hank's balanced salt solution (HBSS) and trifluoroacetic acid (TFA), both from Fischer Scientific (Schwerte, Germany) were used for the experiments. Further, ultrapure water (18.2 MΩ cm−1 at 25 °C) prepared with a Milli-Q water purification system from Merck (Darmstadt, Germany) was used.Pigment suspensions and tattoo inks
A tattoo matrix was simulated through mixing aqueous 20% solutions of glycerol and propylene glycol, ultrapure water and ACN as organic solvent in a volume ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1:2. ACN was used in analogy to the preparation of the calibration mixture and MALDI matrices instead of typical tattoo solvents like EtOH or 2-propanol. Pure pigments were pre-dispersed in ultrapure water in a concentration of 100 mg mL−1. Suspensions were further diluted with the self-mixed tattoo matrix to give final pigment concentrations of 80, 16 and 1.6 mg mL−1, respectively.
:1:1:2. ACN was used in analogy to the preparation of the calibration mixture and MALDI matrices instead of typical tattoo solvents like EtOH or 2-propanol. Pure pigments were pre-dispersed in ultrapure water in a concentration of 100 mg mL−1. Suspensions were further diluted with the self-mixed tattoo matrix to give final pigment concentrations of 80, 16 and 1.6 mg mL−1, respectively.
For mechanical disruption, 1 mL pigment suspension was added to a 2 mL reaction tube (Eppendorf, Hamburg, Germany) together with 2 stainless steel beads (5 mm diameter, Quiagen, Hilden, Germany). Samples were homogenized at 30 Hz for 5 min in a TissueLyser II (Quiagen, Hilden, Germany).
For preparation of commercial tattoo inks, ca. 50–100 μl (1–2 drops) of ink is given into a 1.5 ml reaction tube (Eppendorf, Hamburg, Germany). To precipitate the pigments, 1 ml ultrapure water are added to the ink followed by centrifugation at 500g for 10 min. Before spotting, samples were washed by resuspending the pigment pellet in EtOH and centrifugation using the same parameters. The pellet was taken up in 0.5 mL EtOH.
Preparation of skin samples
Postmortem tattooed porcine skin was used to mimic an extraction from skin matrix. Pig skin was obtained from VION food GmbH (Perleberg, Germany). Therefore, abdominal porcine skin was unhaired postmortem with electrical razor blades and stored at −20 °C until further use. Thawed porcine skin was tattooed with self-mixed tattoo ink. The inks contained each 8–10% w/v pigment dispersed in a mixture of an aqueous 40% solution of 2-propanol, an aqueous 1% PVP solution and ultrapure water. Before tattooing, each ink was further dispersed using a probe sonifier (Bandelin Sonopuls HD 2200, Bandelin Electronic, Berlin, Germany) with an amplitude of 10% at 200 W for 5 min. A Cheyenne hawk thunder tattoo machine with a 17 magnum long taper tattoo needle (thickness 0.35 mm, both from MT.Derm, Berlin, Germany) was used for ink application. Biopsies of 4 mm diameter were taken from postmortem tattooed porcine skin. In the course of our study, 3 pigment extraction methods were tested. At first, sole mechanical disruption was carried out in analogy to the pigment suspensions method by adding 1 mL HBSS to each biopsy. For the enzymatic digestion, the biopsy was lysed using 500 μL of a 1 mg mL−1 collagenase solution in HBSS with an incubation time of at least 16 h at 37 °C. An alternative enzymatic digestion was carried out using 500 μL of a solution containing proteinase K solution, ATL and DPBS in volume ratios of 1:9:200 with a 2 h incubation and shaking at 50 °C. Non-tattooed porcine skin as well as enzyme solutions without biological material served as controls. After incubation, 500 μL HBSS or DPBS were added to the samples and controls, respectively. Following enzymatic digestion, samples were mechanically disrupted as described above. Before spotting, the samples of each treatment were washed 3 times by resuspending the pigment pellet in ultrapure water followed by centrifugation at 500g for 10 min. The pellet was taken up with 0.3 mL of self-mixed tattoo matrix or EtOH.
Sample spotting for MALDI analysis
In addition to EtOH, samples were also applied as dried-droplets using the MALDI matrices CPA and SA. CPA is suitable for the desorption of polycyclic aromatic hydrocarbon16 while SA is commonly used for protein analysis.17 A 1:2 volume ratio mixture of ACN and an aqueous 0.1% TFA solution were used to obtain a saturated solution of the MALDI matrices CPA and SA. Samples were applied as dried-droplets. Each 5 μL of matrix solution or EtOH and 5 μL samples were premixed and 0.5 μL of the resulting solution was spotted on a 384-ground steel target plate (Bruker Daltonik, Bremen, Germany) followed by air-drying.
