
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A facile electrochemical intercalation and microwave assisted exfoliation methodology applied to screen-printed electrochemical-based sensing platforms to impart improved electroanalytical outputs†
Gastón D.
Pierini
ab,
Christopher W.
Foster
 b,
Samuel. J.
Rowley-Neale
b,
Héctor
Fernández
a and
Craig E.
Banks
*b
b,
Samuel. J.
Rowley-Neale
b,
Héctor
Fernández
a and
Craig E.
Banks
*b
aDepartamento de Química, Facultad de Ciencias Exactas, Físico-Químicas y Naturales, Universidad Nacional de Río Cuarto, Agencia Postal No 3, 5800 Río Cuarto, Argentina. E-mail: c.banks@mmu.ac.uk; Web: http://www.craigbanksresearch.com Fax: +44 (0)161-247-6831; Tel: +44 (0)161-247-1196
bFaculty of Science and Engineering, Manchester Metropolitan University, Chester Street, Manchester M1 5GD, UK
First published on 18th May 2018
Abstract
Screen-printed electrodes (SPEs) are ubiquitous with the field of electrochemistry allowing researchers to translate sensors from the laboratory to the field. In this paper, we report an electrochemically driven intercalation process where an electrochemical reaction uses an electrolyte as a conductive medium as well as the intercalation source, which is followed by exfoliation and heating/drying via microwave irradiation, and applied to the working electrode of screen-printed electrodes/sensors (termed EDI-SPEs) for the first time. This novel methodology results in an increase of up to 85% of the sensor area (electrochemically active surface area, as evaluated using an outer-sphere redox probe). Upon further investigation, it is found that an increase in the electroactive area of the EDI-screen-printed based electrochemical sensing platforms is critically dependent upon the analyte and its associated electrochemical mechanism (i.e. adsorption vs. diffusion). Proof-of-concept for the electrochemical sensing of capsaicin, a measure of the hotness of chillies and chilli sauce, within both model aqueous solutions and a real sample (Tabasco sauce) is demonstrated in which the electroanalytical sensitivity (a plot of signal vs. concentration) is doubled when utilising EDI-SPEs over that of SPEs.
Introduction
Over recent decades there has been a focus and acceleration within the research community towards the miniaturisation/simplification of analytical protocols.1 As such, the utilisation of screen-printing technology for the mass production of screen-printed electrodes (SPEs) has allowed low cost bespoke electrochemical sensors to be realised,2,3 allowing researchers to translate sensor technology from the laboratories to the field. To impart improvements in the electroanalytical sensing abilities of screen-printed electrochemical platforms, researchers readily modify SPEs with common nanomaterials such as graphene,4 metallic nanoparticles5 and metal phthalocyanines,6,7 to name just a few. The approach for the surface modification of SPEs involves drop-casting the desired nanomaterials upon the SPEs but can in some instances produce unstable/irreproducible surfaces.6 Another approach to overcome this is to mix the nanomaterial into the bulk of the screen-printing ink for the development of bespoke nanomaterial modified inks.8,9 An alternative approach is to make structural/compositional changes to the SPEs, which could possibly provide increased electroanalytical signals. For