
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Anion–π catalysis: bicyclic products with four contiguous stereogenic centers from otherwise elusive diastereospecific domino reactions on π-acidic surfaces†
Le
Liu
a,
Yoann
Cotelle
ab,
Juliane
Klehr
bc,
Naomi
Sakai
a,
Thomas R.
Ward
bc and
Stefan
Matile
 *ab
*ab
aDepartment of Organic Chemistry, University of Geneva, Geneva, Switzerland. E-mail: stefan.matile@unige.ch; Web: www.unige.ch/sciences/chiorg/matile/ Tel: +41 22 379 6523
bNational Centre of Competence in Research (NCCR) Molecular Systems Engineering, Switzerland Web: www.nccr-mse.ch
cDepartment of Chemistry, University of Basel, Basel, Switzerland
First published on 17th March 2017
Abstract
Anion–π interactions have been introduced recently to catalysis. The idea of stabilizing anionic intermediates and transition states on π-acidic surfaces is a new fundamental concept. By now, examples exist for asymmetric enolate, enamine, iminium and transamination chemistry, and the first anion–π enzyme has been created. Delocalized over large aromatic planes, anion–π interactions appear particularly attractive to stabilize extensive long-distance charge displacements during domino processes. Moving on from the formation of cyclohexane rings with five stereogenic centers in one step on a π-acidic surface, we here focus on asymmetric anion–π catalysis of domino reactions that afford bicyclic products with quaternary stereogenic centers. Catalyst screening includes a newly synthesized, better performing anion–π version of classical organocatalysts from cinchona alkaloids, and anion–π enzymes. We find stereoselectivities that are clearly better than the best ones reported with conventional catalysts, culminating in unprecedented diastereospecificity. Moreover, we describe achiral salts as supramolecular chirality enhancers and report the first artificial enzyme that operates in neutral water with anion–π interactions, i.e., interactions that are essentially new to enzymes. Evidence in support of contributions of anion–π interactions to asymmetric catalysis include increasing diastereo- and enantioselectivity with increasing rates, i.e., asymmetric transition-state stabilization in the presence of π-acidic surfaces and inhibition with the anion selectivity sequence NO3− > Br− > BF4− > PF6−.
Contributions of anion–π interactions1,2 to catalysis were first demonstrated explicitly in 2013.3 Since then, anion–π catalysis has been explored with enolate,4,5 enamine,6 iminium,7 transamination8 and oxocarbenium9 chemistry, and the first anion–π enzyme has been created.5 The idea of stabilizing anionic intermediates and transition states on π-acidic surfaces is a new fundamental concept. Delocalized over large aromatic planes, anion–π interactions appear particularly advantageous to stabilize extensive charge displacements over long distances. For the conventional cation–π catalysis, this advantage is best illustrated with the stabilization of carbocations moving along the emerging rings during the cyclization of terpenes into steroids.10 In anionic tandem, domino or cascade reactions, the key intermediates in need of stabilization are not carbocations but enolates, nitronates, and so on. The most sophisticated domino reaction catalyzed so far with anion–π interactions is the stereoselective formation of a cyclohexane ring from achiral starting materials.7 From there, a continuing increase in sophistication of long-distance cascade charge displacements on π-acidic surfaces naturally leads to bicyclic products on the one hand and quaternary stereogenic centers on the other. For this purpose, we focused first on the addition of cyclohexanedione 1 to nitroolefin 2 (Fig. 1). In the presence of a base, they engage in a domino Michael–Henry reaction to afford bicyclo[3.2.1]octan-8-one 3, which is a bicyclic product with four chiral centers made from achiral substrates.11–13 The first step is the Michael addition of the conjugate enolate base of 1 to acceptor 2 (see transition state TS1, Fig. 1). The resulting nitronate engages in an intramolecular Henry reaction to close the second carbocycle (TS2, Fig. 1). This reaction was attractive for anion–π catalysis because stabilization of the anionic enolate and nitronate intermediates on π-acidic surfaces was conceivable, and the diastereoselectivity reported in the literature for metal-free organocatalysts (up to 12
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) appeared improvable.11–13 One report highlights that poor 1:3 dr originates from epimerization between 3 and 3d in the presence of base catalysts.11 Here, we report that domino catalysis on π-acidic surfaces provides access to diastereospecificity, i.e., the exclusive formation of one diastereomer. This breakthrough with the most sophisticated tandem process realized so far is achieved with three different functional systems, i.e., new anion–π cinchona fusion catalysts, anion–π enzymes and achiral anion–π chirality enhancers. Decisive contributions from anion–π interactions could be deduced from the observed increase in rate and stereoselectivity upon attachment of π-acidic surface, and selective inhibition with anions.