Alkali metal cation attachment
Polar organic compounds will form adduct ions [M + alkali]+ when alkali metal salts are added to the sample.18 For adduct formation, lithium, potassium and sodium salt solutions in concentrations of 0.3–3 g L−1 were used as described elsewhere.19 Each 0.5 μL were added to the 10 μL matrix solution sample mix.MALDI-MS and -MS/MS
MALDI measurements were performed using an UltrafleXtreme MALDI-TOF/TOF (Bruker Daltonik, Bremen, Germany). MS spectra in automatic mode were obtained by averaging 8000 individual spectra using a random walk movement, with a laser rate of 1000 Hz in positive reflector mode in an m/z range from 100 to 4000. The instrument was calibrated with an external ProteoMass MALDI Calibration Kit in the range of m/z 50 to 4000 before each analysis (Sigma Aldrich, Munich, Germany). For MS/MS analysis the instrument's LIFT technique with collision induced dissociation (CID) with argon as collision gas (3.5 bar) was used as described elsewere.20 For MS/MS sum spectra, 2000 parent and 8000 fragment spectra were accumulated with a random walk movement. The parent was assigned automatically and a detector gain boost of 140%, a laser energy boost of 1% and an analog offset of 0.5% were used in fragment mode. The Precursor Ion Selector (PCIS) range was set to 0.45% of the parent mass and the Post LIFT Metastable Suppressor (PLMS) was turned off. Data were processed using flexControl 3.4 and flexAnalysis 3.4 software (Bruker Daltonik, Bremen, Germany). Isotope distributions were calculated using the online tool Isotope Distribution Calculator and Mass Spec Plotter (Scientific Instrument Services, Ringoes, NJ, USA).21Automation of MALDI-MS and -MS/MS acquisition
WARP-LC 1.4 version 1.4.0.0 and ProteinScape version 4.0.0 253 (both from Bruker Daltonik, Bremen, Germany) were used for automation of MALDI-MS and -MS/MS acquisition. Samples were spotted with empty lines in between to analyze each one empty spot after a sample. This was necessary since WARP-LC picks only one of two consecutive samples for MS/MS analysis if same masses appear in both. Therefore, the minimum value for merging sample fractions of 1 needs to be chosen. For the automated work-flow, an AutoXecute run was programmed in WARP-LC. Here, a ProteinScape Experiment ID had to be given to start the measurements. Data generated in ProteinScape were of no interest for the further analysis and were deleted afterwards. Parameters used in the AutoXecute run were set in flexAnalysis. Integration parameters were set to centroid peak detection algorithm, a signal to noise threshold of 10 as well as a relative intensity threshold and minimum intensity threshold of 0%.Initial laser energy started at a lower value of 45% and was automatically increased to 70% if spectra quality was not sufficient for integration. Spectrum acquisition was quit after 15 subsequently failed judgements. A maximum of 400 peaks were integrated with a width of m/z 0.1. The height was set to 80% and TopHat baseline subtraction was used. Peaks were integrated with the set parameters and the generated peak list was automatically compared to a precursor list in WARP-LC. Mass tolerance was set to 0.7 Da, the minimum of 1 fraction was chosen for merging of compounds and background identification was not carried out. The scheduled precursor list for MS/MS acquisition contained the m/z values of the pigments’ precursors determined beforehand. Mass