
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Metal-free direct alkylation of unfunctionalized allylic/benzylic sp3 C–H bonds via photoredox induced radical cation deprotonation†
Rong
Zhou
ab,
Haiwang
Liu
a,
Hairong
Tao
c,
Xingjian
Yu
a and
Jie
Wu
 *a
*a
aDepartment of Chemistry, National University of Singapore, 3 Science Drive 3, 117543, Republic of Singapore. E-mail: chmjie@nus.edu.sg
bCollege of Chemistry and Chemical Engineering, Taiyuan University of Technology, Taiyuan, 030024, China
cCollege of Chemistry, Beijing Normal University, Beijing, China
First published on 28th April 2017
Abstract
Despite notable recent efforts, a catalytic and convenient strategy for the direct alkylation of unactivated allylic or benzylic sp3 C–H bonds remains a formidable challenge facing the synthesis community. We herein report an unprecedented allylic/benzylic alkylation using only an organo-photoredox catalyst, which enables coupling of a broad scope of alkenes/arenes and electron-deficient alkenes in an atom- and redox-economic manner. A photoredox induced alkene/arene radical cation deprotonation is proposed to smoothly generate the key allylic and benzylic radical intermediates. It represents the first C–C bond formation via radical cation deprotonation under visible light conditions. The resulting products can be easily scaled up and directly converted to γ,δ-unsaturated or α,β-diaryl-acids, -esters, -amides, -pyrazoles, -isoxazoles, as well as lactones, which enables this mild and selective sp3 C–H alkylation to rapidly access complex bioactive molecules.
Introduction
Direct C–C bond formation from non-activated C–H bonds has profoundly affected organic synthesis, maximizing atom and step economy and enabling novel disconnections in the retrosynthesis of carbon skeletons. Although substantial progress has been achieved in this research field, the development of general and mild strategies for the engagement of sp3 C–H bonds in C–C bond forming reactions has proved difficult. In this context, allylic and benzylic C–H activations have attracted a special interest because these C–H bonds can be activated relatively more easily than the other sp3 C–H bonds. Even though protocols for oxidative functionalization of allylic and benzylic C–H bonds have been intensively studied (Scheme 1a),1 the engagement of these substrates in C–C bond formation is still limited, which normally involves a nucleophilic or carbenoid counterpart under transition-metal catalysis.2 An orthogonal approach for C–C bond formation of these sp3 C–H bonds is the reaction of allylic/benzylic radicals generated from homolytic hydrogen abstraction with electrophilic Michael acceptors.3 However, reported examples were limited to benzylic alkylations,4 and suffered from narrow substrate scopes, harsh conditions, requiring excess radical initiators, or use of harmful and equipment-demanding UV light.3 Nonetheless, a metal-free, mild and broadly applicable strategy for both allylic and benzylic alkylation remains underdeveloped but is highly desirable.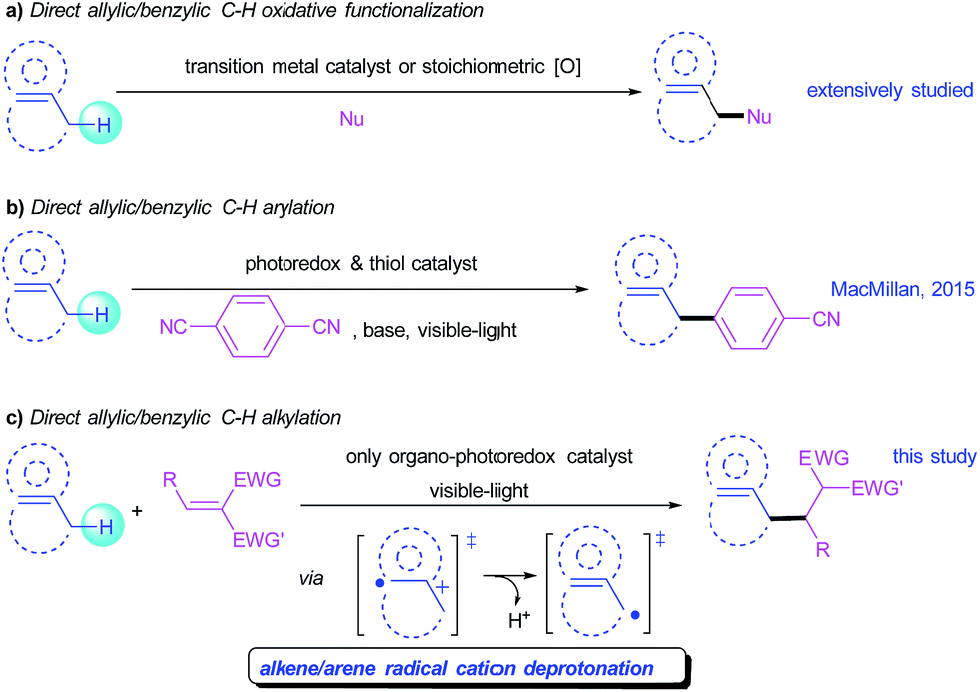 | ||
| Scheme 1 Direct functionalization of allylic/benzylic sp3 C–H bonds. | ||
Visible-light-mediated photoredox catalysis has attracted increasing attention over the past decade because of its unique reactivity patterns and high efficiency, enabling many previously unattainable transformations via single-electron transfer (SET) process.5 A series of new C–C bond forming reactions from native C–H bonds have been developed using this novel activation mode. In particular, the MacMillan group recently reported a direct arylation of allylic/benzylic sp3 C–H bonds through the combination of a photoredox and a thiol organic catalysis that promotes a hydrogen atom transfer (HAT) to access allylic/benzylic radicals (Scheme 1b).6