example, Washe et al.10 reported a solvent-based modification, which is postulated to alter/remove the binder within the screen-printing ink thus creating increased access to the graphitic flakes within the ink and hence an associated increase in the electrochemically active area of up to 57 times. However, recent work has questioned such elaborate increments.11 Other studies have sought to explore the polishing of SPEs upon polishing pads, which is generally undertaken only for solid electrode substrates (such as glassy carbon) which gives rise to changes in the SPE's surface morphology and carbon–oxygen functionalities11,12 resulting in significant improvements in sensor performance.11An approach used in the academic literature to increase the area of carbon materials involves electrochemical techniques, which offer intercalation with acidic intercalants such as sulphuric, perchloric, nitric and trifluoroacetic acids.13–16 Electrochemical Interaction Reactions (EIRs) are utilised to exfoliate graphite. In these approaches, guest ions (cations, M+ or anions, X−) intercalate under electrochemical perturbation into the Van der Waal's gaps between carbon layers and enlarge the inter-layer distance.17 These EIRs can be summarised as:
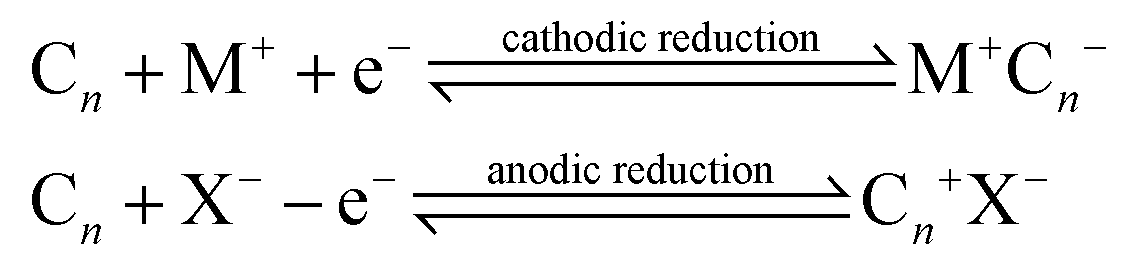
In such cases, the Cn needs to be electrochemically active and the host carbon structure should be capable of accepting, not only sterically but also electronically, the guest ions via a suitable energy band structure.17 Interaction reactions identified above are reversible and topotactic.17 Another interesting scenario is the creation of ternary phases, via:
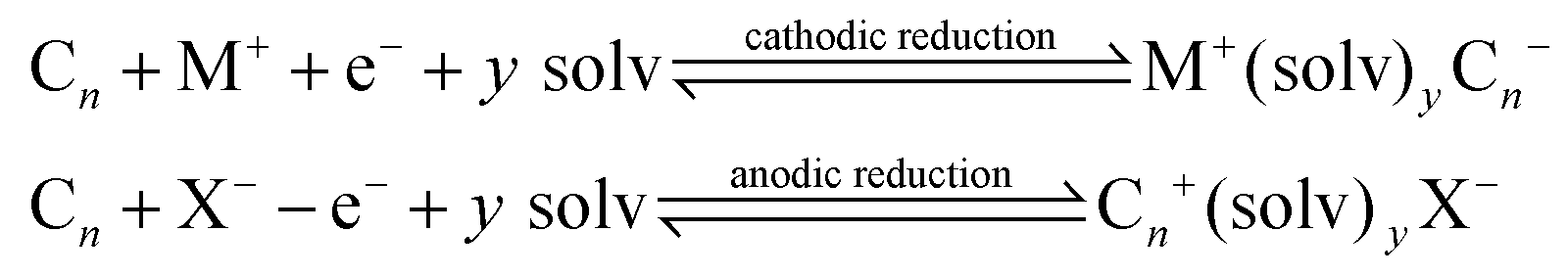
Some examples reported in the academic literature utilising the above approaches include the work of Morales et al.18 who reported the production of few-layer graphene via electrochemically produced graphite intercalation in aqueous perchloric acid solutions and expansion by microwave radiation, which contributes to the removal of the trapped solvent causing further expansion.19 While the approaches above are generally utilised to produce expanded graphite nanoplatelets19 or few-layer graphene,18 Zhang et al.20 have utilised this graphite expansion method to produce a potential anode within a Li-ion battery, where the expansion of the graphitic structure allows for an increased number of reaction sites with the electrolyte, leading to improved energy capabilities.20 The utilisation of the above approaches to screen-printed electrodes has not yet been, to the best of our knowledge, reported which can potentially be utilised to increase the electrochemical active surface area to impart improvements in electroanalytical performance.
Herein, we explore the feasibility of an anodically driven electrochemically intercalation process, where an electrochemical reaction uses an electrolyte as a conductive medium and as an intercalation source, which is followed by exfoliation and heating/drying via microwave irradiation, applied to the working electrode of screen-printed electrodes/sensors (EDI-SPEs) for the first time. In this approach, perchloric and formic acids are utilised as both the electrolyte and the intercalation compounds and the EDI-SPEs are evaluated towards redox probes and commonly benchmarked analytically useful analytes: sodium nitrite, dopamine, uric acid and capsaicin.