tolerance was set to ±0.2 Da. For MALDI-MS/MS acquisition the signal to noise threshold was set to 1, minimal mass distance to co-elution compounds to 1 Da, maximum number of 100 MS/MS per fraction with a descending acquisition order and include compounds only. For integration of MS/MS spectra, the centroid peak detection algorithm, a signal to noise threshold of 3, a relative intensity threshold of 0% and a minimum intensity threshold of 0% were chosen. Laser intensity was automatically increased from 60% to 90% if spectra quality was not sufficient for integration. A maximum of 200 peaks were integrated with a width of m/z 0.2, a height of 80% and TopHat baseline subtraction was used. Integration and smoothing of the MS and MS/MS spectra were performed with a macro each. Both, the acquired MALDI-MS and -MS/MS spectra were exported as Mascot Generic Format (*.MGF) via these macros (ESI Tables S1 and S2†).
Pigment identification
Acquired MALDI-MS and -MS/MS spectra were renamed from *.MGF into *.MSP format with the program Bulk rename utility version 3.0.0.1 (TGRMN Software, Fullarton, Australia). Pigment identification was performed with NIST MS Search 2.0 (National Institute of Standards and Technology, Gaithersburg, MD, USA) using 3 self-made libraries containing MS, lithium-MS and MS/MS reference spectra of pure pigments. Library search options for MS spectra were set to normal identity spectrum search type with penalizing of rare compounds and default presearch. The molecular weight was constrained from m/z 250 to 2000 and a minimum abundance of 3. MS/MS library search was performed with an m/z tolerance for the precursor of ±1 and for the product of ±2. Evaluation of pigment identity was carried out using a developed decision tree.Results & discussion
Method overview
For pigment identification the tattoo inks or tattooed skin samples are either diluted or enzymatically digested for purification of the used colorant (Scheme 1). Precipitated colorants are spotted in two different matrices and EtOH to cope for discrimination of single pigments or fragment ions. Automated MS acquisition and peak evaluation is carried out with subsequent MS/MS analysis if masses of known pigments are present. Corresponding MS and MS/MS spectra are screened using the NIST program and a self-made pigment library. Identification is carried out using a decision tree which includes a comparison of original high resolution spectra of samples and library pigments. Whenever pigment identification is insufficient, cation attachment with Li+ may be used to increase signal to noise ratio and verify mass shifts for pigments that can be ionized this way.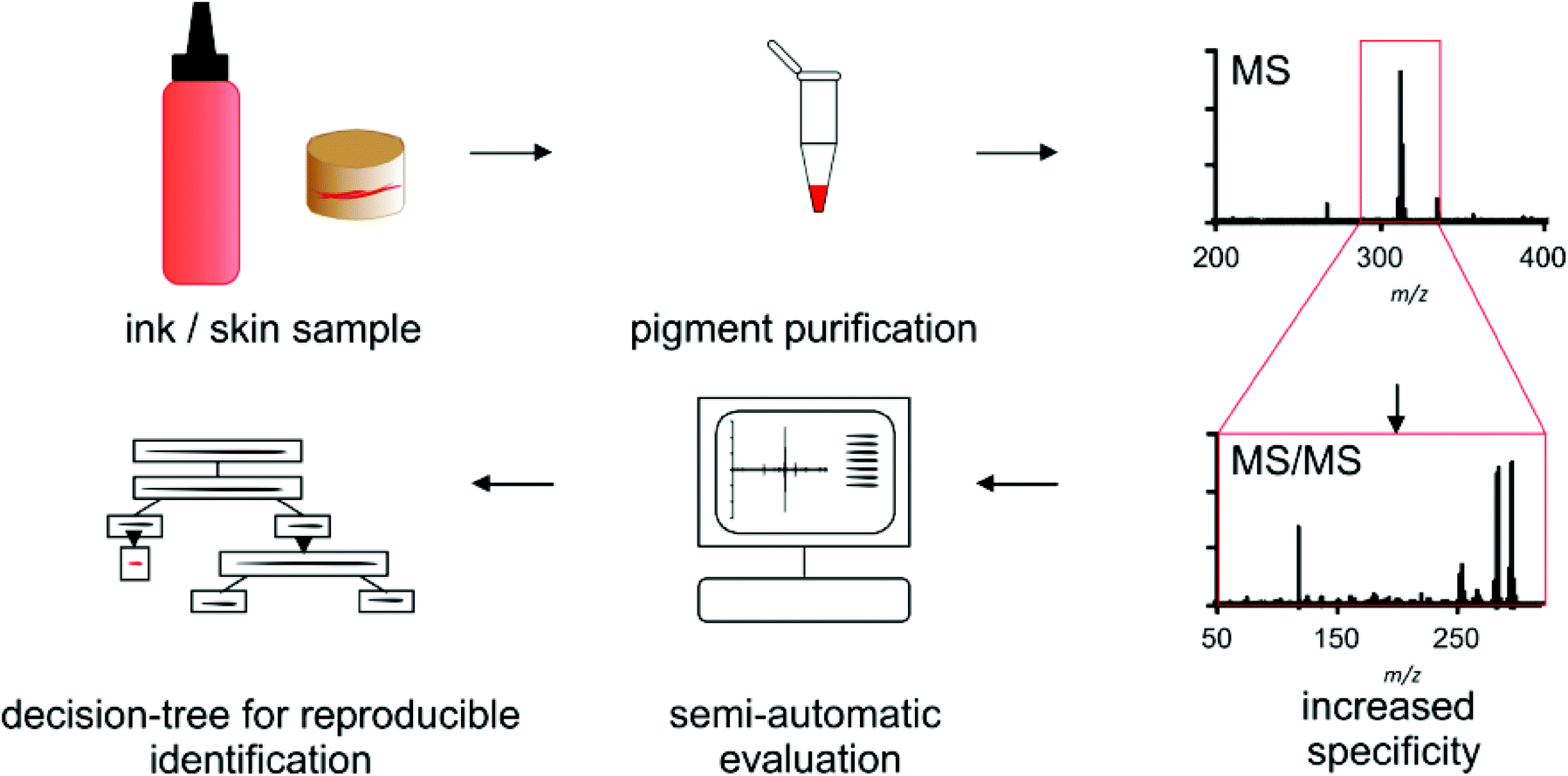 | ||
| Scheme 1 Highly specific identification of water-insoluble organic pigments is facilitated by automated matrix-assisted laser desorption/ionization tandem mass spectrometry. Semi-automated evaluation following a decision-tree allows for reproducible identification in complex matrices for market surveys and research of tattoo-derived side effects. | ||
Optimization of sample preparation