Among visible-light-mediated photoredox reactions, radical cation species effectively generated through reductive quenching of the excited photocatalyst via an SET process, have frequently been involved to achieve important transformations. For example, alkene radical cations have been successfully applied to [2 + 2],7 [4 + 2] cycloadditions,8 and anti-Markovnikov alkene hydrofunctionalization.9 Moreover, arene radical cations have been accessed for amination without the need for prefunctionalization of the aromatic component, as described by the Nicewicz group.10 Deprotonation is a typical reaction pathway for π-C–H+˙ type radical cations, because the acidity is inherently augmented.11 This process is appealing for the convenient generation of allylic or benzylic radicals. However, useful synthetic transformations based on deprotonation of radical cations generated by photo-induced SET are scarce. This scarcity is mainly a consequence of the inefficient deprotonation that competes unfavorably with the radical ion pair induced back electron transfer.12 Other challenges for a practical radical cation deprotonation process include side-reactions such as dimerization or further oxidation of the generated radicals.11,12
Results and discussion
Designing strategy
Considering the ease of accessing π-C–H+˙ type radical cations under photoredox condition, we envisioned that an alkene/arene radical cation generated via the visible-light induced SET might accomplish a smooth deprotonation to form an allylic/benzylic radical, which would subsequently undergo C–C bond formations with Michael acceptors, thereby achieving direct alkylation of allylic/benzylic sp3 C–H bonds. However, to realize such allylic/benzylic radical formation, the photo-catalyst should be capable of oxidizing a wide range of alkenes and arenes, can minimize the unproductive back electron transfer, and does not interact with the generated allylic/benzylic radical.Recent reports indicate organo-photoredox catalyst 9-mesityl-10-methylacridinium perchlorate [Acr+-Mes]ClO4 (4) may be a good photooxidant that fulfils all of the aforementioned requirements. Both alkenes and arenes can be conveniently oxidized to radical cation I by 4* because of its high excited state oxidizing power (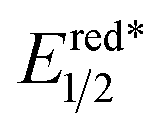 = +2.06 V vs. SCE in MeCN) (Fig. 1).9,10 The reduced form of the acridinium catalyst 4′ would not be actively involved in the unproductive back electron transfer or interact with radical cation I because it exists in the neutral radical form with a sterically bulky substituent.9b Both Fukuzumi and Lei's studies have demonstrated that benzyl radical cations generated by this catalyst gave the corresponding benzyl radicals via deprotonation, which were subsequently oxidized by O2 to deliver aldehydes and ketones.13 However, C–C bond formation has never been achieved via this mild radical generation pathway. We proposed that radical cation I would favor deprotonation to deliver radical II, which is in equilibrium with radical III. Nucleophilic addition of the less-hindered allylic/benzylic radical III to Michael acceptors such as methylene-malononitriles 2 would furnish alkyl radical IV. Single-electron reduction of this electron-deficient radical IV by the reduced form of the acridinium catalyst 4′ (Eox1/2 = −0.57 V vs. SCE in MeCN) would deliver alkylation product 3 after protonation, while regenerating photocatalyst 4.
= +2.06 V vs. SCE in MeCN) (Fig. 1).9,10 The reduced form of the acridinium catalyst 4′ would not be actively involved in the unproductive back electron transfer or interact with radical cation I because it exists in the neutral radical form with a sterically bulky substituent.9b Both Fukuzumi and Lei's studies have demonstrated that benzyl radical cations generated by this catalyst gave the corresponding benzyl radicals via deprotonation, which were subsequently oxidized by O2 to deliver aldehydes and ketones.13 However, C–C bond formation has never been achieved via this mild radical generation pathway. We proposed that radical cation I would favor deprotonation to deliver radical II, which is in equilibrium with radical III. Nucleophilic addition of the less-hindered allylic/benzylic radical III to Michael acceptors such as methylene-malononitriles 2 would furnish alkyl radical IV. Single-electron reduction of this electron-deficient radical IV by the reduced form of the acridinium catalyst 4′ (Eox1/2 = −0.57 V vs. SCE in MeCN) would deliver alkylation product 3 after protonation, while regenerating photocatalyst 4.
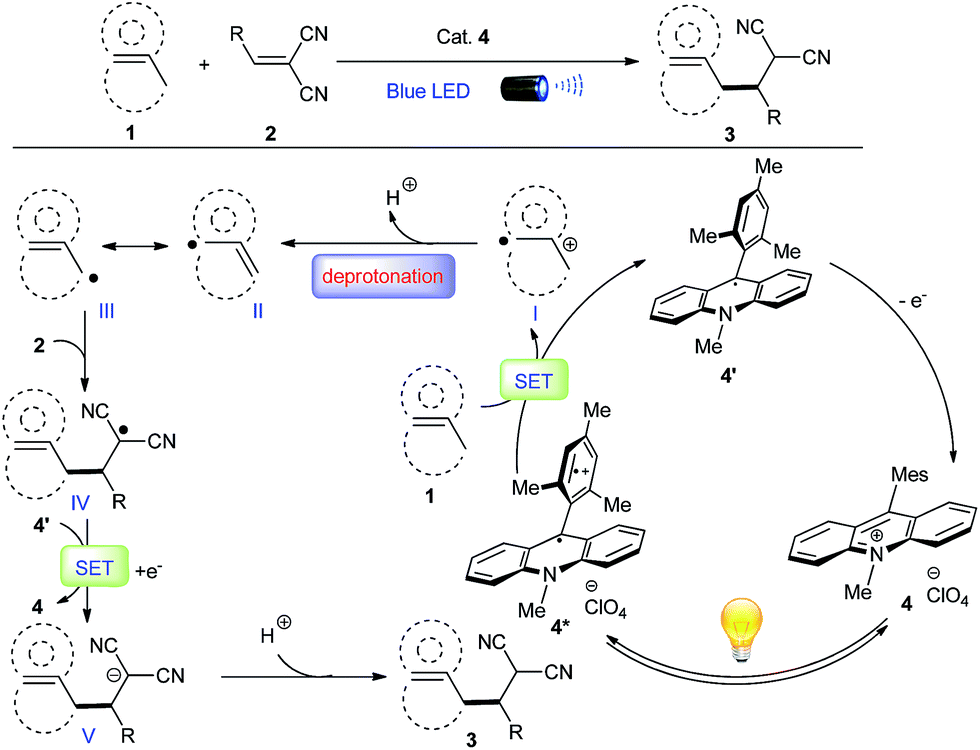 | ||
| Fig. 1 Proposed radical cation deprotonation for alkylation of allylic/benzylic C–H bonds. | ||
Optimization of reaction conditions
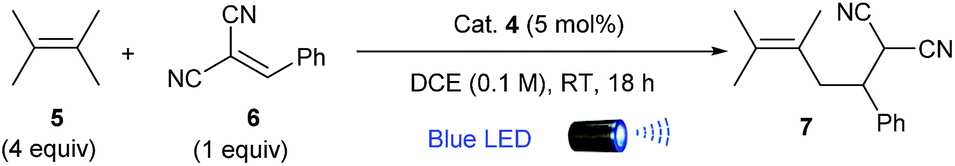
We initially attempted the proposed alkylation protocol by exposing tetramethylethylene (5) and benzylidenemalononitrile (6) to blue LED light (λmax = 425 nm) in the presence of 4 (5 mol%) in DCE (0.1 M) for 18 h at ambient temperature (Table 1, entry 1), which afforded alkylation product 7 in 72% isolated yield, with no detection of [2 + 2] cycloaddition byproducts. An excess amount of tetramethylethylene (4 equiv.) was required to achieve a full conversion of the Michael acceptor. Examination of a range of photocatalysts and solvents revealed that 4 and DCE was the most effective combination (entries 2–7). Remarkably, only the strong photo-oxidants 4 and 2,4,6-tri(p-tolyl)pyrylium tetrafluoroborate gave alkylation product 7 in good yields (entries 1 and 2), as no reaction occurred with weaker oxidants (entries 3 and 4); this result is consistent with our hypothesis because tetramethylethylene 5 (Eox1/2 = +1.22 V vs. Ag/AgNO3 in MeCN)14 requires a relatively strong oxidant for the generation of its radical cation. A higher yield (91%) was obtained when the reaction concentration was diluted to 0.05 M and the catalyst loading was reduced to 2.5 mol% (entry 8). When this visible-light-mediated reaction was conducted in our recently developed “stop-flow” micro-tubing (SFMT) reactors instead of in batch flasks,15 the required reaction time was substantially reduced from 18 h to 5 h and a similar yield was obtained (entry 9). This result underscores the remarkable light-irradiation efficiency achieved through the use of the SFMT reactors. Finally, no product formation was detected in the absence of either photocatalysts or light, demonstrating the need for all of these components (Table S1, ESI†).|
|
||
|---|---|---|
| Entry | Modifications | Yielda (%) |
| a Isolated yield; na = no reaction. | ||
| 1 | None | 72 |
| 2 | 2,4,6-Tri(p-tolyl)pyrylium tetrafluoroborate instead of 4 | 70 |
| 3 | Ir(ppy)2(dtbbpy)PF6, Ir(ppy)3, Eosin Y instead of 4 | na |
| 4 | Ru(bpy)3Cl2 instead of 4 | Trace |
| 5 | DCM instead of DCE | 54 |
| 6 | Acetone instead of DCE | 21 |
| 7 | MeCN, DMSO, DMF instead of DCE | Trace |
| 8 | 0.05 M concentration, 2.5 mol% 4 | 91 |
| 9 | 0.05 M concentration, 2.5 mol% 4, SFMT reactor, 5 h | 90 |
Scope of Michael acceptors for allylic alkylation
Adopting the optimized batch conditions described in entry 8 (Table 1), we sought to determine the generality of the allylic alkylation by evaluating various methylene-malononitriles (Scheme 2a). “Stop-flow” conditions (Table 1, entry 9) were applied in cases where conversions were low in batch reactors. Experiments probing the scope of methylene-malononitriles revealed that a variety of electron-rich (8 to 11) and electron-deficient arenes (12 to 20) on the Michael acceptor were well tolerated, even with functional groups such as phenols (11), aryl halides (12 to 16), and acids (18). Sterically demanding arenes (21) and heterocycles such as furan (22), thiophene (23), and benzothiadiazole (24) were readily accommodated. Substrates with alkyl instead of aryl substituents on the methylene-malononitrile participated in this coupling reaction smoothly (25 and 26). The change of one nitrile group in malononitrile moiety to ester was feasible (27). However, other Michael acceptors such as an unsaturated diketone afforded only sluggish reactivity (28). Interestingly, the alkylation using the Michael acceptor with a terminal alkene moiety went smoothly (29). Notably, diene Michael acceptors can be successfully applied to this transformation to give 1,6-addition type products (30 and 31).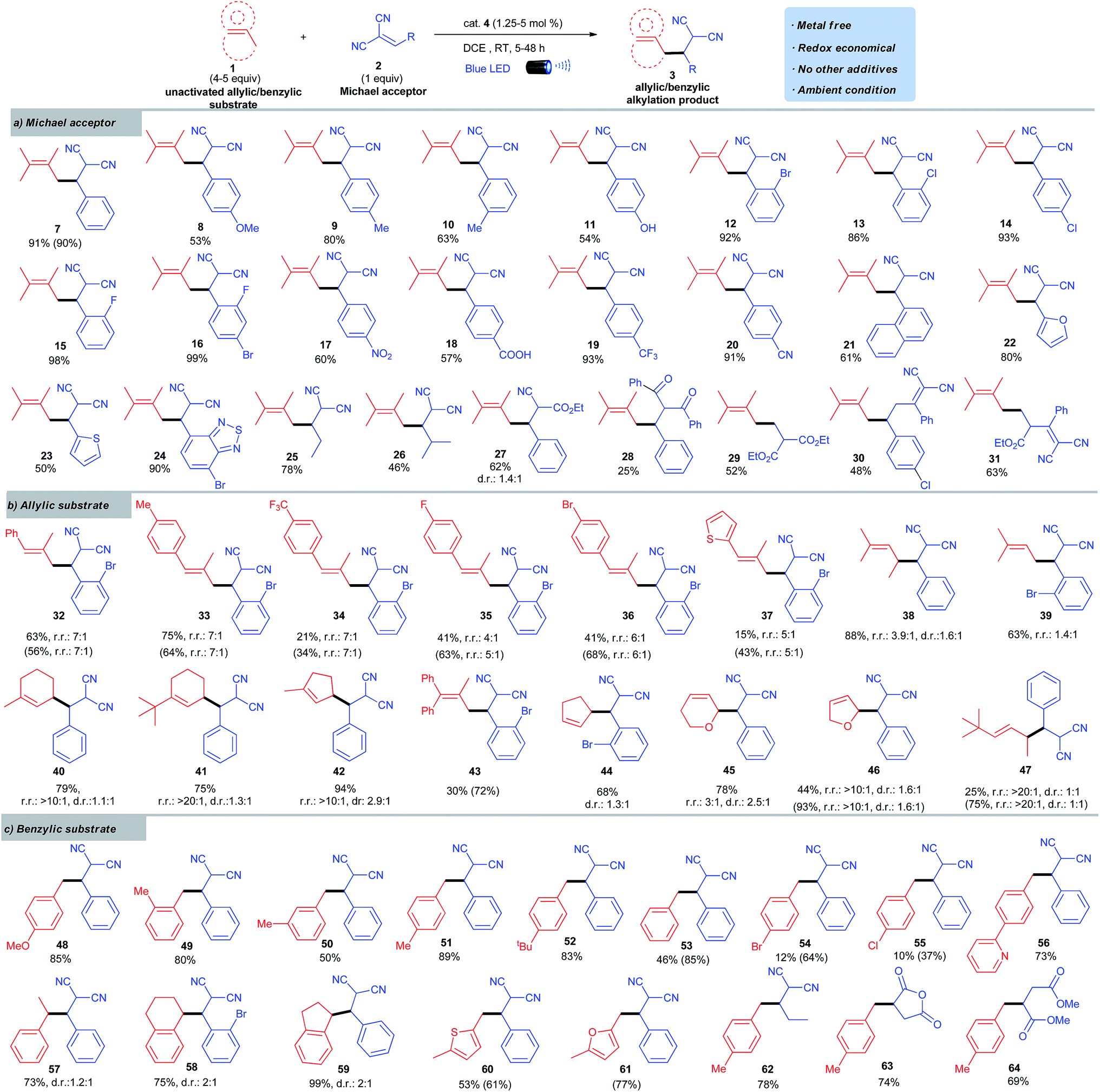 | ||
| Scheme 2 Run in a 0.2 mmol scale with respect to the Michael acceptor. Isolated yields. Regio- and diastereoselectivity were based on 1H NMR analysis. Regioselectivity shown as the major isomer vs. sum of minor isomers. Diastereoselectivity only shown for the major regioisomer. Results shown in parentheses were obtained for reactions in SFMT reactors. | ||
Scope of allylic compounds
We next examined the scope of allylic substrates for this convenient alkylation reaction. As shown in Scheme 2b, a wide range of tri-substituted alkenes could be used in this photoredox process because they were sufficiently electron-rich to be oxidized by 4 (32 to 42). Both acyclic and cyclic olefins were readily accommodated. A series of 2-methyl arylpropenes provided high alkylation efficiency while exhibiting good regioselectivity (32 to 37). The use of 2-methyl-2-pentene led predominantly to the formation of the branched alkylation product (38) with moderate selectivity. The regioselectivity with 2-methyl-2-butene (39) was low compared to that achieved with 2-methyl-2-pentene (38). Remarkably, cyclic substrates bearing alkyl substituents served as excellent allylic coupling partners with excellent regioselectivity favoring the less hindered cyclic methylene (40 to 42).16 Tetra-substituted alkenes with unhindered allylic C–H bonds could be oxidized effectively using the SFMT reactors under visible light followed by C–C bond formation (43). In batch reactors, the reaction was slow, most likely due to the increased steric hindrance. Disubstituted olefins, including cyclic (44 to 46) and acyclic alkenes (47), can also participate in the photoredox alkylation. Notably, despite the high oxidation potential of cyclopentene (+2.32 V vs. SCE in MeCN),17 it afforded product 44 in a good yield. Overall, the observed regioselectivity was attributed to both electronic and steric effects. The most stable allylic radical would be preferentially formed through deprotonation of the selectively generated radical cation intermediate, which subsequently underwent alkylation with its least hindered resonance. To the best of our knowledge, this study represents the first example of effective alkylation of allylic sp3 C–H bonds with electron-deficient alkenes.4Scope of benzylic compounds
This alkylation protocol was further extended to benzylic substrates (Scheme 2c). Toluene moieties with electron-donating substituents were good candidates for the alkylation (48 to 52). Electron-neutral and electron-poor arenes reacted much slower than electron-rich arenes because of their increased oxidative potential (e.g. Eox1/2toluene = +2.36 V vs. SCE in MeCN).17 However, the use of the SFMT reactor could accelerate the reaction and give satisfactory conversions (53 to 55). Notably, 2-(p-tolyl)pyridine uneventfully afforded its alkylation product in good yield (56). In addition to toluenes, ethylbenzene, tetralin, and indane were all suitable candidates and the reactions proceeded smoothly to give alkylation products in good to excellent yields (57 to 59). Methyl-substituted heterocycles such as thiophenes and furans were alkylated in good yields with the assistance of the SFMT reactor (60, 61). Incorporation of an alkyl substituent instead of the aryl substituent in methylene-malononitriles, or changing of the Michael acceptor to maleic anhydride and dimethyl maleate, was also feasible (62 to 64). This photoredox induced allylic/benzylic alkylation therefore represents a metal-free, atom-, and redox-economical C–H functionalization protocol.Synthetic utilities of alkylation products
To further demonstrate the synthetic utility of this methodology, the alkylations were amenable to scale-up to gram quantities in both batch reactors and continuous-flow reactors (Scheme 3a). The resulted alkylation products can be directly transformed to γ,δ-unsaturated or α,β-diaryl acids (66, 67),18 esters (68, 69),19 amides (70, 71),20 and lactones (72) in a convenient manner (Scheme 3b). These building blocks are core structures for a variety of drug molecules, such as nafronyl (76),21 racecadotril (77),22 and DX-9065a (78)23 (Scheme 3c), enabling a convenient way to synthesize these pharmaceutical compounds and their derivatives. The opportunities to effect synthetic streamline with this general C–H reactivity are further illustrated in a facile synthesis of 75 (Scheme 3b), a representative compound of a group of M1 antagonists.24 In addition, the malononitrile moieties can be directly converted to diaminopyrazoles (73), that are important intermediates for preparing therapeutically interesting pyrazolo[1,5-α]pyrimidines,25 and aminoisoxazoles (74) with potential hypoglycemic activity.26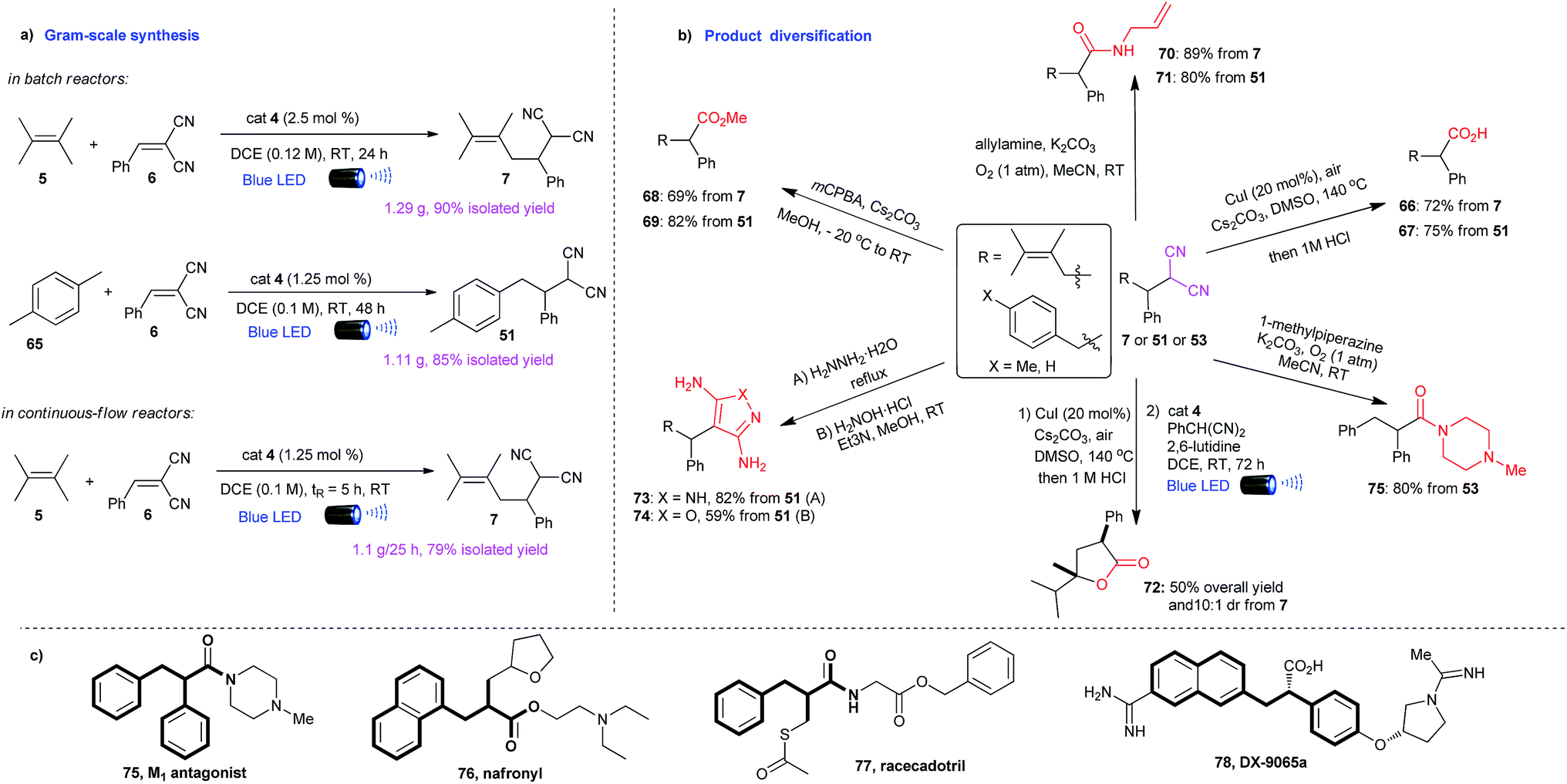 | ||