:1) appeared improvable.11–13 One report highlights that poor 1:3 dr originates from epimerization between 3 and 3d in the presence of base catalysts.11 Here, we report that domino catalysis on π-acidic surfaces provides access to diastereospecificity, i.e., the exclusive formation of one diastereomer. This breakthrough with the most sophisticated tandem process realized so far is achieved with three different functional systems, i.e., new anion–π cinchona fusion catalysts, anion–π enzymes and achiral anion–π chirality enhancers. Decisive contributions from anion–π interactions could be deduced from the observed increase in rate and stereoselectivity upon attachment of π-acidic surface, and selective inhibition with anions.
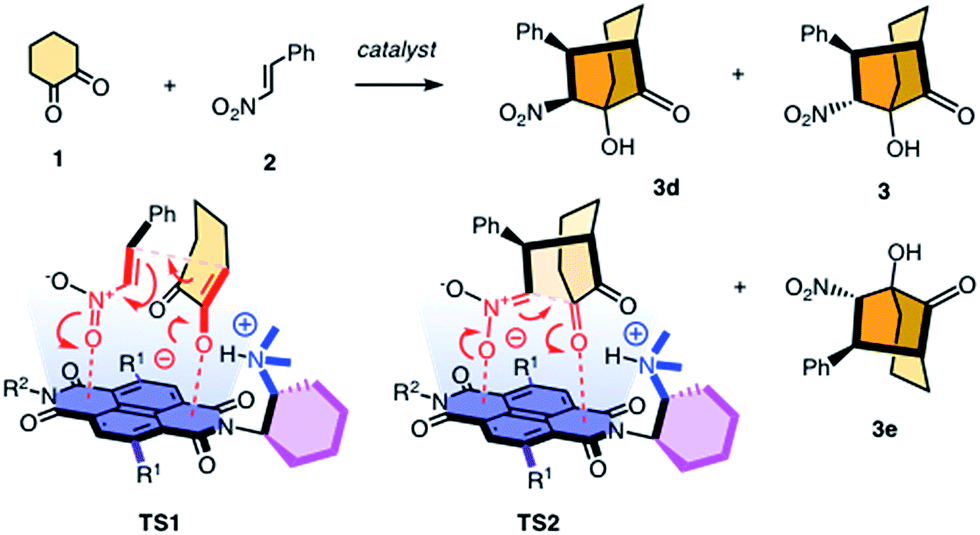 | ||
| Fig. 1 The reaction selected for anion–π catalysis of cascade reactions to bicyclic products, with notional structures for the stabilization of the anionic transition state TS1 and TS2 on the π-acidic surface of naphthalenediimides; for R1 and R2, see Fig. 2. | ||
To elaborate on the formation of bicyclic products from achiral diketones on π-acidic surfaces, catalysts and controls 4–14 were prepared (Fig. 2). Most were accessible following reported procedures; details can be found in the ESI (Schemes S1–S5†). Anion–π catalyst 4 has been designed around the π-acidic surface of a naphthalenediimide (NDI).2 NDIs offer a privileged platform in anion–π catalysis because their intrinsic quadrupole moment perpendicular to the π surface is very high and can be easily modulated with substituents in the core.2–8 In anion–π catalyst 4, this π surface was connected to a tertiary amine catalyst via a fixed Leonard turn.4 These turns have been introduced to assure that the reactions really occur on the π surfaces and benefit best from anion–π interactions, even when they are not particularly strong.4 The imide on the other side of the π-acidic surface continues with a simple solubilizing group.