As a first step to establish a useful pigment identification method, the sample preparation was optimized. Mass accuracy in MALDI-MS acquisition is dependent on homogeneous thin sample spotting. Pigment agglomeration leads to an increased nonuniform distance from sample to target plate and thus a loss of mass accuracy. Therefore, samples were mechanically disrupted. Hereby, pigment agglomerates were broken down leading to a clear improvement of the pigment size homogeneity which was visible by a reduced number of large agglomerates on the target spots visible via the camera in the MALDI system.Spotting in EtOH was feasible since most pigments are well ionized without matrix addition. EtOH was used to circumvent matrix interference with small pigments and eases sample preparation. Samples were also spotted as dried droplets using the matrices CPA and SA since the ionization efficiency of pigments varies in different matrices (ESI Fig. S1†). For some pigments, characteristic fragment ions could already be detected in addition to the molecular ion peak in the MS spectrum.
Sample preparation was also optimized for the identification of tattoo pigments in biological samples. This complex matrix was simulated with porcine skin tattooed postmortem with the 8 different pigments P.B.15, P.G.7, P.G.36, P.O.13, P.R.170, P.R.254, P.V.19 and P.Y.138. The pigments represent the six most important pigment classes phthalocyanines, diazo pigments, naphthol AS pigments, diketopyrrolopyrroles, quinacridones and quinophthalones (Fig. 1, ESI Fig. S2†). In total, 3 different digestion strategies were tested comprising only mechanical disruption, enzymatic digestion with collagenase or proteinase K each followed by mechanical disruption. The treatment with collagenase combined with mechanical disruption led to the best dissolution of the skin and therefore released the highest amounts of pigments embedded in the dermis which were subsequently available for identification by MALDI. Recorded spectra of pigments in pig skin are comparable to pure pigments spotted in EtOH or different matrices for most pigments. However, in the case of P.O.13 and P.R.254 additional peaks from skin or collagenase treatment appear in the spectra (Fig. 1B and C). Low abundant pigments might be overlooked MS spectra with high abundant matrix molecules. By using the MALDI-MS/MS method described in the following, all pigments can be unequivocally identified despite possible skin matrix interference.
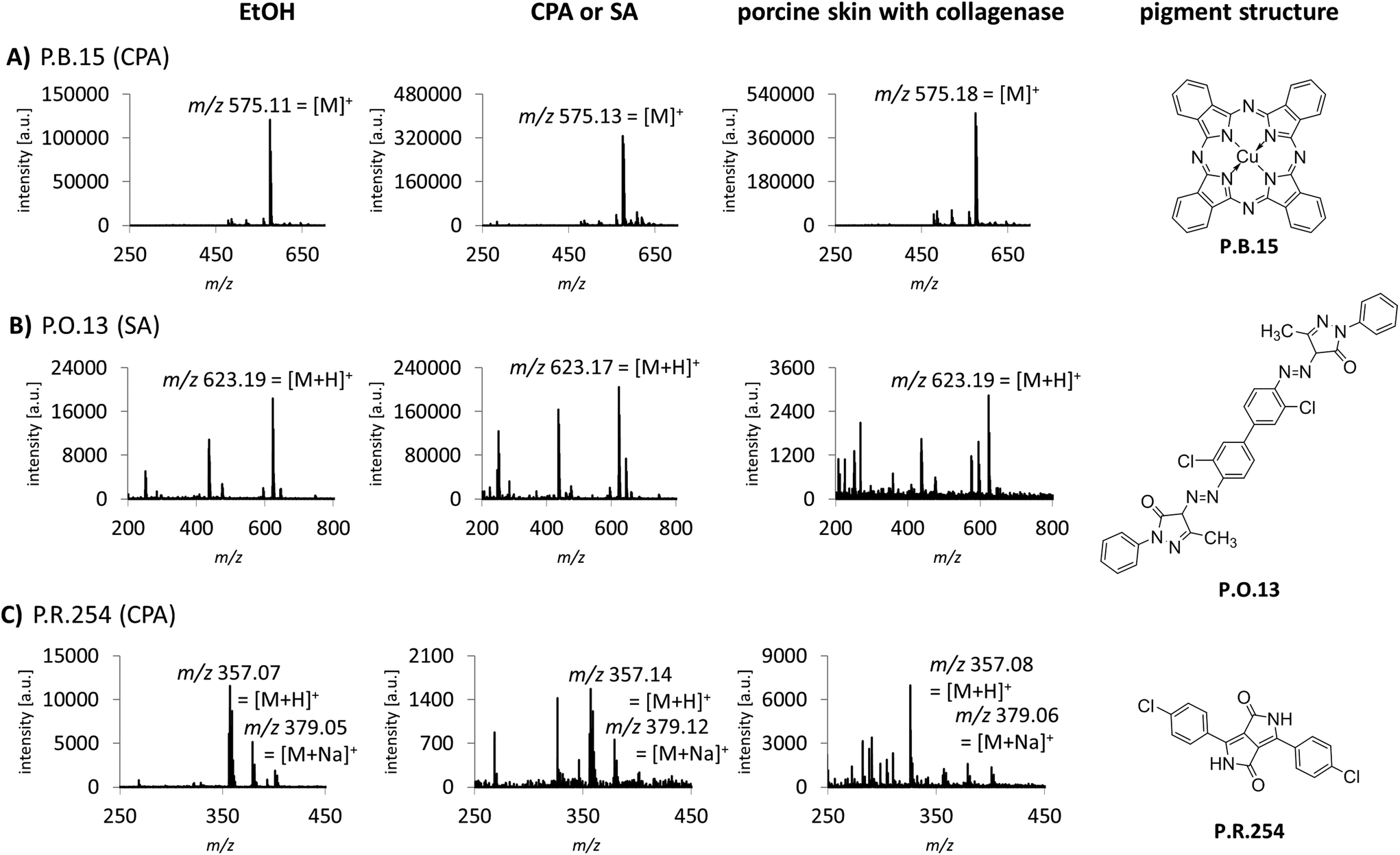 | ||
| Fig. 1 Comparison of spectra of pure pigments dispersed in the tattoo matrix and pigments extracted from porcine skin. Optimized preparation of biological samples included enzymatic digestion with collagenase followed by mechanical disruption. Spectra of pure pigments in EtOH (left) or matrix (middle) are displayed for each pigment in (A)–(C), respectively. Spectra of pigments extracted from pig skin are shown on the right. The matrix used for each pigment is given in brackets. | ||
MALDI-MS/MS
The identification of organic pigments without specific isotope and fragment patterns by standard MALDI-MS methods is based on the assumption that no other molecule with the same combination of atoms is present in the sample. Especially regarding complex matrices like lysed skin, this assumption might not be generally true. In a recent study, we found P.O.13 with only low abundancy of the molecular peak in human skin samples.15 Additionally, fragment ions induced by laser ionization are sometimes suppressed by the tissue matrix. Low pigment abundancies might also lead to equivocal MS spectra which are not sufficient to clearly identify the corresponding pigments in tissue samples. Therefore we needed to improve specificity of pigment identification in tissue samples. We