| Scheme 3 Synthetic applications of alkylation products. | ||
Control experiments to support the proposed mechanism
Several control experiments were performed to gain further insights into the reaction pathway (for details, see ESI†). First, no alkylation products were observed when the reaction was performed in the presence of radical scavengers such as TEMPO and hydroquinone, suggesting a radical process was involved. Furthermore, as indicated by the luminescence quenching experiments, both alkene 5 and 4-methylanisole can quench the excited state of acridinium (4*), whereas methylene-malononitrile 6 cannot; this result is in agreement with our proposed mechanism (Fig. 1). Several deuterium-labeling experiments were investigated, revealing that the anion V would most likely abstract a proton from the solvent rather than from substrate 1. The intermolecular competition experiment established that the magnitude of the kinetic isotopic effect (KIE) given by kH/kD was 3.3, indicating that the deprotonation might be involved in the rate-limiting step. We also questioned whether the alkene/arene radical cation I generated via photoredox mediated SET process could accomplish an intermolecular H-atom abstraction from another molecule of 1; in this case, the radical cation itself served as a HAT catalyst.27 However, the quantum chemistry calculation using a composite CBS-QB3 method, did not support this process (Schemes S1 and S2, ESI†).Conclusions
Overall, we have developed a visible-light-mediated alkylation of unactivated allylic/benzylic sp3 C–H bonds, which represents one of the most green and sustainable protocols featuring merits such as metal- and additive-free, atom- and redox-economical, and works effectively with a wide range of substrates (>55 examples). Use of SFMT reactors effectively enhanced the efficiency of light-promoted transformations and broadened the substrate scope. The alkylation products can be conveniently converted into γ,δ-unsaturated or α,β-diaryl-acids, -esters, -amides, -pyrazoles, -isoxazoles, as well as lactones for further elaborating valuable pharmaceutical molecules. This visible-light photoredox induced radical cation deprotonation process represents a powerful strategy and will likely find more applications for selective activation of native C–H bonds as latent nucleophilic handles.Acknowledgements
We are grateful for the financial support provided by Singapore A*STAR SERC Public Sector Research Funding (PSF; R-143-000-566-305), the National University of Singapore (R-143-000-645-112, R-143-000-665-114), and GSK-EDB (R-143-000-492-592).Notes and references
- For selected reviews, see: (a) S. E. Mann, L. Benhamou and T. D. Sheppard, Synthesis, 2015, 47, 3079 CrossRef CAS; (b) T. Wang and N. Jiao, Acc. Chem. Res., 2014, 47, 1137 CrossRef CAS PubMed; (c) B.-J. Li and Z.-J. Shi, Chem. Soc. Rev., 2012, 41, 5588 RSC; (d) J. Eames and M. Watkinson, Angew. Chem., Int. Ed., 2001, 40, 3567 CrossRef CAS; (e) M. Johannsen and K. A. J∅rgensen, Chem. Rev., 1998, 98, 1689 CrossRef CAS PubMed.
- For selected recent examples of direct alkylation of allylic or benzylic C–H bonds, see: (a) C. Qin and H. M. L. Davies, J. Am. Chem. Soc., 2014, 136, 9792 CrossRef CAS PubMed; (b) J. M. Howell, W. Liu, A. J. Young and M. C. White, J. Am. Chem. Soc., 2014, 136, 5750 CrossRef CAS PubMed; (c) S. Lin, C.-X. Song, G.-X. Cai, W.-H. Wang and Z.-J. Shi, J. Am. Chem. Soc., 2008, 130, 12901–12903 CrossRef CAS PubMed; (d) A. J. Young and M. C. White, J. Am. Chem. Soc., 2008, 130, 14090 CrossRef CAS PubMed.
- For selected recent examples, see: (a) H. Qrareya, D. Ravelli, M. Fagnoni and A. Albini, Adv. Synth. Catal., 2013, 355, 2891 CrossRef CAS; (b) M. Ueda, E. Kondoh, Y. Ito, H. Shono, M. Kakiuchi, Y. Ichii, T. Kimura, T. Miyoshi, T. Naito and O. Miyata, Org. Biomol. Chem., 2011, 9, 2062 RSC.
- To the best of our knowledge, only one single example has been reported for allylic sp3 C–H bond alkylation with an electron-deficient alkene, which is conducted under UV light conditions with a very low yield (29%): S. Kamijo, G. Takao, K. Kamijo, T. Tsuno, K. Ishiguro and T. Murafuji, Org. Lett., 2016, 18, 4912 CrossRef CAS PubMed.
- For selected reviews, see: (a) Q. Qin, H. Jiang, Z. Hu, D. Ren and S. Yu, Chem. Rec., 2017 DOI:10.1002/tcr.201600125; (b) N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075 CrossRef CAS PubMed; (c) D. Ravelli, S. Protti and M. Fagnoni, Chem. Rev., 2016, 116, 9850 CrossRef CAS PubMed; (d) K. L. Skubi, T. R. Blum and T. P. Yoon, Chem. Rev., 2016, 116, 10035 CrossRef CAS PubMed; (e) M. D. Kärkäs, J. A. Porco Jr and C. R. J. Stephenson, Chem. Rev., 2016, 116, 9683 CrossRef PubMed; (f) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322 CrossRef CAS PubMed; (g) J. Xuan and W.-J. Xiao, Angew. Chem., Int. Ed., 2012, 51, 6828 CrossRef CAS PubMed; (h) J. M. R. Narayanam and C. R. J. Stephenson, Chem. Soc. Rev., 2011, 40, 102 RSC.
- J. D. Cuthbertson and D. W. C. MacMillan, Nature, 2015, 519, 74 CrossRef CAS PubMed.
- M. A. Ischay, Z. Lu and T. P. Yoon, J. Am. Chem. Soc., 2010, 132, 8572 CrossRef CAS PubMed.
- S. Lin, M. A. Ischay, C. G. Fry and T. P. Yoon, J. Am. Chem. Soc., 2011, 133, 19350 CrossRef CAS PubMed.
- (a) D. J. Wilger, J.-M. M. Grandjean, T. R. Lammert and D. A. Nicewicz, Nat. Chem., 2014, 6, 720 CAS; (b) D. S. Hamilton and D. A. Nicewicz, J. Am. Chem. Soc., 2012, 134, 18577 CrossRef CAS PubMed.
- N. A. Romero, K. A. Margrey, N. E. Tay and D. A. Nicewicz, Science, 2015, 349, 1326 CrossRef CAS PubMed.
- (a) M. Schmittel and A. Burghart, Angew. Chem., Int. Ed., 1997, 36, 2550 CrossRef; (b) A. Albini, M. Mella and M. Freccero, Tetrahedron, 1994, 50, 575 CrossRef CAS.
- (a) M. Vanossi, M. Mella and A. Albini, J. Am. Chem. Soc., 1994, 116, 10070 CrossRef CAS; (b) R. M. Borg, D. R. Arnold and T. S. Cameron, Can. J. Chem., 1984, 62, 1785 CrossRef CAS.
- (a) H. Yi, C. Bian, X. Hu, L. Niu and A. Lei, Chem. Commun., 2015, 51, 14046 RSC; (b) K. Ohkubo, K. Mizushima, R. Iwata, K. Souma, N. Suzuki and S. Fukuzumi, Chem. Commun., 2010, 46, 601 RSC.
- T. Yamashita, J. Itagawa, D. Sakamoto, Y. Nakagawa, J. Matsumoto, T. Shiragami and M. Yasuda, Tetrahedron, 2007, 63, 374 CrossRef CAS.
- F. Xue, H. Deng, C. Xue, M. K. D. Mohamed, Y. K. Tang and J. Wu, Chem. Sci. 10.1039/c7sc00408g.
- CCDC 1506724 (41-major diastereomer) contains the supplementary crystallographic data for this paper. For ORTEP representation of the structure, see ESI.†.
- H. G. Roth, N. A. Romero and D. A. Nicewicz, Synlett, 2016, 27, 714 CAS.
- D. Yang, H. Yang and H. Fu, Chem. Commun., 2011, 47, 2348 RSC.
- S. Förster, O. Tverskoy and G. Helmchen, Synlett, 2008, 18, 2803 Search PubMed.
- J. Li, M. J. Lear and Y. Hayashi, Angew. Chem., Int. Ed., 2016, 55, 9060 CrossRef CAS PubMed.
- S. A. R. Aly, M. Hossain, M. A. Bhuiyan, T. Nakamura and T. Nagatomo, J. Pharmacol. Sci., 2009, 110, 445 CrossRef.
- A. J. Matheson and S. Noble, Drugs, 2000, 59, 829 CrossRef CAS PubMed.
- (a) S. Komoriya, N. Kanaya, T. Nagahara, A. Yokoyama, K. Inamura, Y. Yokoyama, S. Katakura and T. Hara, Bioorg. Med. Chem., 2004, 12, 2099 CrossRef CAS PubMed; (b) T. Nagahara, Y. Yokoyama, K. Inamura, S. Katakura, S. Komoriya, H. Yamaguchi, T. Hara and M. Iwamoto, J. Med. Chem., 1994, 37, 1200 CrossRef CAS PubMed.
- M. S. Poslusney, C. Sevel, T. J. Utley, T. M. Bridges, R. D. Morrison, N. R. Kett, D. J. Sheffler, C. M. Niswender, J. S. Daniels, P. J. Conn, C. W. Lindsley and M. R. Wood, Bioorg. Med. Chem., 2012, 22, 6923 CrossRef CAS PubMed.
- X. Wang, S. Magnuson, R. Pastor, E. Fan, H. Hu, V. Tsui, W. Deng, J. Murray, M. Steffek, H. Wallweber, J. Moffat, J. Drummond, G. Chan, E. Harstad and A. J. Ebens, Bioorg. Med. Chem. Lett., 2013, 23, 3149 CrossRef CAS PubMed.
- (a) T. Liu, X. Dong, N. Xue, R. Wu, Q. He, B. Yang and Y. Hu, Bioorg. Med. Chem., 2009, 17, 6279 CrossRef CAS PubMed; (b) W. J. Fanshawe, V. J. Bauer and S. R. Safir, J. Org. Chem., 1965, 30, 2862 CrossRef CAS.
- For using amine radical cation as a HAT catalyst, see: (a) M. H. Shaw, V. W. Shurtleff, J. A. Terrett, J. D. Cuthbertson and D. W. C. MacMillan, Science, 2016, 352, 1304 CrossRef CAS PubMed; (b) J. L. Jeffrey, J. A. Terrett and D. W. C. MacMillan, Science, 2015, 349, 1532 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterization data and NMR spectra of all new compounds; ORTEP drawing of compound 41 (major diastereomer). CCDC 1506724. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7sc00953d |
| This journal is © The Royal Society of Chemistry 2017 |