Experimental section
All chemicals used were obtained from Sigma-Aldrich at analytical grade and were used without any further purification. All solutions were prepared with deionised water of resistivity not less than 18.2 MΩ cm. Phosphate buffer solution (PBS) of pH 7 was prepared using disodium hydrogen phosphate (0.025 M) and potassium dihydrogen phosphate (0.025 M). Perchloric acid and phosphoric acid were diluted with water to obtain the desired concentration. Voltammetric measurements were carried out using an Emstat (Palmsens Instruments BV, The Netherlands) potentiostat and controlled by PS Trace 4.4 software. When it was necessary an external electrode Ag/AgCl, 3 M KCl (BAS, RE-5B) and a platinum wire were used as reference and counter electrodes respectively. SEM images were obtained with a JEOL JSM-5600LV model. The X-ray photoelectron spectroscopy (XPS) data was acquired using a AXIS Supra with a Al X-ray source (1486.6 eV) operating at 300 W for survey scans and 450 W for narrow scans. Al X-rays were monochromated using a 500 mm Rowland circle quartz crystal X-ray mirror. The angle between X-ray source and analyser was 54.7°.Procedure to fabricate the EDI-SPEs
The SPEs fabricated herein have been extensively characterised via Raman and XPS analysis and have been published within recent literature;21 this information is re-reported here for the convenience of the reader. Screen-printed graphite electrodes were fabricated in-house with appropriate stencil designs using a microDEK 1760RS screen-printing machine (DEK, Weymouth, UK). For each of the screen-printed electrodes, a carbon–graphite ink formulation was first screen-printed onto a polyester flexible film (Autostat, 250 μm thickness). This layer was cured in a box fan oven with extraction at 60 °C for 30 min. Next, a silver/silver chloride (60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :40) reference electrode was applied by screen-printing Ag/AgCl paste (Product Code: C2040308P3; Gwent Electronic Materials Ltd, UK) onto the plastic substrate. This layer was once more cured in an oven at 60 °C for 30 min. Finally, an insulating dielectric paste ink (Product Code: D2070423D5; Gwent Electronic Materials Ltd, UK) was printed to cover the connections and define the 3 mm diameter graphite working electrode. After curing at 60 °C for 30 min the screen-printed electrodes are ready to use and have been thoroughly benchmarked previously.22,23
:40) reference electrode was applied by screen-printing Ag/AgCl paste (Product Code: C2040308P3; Gwent Electronic Materials Ltd, UK) onto the plastic substrate. This layer was once more cured in an oven at 60 °C for 30 min. Finally, an insulating dielectric paste ink (Product Code: D2070423D5; Gwent Electronic Materials Ltd, UK) was printed to cover the connections and define the 3 mm diameter graphite working electrode. After curing at 60 °C for 30 min the screen-printed electrodes are ready to use and have been thoroughly benchmarked previously.22,23
The procedure for the expansion of the electrochemical active area consists of two facile steps: Step 1, electrochemical intercalation step:
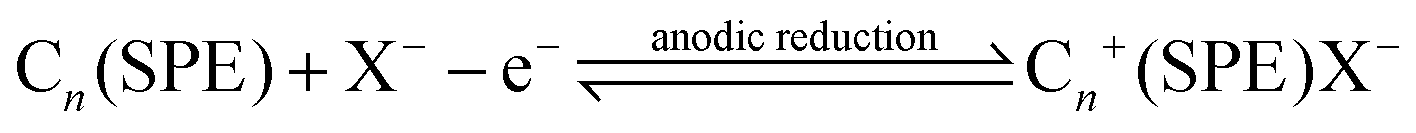
Step 2, expansion step: where the electrochemically driven intercalation compounds are removed via heating, in this case, microwave irradiation (Argos Stores, UK Simple Value, UK, Model: MM7 17CNF (F)-PM), which will also remove the trapped species, but assist with further expansion of the graphite. In this work, we also considered heating via a fan oven (see later).
Electrochemical characterisation (see the ESI†) of the electrodes utilised throughout this work was via the effective electroactive area (Aeff)/electrode area. To evaluate this parameter (Aeff) we utilised the redox probe 1 mM hexaammineruthenium(III) chloride in 0.1 M KCl, and the Randles–Ševčík equation (at 298 K):26IpQuasi = 2.65 × 105n3/2D1/2v1/2CAeff where n is the number of electrons involved in the electrochemical process, D is the diffusion coefficient (9.1 × 10−6 cm2 s−1) of the electrochemical redox probe used,27ν is the applied voltammetric scan rate, C is the concentration of the electroactive substance, and Aeff is the effective electrode area.
To fully understand and benchmark these sensing platforms an array of scan rates was utilised (0.05 to 0.5 V s−1) to estimate Aeff. To determine if the expansion causes an improvement in the surface's electron transfer behaviour, we evaluated the effect of the standard heterogeneous rate constant (k0), using the Nicholson method:23,28k0 = [2.18(DαnFv/RT)0.5]exp[−(α2nF/RT)×ΔEp], where the constants are the same as described previously, apart from the transfer coefficient α, which is assumed to correspond to 0.5, F is the Faraday constant, R the universal gas constant, and the experiments are performed at a temperature T. ΔEp is peak-to-peak separation.
In the case of the determination of capsaicin in hot chilli sauce, ∼2 g of Tabasco hot sauce was accurately weighed in a small conical flask and diluted with methanol (typically mL). The samples were ultra-sonicated for 20 minutes in order to extract the capsaicin, via liquid–liquid extraction from the chilli sauce. After this step, the samples were centrifuged for 10 min. The real sample was then filtered via gravitation into a 20 mL volumetric flask and diluted with methanol. Finally, 1.0 mL of the final solution was placed within an electrochemical cell and diluted further with 9 mL of 0.1 M H3PO4, which acts as the supporting electrolyte.
Results and discussion
We explore the use of screen-printed electrodes (SPEs) that have been subjected to the anodic electrochemically driven intercalation process as detailed in the Experimental section, where the electrolyte serves as a conductive medium to undertake electrochemical experiments while also acting as the intercalation source. This is followed by a further exfoliation and heating/drying step via microwave irradiation, all applied to the working electrode of screen-printed electrodes/sensors, for the first time (see Fig. 1). The microwave irradiation power, time and electrolytes have been explored and are summarized in the ESI.† The electroanalytical performance of the EDI-SPEs is considered and compared with that of the unmodified SPEs, towards the electroanalytical sensing of various electrochemical redox probes and electroanalytical targets, to allow direct comparison with prior published literature work. Herein, the redox probes hexaammineruthenium(III) chloride/0.1 M KCl and ammonium iron(II) sulfate/0.2 M HClO4 are used to electrochemically benchmark the EDI-SPEs with the academic literature as well as to explore electroanalytically useful probes. This data is presented in the ESI† and demonstrate that both the intercalation and expansion steps do not induce any significant oxygenated species upon the electrode surface and additionally that there has been no significant electronic change in the electrode/material structure, such results have been corroborated with comparative XPS analysis of unmodified and EDI-SPEs, presented in ESI Fig. 5.†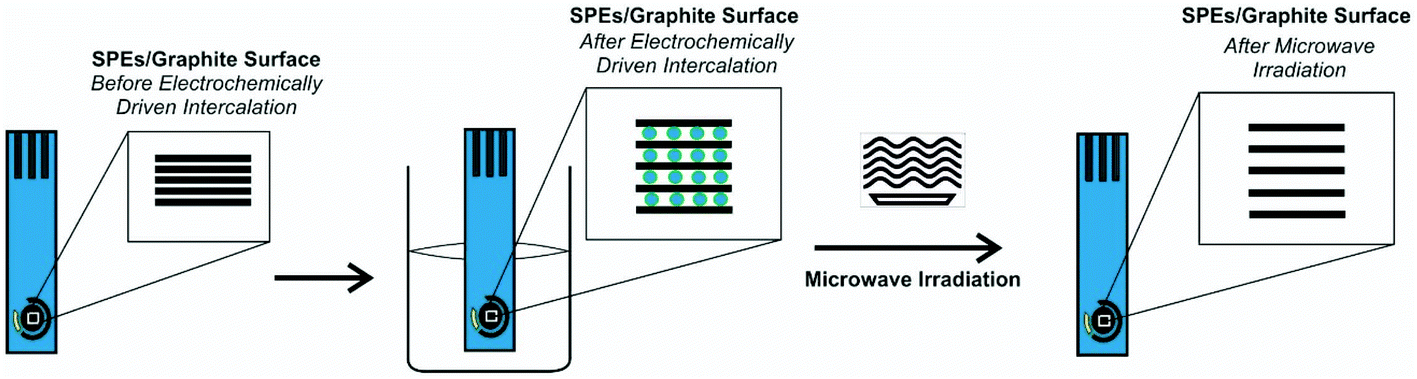 | ||
| Fig. 1 Schematic overview of the modification protocol of the microwave assisted electrochemically driven intercalated screen-printed electrodes (EDI-SPEs). | ||
First, as depicted in Fig. 2 and Table 1, the electroanalytical performance of the EDI-SPEs is benchmarked towards sodium nitrite, dopamine, uric acid and capsaicin. In all cases, utilising the unmodified and EDI-SPE electrochemical analysis over the concentration range is linear, however it is apparent that the EDI-SPE does not significantly improve the sensitivity of the EDI-SPEs/sensor towards the detection of dopamine, sodium nitrite and uric acid. Nonetheless, there are slight analytical improvements (when utilising the EDI-SPEs) within the limits of detection (LOD, 3σ) of each analyte (sodium nitrite, dopamine and uric acid), but the response is hindered due to the increased capacitive current. The electrochemical detection of capsaicin (presented in Fig. 2G and H) reveals that the EDI-SPEs result in an improved sensitivity (0.126 and 0.239 μA μM−1 for the SPEs and the EDI-SPE respectively) and LOD (1.76 and 0.36 μM for the SPEs and the EDI-SPE respectively) when utilising the EDI-SPEs over that of SPEs.
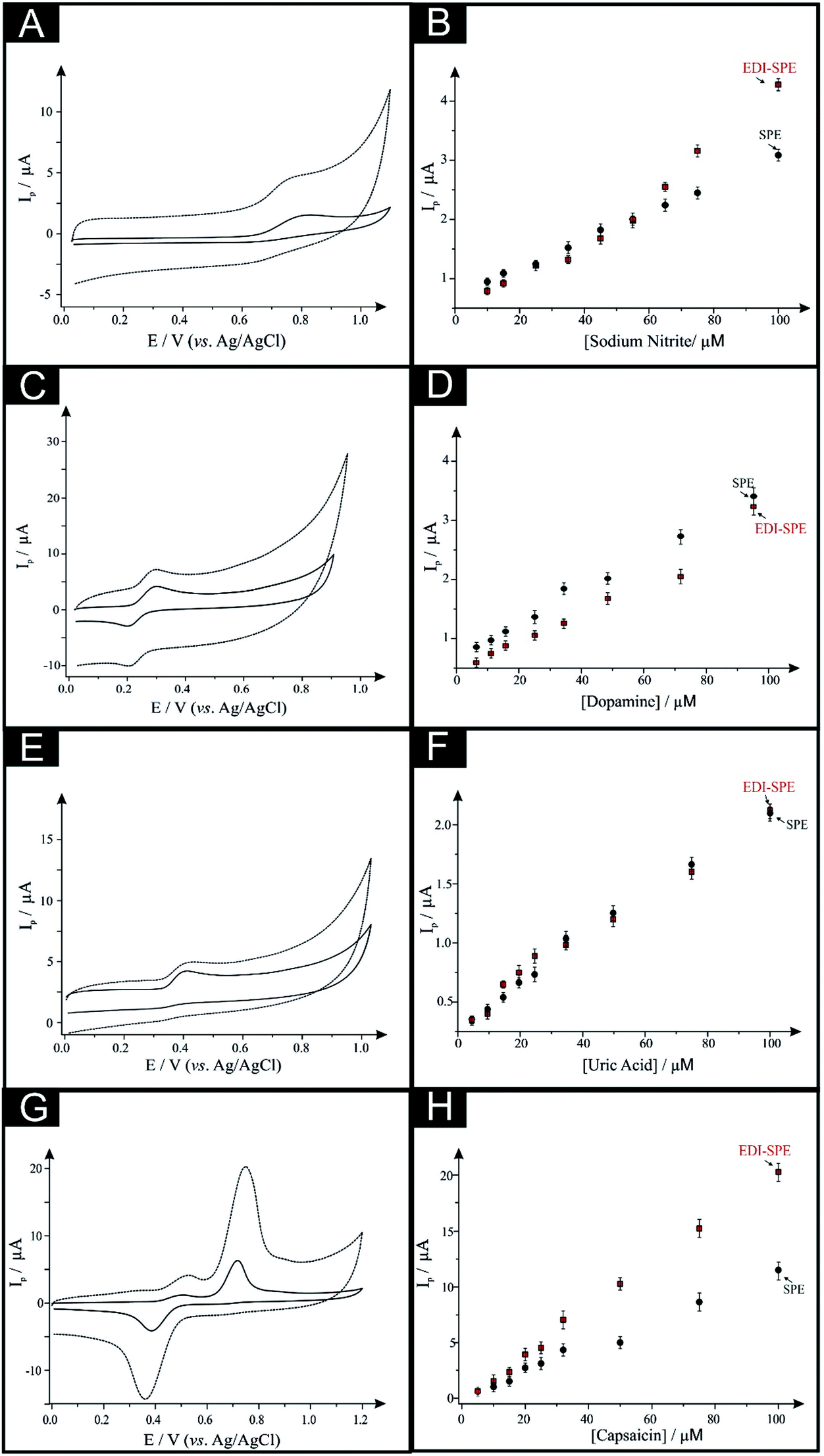 | ||
| Fig. 2 Cyclic voltammograms utilising EDI-SPEs explored towards the electroanalytical detection of: (A) 50 μM nitrite, (C) 50 μM dopamine, (E) 50 μM uric acid and (G) 50 μM capsaicin obtained for SPEs (solid line) and EDI-SPEs (checked lines). Resulting calibration plots for (B) nitrite, (D) dopamine, (F) uric acid and (H) capsaicin, obtained for SPEs (circles) and EDI-SPEs (squares). In all cases, scan rate: 0.1 V s−1. | ||
| Analyte | Methodology | Sensitivity (μA μM−1) | LOD (μM) | Linear range (μM) |
|---|---|---|---|---|
| Nitrite | SPEs | 0.024 | 1.66 | 10–100 |
| EDI-SPEs | 0.038 | 6.32 | 10–100 | |
| Dopamine | SPEs | 0.032 | 0.92 | 5–100 |
| EDI-SPEs | 0.031 | 4.91 | 5–100 | |
| Uric acid | SPEs | 0.024 | 1.70 | 5–100 |
| EDI-SPEs | 0.020 | 2.47 | 5–100 | |
| Capsaicin | SPEs | 0.111 | 4.76 | 5–100 |
| EDI-SPEs | 0.209 | 2.77 | 5–100 |
Thus, we can conclude that this modification technique/process does not improve the electroanalytical performance of analytes that undergo diffusional or mixed diffusional-adsorptive reaction mechanisms using the EDI-SPEs. However, it is clear that analytes that require a strong adsorptive mechanism, as is the case with capsaicin, demonstrate an improved electroanalytical performance when utilising EDI-SPEs. The observed electrochemical behaviour agrees with independent work by Compton et al.29 who demonstrate that the electrochemical mechanism of capsaicin is via an adsorption mechanism (rather than diffusion). Hence, they utilise this adsorption (via adsorption stripping voltammetry) using multi-walled carbon nanotubes (MWCNT) modified electrodes where the adsorption is favoured strongly by pi–pi interactions between the aromatic moieties in capsaicin and the surface of the carbon nanotubes (CNTs). Thus in our work, the exfoliation increases the surface area of the basal sheets of graphite providing more aromatic surfaces for capsaicin to adsorb onto; hence the observed increase in the sensitivity (∼×2).29
Next, we consider the applicability of the EDI-SPEs towards the detection of capsaicin within a real sample, Tabasco sauce. Fig. 3 demonstrates linear sweep voltammograms directly from five spiked Tabasco sauce samples. However, the content of capsaicin in the real sample was found to be under the detection limit of this analytical procedure, therefore recovery assays were employed in order to validate the methodology, with an average recovery of 95% utilising the EDI-SPEs (see Table 2); in summary we can conclude that the EDI-SPEs are useful for the electroanalytical determination of capsaicin within real chilli sauce.
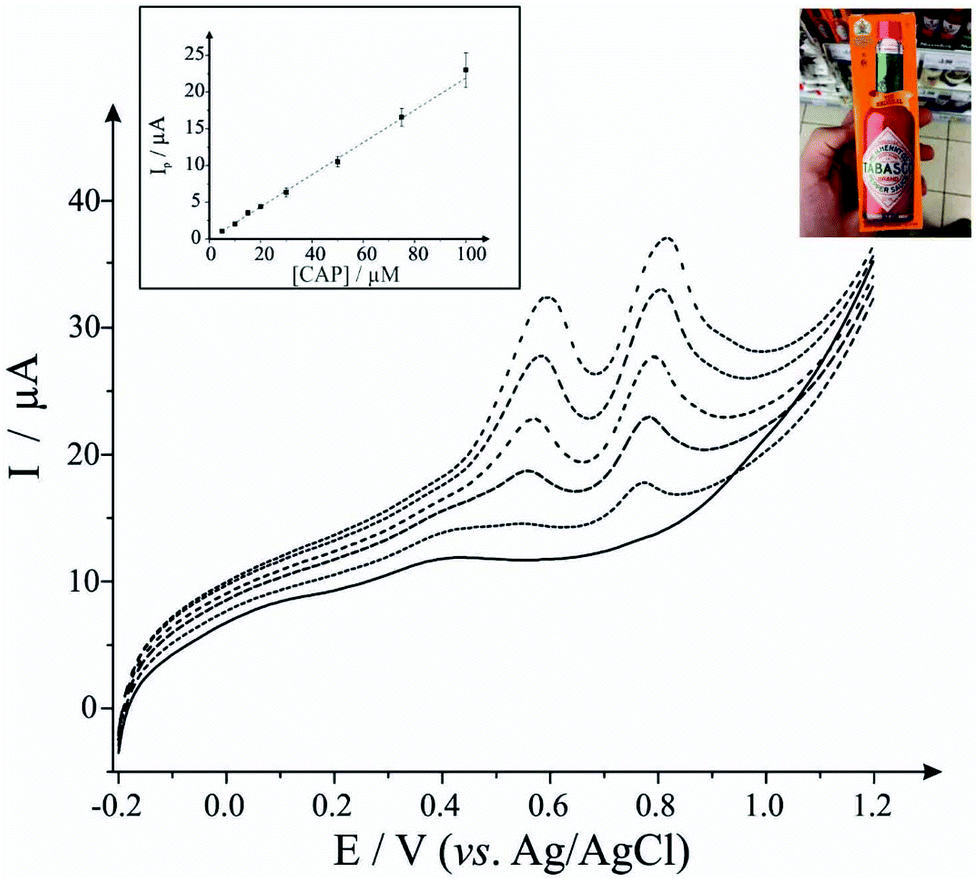 | ||
| Fig. 3 Calibration curves for the electroanalytical detection of capsaicin using EDI-SPEs. The insert shows the analysis of addition of capsaicin to pH 1.2 phosphate buffer solution using linear sweep voltammetry (plot of peak current vs. capsaicin concentration with the average and standard deviation presented). The main figure shows the linear sweep voltammograms obtained for different additions of capsaicin made into the chilli sauce (dotted lines) and the blank response (black solid line) prior to capsaicin addition. See experimental section for more details. | ||
| Samples | Added/μM | Recovery/μM | Recovery/% | Relative error/% |
|---|---|---|---|---|
| S1 | 10 | 9.41 | 94.1 | −5.9 |
| S2 | 20 | 18.64 | 93.2 | −6.8 |
| S3 | 30 | 30.35 | 101.2 | +1.2 |
| S4 | 40 | 40.06 | 100.1 | +0.1 |
| S5 | 50 | 45.55 | 91.1 | −8.9 |
| Recovery analysis | ||||
| Mean recovery (Rexp) | 95.94 | |||
| Standard deviation of recoveries (SR) | 4.45 | |||
| Number of samples (N) | 5 | |||
| t exp | 2.0 | |||
| t (0.025, 4) | 2.8 | |||
Conclusions
We have explored an electrochemically driven intercalation process where the electrochemical reaction uses an electrolyte as a conductive medium as well as the intercalation source, which is followed by exfoliation and heating/drying via microwave irradiation, applied to the working electrode of screen-printed electrodes/sensors for the first time. We demonstrate proof-of-concept that the protocol can be used beneficially to increase the surface area of electrochemical sensors of adsorption controlled electrochemically active analytes. We demonstrate that the electroanalytical detection of capsaicin using the EDI-SPEs exhibits a double increase in the electroanalytical sensitivity with a lower limit of detection in comparison with unmodified SPEs. We extended this exciting work to the determination of capsaicin in real hot chilli sauce demonstrating that EDI-SPEs can be employed as an interesting alternative for the determination of capsaicin in chilli hot sauce with improved analytical sensitivity. This approach has generic advantages for electrochemically active probes that occur via an adsorptive mechanism and should be considered further allowing improvements in the electroanalytical sensitivity.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the support of the Analytical Chemistry Trust Fund of the Royal Society for a Developing World Scholarship Grant for BT. Funding from the Engineering and Physical Sciences Research Council (Reference: EP/N001877/1) and a British Council Institutional Grant Link (No. 172726574) is acknowledged. G. D. Pierini is grateful to CONICET for a postdoctoral fellowship and for the “Financiamiento Parcial para Estadías breves en el exterior” fellowship. The authors would also like to thank Kratos (Manchester, UK), for their XPS expertise.References
- S. A. Wring and J. P. Hart, Analyst, 1992, 117, 1215–1229 RSC.
- A. P. Ruas de Souza, M. Bertotti, C. W. Foster and C. E. Banks, Electroanalysis, 2015, 27, 2295–2301 CrossRef.
- K. Senthilkumar, T. N. Sithini, N. Thiyagarajan, S. Baskar and J.-M. Zen, Electrochem. Commun., 2015, 60, 113–116 CrossRef.
- F. Arduini, L. Micheli, D. Moscone, G. Palleschi, S. Piermarini, F. Ricci and G. Volpe, TrAC, Trends Anal. Chem., 2016, 79, 114–126 CrossRef.
- R. A. Farghali and R. A. Ahmed, Int. J. Electrochem. Sci., 2015, 10, 11 Search PubMed.
- C. Foster, J. Pillay, J. Metters and C. Banks, Sensors, 2014, 14, 21905 CrossRef PubMed.
- M. A. T. Gilmartin, J. P. Hart and B. J. Birch, Analyst, 1994, 119, 243–252 RSC.
- S. J. Rowley-Neale, C. W. Foster, G. C. Smith, D. A. C. Brownson and C. E. Banks, Sustainable Energy Fuels, 2017, 1, 74–83 Search PubMed.
- E. P. Randviir, D. A. C. Brownson, J. P. Metters, R. O. Kadara and C. E. Banks, Phys. Chem. Chem. Phys., 2014, 16, 4598–4611 RSC.
- A. P. Washe, P. Lozano-Sánchez, D. Bejarano-Nosas and I. Katakis, Electrochim. Acta, 2013, 91, 166–172 CrossRef.
- E. Blanco, C. W. Foster, L. R. Cumba, D. R. do Carmo and C. E. Banks, Analyst, 2016, 141, 2783–2790 RSC.
- J. Lee, D. W. M. Arrigan and D. S. Silvester, Sens. Bio-Sensing Res., 2016, 9, 38–44 CrossRef.
- M. S. Dresselhaus and G. Dresselhaus, Adv. Phys., 2002, 51, 1–186 CrossRef.
- M. Noel and R. Santhanam, J. Power Sources, 1998, 72, 53–65 CrossRef.
- A. Yoshida, Y. Hishiyama and M. Inagaki, Carbon, 1991, 29, 1227–1231 CrossRef.
- M. Inagaki, J. Mater. Res., 2011, 4, 1560–1568 CrossRef.
- J. O. Besenhard and H. P. Fritz, Angew. Chem., Int. Ed. Engl., 1983, 22, 950–975 CrossRef.
- G. M. Morales, P. Schifani, G. Ellis, C. Ballesteros, G. Martínez, C. Barbero and H. J. Salavagione, Carbon, 2011, 49, 2809–2816 CrossRef.
- L. M. Viculis, J. J. Mack, O. M. Mayer, H. T. Hahn and R. B. Kaner, J. Mater. Chem., 2005, 15, 974–978 RSC.
- H. Zhang and M. M. Lerner, Inorg. Chem., 2016, 55, 8281–8284 CrossRef PubMed.
- A. Gomis-Berenguer, M. Gomez-Mingot, V. Montiel, A. Canals, T. Thiemann, R. O. Kadara, C. E. Banks and J. Iniesta, RSC Adv., 2012, 2, 7735–7742 RSC.
- F. E. Galdino, C. W. Foster, J. A. Bonacin and C. E. Banks, Anal. Methods, 2015, 7, 1208–1214 RSC.
- C. W. Foster, A. P. de Souza, J. P. Metters, M. Bertotti and C. E. Banks, Analyst, 2015, 140, 7598–7612 RSC.
- J. Jiang, Carbon, 1993, 31, 663–666 CrossRef.
- F. Kang, Y. Leng and T. Y. Zhang, Carbon, 1997, 35, 1089–1096 CrossRef.
- D. A. C. Brownson, C. W. Foster and C. E. Banks, Analyst, 2012, 137, 1815–1823 RSC.
- C. E. Banks, R. G. Compton, A. C. Fisher and I. E. Henley, Phys. Chem. Chem. Phys., 2004, 6, 3147–3152 RSC.
- I. Lavagnini, R. Antiochia and F. Magno, Electroanalysis, 2004, 16, 505–506 CrossRef.
- R. T. Kachoosangi, G. G. Wildgoose and R. G. Compton, Analyst, 2008, 133, 888–895 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7an01982c |
| This journal is © The Royal Society of Chemistry 2018 |