tested an MALDI-MS/MS approach with an argon based collision looking for characteristic fragment ions for pigment identification. P.V.19 displays a good example for a relatively unspecific MALDI-MS spectrum. Mainly its molecular mass peak [M + H]+ with m/z 313 and minor amounts of its corresponding sodium ion adducts [M + Na]+ with m/z 335 are present (Fig. 2A and B). In contrast, the MS/MS spectrum contains several fragment ions that are cleaved from the pigment in a structure dependent manner (Fig. 2C). In accordance to common cleavage products from pyrolysis,9 possible decomposition products of P.V.19 were postulated (Fig. 2D and E). The presence of nearly equally high peaks with a mass difference of m/z 1 leads to the assumption that a mixture of fragment ions with hydroxy or keto groups might be present simultaneously. The addition of the calculated isotope distributions of both fragment ions results in a pattern close to the one observed in the spectra (Fig. 2D and E). Most azo pigments already show fragmentation patterns in standard MS acquisition which lead to higher specificity in pigment identification if matrix interferences are low and pigment concentrations are high, e.g. in purified tattoo inks.12 In tissue samples, the use of the MS/MS technique helps to make the identification of these pigment incontrovertible if the comparison of MS spectra is unsatisfying.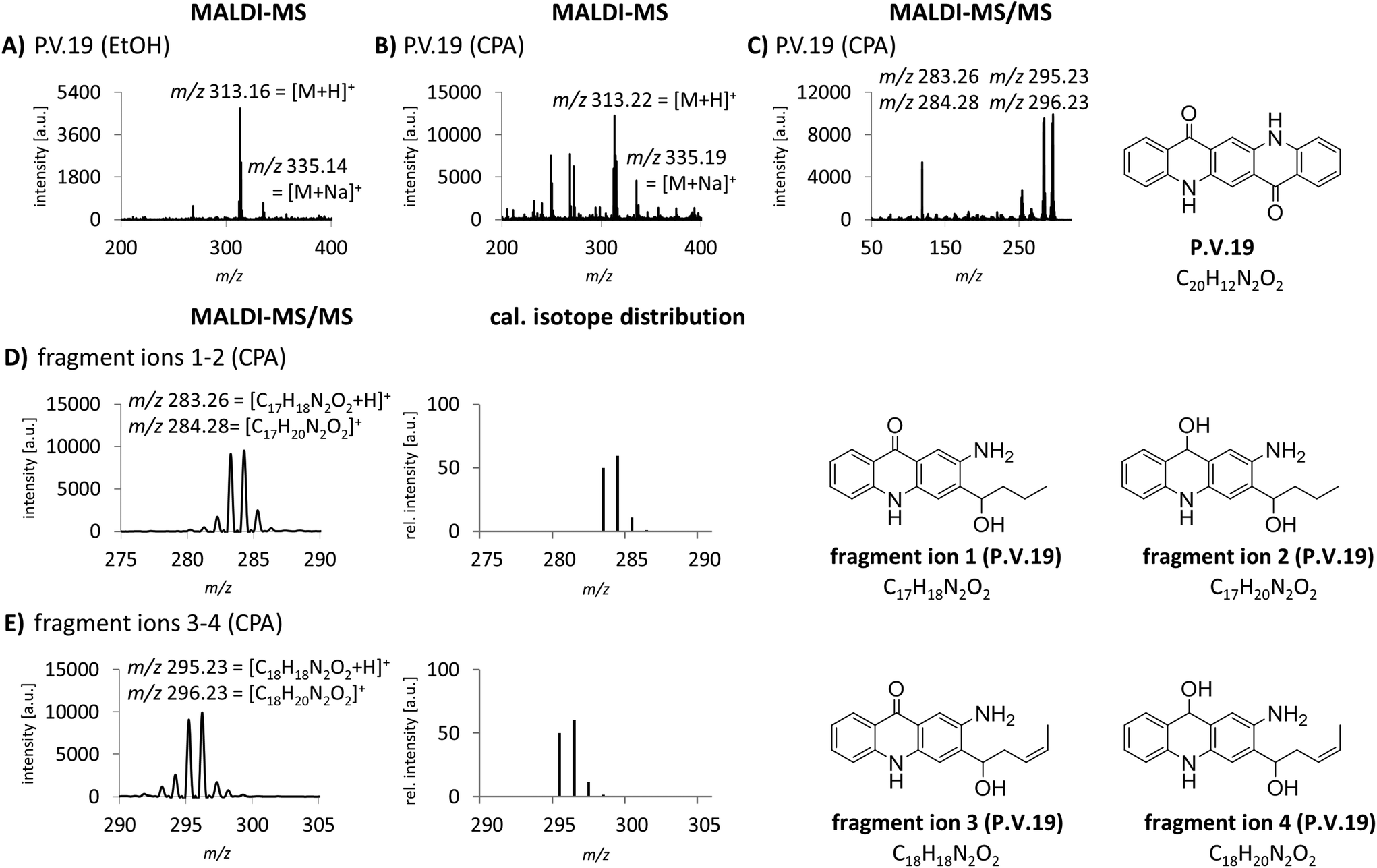 | ||
| Fig. 2 Increase of specificity in the analysis of P.V.19 by MALDI-MS/MS. (A) MALDI-MS spectrum of P.V.19 spotted with EtOH. (B) MALDI-MS spectrum of P.V.19 spotted with CPA. (C) MALDI-MS/MS spectrum with the precursor ion m/z 313 (left) and the structural formula of P.V.19 (right). MS/MS analysis was performed with CPA matrix to improve ionization of the pigment. (D) MS/MS spectrum of the fragment ions 1 and 2 (left), addition of the calculated isotope distributions of fragment ion 1 and 2 (center) with the proposed structural formulas (right). (E) MS/MS spectrum of the fragment ions 3 and 4 (left), addition of the calculated isotope distributions of fragment ion 3 and 4 (center) with the proposed structural formulas (right). Abbreviations: cal. = calculated, rel. = relative. | ||
MALDI-MS and -MS/MS analysis with alkali metal cation attachment
Cation attachment is a possible approach to verify the molecular peaks and possible adduct ions obtained in the MS spectra. Here, the formation or the switch to specific adduct ions [M + alkali]+ is forced by adding an excessive amount of the chosen alkali salt to the sample. This will lead to the corresponding m/z shift in the spectrum compared to the control or pure sample. Alkali salts like sodium are likely present in complex mixtures such as tattoo inks or biological samples. In the case of P.R.170, a peak of m/z 477 corresponding to the sodium adduct [M + Na]+ with an additional fragment peak at m/z 318 are seen in the MS spectrum (Fig. 3A).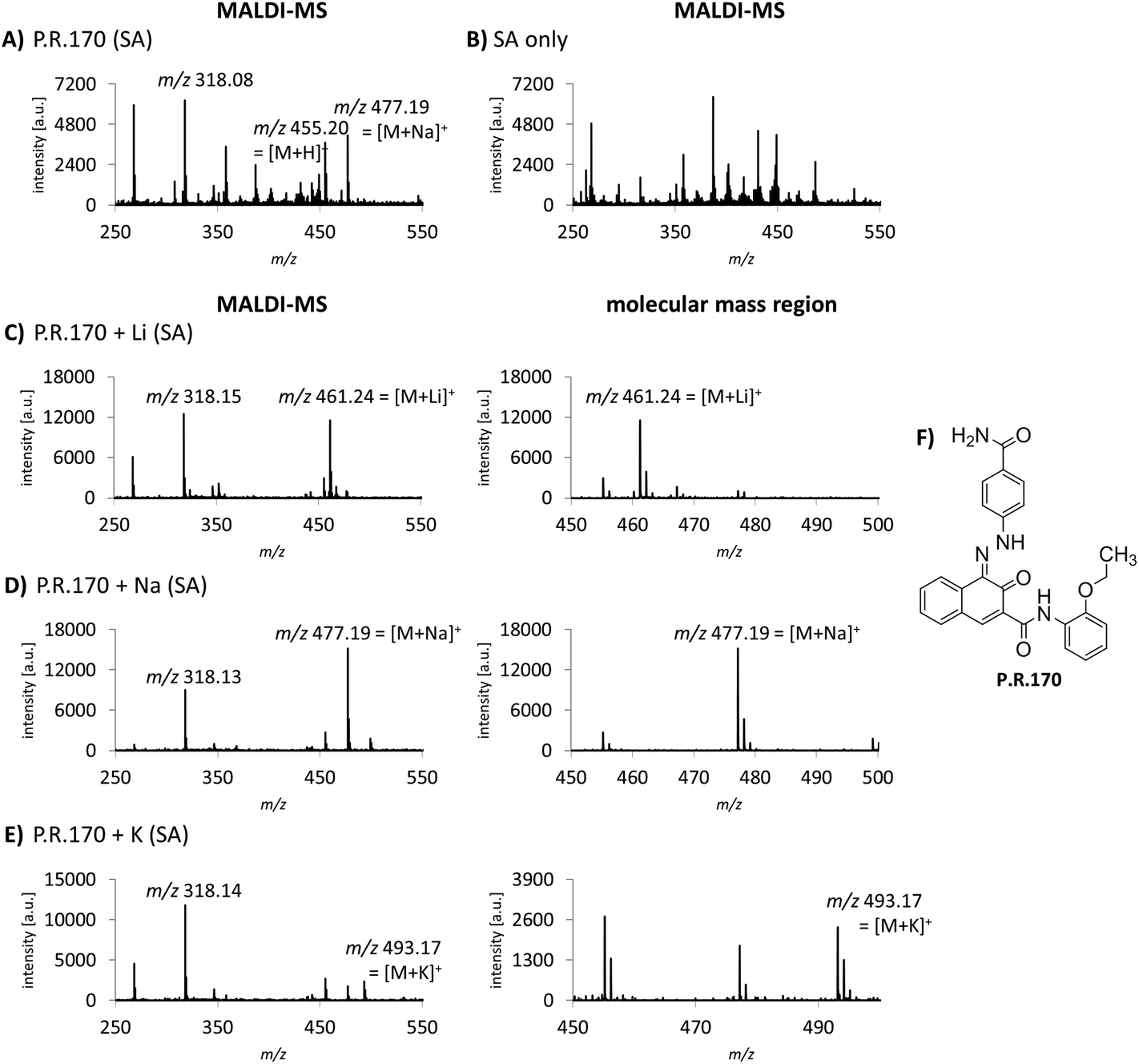 | ||
| Fig. 3 MALDI-MS and -MS/MS analysis of P.R.170 with alkali metal cation attachment. (A) MALDI-MS spectrum of P.R.170. (B) Background MALDI-MS spectrum of the matrix SA in tattoo matrix without pigment. (C)–(E) MALDI spectra of P.R.170 after lithium, sodium and potassium addition, respectively. The altered mass shift from m/z 455 [M+H]+ to m/z 461 [M+Li]+ and m/z 493 [M+K]+ have a higher abundance compared to the molecular peak and the sodium adduct m/z 477 [M+Na]+ in (A). For each cation, the MALDI-MS spectrum (left) and the magnified molecular mass region (right) are displayed. (F) Molecule structure of P.R.170. | ||