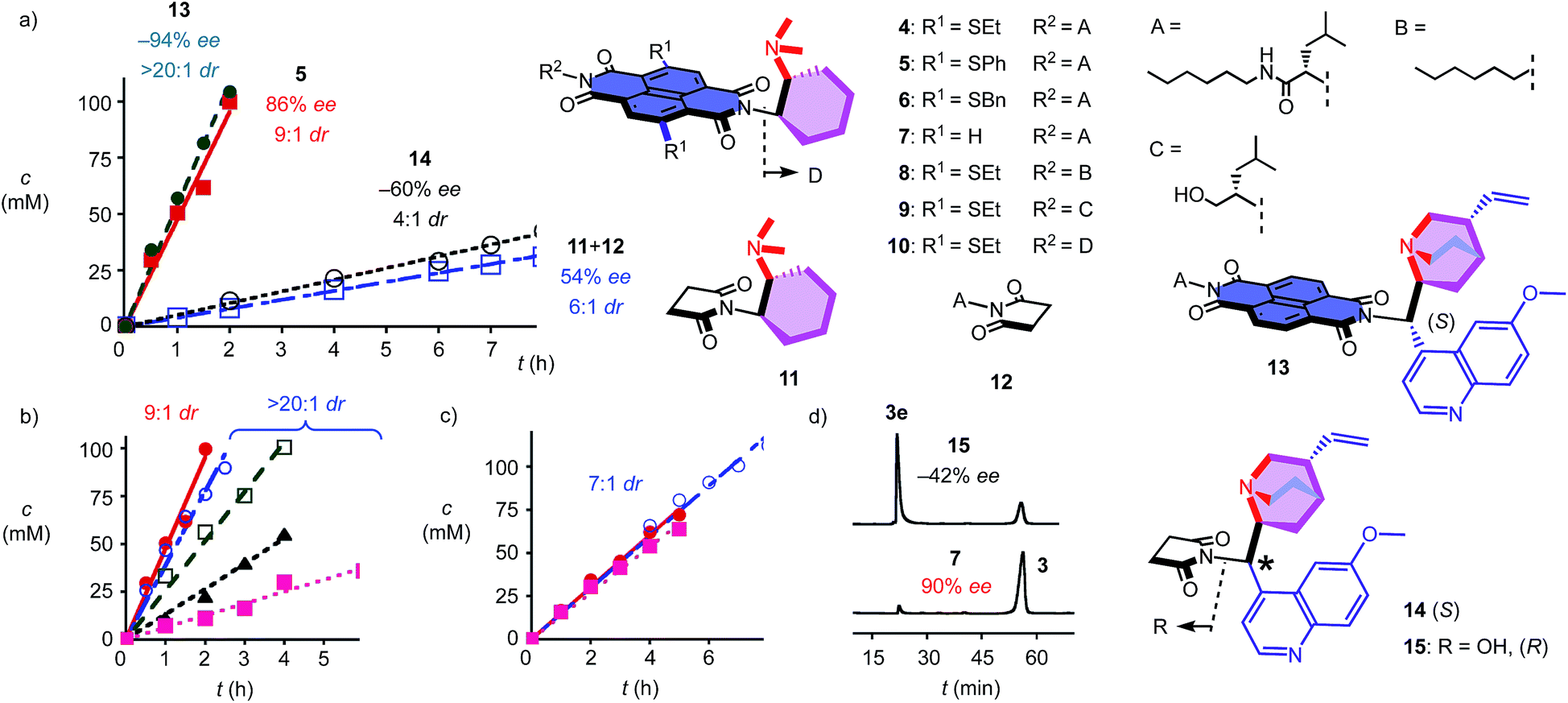 | ||
| Fig. 2 Structure of original anion–π catalysts and controls, and product concentration as a function of time (a) in the presence of catalysts 13 (●), 5 (■), 14 (○) and 11 + 12 (□), (b) with 5 (●) and 1.2 M TBAPF6 (○), TBABF4 (□), TBABr (▲) or TBANO3 (■) and (c) with 11 + 12 (●) and 1.2 M TBAPF6 (■) or TBANO3 (○). (d) Chiral HPLC analyses for product enantiomers 3e (first peak) and 3 (second) obtained with 15 (top) and 7 (bottom). | ||
In the presence of 10 mol% of anion–π catalyst, the formation of bicyclo[3.2.1]octan-8-one 3 occurred within 1–3 days at ambient temperature. The known absolute configuration of product 3e obtained with quinine 15 allowed us to assign the absolute configuration of enantiomer 3 obtained with 4–12 (Fig. 2d).12 Solvent screening with 4 gave best results in C6F6, i.e. 10:1 dr and 80% ee (Table 1, entries 12–15). With 20% C6D6, a slight increase to 13:1 dr coincided with a slight decrease to 77% ee (Table 1, entry 7). Further decreasing π acidity of the solvent gradually decreased stereoselectivity down to 6:1 dr and 66% ee in C6D6 (Table 1, entry 4). These trends were interesting because they supported contributions from synergistic anion–π interactions with polarization of the NDI plane induced by the solvent.14 From C6F6, enantioselectivities could be further increased with co-solvents, best with 88% ee in C6F6/CDCl3 2:1, but diastereoselectivity dropped to 7:1 dr under these conditions (Table 1, entry 9).
| Entry | Cata (mol%) | Conditionsb | η (%) | ee (%) | dr | |
|---|---|---|---|---|---|---|
| a Catalysts (see Fig. 2) and loading. b 1 (entries 1–34: 0.4 M; entries 35–40: 6.7 mM), 2 (entries 1–34: 0.8 M; entries 35–40: 16.6 mM), 2.5–10 mol% catalyst, 20 °C unless stated, 1–3 days, modifications from standard conditions are indicated at first appearance. c Yield based on crude 1H NMR spectroscopy with dibromomethane as an internal standard. d Enantiomeric excess; positive values refer to 3, negative values to 3e, Fig. 1. e Diastereomeric ratio, 3vs.3d or 3evs.3ed (Fig. 1). f 1.2 M TBA salts. g 16 bound to WT or mutant streptavidin as a catalyst (5 mol%). h 33 mM Bis–Tris, 33% MeCN. | ||||||
| 1 | 4 (10) | CDCl3 | 86 | 67 | 8:1 |
|
| 2 | 4 (10) | THF-d8 | 73 | 79 | 5:1 |
|
| 3 | 4 (10) | CD3CN | 60 | 80 | 7:1 |
|
| 4 | 4 (10) | C6D6 | 75 | 66 | 6:1 |
|
| 5 | 4 (10) | Toluene-d8 | 83 | 68 | 10:1 |
|
| 6 | 4 (10) | C6H5NO2 | 74 | 75 | 10:1 |
|
| 7 | 4 (10) | C6F6/C6D6 4:1 |
88 | 77 | 13:1 |
|
| 8 | 4 (10) | C6F6/CD3CN 4:1 |
80 | 80 | 9:1 |
|
| 9 | 4 (5) | C6F6/CDCl3 2:1 |
92 | 88 | 7:1 |
|
| 10 | 4 (5) | C6F6/CDCl3 3:1 |
95 | 86 | 8:1 |
|
| 11 | 4 (5) | C6F6/CDCl3 4:1 |
91 | 84 | 9:1 |
|
| 12 | 4 (10) | C6F6 | 93 | 80 | 10:1 |
|
| 13 | 4 (5) | C6F6 | 92 | 78 | 10:1 |
|
| 14 | 4 (2.5) | C6F6 | 92 | 78 | 10:1 |
|
| 15 | 4 (5) | C6F6, 5 °C | 95 | 79 | 13:1 |
|
| 16 | 5 (5) | C6F6 | 91 | 83 | 13:1 |
|
| 17 | 6 (5) | C6F6 | 86 | 78 | 7:1 |
|
| 18 | 7 (5) | C6F6 | 95 | 90 | 7:1 |
|
| 19 | 9 (5) | C6F6 | 86 | 73 | 11:1 |
|
| 20 | 11 + 12 (5) | C6F6 | 86 | 54 | 7:1 |
|
| 21 | 8 (5) | C6F6/CDCl3 2:1 |
94 | 89 | 9:1 |
|
| 22 | 10 (5) | C6F6/CDCl3 2:1 |
80 | 82 | 6:1 |
|
| 23 | 13 (5) | C6F6/CDCl3 4:1 |
89 | −94 | >20:1 |
|
| 24 | 14 (5) | C6F6/CDCl3 4:1 |
90 | −60 | 4:1 |
|
| 25 | 15 (5) | Toluene-d8 | 83 | −42 | 4:1 |
|
| 26 | 5 (5) | C6F6/CDCl3 4:1 |
91 | 86 | 9:1 |
|
| 27 | 5 (5) | +PF6–f | 90 | 90 | >20:1 |
|
| 28 | 5 (5) | +BF4–f | 89 | 85 | >20:1 |
|
| 29 | 5 (5) | +Br–f | 94 | 85 | >20:1 |
|
| 30 | 5 (5) | +NO3–f | 89 | 78 | >20:1 |
|
| 31 | 13 (5) | +NO3–f | 92 | −94 | >20:1 |
|
| 32 | 11 + 12 (10) | C6F6/CDCl3 4:1 |
95 | 54 | 7:1 |
|
| 33 | 11 + 12 (10) | +PF6–f | 94 | 30 | 10:1 |
|
| 34 | 11 + 12 (10) | +NO3–f | 92 | 52 | 8:1 |
|
| 35 | 16 + WTg | Buffer pH 6.5h | 53 | −45 | >20:1 |
|
| 36 | 16 + K121Ag | 39 | 0 | |||
| 37 | 16 + K121Rg | 47 | −20 | >20:1 |
||
| 38 | 16 + S112Yg | 47 | −53 | >20:1 |
||
| 39 | 16 + S112Wg | 51 | −76 | >20:1 |
||
| 40 | 16 + S112Wg | +NO3– | 33 | −8 | >20:1 |
|
In C6F6, catalyst loadings could be reduced to 2.5 mol% without significant losses in stereoselectivity (Table 1, entries 12–14). At 5 mol% in C6F6, reduction of the temperature to 5 °C further increased diastereoselectivity to 13:1 dr, whereas enantioselectivity did not change significantly (Table 1, entry 15).
Variation of the core substituents did not affect the activity of catalysts 4–7 significantly, also because the π acidity of the π surface was not much changed (Table 1, entries 13 and 16–18).15 The best activity was obtained for 5 with phenylsulfides in the core, i.e., 83% ee and 13:1 dr for 5 mol% in C6F6 (Table 1, entry 16). The 90% ee obtained at maximal π acidity without core substituents in 7 was accompanied by a decrease in diastereoselectivity to 7:1 dr (Table 1, entry 18). Similarly insignificant were variations of the second imide substituent, i.e. R2 opposite to the Leonard-turned amine in catalysts 8–10 (Table 1, entries 19, 21 and 22). This trend suggested that these anion–π catalysts are formally bifunctional, but not trifunctional (Fig. 1).
Cinchona alkaloids are most popular in amine-based asymmetric organocatalysis.11,12,16 In the newly designed catalyst 13, this bicyclic amine was attached to a π-acidic surface via a Leonard turn, similar to the one introduced in catalysts 4–10 (Fig. 2). The synthesis of the new anion–π cinchona fusion catalyst 13 was very straightforward (Scheme S4†). In C6F6/CDCl3 4:1, 5 mol% of cinchona catalyst 13 produced bicycle 3e in 89% yield with −94% ee and only one detectable diastereomer, i.e., >20:1 dr (Table 1, entry 23). This shift from diastereoselectivity to diastereospecificity is unprecedented for this domino process; previous records in the literature, achieved with cinchona catalysts interfaced with conventional thioureas, stopped at 12:1 dr.12 The diastereospecificity obtained with cinchona catalyst 13 suggested that on π-acidic surfaces, either the epimerization between 3e and 3ed is suppressed or the protonation of the nitronate intermediate is stereospecific.
Cinchona controls 14 and 15 without a π-acidic surface gave only −60% ee (4:1 dr) and −42% ee (lit. −17% ee,12Table 1, entries 24 and 25), respectively. The increase in stereoselectivity upon interfacing of cinchona alkaloids with a π-acidic surface in cinchona catalyst 13 coincided with a rate enhancement of v/v0 = 10 (Fig. 2a). This acceleration corresponded to a transition-state stabilization (or ground-state destabilization) of ΔEa = −5.7 kJ mol−1. For comparison with Leonard catalyst 5, controls 11 and 12 without π-acidic surface also gave much poorer stereoselectivity (54% ee, 7:1 dr) at a slower rate (Fig. 2a, Table 1, entries 16 and 20). The rate enhancement v/v0 = 12 corresponded to ΔEa = −6.1 kJ mol−1. The similar transition-state stabilization obtained with the conventional Leonard catalyst 5 and new fusion catalyst 13 was meaningful considering the similarity of the π acidity of the respective π surfaces. Rate enhancements coinciding with increased stereoselectivity in the presence of π-acidic surfaces provided strong support for contributions of anion–π interactions to asymmetric catalysis.3–8
Corroborative support for this important conclusion was obtained by inhibition with inorganic anions. Consistent with competitive anion–π interactions, the velocity of Leonard catalyst 5 decreased in the presence of various tetrabutylammonium (TBA) salts with a selectivity sequence PF6− < BF4− < Br− < NO3− (Fig. 2b; Table 1, entries 27–30). A half maximal inhibitory concentration (IC50) of nitrate was calculated to be 0.70 M for catalyst 5 (Fig. S17†). For cinchona catalyst 13, a similar IC50 = 0.75 M was obtained (Fig. S18†; Table 1, entry 31). Controls 11 and 12 were insensitive to the presence of anions (Fig. 2c, Table 1, entries 32–34). Interestingly, the diastereoselective Leonard catalyst 5 became diastereospecific in the presence of various TBA salts (Table 1, entries 26–30). The independence of this trend on the anion involved suggested that the TBA cations are responsible. A plausible explanation for this finding was that TBA cations induce the polarization of the π-acidic NDI core, comparable to the expected function of C6F6.14 Indeed, the observed acceleration of reactions with low concentrations of TBANO3 salts was consistent with this explanation (Fig. S16†). With cinchona catalyst 13 already producing bicycle 3e diastereospecifically, the discovery of achiral salts as unprecedented supramolecular anion–π chirality enhancers for Leonard catalyst 5 suggested that both enantiomers 3 and 3e could be obtained diastereospecifically on π-acidic surfaces.
To position anion–π catalysts within the chiral space of proteins, anion–π catalyst 4 has been coupled with a biotin.5 Binding of the resulting conjugate 16 to streptavidin then afforded an artificial anion–π enzyme, which operates with an essentially unknown interaction to biological enzymes (Fig. 3a).5 Interfacing of anion–π catalysts with proteins is attractive because access to mutant screening allows performance to be readily optimized. This screening approach has previously afforded anion–π enzymes that catalyze, at pH 3.0, the addition of malonic acid half thioesters to enolate acceptors with 95% ee and unprecedented chemospecificity with regard to the intrinsically favored decarboxylation.5 A focused mutant screening for the domino reaction to bicycle 3 in the presence of 16 gave best results for S112W at pH 6.5 (−76% ee, Table 1, entry 39). Mutant S112Y, the best for the addition of malonic acid half thioesters, was with −53% ee slightly better than the wild-type (WT) protein with −45% ee (Table 1, entries 35 and 38). K121 was confirmed as essential, presumably to keep the tertiary amine base in 16 from protonation under experimental conditions (Table 1, entries 36 and 37). In the presence of nitrate, the enantioselectivity decreased, with IC50 = 0.34 M (Fig. 3b, Table 1, entry 40). This finding provided experimental support for operational anion–π interactions, i.e. the existence of anion–π enzymes. Interestingly, all anion–π enzymes obtained with 16 gave enantiomer 3e with a maximum ee of −76% as the main product in nearly neutral water/MeCN 2:1, whereas the protein-free analog 4 gave the opposite enantiomer 3 with a maximum ee of 88% as the main product in C6F6/CDCl3 4:1. It is perhaps this inversion of the intrinsic enantioselectivity induced by the fixed Leonard turn derived from 1R,2R-diaminocyclohexane in both 4 and 16 that hindered access to higher ee’s with anion–π enzymes.
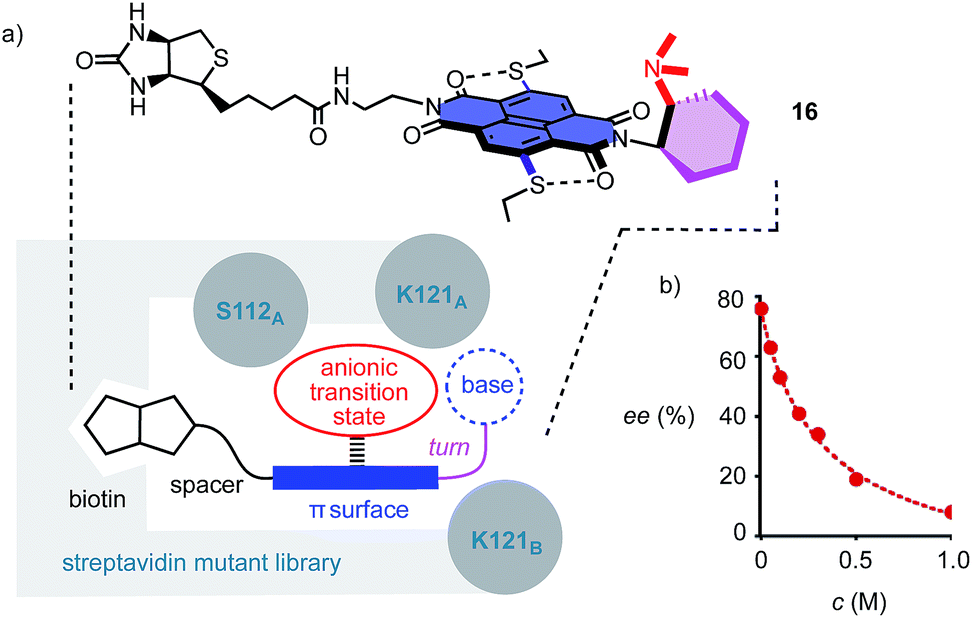 | ||
| Fig. 3 a) The concept of anion–π enzymes, with indication of the structure of the anion–π biotin conjugate 16 interface used with streptavidin mutants and the position of the mutated key residues from monomers A and B of the streptavidin tetramer, and (b) the dependence of the ee of 3e produced by the S112W mutant on the concentration of NO3– (NaNO3). | ||
In summary, this study drives the development of anion–π catalysts to unprecedented sophistication with regard to anionic domino reactions that take place on π-acidic surfaces. The most important findings are anion–π cinchona fusion catalysts that clearly exceed the performance of conventional metal-free organocatalysts for the first time, the first example for diastereospecificity, anion–π enzymes that operate in neutral water and the discovery of achiral tetraalkylammonium salts as supramolecular chirality enhancers. The discovery of the best anion–π catalysts with the most sophisticated domino reaction supports the important expectation that anion–π interactions, delocalized over large aromatic planes, will be most advantageous in stabilizing extensive long-distance charge displacements during multiple coupled anionic intermediates and transition states. Long-term perspectives include the discovery of otherwise inaccessible reactions with anion–π catalysis, the integration into more complex systems,5,17 and the introduction of other unorthodox interactions to catalysis.3,18
Acknowledgements
We thank the NMR and the Mass Spectrometry platforms for services, and the University of Geneva, the Swiss National Centre of Competence in Research (NCCR) Molecular Systems Engineering, the NCCR Chemical Biology and the Swiss NSF for financial support.Notes and references
- (a) A. Frontera, P. Gamez, M. Mascal, T. J. Mooibroek and J. Reedijk, Angew. Chem., Int. Ed., 2011, 50, 9564–9583 CrossRef CAS PubMed; (b) M. Giese, M. Albrecht and K. Rissanen, Chem. Commun., 2016, 52, 1778–1795 RSC; (c) H. T. Chifotides and K. R. Dunbar, Acc. Chem. Res., 2013, 46, 894–906 CrossRef CAS PubMed; (d) D.-X. Wang and M.-X. Wang, Chimia, 2011, 65, 939–943 CrossRef CAS PubMed; (e) P. Ballester, Acc. Chem. Res., 2013, 46, 874–884 CrossRef CAS PubMed; (f) Q. He, Y.-F. Ao, Z.-T. Huang and D.-X. Wang, Angew. Chem., Int. Ed., 2015, 54, 11785–11790 CrossRef CAS PubMed.
- (a) M. Al Kobaisi, S. V. Bhosale, K. Latham, A. M. Raynor and S. V. Bhosale, Chem. Rev., 2016, 116, 11685–11796 CrossRef PubMed; (b) J.-Z. Liao, C. Wu, X.-Y. Wu, S.-Q. Deng and C.-Z. Lu, Chem. Commun., 2016, 52, 7394–7397 RSC; (c) J.-J. Liu, Y.-F. Guan, C. Jiao, M.-J. Lin, C.-C. Huang and W.-X. Dai, Dalton Trans., 2015, 44, 5957–5960 RSC; (d) S. Kumar, M. R. Ajayakumar, G. Hundal and P. Mukhopadhyay, J. Am. Chem. Soc., 2014, 136, 12004–12010 CrossRef CAS PubMed; (e) S. T. Schneebeli, M. Frasconi, Z. Liu, Y. Wu, D. M. Gardner, N. L. Strutt, C. Cheng, R. Carmieli, M. R. Wasielewski and J. F. Stoddart, Angew. Chem., Int. Ed., 2013, 52, 13100–13104 CrossRef CAS PubMed.
- Y. Zhao, Y. Domoto, E. Orentas, C. Beuchat, D. Emery, J. Mareda, N. Sakai and S. Matile, Angew. Chem., Int. Ed., 2013, 52, 9940–9943 CrossRef CAS PubMed.
- Y. Cotelle, S. Benz, A.-J. Avestro, T. R. Ward, N. Sakai and S. Matile, Angew. Chem., Int. Ed., 2016, 55, 4275–4279 CrossRef CAS PubMed.
- Y. Cotelle, V. Lebrun, N. Sakai, T. R. Ward and S. Matile, ACS Cent. Sci., 2016, 2, 388–393 CrossRef CAS PubMed.
- (a) Y. Zhao, Y. Cotelle, A.-J. Avestro, N. Sakai and S. Matile, J. Am. Chem. Soc., 2015, 137, 11582–11585 CrossRef PubMed; (b) K. S. Lee and J. R. Parquette, Chem. Commun., 2015, 51, 15653–15656 RSC.
- L. Liu, Y. Cotelle, A.-J. Avestro, N. Sakai and S. Matile, J. Am. Chem. Soc., 2016, 138, 7876–7879 CrossRef CAS PubMed.
- L. Liu and S. Matile, Supramol. Chem., 2017 DOI:10.1080/10610278.2016.1258118.
- A. Berkessel, S. Das, D. Pekel and J.-M. Neudörfl, Angew. Chem., Int. Ed., 2014, 53, 11660–11664 CrossRef CAS PubMed.
- (a) C. R. Kennedy, S. Lin and E. N. Jacobsen, Angew. Chem., Int. Ed., 2016, 55, 12596–12624 CrossRef CAS PubMed; (b) Y. Zhao, Y. Cotelle, N. Sakai and S. Matile, J. Am. Chem. Soc., 2016, 138, 4270–4277 CrossRef CAS PubMed; (c) T. M. Bräuer, Q. Zhang and K. Tiefenbacher, Angew. Chem., Int. Ed., 2016, 55, 7698–7701 CrossRef PubMed.
- M. Rueping, A. Kuenkel and R. Fröhlich, Chem.–Eur. J., 2010, 16, 4173–4176 CrossRef CAS PubMed.
- D. Ding, C.-G. Zhao, Q. Guo and H. Arman, Tetrahedron, 2010, 66, 4423–4427 CrossRef CAS PubMed.
- F. Felluga, C. Forzato, P. Nitti, G. Pitacco, E. Valentin and E. Zangrando, J. Heterocyclic. Chem., 2010, 47, 664–670 CAS.
- (a) A. Frontera, D. Quiñonero and P. M. Deyà, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2011, 1, 440–459 CrossRef CAS; (b) A. Lattanzi, C. De Fusco, A. Russo, A. Poater and L. Cavallo, Chem. Commun., 2012, 48, 1650–1652 RSC.
- F. N. Miros and S. Matile, ChemistryOpen, 2016, 5, 219–226 CrossRef CAS PubMed.
- (a) C. M. R. Volla, I. Atodiresei and M. Rueping, Chem. Rev., 2014, 114, 2390–2431 CrossRef CAS PubMed; (b) A. Moyano and R. Rios, Chem. Rev., 2011, 111, 4703–4832 CrossRef CAS PubMed; (c) D. Enders, C. Grondal and M. R. M. Hüttl, Angew. Chem., Int. Ed., 2007, 46, 1570–1581 CrossRef CAS PubMed.
- (a) B. Baumeister, N. Sakai and S. Matile, Org. Lett., 2001, 3, 4229–4232 CrossRef CAS PubMed; (b) N. Sakai and S. Matile, J. Am. Chem. Soc., 2011, 133, 18542–18545 CrossRef CAS PubMed; (c) B. A. F. Le Bailly, L. Byrne and J. Clayden, Angew. Chem., Int. Ed., 2016, 55, 2312–2316 CrossRef PubMed.
- (a) S. Benz, J. López-Andarias, J. Mareda, N. Sakai and S. Matile, Angew. Chem., Int. Ed., 2017, 56, 812–815 CrossRef CAS PubMed; (b) D. Bulfield and S. M. Huber, Chem.–Eur. J., 2016, 22, 14434–14450 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed procedures and results for all reported experiments. See DOI: 10.1039/c7sc00525c |
| This journal is © The Royal Society of Chemistry 2017 |