Using lithium, potassium and sodium salts, the expected m/z shifts of the molecular mass ion of P.R.170 occurred in the MS spectra (Fig. 3C–E). However, adduct formation was less efficient with potassium leading to a lower abundance of the molecular peak ion (Fig. 3E).
Alkali metal cation attachment can be used as a control step for the identification of the pigment's molecular ion in complex matrices. In general, pigments that can be ionized without protonation such as copper phthalocyanines and P.V.23 did not show cation attachment. Corresponding cation attachment spectra should therefore not be altered from the standard acquisition spectra. Lithium ions display the best choice since they lead to high peak intensities and are less likely to occur in the samples. Alternatively, Niederer et al. used standard addition to confirm the pigments’ identity.12
Automated acquisition and spectral evaluation for pigment identification
Time-efficient processing of data acquisition is strongly required for routine use of tattoo pigment identification. Therefore, we combined processing and analysis of MALDI-MS and -MS/MS acquisitions in an automated work-flow.Since there is currently no program available that is fully capable of the technical requirements of such a work-flow, a custom made solution was established. The commonly available programs WARP-LC and ProteinScape were used which originate from proteomics analyses. A precursor list was created in WARP-LC containing m/z values for MS/MS analysis of all pigments of interest (ESI Table S3†). During the automated work-flow, the entries of the generated peak list of the MS spectrum were compared with the m/z values in the precursor list in WARP-LC. In case of a match, the order for an MS/MS acquisition was given automatically for this sample using the correspondent m/z value as precursor. Furthermore, the laser energy was automatically increased both in the MS and the MS/MS spectra acquisition for samples where the spectra quality was not sufficient for peak integration. Hence, ionization will be increased in the presence of an interfering matrix.
For pigment identification, MS, lithium-MS and MS/MS libraries with reference spectra of the pure pigments in different matrices were created using the NIST MS Search program. The NIST program is used to have an automated spectral comparison with a probability measure. For evaluation of pigment identity and to prevent false positive findings, a decision tree was developed for data analysis (Fig. 4). For the MS spectra, the 10 best hits (Fig. 4A) and for the MS/MS spectra the first 5 hits are taken into account as possible pigments (Fig. 4B). Both the acquired unknown MS and MS/MS spectra and the suggested reference spectra will be compared in flexAnalysis for final identification. Sometimes no MS/MS spectrum was acquired for pigments later suggested by the MS spectral comparison. In that case, the MS/MS acquisition has to be repeated manually using the m/z values of the precursors of the suspected pigments. The probability of the MS/MS spectral match should be 70% or higher. If multiple similar spectra of the same pigment appear as best hits, the probability might be lower in rare cases. Thus, the acceptance of lower matches has to be evaluated case by case.
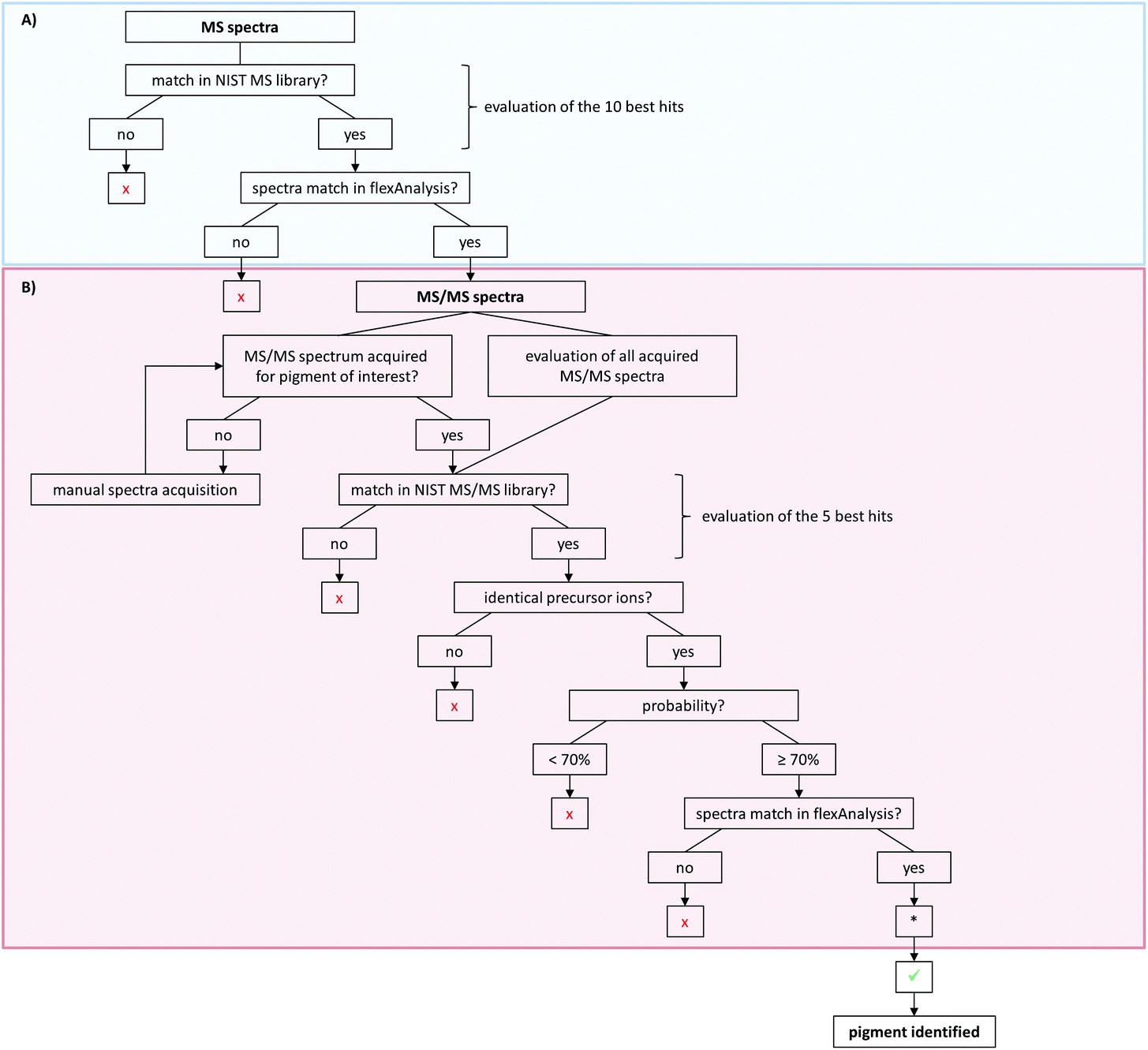 | ||
| Fig. 4 Decision tree for pigment identification with NIST MS Search. (A) Evaluation of MS spectra. Spectra are considered to match if the molecular peak of the suggested library pigment or a high abundant fragment peak is present in the sample spectrum. (B) Analysis of MS/MS spectra upon successful or equivocal MS evaluation. If multiple similar spectra of the same pigment appear as best hits, the probability might be lower than 70% in rare cases. Evaluation of lithium-MS spectra is carried out analogously to MS analysis. x = pigment suggestion has to be turned down; ✓ = pigment suggestion can be accepted. *In cases where the MS/MS spectra analysis results in the suggestion of a pigment that was not found following the path of the MS spectra analysis, the MS spectrum has to be manually inspected for the corresponding pigment peaks. | ||
For further confirmation, samples can also be analyzed with alkali metal cation attachment using the lithium-MS library for data evaluation. Here, the same criteria have to be met as for the MS analysis given in the decision tree (Fig. 4A).
However, the identification of pigments with low ionization efficiencies remains an issue for both the MALDI-MS and -MS/MS approach. Signal intensities of molecular ions of different pigments may vary within the same matrix in the MALDI-MS spectra since the ionization efficiency of each pigment varies. These individual ionization properties might lead to detection difficulties in samples containing a mixture of pigments. A badly ionizing pigment might be concealed by a highly efficiently ionizing pigment in the MS spectrum and will therefore consequently not be available for the MS/MS analysis either. Furthermore, tattoo inks are complex mixtures also including binders, solvents and additives.6 These substances can also interfere with the ionization of the pigments as already shown for the non-ionic surfactant Triton X-100.12 Similarly, the biological matrix of skin samples might also contain different compounds and ingredients that can reduce the ionization efficiency of organic pigments. Therefore, the choice of integration parameters plays a pivotal role in the automated MS and MS/MS acquisition in order to record all pigment related peaks. To overcome these limitations, methods such as LC-MS may serve as complementary techniques for soluble pigments to facilitate their identification in complex mixtures.12,14
Method validation
The MALDI-MS/MS method was only validated in terms of mass precision of pigment peaks and accuracy of pigment identification since it is only a qualitative method.The instruments’ mass accuracy for internally calibrated spectra is below 1 ppm (e.g. <0.001 Da for a 1000 Da substance). However, mass precision was strongly reduced upon analyzing pigment standards and was depending on the ionization matrix, pigment agglomeration, presence of tissue matrix and varied between 0.01 and 0.25 Da. The variations are in the same range as the observed 0.1 Da mass shift at higher laser energies by Niederer and collegues.12 Therefore, substance identification by the high resolution MS technique is limited. As stated above, interfering peaks and low abundant pigments in human samples additionally lead to insecurities in these samples in contrast to tattoo inks.
The accuracy of pigment identification by MALDI-MS/MS and our data evaluation strategy was cross validated by analyzing 36 pigments in self-made tattoo matrix with the automated MALDI-MS/MS method in accordance to the decision tree (Fig. 4). If only MS spectra were evaluated, 33 of 36 pigments were correctly identified. This is, since some pigment peaks might be hidden when mixed with pigments that give the same m/z in the spectra. Especially the identification of P.Y.74 was not sufficient when using only MS spectra due to its low abundance in the spectra in the used concentration. If MS and MS/MS spectra were evaluated with the decision tree, all 36 pigments were correctly identified. The correct identification of pigments with low abundance in the spectra is highly dependent on the integration parameters used to create the MS/MS list. Sensitive parameters therefore help to find pigments in MS/MS mode which might have been overlooked during NIST MS spectra evaluation.
The method was also tested with commercial tattoo inks. In the majority of inks, declared pigments have been fully or partly identified. In other cases pigments that were not declared but identified by MALDI-MS/MS have also been identified by py-GC/MS and are therefore regarded as correct. However, next to often false declared pigments, concentrations of pigments are unknown and therefore may fall below the limit of detection (LOD) of pigments.
The LOD was strongly influenced by the applied matrix for ionization and varied for each pigment. LOD was also impaired if a pigment with high ionization efficiency such as P.Y.154 was present in an ink mixture which reduced the abundance of other pigments. Addition of cations such as Li+ or Na+ can increase signal to noise ratios for some pigments (Fig. 3). Niederer et al. also report lower LOD upon mixture with carbon black or titanium dioxide which can be compensated by increasing laser intensity.12 Since our automated acquisition step wise increases laser energy if the spectra do not meet integration criteria, these effects will likely be automatically reduced by our method.
The method was also tested in an intra-laboratory comparison by two individuals in terms of data acquisition and evaluation and resulted in the identification of the same pigments. Hence, the robustness of the method is considered high since data acquisition is fully automated and data evaluation is conducted following a decision tree. However, basic knowledge of pigment chemistry or detailed comments in the spectral libraries may be needed since multiple azo pigments may have the same fragment ion in the MS spectra, e.g. P.R.9 and P.Br.25 (cf. ESI Table S3†).
Conclusion
The extension of the standard MALDI-MS method by an MS/MS approach established in this paper can strengthen the reliability of pigment identification based on specific pigment fragment ions. The method helps to identify the target compounds with high specificity even in the presence of possible interfering molecules from biological matrices. The automated MS/MS acquisition of known pigment ions may increase sensitivity since interfering matrix peaks can impair a successful search in the NIST library. For some pigments, further confirmation can also be gained through the proposed alkali metal cation attachment.Our optimized sample preparation for biological samples is the basis for a successful application of MALDI-MS and -MS/MS pigment analysis. For routine deployment of an analytical method, also time efficiency and the applicability at lager scales are decisive. Through our automated approach of the MALDI-MS and -MS/MS spectra acquisition, numerous different samples can be analyzed in one run. Using a decision tree, reproducible identification of organic pigments can be achieved.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the intramural research project (SFP #1322-604) at the German Federal Institute for Risk Assessment (BfR).References
- S. Fickweiler, R.-M. Szeimies, W. Bäumler, P. Steinbach, S. Karrer, A. E. Goetz, C. Abels, F. Hofstädter and M. Landthaler, J. Phys. Chem. B, 1997, 38, 178–183 Search PubMed.
- P. Laux, T. Tralau, J. Tentschert, A. Blume, S. A. Dahouk, W. Bäumler, E. Bernstein, B. Bocca, A. Alimonti, H. Colebrook, C. de Cuyper, L. Dähne, U. Hauri, P. C. Howard, P. Janssen, L. Katz, B. Klitzman, N. Kluger, L. Krutak, T. Platzek, V. Scott-Lang, J. Serup, W. Teubner, I. Schreiver, E. Wilkniß and A. Luch, Lancet, 2016, 387, 395–402 CrossRef.
- N. Sonoda, Stud. Conserv., 1999, 44, 195–208 Search PubMed.
- P. Laux and A. Luch, in Tattooed Skin and Health. Current Problems in Dermatology, ed. J. Serup, N. Kluger and W. Bäumler, Karger, Basel, 2015, vol. 48, pp. 196–200 Search PubMed.
- S. Wenzel, J. Welzel, C. Hafner, M. Landthaler and W. Bäumler, Contact Dermatitis, 2010, 63, 223–227 CrossRef PubMed.
- M. Dirks, in Tattooed Skin and Health. Current Problems in Dermatology, ed. J. Serup, N. Kluger and W. Bäumler, Karger, Basel, 2015, vol. 48, pp. 118–127 Search PubMed.
- A. Vila and J. F. García, Anal. Lett., 2012, 45, 1274–1285 CrossRef.
- K. W. C. Poon, I. R. Dadour and A. J. McKinley, J. Raman Spectrosc., 2008, 39, 1227–1237 CrossRef.
- I. Schreiver, C. Hutzler, S. Andree, P. Laux and A. Luch, Arch. Toxicol., 2016, 90, 1639–1650 CrossRef PubMed.
- O. Chiantore, D. Scalarone and T. Learner, Int. J. Polym. Anal. Charact., 2003, 8, 67–82 CrossRef.
- E. Engel, F. Santarelli, R. Vasold, H. Ulrich, T. Maisch, B. König, M. Landthaler, N. V. Gopee, P. C. Howard and W. Bäumler, Anal. Chem., 2006, 78, 6440–6447 CrossRef PubMed.
- M. Niederer, U. Hauri, L. Kroll and C. Hohl, F1000Research, 2017, 6, 2034 Search PubMed.
- J. J. Boon and T. Learner, J. Anal. Appl. Pyrolysis, 2002, 64, 327–344 CrossRef.
- U. Hauri, Inks for tattoos and permanent make-up/pigments, preservatives, aromatic amines, polyaromatic hydrocarbons and nitrosamines. Swiss National Investigation Campaign 2014. http://www.kantonslabor.bs.ch/dam/jcr:d12e5456-c71d-4e59-8f29-4a7d8c38d15d/Tattoo_PMU_2014_EN(UK).pdf. Accessed 16 April 2018, Department of Health, Kanton Basel-Stadt.
- I. Schreiver, B. Hesse, C. Seim, H. Castillo-Michel, J. Villanova, P. Laux, N. Dreiack, R. Penning, R. Tucoulou, M. Cotte and A. Luch, Sci. Rep., 2017, 7, 11395 CrossRef PubMed.
- M. George, J. M. Y. Wellemans, R. L. Cerny and M. L. Gross, J. Am. Soc. Mass Spectrom., 1994, 5, 1021–1025 CrossRef PubMed.
- R. C. Beavis and B. T. Chait, Rapid Commun. Mass Spectrom., 1989, 3, 432–435 CrossRef PubMed.
- M. Hesse, H. Meier and B. Zeeh, Spektroskopische Methoden in der organischen Chemie (article in German), Thieme, Stuttgart, 2005 Search PubMed.
- S. Trimpin, P. Eichhorn, H. J. Räder, K. Müllen and T. P. Knepper, J. Chromatogr. A, 2001, 938, 67–77 CrossRef PubMed.
- D. Suckau, A. Resemann, M. Schuerenberg, P. Hufnagel, J. Franzen and A. Holle, Anal. Bioanal. Chem., 2003, 376, 952–965 CrossRef PubMed.
- D. Corry, P. Kulkarni and M. F. Libscomb, Am. J. Pathol., 1984, 115, 321–328 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8an00702k |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |