DOI:
10.1039/C7RA06796H
(Paper)
RSC Adv., 2017,
7, 35905-35916
Syntheses, crystal structures and catalytic activities of two solvent-induced homotrinuclear Co(II) complexes with a naphthalenediol-based bis(Salamo)-type tetraoxime ligand†
Received
19th June 2017
, Accepted 13th July 2017
First published on 19th July 2017
Abstract
Two homotrinuclear Co(II) complexes, [Co3(L)(OAc)2(CH3OH)2] 2CHCl3 (1) and [Co3(L)(OAc)2(H2O)2] (2) based on a naphthalenediol-based acyclic bis(Salamo)-type tetraoxime ligand were synthesized. 1 and 2 were influenced by the coordinated methanol and water molecules, respectively, and their X-ray crystal structures revealed that they have similar molecular structures. Two terminal Co(II) ions, located at the N2O2 coordination spheres of the Salamo moieties, are both in distorted trigonal-bipyramidal geometries, while the third Co(II) ion in the central O4 cavity shows an octahedral geometry with two solvent molecules placed in the apical sites. Different solvent molecules lead to different supramolecular structures. The catecholase activities of 1 and 2 were examined using 3,5-di-tert-butylcatechol (3,5-DTBC) in acetonitrile solution under completely aerobic conditions. The catalytic reaction follows Michaelis–Menten enzymatic reaction kinetics with turnover numbers (Kcat) of 14.72 h−1 and 26.39 h−1 for 1 and 2, respectively.
Introduction
The Salen-type ligand and its analogues continue to play significant roles in modern coordination chemistry.1 Their metal complexes have been explored to construct supramolecular structures2 and coordination polymers.1e Numerous investigations on these compounds, such as their luminescent properties,3 biological activities,4 catalysis activity,5 magnetic properties,6 electrochemical field7 and molecular recognition8 etc., also have been examined. Recently, many appropriate classes of Salen-type bisoxime compounds (Salamo and its derivatives) have been reported using o-alkyloxime units (–CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–O–(CH2)n–O–NCH–) rather than the non o-alkyloxime (–CHN–(CH2)n–NCH–) units, and the large electronegativity of oxygen atoms is expected to strongly affect the electronic property of the N2O2 coordination sphere, which can lead to novel structures and different properties of the resulted complexes. Thus, Salamo-type ligands and their complexes have also been explored by Nabeshima group and ours,9 however, the bis(Salamo)-type metal complexes were rarely reported.10
N–O–(CH2)n–O–NCH–) rather than the non o-alkyloxime (–CHN–(CH2)n–NCH–) units, and the large electronegativity of oxygen atoms is expected to strongly affect the electronic property of the N2O2 coordination sphere, which can lead to novel structures and different properties of the resulted complexes. Thus, Salamo-type ligands and their complexes have also been explored by Nabeshima group and ours,9 however, the bis(Salamo)-type metal complexes were rarely reported.10
Over the decades, Salen-type complexes11 have been widely utilized as catalysts in the industry, and functions in many important chemical reactions such as oxidation, reduction and polymerization.12 However, studies on the participation of Salamo-type complexes in the catalytic process is yet to be explored. It has been known that the ubiquitous bio-enzyme catechol oxidase catalyzes exclusively the oxidation of catechols to the corresponding o-quinones in the presence of molecular oxygen. The active site of the catechol oxidase consists of a hydroxobridged dicopper(II) centre where each copper(II) centre is coordinated to three histidine nitrogens and adopts a trigonal pyramidal coordination geometry with one nitrogen in the apical site.13 Up to now, numerous researchers are designing new molecules that can give promising model complexes as effective replacements for catechol oxidase. The design of ligands has to satisfy a number of conditions such as metal–metal distance, steric effects,14 electronic and bridging ligand features,15 redox potentials,16 and pH.17 Several model complexes have been reported to elucidate the structure–function relationship of this biocatalytic catechol–quinone conversion. Although most functional mimics of catechol oxidase are mono- and dinuclear Cu(II) complexes, other transition metal ions such as Mn(II/III/IV), and Co(III) can also serve as catalytically active complexes.18
And as far as we know, the current work is to first report the Salamo-type complex as a catechol oxidase model complex, which is a Salamo-type complex application provides a new idea.
As important raw materials and intermediates in organicchemical, catechins have a wide range of applications in agriculture, pharmaceutical, photographic materials and other fields. However, little research on Co(II) complexes has been done in regards to catecholase activity. Herein, we designed and synthesized a new acyclic naphthalenediol-based bis(Salamo)-type tetraoxime ligand (H4L) containing two Salamo moieties, which is likely to form multinuclear metal complexes. Two homotrinuclear Co(II) complexes (solv: MeOH and H2O, respectively) were synthesized by the reaction of H4L with Co(II) acetate tetrahydrate in different solvents. Due to the unique structure of Salamo-type complexes, a study shown that it is at least 104 times more stable than Salen-type complexes.19 It suggests that Salamo-type complexes have a unique advantage in catalysis. As far as we know, the present work is the first to report on the Salamo-type complexes as catechol oxidase model complexes, which provide a new idea for the application of Salamo-type complexes.
Experimental
Materials and instruments
All materials used were of high commercially available grade and were used as purchased. 3,5-Dibromosalicylaldehyde, methyl trioctyl ammonium chloride, pyridiniumchlorochromate and borontribromide were purchased from Alfa Aesar. Hydrobromic acid 33 wt% solution in acetic acid was purchased from J&K Scientific Ltd and 3,5-di-tert-butylcatechol was bought from Tokyo Chemical Industry. The other reagents and solvents were from Tianjin Chemical Reagent Factory.
Elemental (carbon, hydrogen, and nitrogen) analyses were performed on GmbH VariuoEL V3.00 automatic elemental analyzer. Elemental analysis for Co was detected by an IRIS ER/SWP-1 ICP atomic emission spectrometer. Melting points were measured by the use of a microscopic melting point apparatus made in Beijing Taike Instrument Limited Company. FT-IR spectra (400–4000 cm−1) were recorded on a VERTEX70 FT-IR spectrophotometer using KBr as medium. UV-vis absorption spectra were recorded on a Hitachi U-3900H spectrophotometer. 1H NMR spectra were determined by German Bruker AVANCE DRX-400/600 spectroscopy.
Synthesis of the ligand H4L
1,2-Bis(aminooxy)ethane, 2-[O-(1-ethyloxyamide)]oxime-4,6-dibromophenol19 and 2,3-dihydroxynaphthalene-1,4-dicarbaldehyde20 were synthesized according to the analogous reported procedures. 2,3-Dihydroxynaphthalene-1,4-dicarbaldehyde (0.354 g, 1.0 mmol) was added to the ethanol solution (30 mL) of 2-[O-(1-ethyloxyamide)]oxime-4,6-dibromophenol (0.540 g, 2.5 mmol). The suspension solution was stirred and refluxed at 45 °C for 4 h and then faint yellow solid of bis(Salamo)-type tetraoxime ligand (H4L1) was obtained, which was collected by suction filtration at 45 °C immediately, and washed with hot ethanol solution (3 × 10 mL, 45 °C) to give a yellow powder. Yield, 0.75 g (85%). Mp 205–207 °C. Anal. calc. for C30H24Br4N4O8: C, 40.57; H, 2.72; N, 6.31. Found: C, 40.54; H, 2.74; N, 6.38%. 1H NMR (400 MHz, DMSO-d6, δ, ppm): 4.56 (s, 8H, OCH–H), 7.38–7.40 (m, 2H, Ar-H), 7.69 (s, 2H, Ar-H), 7.81 (s, 2H, Ar-H), 8.47–8.49 (m, 2H, Ar-H), 8.51 (s, 2H, NC–H), 9.10 (s, 2H, NC–H), 10.59 (s, 4H, O–H). IR (KBr, cm−1): 3307 (m), 3059 (m), 2927 (m), 2904 (m), 2854 (m), 1605 (m), 1433 (s), 1256 (m), 1212 (m). UV-vis (CHCl3/CH3OH 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 v/v): λmax (εmax): 341, 362 and 380 nm (1.88 × 104, 1.57 × 104 and 1.66 × 104 M−1 cm−1).
:2 v/v): λmax (εmax): 341, 362 and 380 nm (1.88 × 104, 1.57 × 104 and 1.66 × 104 M−1 cm−1).
Syntheses of the Co(II) complexes 1 and 2
[Co3(L)(OAc)2(CH3OH)2]·2CHCl3 (1). A methanol solution (3 mL) of Co(OAc)2·4H2O (3.72 mg, 0.015 mmol) was added to the suspended solution of H4L (4.40 mg, 0.005 mmol) in chloroform (2 mL) and stirred for 2 min. The solution turned to transparent and then filtered. Diffraction quality single crystals were obtained after few days on slow evaporation of the solution in open atmosphere. Brown blocks. Yield, 4.77 mg (64%). The experiments on larger scale also was performed under similar conditions. Anal. calc. for [Co3(L)(OAc)2(CH3OH)2] 2CHCl3 (C38H36N4O14Br4Cl6Co3): C, 30.80; H, 2.45; N, 3.78; Co, 11.93. Found: C, 30.85; H, 2.41; N, 3.73; Co, 11.90%. IR (KBr, cm−1): 3324 (m), 3066 (m), 2971 (m), 2896 (m), 1587 (m), 1440 (s), 1258 (m). UV-vis (CHCl3/CH3OH 3:2 v/v): λmax (εmax) 371 and 415 nm (1.70 × 104 and 1.52 × 104 M−1 cm−1). UV-vis (MeCN): λmax (εmax) 376 and 416 nm (1.82 × 104 and 1.53 × 104 M−1 cm−1).
[Co3(L)(OAc)2(H2O)2] (2). An i-PrOH/H2O solution (3:0.5 v/v, 3.5 mL) of Co(OAc)2·4H2O (5.20 mg, 0.021 mmol) was added to the suspended solution of H4L (6.16 mg, 0.007 mmol) in (CH3)2CO/CHCl3 (2:1 v/v, 3 mL) and stirred for 20 min. The solution turned to transparent and then filtered. Diffraction quality single crystals were obtained after few days on slow evaporation of the solution in open atmosphere. Brown needles. Yield, 4.61 mg (54%). The experiments on larger scale also was performed under similar conditions. Anal. calc. for [Co3(L)(OAc)2(H2O)2] (C34H30Br4N4O14Co3): C, 33.61; H, 2.49; N 4.61; Co, 14.55. Found C, 33.67; H, 2.44; N, 4.57; Co, 14.49%. IR (KBr, cm−1): 3420 (m), 3072 (m), 2922 (m), 2850 (m), 1590 (m), 1438 (s), 1257 (m). UV-vis (CHCl3/CH3OH 3:2 v/v): λmax (εmax) 371 and 412 nm (1.55 × 104 and 1.32 × 104 M−1 cm−1). UV-vis (MeCN): λmax (εmax) 373 and 413 nm (1.48 × 104 and 1.29 × 104 M−1 cm−1).
Catalytic oxidation of 3,5-DTBC by 1 and 2 under complete aerobic condition
6.72 mg (0.0045 mmol) of 1 and 1.00 g (4.54 mmol, 1000 equiv. of 1) of 3,5-DTBC were dissolved in 20 mL of MeCN. The solution was stirred for 48 h at room temperature and color was changed remarkably from light brown to deep red. The product was purified by column chromatography and recrystallized in diethyl ether to yield 3,5-DTBQ as red block-like crystals. 2 was also tested by the same methods.
Crystal structure determination
Intensity data for 1 and 2 were collected on a Bruker Smart 1000 CCD area detector with Mo-Kα radiation (λ = 0.71073 Å). Reflection data were corrected for Lorentz and polarization factors and for absorption using the multi-scan method. The structures were solved by using the program SHELXL-2016 (ref. 21) and Fourier difference techniques, and refined by the full-matrix least-squares method based on F2.22 The hydrogen atoms (O13 and O14) of 1 were located in Fourier-difference maps and all the other hydrogen atoms were inserted in calculated positions with isotropic thermal parameters and refined. Non-hydrogen atoms were refined with anisotropic displacement parameters during the final cycles. All hydrogen atoms were placed in calculated positions with isotropic displacement parameters set to 1.2 × Ueq of the attached atom. In addition, in complex 2 existence carbon atoms in the aromatic rings have high thermal motion (C25–C30) and disordered bromine atoms (Br3 and Br4), we use the command in the refinement process by ISOR restraint. The crystallographic data are summarized in Table 1. CCDC – 1508735 (1) and 1506120 (2).†
Table 1 X-ray crystallographic data for 1 and 2
| Complex |
1 |
2 |
| R1 = Σ||Fo| − |Fc||/Σ|Fo||. wR2 = {Σw(Fo2 − Fc2)2/Σ[w(Fo2)]2}1/2. |
| Formula |
C38H36N4O14Br4Cl6Co3 |
C34H30Br4N4O14Co3 |
| Formula weight |
1481.84 |
1215.05 |
| Wavelength (Å) |
0.71073 |
0.71073 |
| Crystal system |
Triclinic |
Monoclinic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P21/c |
| a (Å) |
14.5421(10) |
19.182(2) |
| b (Å) |
14.8347(13) |
18.751(2) |
| c (Å) |
17.3221(18) |
18.722(2) |
| α (deg) |
113.715(9) |
90.00 |
| β (deg) |
90.013(7) |
113.6144(18) |
| γ (deg) |
108.037(7) |
90.00 |
| V (Å3) |
3137.4(2) |
6169.9(12) |
| Z, Dcalc (g cm−3) |
2, 1.529 |
4, 1.308 |
| θ range (deg) |
3.529–26.022 |
1.588–25.499 |
| F(000) |
1454 |
2380 |
| Crystal size (mm) |
0.21 × 0.22 × 0.31 |
0.22 × 0.25 × 0.28 |
| Index ranges |
−17 ≤ h ≤ 17 |
−16 ≤ h ≤ 23 |
| −17 ≤ k ≤ 18 |
−22 ≤ k ≤ 20 |
| −19 ≤ l ≤ 21 |
−22 ≤ l ≤ 22 |
| Reflections collected/unique |
21805/12621 [Rint = 0.0601] |
35080/11495 [Rint = 0.0527] |
| Completeness (%) (θ) |
99.5 (26.022) |
99.9 (26.022) |
| Data/restraints/parameters |
12621/6/632 |
11491/12/610 |
| Goodness-of-fit on F2 |
0.913 |
1.077 |
| R1a/wR2b [I > 2σ(I)] |
0.0721/0.1693 |
0.0568/0.1519 |
Catecholase activity measurement
UV-vis absorption spectra were measured on a Hitachi U-3900H spectrophotometer at 25 °C. Solutions of 1 and 2 (2.5 × 10−5 M) and 3,5-DTBC (in the range of 0.188 M to 1.88 M) were prepared in MeCN (99.99%). Different concentrations of 3,5-DTBC were added (2 μL at a time) to 3 mL complex (1 or 2) solution to obtain mixed solution (final concentration of 3,5-DTBC ranges from 2.5 × 10−3 to 2.5 × 10−2 M). The absorption at λmax = 400 nm (ε = 1900 M−1 cm−1), which is characteristic of the formed quinone, was measured as a function of time. A 2.5 × 10−5 M solution of 1 with 100 equiv. of 3,5-DTBC was used and the solution saturated under complete aerobic condition. Under these conditions, catechol was hardly oxidized in the absence of complexes. The kinetic parameters were determined for 2.5 × 10−5 M solutions of 1 and 2 and 2.5 × 10−3 to 2.5 × 10−2 M solutions of the substrate.
Results and discussion
Formations and characterizations of H4L and its Co(II) complexes
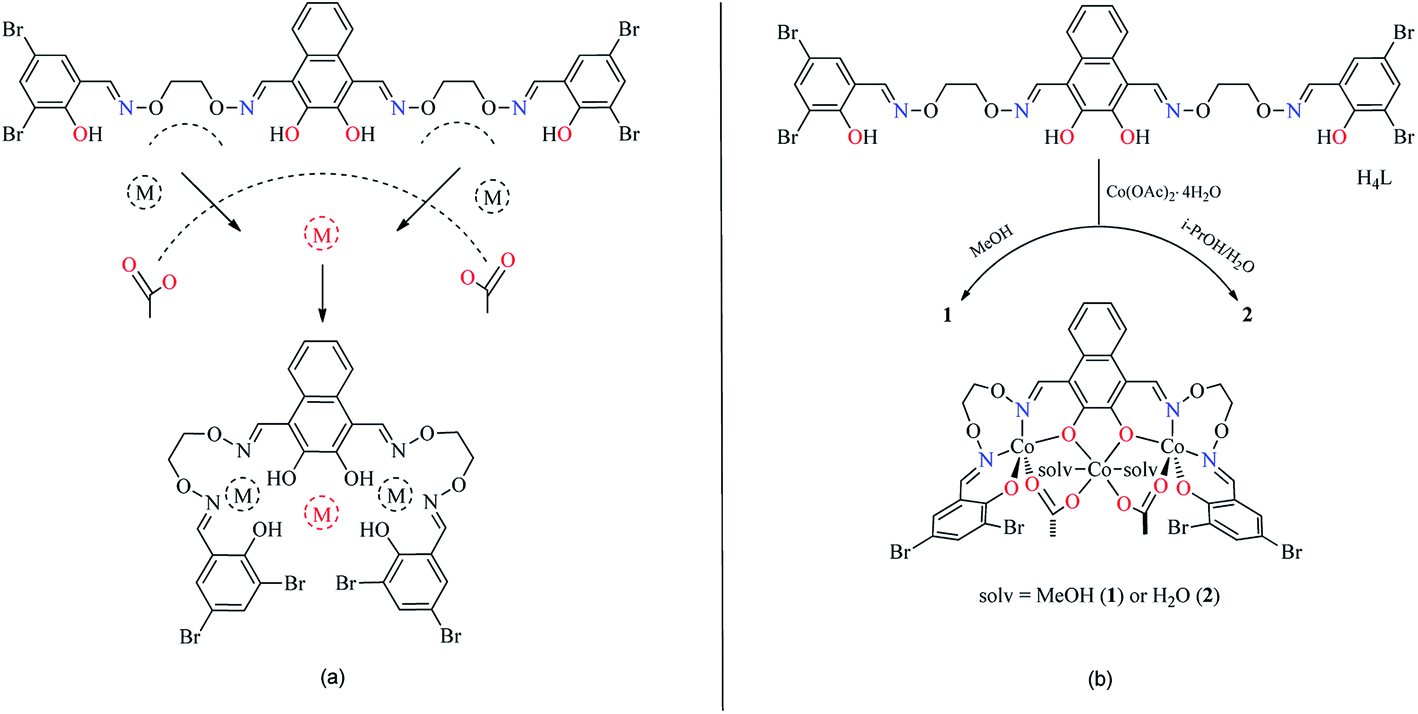
The reaction of 2,3-dihydroxynaphthalene-1,4-dicarbaldehyde with 2-[O-(1-ethyloxyamide)]oxime-4,6-dibromophenol in ethanol gave the acyclic bis(Salamo)-type tetraoxime ligand (H4L), which was collected by suction filtration. When H4L combines with two equivalents of Co(II) ions, the acyclic structural form may be change into a C-shaped form to give a central cavity (Scheme 1a). The intermediate complex is expected to have a high affinity toward another metal ion, since the donor atoms are arranged in a cyclic fashion and negatively charged, which tends to form trinuclear metal complexes and have been proven by UV-vis titration experiment.
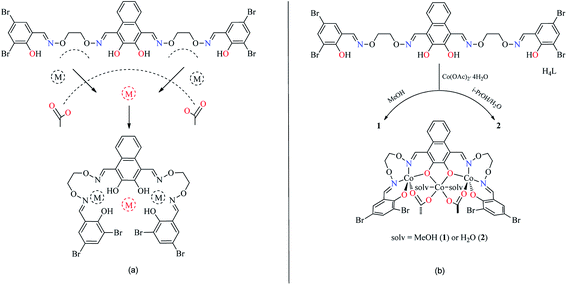 |
| | Scheme 1 Possible coordination pattern (a) and formations of 1 and 2 (b). | |
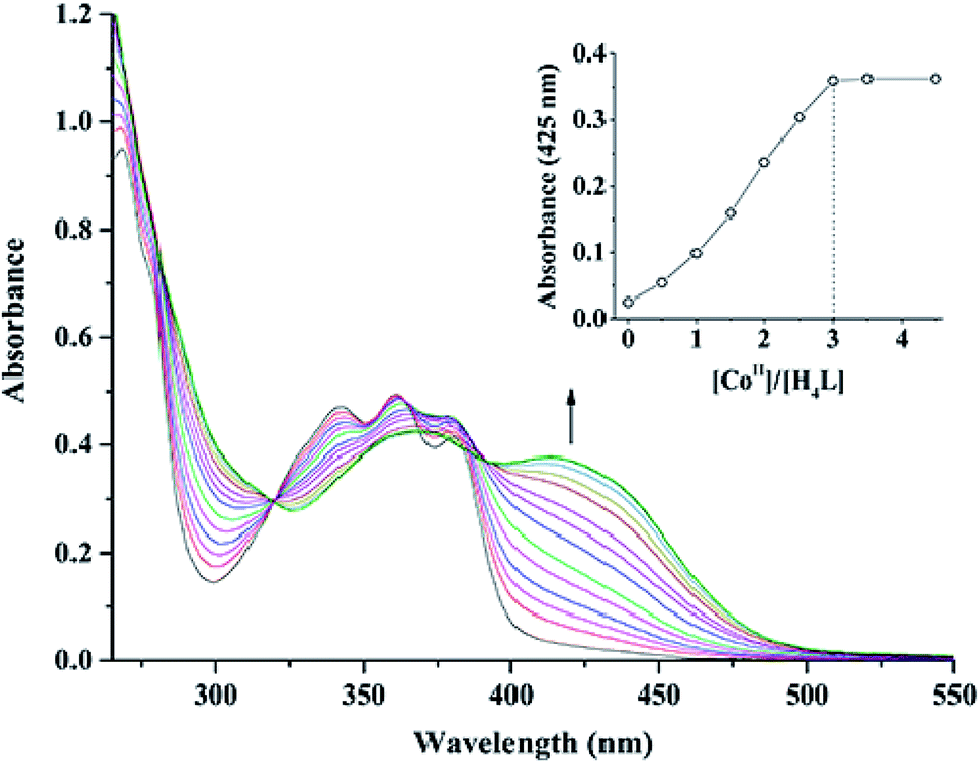
In the UV-vis titration experiment of 1, the color of a solution of H4L in CHCl3/MeOH (3:2 v/v, at this condition, the single crystals of 1 were obtained) gradually changed from colorless to light brown upon addition of a solution of Co(II) acetate. For H4L, the peaks at 342, 361 and 380 nm can be assigned to the intra-ligand π–π* transitions. Upon addition of Co(II) ion, these peaks gradually disappeared with the emergence of new peaks at 369 and 414 nm. The former absorption peak at 369 nm can be attributed to π–π* transitions,9d while the latter one at near 414 nm in the low energy region may be due to L → M charge-transfer transition, which are characteristic of the transition metal complexes with N2O2 coordination spheres.23 However, the absorbance of the solution hardly changed after Co(II) ion was added up to 3 equiv. (Fig. 1). Spectroscopic titration clearly indicates the reaction stoichiometry ratio of 1:3 as shown in the homotrinuclear Co(II) complex, although the ligand H4L only has two Salamo chelate moieties.
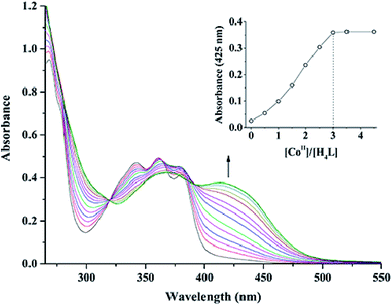 |
| | Fig. 1 UV-vis spectral changes of H4L by the addition of Co(OAc)2 (CHCl3/CH3OH 3:2 v/v, [H4L] = 2.5 × 10−5 M). The inset shows the plot of absorbance at 425 nm versus the molar ratio [CoII]/[H4L]. | |
In order to obtain different Co(II) complexes influenced by the coordinated solvents, various solvents were used as reaction mediums. 1 and 2 were obtained by the addition of different solutions of Co(II) acetate to a suspended solution of H4L at room temperature, and the solutions changed from turbid to transparent. Slow evaporation of the solutions in open atmosphere resulted in the formation of brown polyhedral crystals of 1 and 2 (solv: MeOH and H2O, respectively). It is worth noting that the reaction of Co(II) acetate with H4L in the presence of methanol in the 3:1 molar ratio, respectively, resulted in the coordination of methanol molecules, however, when Co(II) acetate and H4L reacted in the presence of i-PrOH/H2O (3:0.5 v/v), it was water molecules that coordinated to the central Co(II) rather than isopropanol molecules (Scheme 1b).
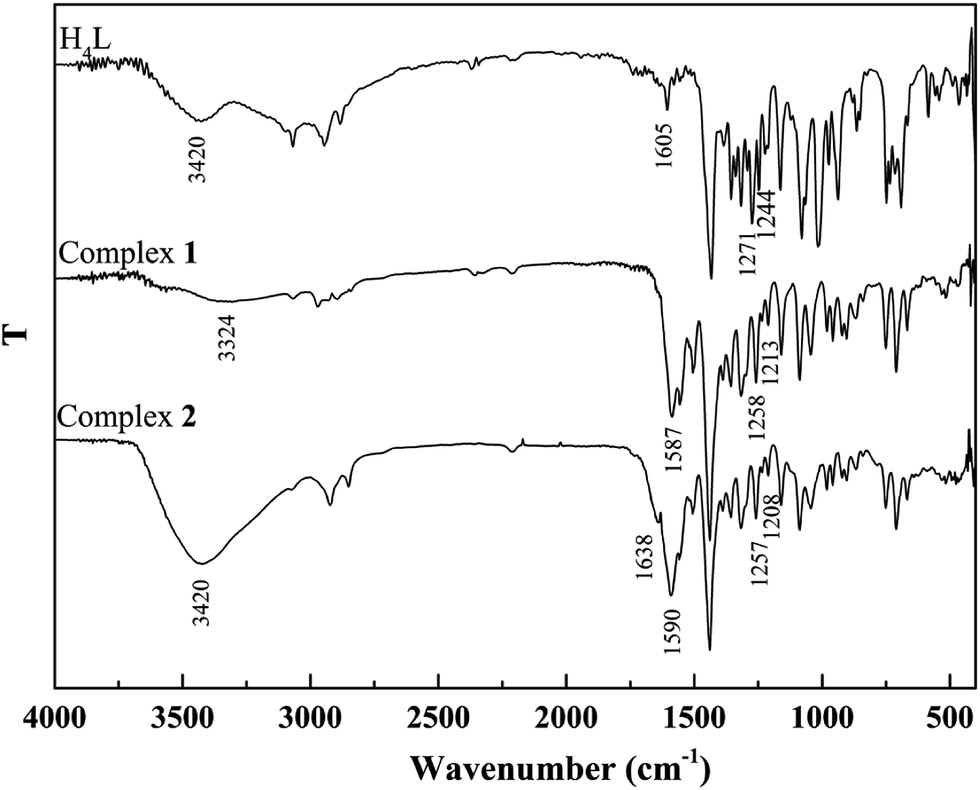
The binding modes were determined by comparing FT-IR spectra of the ligand H4L with those of its complexes (Fig. 2). The free ligand H4L exhibits characteristic CN and Ar–O stretching bands at ca. 1605 and 1244 cm−1, respectively. Upon complexation, the CN and Ar–O bonds shift to the lower frequencies and are therefore a proof of the coordination of azomethine nitrogen and phenolic oxygen to Co(II).24 The free ligand H4L shows the expected absorption band at ca. 3420 cm−1, evidence for the vibration of phenolic OH groups. The expected absorption bands in 1 and 2 are observed at ca. 3324 and 3420 cm−1, indicating the presence of coordinated methanol and water molecules, respectively. In addition, the bending and wagging modes of coordinated water molecules appears at ca. 1638 and 530 cm−1 in 2, which is not found in H4L and 1. This phenomenon indicates that water molecules are involved in the coordination of 2.25
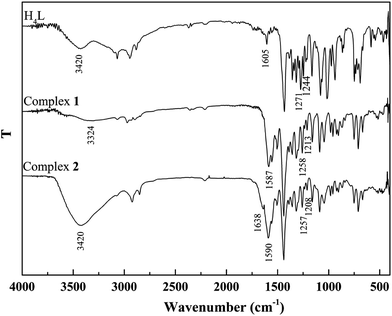 |
| | Fig. 2 FT-IR spectra of the ligand and its corresponding complexes. | |
Description of crystal structures
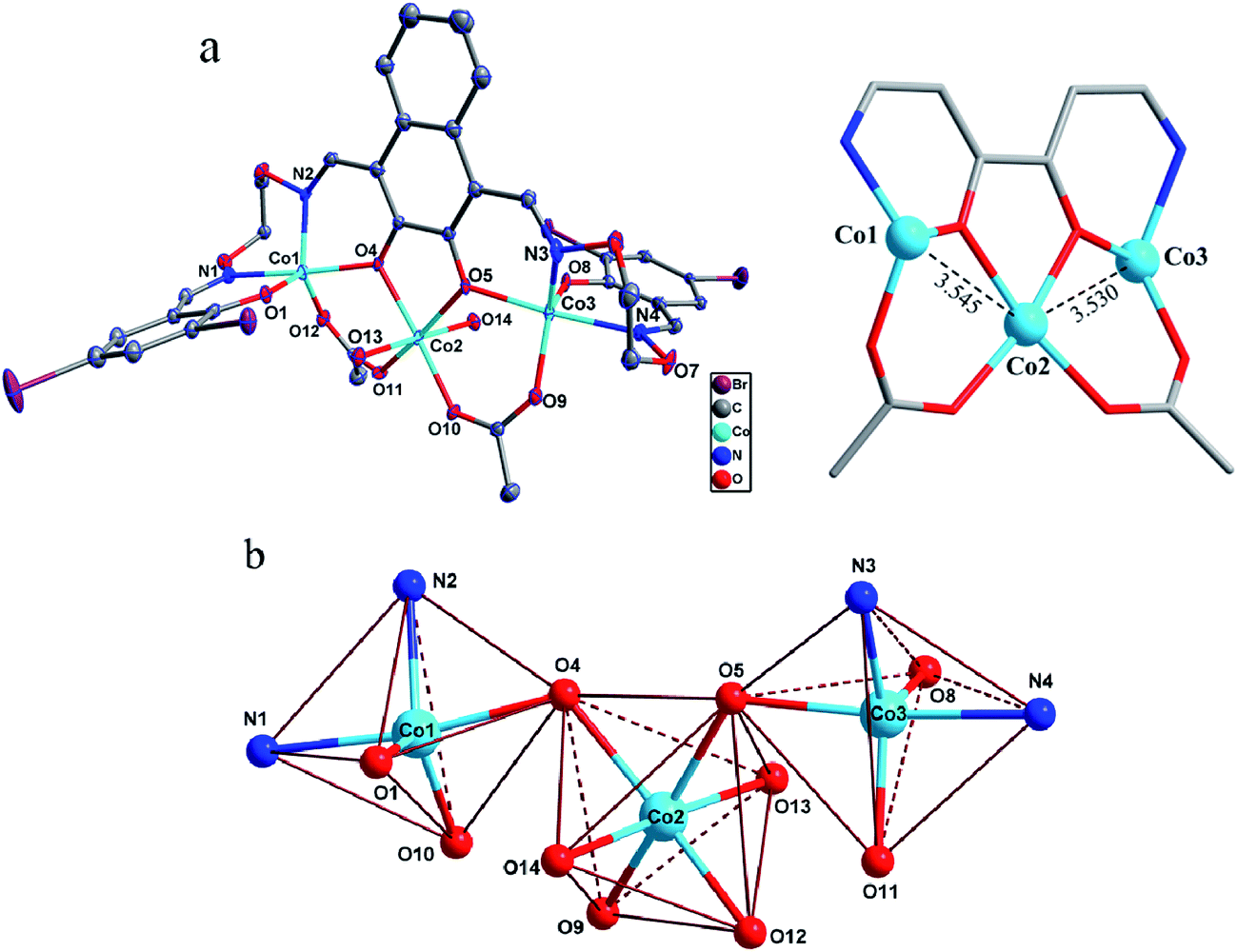
The asymmetric unit of the homotrinuclear 1 is shown in Fig. 3, and their bond lengths and bond angles are listed in Table 2.
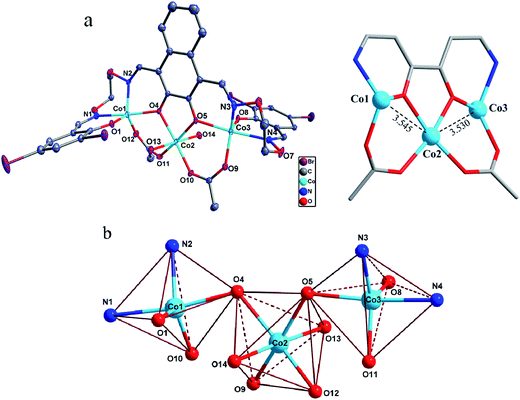 |
| | Fig. 3 (a) Molecule structure and atom numberings of 1 (thermal ellipsoids at 30% probability, hydrogen atoms and the solvated chloroform molecules have been omitted for clarity). (b) The coordination polyhedrons and the spatial relationship of Co(II) centres. | |
Table 2 Selected bond distances (Å) and angles (°) for 1 and 2
| |
1 |
2 |
|
1 |
2 |
| Co1–O1 |
1.938(6) |
1.942(3) |
Co2–O4 |
2.097(5) |
2.101(3) |
| Co1–O4 |
2.083(5) |
2.056(3) |
Co2–O5 |
2.084(5) |
2.063(3) |
| Co1–O10 |
1.956(5) |
1.971(4) |
Co2–O9 |
2.067(5) |
2.092(3) |
| Co1–N1 |
2.120(6) |
2.134(4) |
Co2–O12 |
2.049(6) |
2.051(4) |
| Co1–N2 |
2.039(7) |
2.020(4) |
Co2–O13 |
2.132(6) |
2.068(3) |
| Co3–O5 |
2.071(5) |
2.066(3) |
Co2–O14 |
2.117(6) |
2.125(3) |
| Co3–O8 |
1.938(6) |
1.935(3) |
Co3–N3 |
2.041(5) |
2.025(5) |
| Co3–O11 |
1.975(6) |
1.994(4) |
Co3–N4 |
2.129(7) |
2.132(4) |
| O1–Co1–O4 |
95.1(2) |
96.40(13) |
O4–Co2–O5 |
77.60(18) |
77.61(13) |
| O1–Co1–O10 |
121.3(3) |
124.59(16) |
O4–Co2–O9 |
96.6(2) |
95.91(13) |
| O1–Co1–N1 |
87.6(3) |
86.63(14) |
O4–Co2–O12 |
168.7(2) |
171.33(14) |
| O1–Co1–N2 |
117.0(3) |
111.82(16) |
O4–Co2–O13 |
97.3(2) |
96.52(13) |
| O4–Co1–O10 |
94.0(2) |
94.90(14) |
O4–Co2–O14 |
85.0(2) |
85.04(13) |
| O4–Co1–N1 |
172.0(2) |
175.95(15) |
O5–Co2–O9 |
167.2(2) |
166.97(13) |
| O4–Co1–N2 |
82.9(2) |
84.31(14) |
O5–Co2–O12 |
97.75(19) |
99.61(14) |
| O10–Co1–N1 |
91.1(2) |
85.55(15) |
O5–Co2–O13 |
83.51(19) |
85.91(13) |
| O10–Co1–N2 |
121.7(3) |
123.22(15) |
O5–Co2–O14 |
98.0(2) |
94.55(13) |
| N1–Co1–N2 |
89.1(3) |
92.07(16) |
O5–Co3–N3 |
84.5(2) |
83.62(15) |
| O5–Co3–O8 |
93.9(2) |
94.39(13) |
O5–Co3–N4 |
173.2(2) |
173.99(16) |
| O5–Co3–O11 |
95.5(2) |
97.85(14) |
O9–Co2–O12 |
90.0(2) |
88.37(14) |
| O8–Co3–O11 |
118.6(2) |
118.57(16) |
O9–Co2–O13 |
86.0(2) |
83.60(13) |
| O8–Co3–N3 |
114.0(2) |
112.52(17) |
O9–Co2–O14 |
92.8(2) |
96.14(14) |
| O8–Co3–N4 |
87.5(3) |
87.06(14) |
O12–Co2–O13 |
92.4(2) |
91.44(14) |
| O11–Co3–N3 |
127.3(2) |
128.55(17) |
O12–Co2–O14 |
85.5(2) |
87.02(14) |
| O11–Co3–N4 |
89.7(3) |
86.54(15) |
O13–Co2–O14 |
177.6(2) |
178.44(15) |
| N3–Co3–N4 |
88.8(2) |
90.44(16) |
|
|
|
The single-crystal X-ray structures revealed the differences between methanol and water solvates are relatively minor, which also very similar to the analogous homotrinuclear Co(II) complex coordinated by ethanol in the central position, designed and reported by Nabeshima group.10 Two terminal Co(II) ions (Co1 and Co3) are located at the N2O2 coordination spheres of Salamo moieties, while the third one (Co2) is located in the O2 sites (O4 and O5).10 Co2 is bridged by Co1 and Co3 through two deprotonated phenoxo donors O4 and O5, respectively, as well as two acetate ions in a μ2-η1:η1 fashion. The bond distances between Co1 and Co2 (complex 1: 3.545(2) Å and complex 2: 3.530(1) Å) are longer than that between Co2 and Co3 (complex 1: 3.530(1) Å and complex 2: 3.470(1) Å). Interestingly, Co1 and Co3 in both complexes are penta-coordinated and so, would be expected to experience the same coordination environment. Resulting geometries of Co1 and Co3 are both distorted trigonal-bipyramidal geometries (complex 1: τ/χ = 0.838/0.927 (Co1), 0.765/0.840 (Co3) and complex 2: τ/χ = 0.882/0.900 (Co1), 0.750/0.877 (Co3)),26 whose axial positions are occupied by the N1–O4 and N4–O5 donors, respectively. The central Co2 has an octahedral geometry with six oxygen-donor atoms including two μ2-acetato ligands and two solvent molecules. Oxygen-donor atoms of (L)4− and μ2-acetato ligands form the equatorial plane and the oxygen-donor atoms of the solvent molecules are placed in the apical sites. Owing to these coordination geometries, four 6-membered rings and one central 5-membered ring are constructed, which are connected through the Co(II) centers and display a polycyclic geometry (Fig. 3a). The coordination polyhedrons and the spatial relationship of Co(II) centers are shown in Fig. 3b. A comparison between the isostructural complexes 1 and 2 (Fig. 4) shows that the CoN2O2 fragments of 1 are more distorted than those of 2. Terminal salicylidene moieties of 1 and 2 are dangling in the opposite side, and the dihedral angles between the basal planes (naphthalene ring) and the phenyl rings are shown in Table 2. Compared with 2, the terminal salicylidene moieties of 1 are more deviated from the basal planes, which is likely to be caused by steric hindrance effect of coordinated solvent molecules.
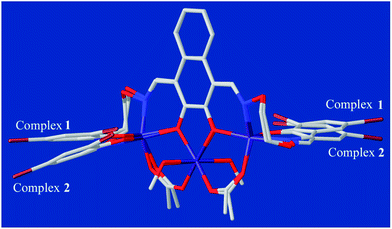 |
| | Fig. 4 Comparison of the molecular structures of 1 and 2 by overlay map (naphthalene rings as the basal planes). | |
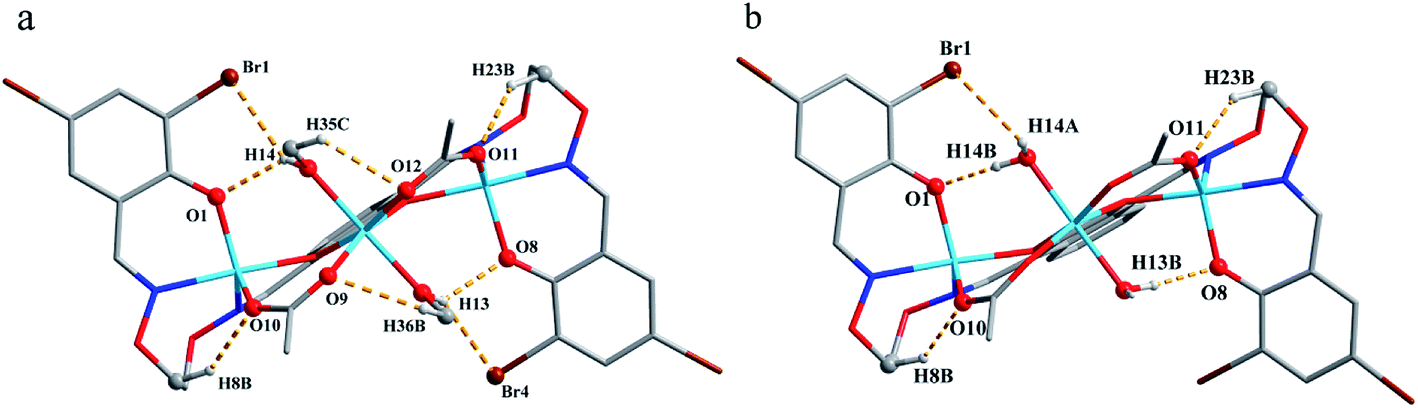
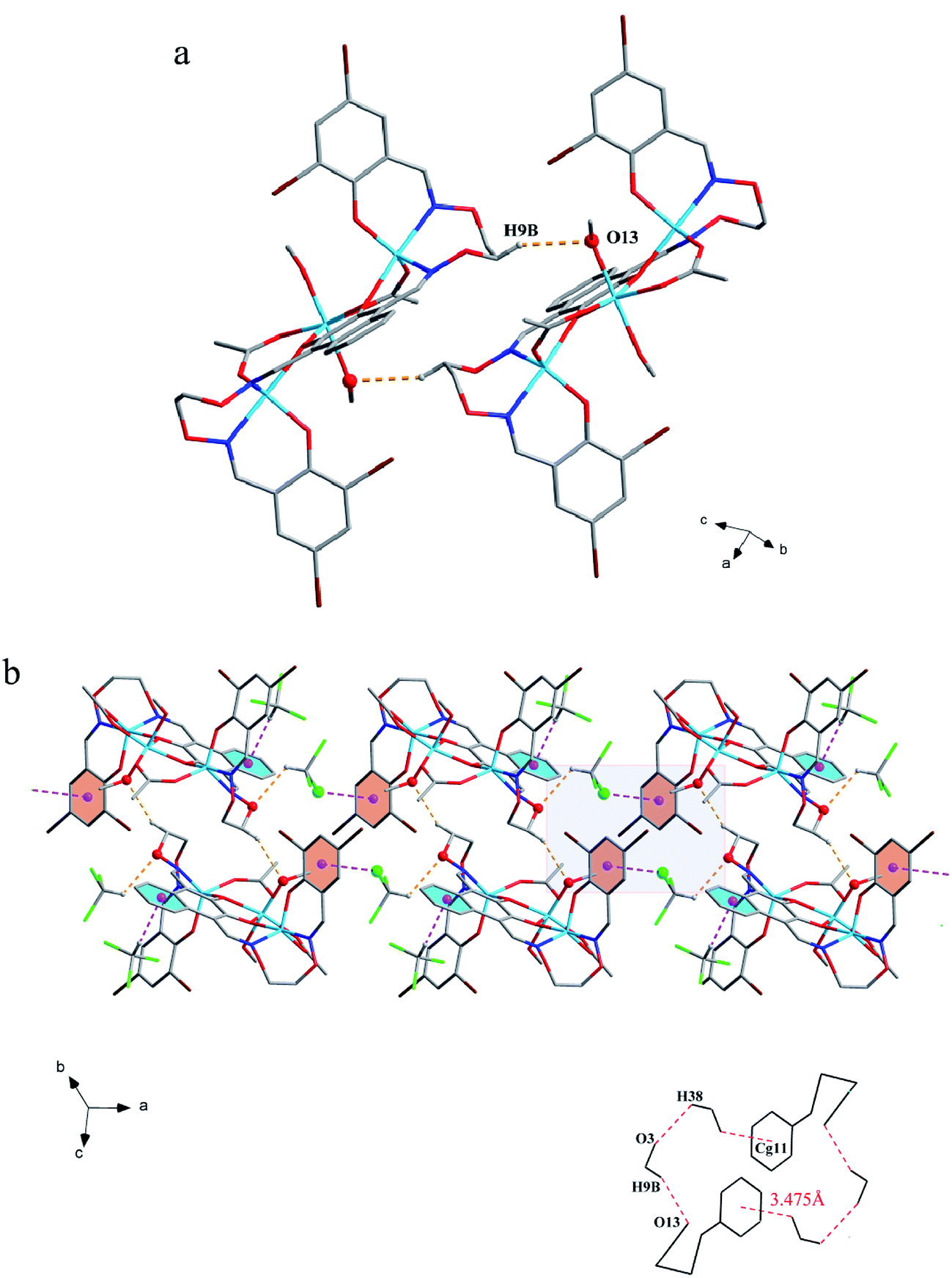
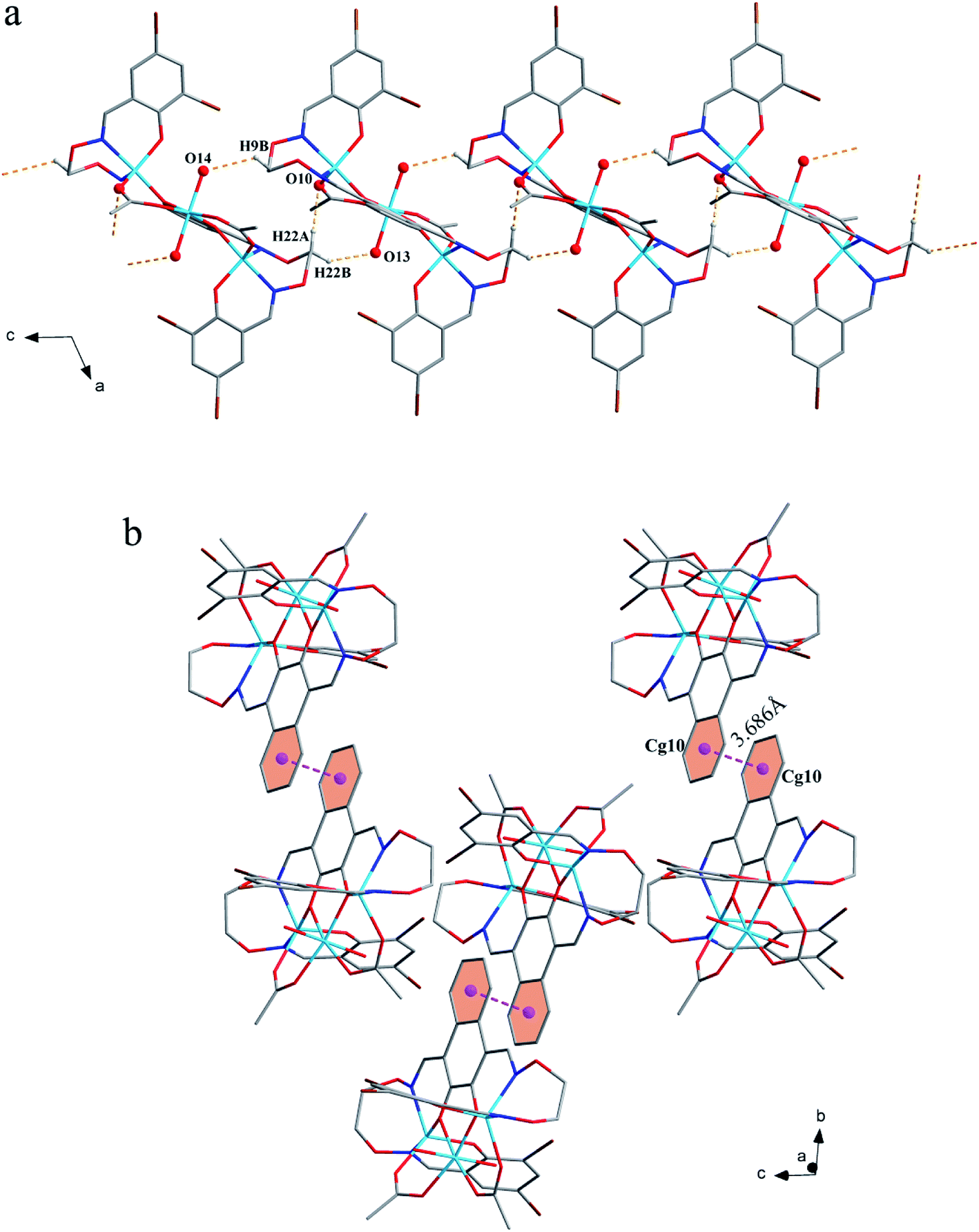
The corresponding interactions of 1 and 2 are summarized in Table 3, where the coordinated solvent molecules play important roles in the construction of supramolecular structures owing to the formation of abundant intra- and intermolecular hydrogen bonds. For 1, hydrogen atoms from two coordinated methanol molecules (–O14H14, –C35H35C, –O13H13 and –C36H36B) form hydrogen bonds with phenoxo (O1 and O8), bromine (Br1 and Br4) atoms and oxygen atoms of acetate ions (O12 and O9). Oxygen atoms (O10 and O11) of acetate ions act as acceptor to form two hydrogen bonds with the protons (–C8H8B and –C23H23B) of ethylenedioxime carbons in each molecule (Fig. 5a). In addition, complex molecules form a dimer structure by a pair of intermolecular hydrogen bonds (C9–H9B⋯O13) (Fig. 6a). Each dimer is connected to two uncoordinated chloroform molecules and each chloroform molecule is connected to two dimers via intermolecular interactions (C38–H38⋯O3 and C38–Cl5⋯Cg11).27 Interestingly, a series of circular units are constructed by the above-mentioned intermolecular interactions, forming a 1D supramolecular chain (Fig. 6b). For 2, one of the two coordinated water hydrogen atoms (–O14H14A and –O14H14B) forms hydrogen bonds with phenoxo and bromine atoms (O1 and Br1), whereas another water hydrogen atom (–O13H13A) just forms one hydrogen bond with phenoxo atom (O8) probably because of the long distances (H13A⋯Br4, 2.960 Å and H13B⋯Br4, 3.809 Å). In addition, acetate ions (O10 and O11) act as acceptor to form two hydrogen bonds with the protons (–C8H8B and –C23H23B) of ethylenedioxime carbons in each molecule (Fig. 5b). Furthermore, two adjacent molecules are interlinked by three intermolecular hydrogen bonds (C9–H9B⋯O14, C22–H22A⋯O10 and C22–H22B⋯O13) to form a 1D chain supramolecular structure, and adjacent chains are further connected to each other via the π⋯π stacking interactions (Cg10⋯Cg10), resulting in a 2D supramolecular structure (Fig. 7).28
Table 3 Hydrogen bond, C–H⋯π, C–Cl⋯π and π⋯π stacking interactions in 1 and 2 (Å, °)
| D–X |
d(D–X) |
d(X⋯A) |
d(D⋯A) |
∠D–X⋯A |
A |
Symmetry codes |
| Cg10 = C15–C20. Cg11 = C25–C30. Cg10 = C15–C20. |
| Complex 1 |
| O13–H13 |
0.86 |
2.67 |
3.465(12) |
155 |
Br4 |
|
| O13–H13 |
0.86 |
2.15 |
2.714(15) |
123 |
O8 |
|
| O14–H14 |
0.86 |
2.6 |
3.387(13) |
154 |
Br1 |
|
| O14–H14 |
0.86 |
2.27 |
2.762(17) |
116 |
O1 |
|
| C8–H8B |
0.97 |
2.29 |
3.14(2) |
146 |
O10 |
|
| C23–H23B |
0.97 |
2.44 |
3.274(19) |
143 |
O11 |
|
| C35–H35C |
0.96 |
2.49 |
3.08(3) |
119 |
O12 |
|
| C36–H36B |
0.96 |
2.59 |
3.17(3) |
120 |
O9 |
|
| C9–H9B |
0.97 |
2.52 |
3.408(10) |
153 |
O13 |
[1 − x, 2 − y, −z] |
| C38–H38 |
0.98 |
2.59 |
3.183(16) |
119 |
O3 |
[1 + x, y, z] |
| C37–H37 |
0.98 |
2.57 |
3.440(16) |
148 |
Cg10a |
[1 − x, 2 − y, 1 − z] |
| C38–Cl5 |
1.68(2) |
3.475(7) |
4.889(16) |
140.6(6) |
Cg11b |
[x, y, z] |
|
| Complex 2 |
| O13–H13A |
0.85 |
2.11 |
2.766(5) |
134 |
O8 |
|
| O14–H14A |
0.96 |
2.85 |
3.390(3) |
117 |
Br1 |
|
| O14–H14B |
0.96 |
2.04 |
2.748(5) |
129 |
O1 |
|
| C8–H8B |
0.97 |
2.36 |
3.153(7) |
139 |
O10 |
|
| O23–H23B |
0.97 |
2.38 |
3.191(7) |
141 |
O11 |
|
| C9–H9B |
0.97 |
2.54 |
3.188(6) |
124 |
O14 |
[x, 3/2 − y, 1/2 + z] |
| C22–H22A |
0.97 |
2.52 |
3.448(7) |
160 |
O10 |
[x, 3/2 − y, −1/2 + z] |
| C22–H22B |
0.97 |
2.44 |
3.285(7) |
147 |
O13 |
[x, 3/2 − y, −1/2 + z] |
| Cg10c |
|
|
3.686(4) |
|
Cg10c |
[1 − x, 1 − y, −z] |
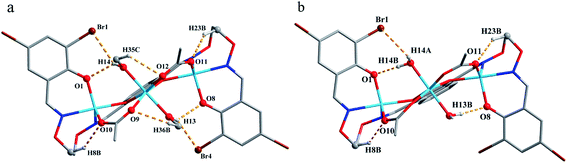 |
| | Fig. 5 View of the abundant intramolecular hydrogen bonds of 1 (a) and 2 (b). | |
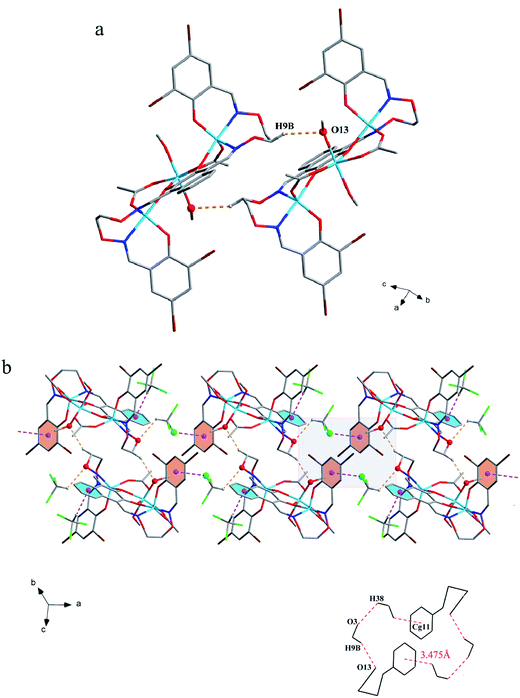 |
| | Fig. 6 (a) View of the dimer structure by intermolecular C–H⋯O interactions. (b) View of the 1D supramolecular chain by intermolecular C–H⋯O and C–Cl⋯π interactions. | |
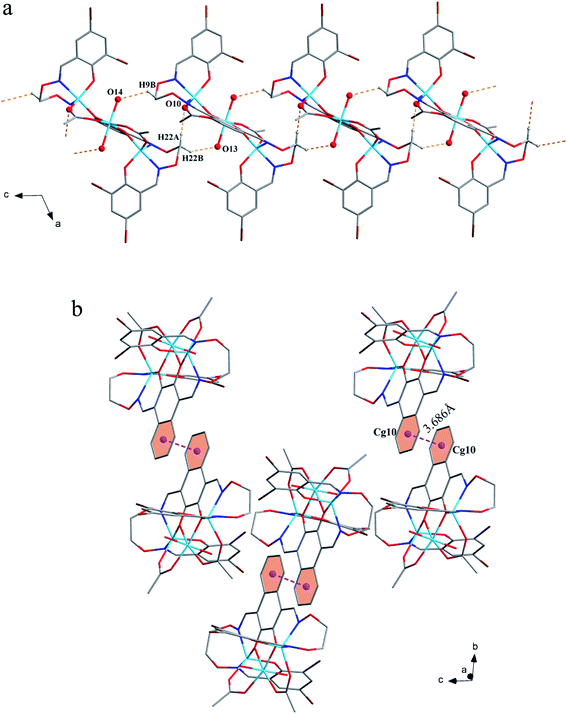 |
| | Fig. 7 (a) View of the 1D structure by intermolecular C–H⋯O interactions. (b) View of the 2D supramolecular layered by intermolecular π⋯π interactions. | |
Catecholase activity: spectrophotometric study
Though analogous bis(Salamo)-type Co(II) complex has been also synthesized by Nabeshima group, the study on catalytic activity of Salamo-type compounds has not been reported up to now.10 In order to initiate a new field for the Salamo-type compounds, we studied the catecholase activities of the two Co(II) complexes and utilized the most commonly used substrate, 3,5-di-tert-butylcatechol (3,5-DTBC) due to its low reduction potential for the quinone–catechol couple, which makes it easy to be oxidized into 3,5-di-tert-butylquinone (3,5-DTBQ).29 Before embarking on detailed kinetic study, we first investigated the ability of the complexes in acetonitrile solvent to mimic the active site of catechol oxidase by treating 2.5 × 10−5 M solutions of the complexes with 2.5 × 10−3 M (100 equivalents) of 3,5-DTBC under aerobic condition (Scheme 2).
 |
| | Scheme 2 Catalytic oxidation of 3,5-DTBC to 3,5-DTBQ in MeCN solvent. | |
 |
| | Scheme 3 Structures of the ligands mentioned in Table 5. | |
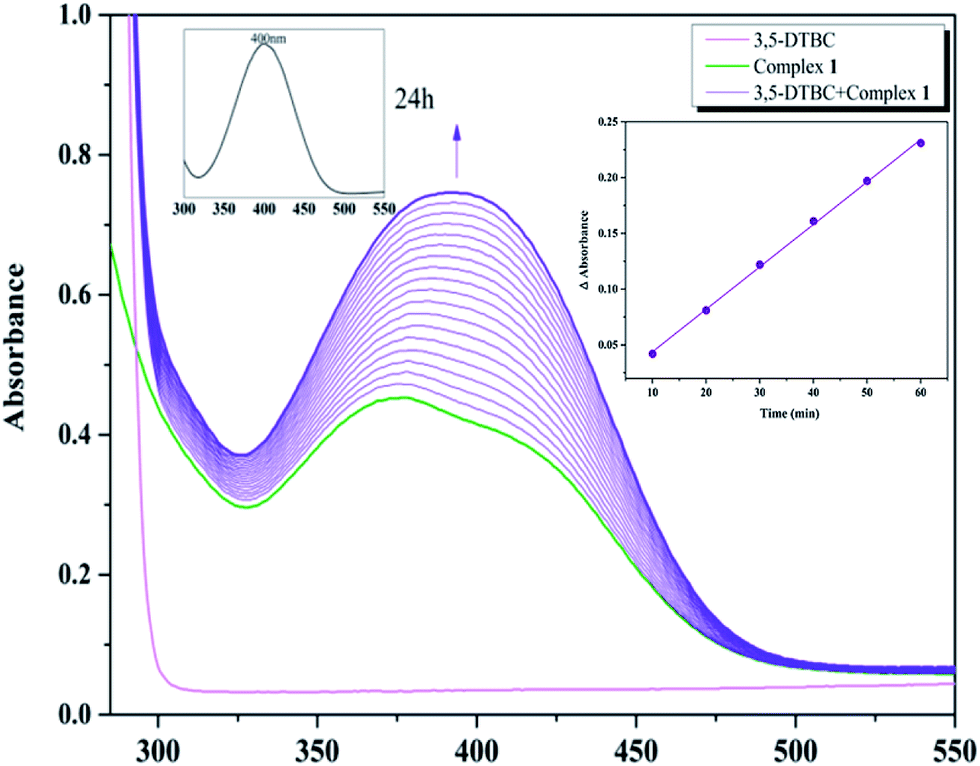
With 1, for instance, the time-dependent UV-vis spectra scan was recorded in pure acetonitrile solution under aerobic condition at room temperature. The course of the reaction was recorded per 5 min (Fig. 8). Spectra bands of 1 appears at 375 and 411 nm, whereas 3,5-DTBC showed single band at 278 nm. It is generally known that 3,5-DTBQ shows maximum absorption in UV-vis spectrum at 400 nm in pure acetonitrile solution. After the addition of 3,5-DTBC, the time dependent spectra scan showed very smooth growing of quinone band and finally a peak at 400 nm is formed after 24 h which is very close to the value reported by Krebs et al.30 and the spectra bands of 1 was hidden by the new absorption, which indicated the formation of the quinine derivative, 3,5-DTBQ. The difference in absorbance (ΔA) at 400 nm was plotted against time (min) to obtain the initial rate for that particular catalyst to substrate concentration ratio. A first-order catalytic reaction was observed, with initial rate of 3.81 × 10−3 min−1. The product was purified and identified by 1H NMR spectroscopy.
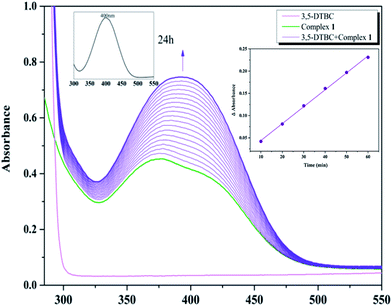 |
| | Fig. 8 Changes observed in UV-vis spectra of 1 in MeCN up to 90 minutes (conc. 2.5 × 10−5 M) upon addition of 100-fold 3,5-DTBC (2.5 × 10−3 M). The inset shows the absorption after 24 h, and plot of the difference in absorbance (ΔA) versus time to evaluate the initial rate of the catalytic oxidation of 3,5-DTBC by 1 in MeCN. | |
To investigated the autooxidation ability of the substrate, control experiments without Co catalysts were also performed. Solution of 3,5-DTBC (2.5 × 10−3 M) was prepared in MeCN and the time-dependent UV-vis spectra scan was also recorded per 5 min under the same condition. As shown in Fig. S1,† there was almost no changes observed in UV-vis spectra up to 60 minutes, which proved that our complexes as catalyst could really improve the oxidation efficiency.
Kinetics study
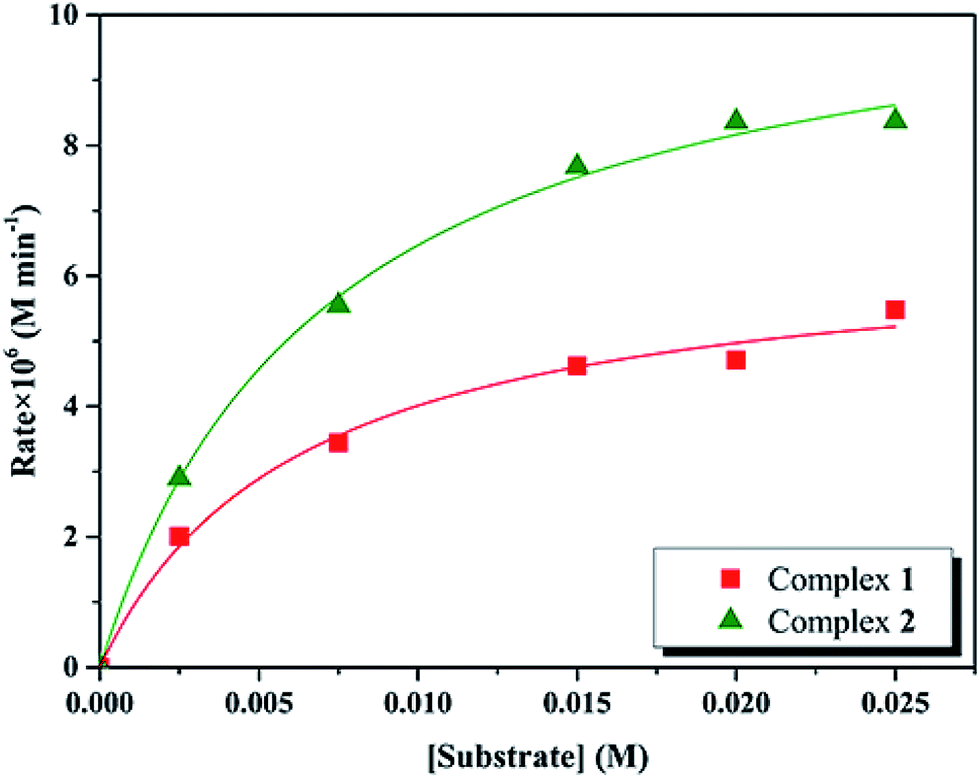
Kinetic studies of the catecholase activities of the catalysts were determined by treating 2.5 × 10−5 M solutions of the complexes and monitoring the increase of the concentration of 3,5-DTBC, ranging from 2.5 × 10−3 to 2.5 × 10−2 M. The rate constants for the catalyst complexes were determined from that plot by initial rate method. Assuming rate (M min−1) is the initial rate, plot of rate versus substrate concentration ([substrate]) for the oxidation reaction catalyzed by 1 and 2 is drawn in Fig. 9. It is found that rate followed saturation kinetics with respect to [substrate]. At sufficiently low [substrate], plot of rate versus [substrate] is linear. When [substrate] becomes sufficiently high, the catalyst (here is model complexes) is saturated, the rate of reaction is zero-order and tends toward a limiting value, maximum velocity (Vmax). Both complexes show saturation kinetics and a treatment based on the Michaelis–Menten model seems to be appropriate.
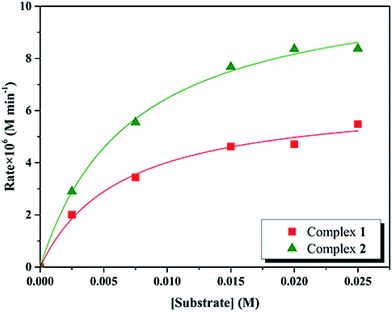 |
| | Fig. 9 Plot of initial rates versus substrate concentration for the oxidation reaction catalyzed by 1 and 2 in MeCN. | |
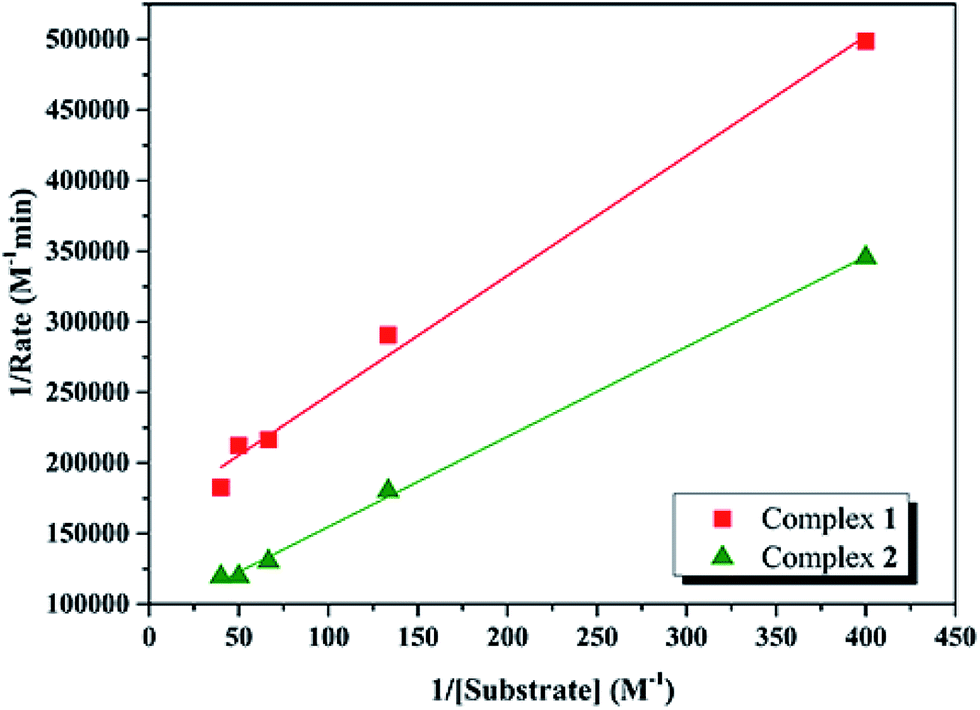
In order to calculate the values of binding constant (KM), maximum velocity (Vmax), and rate constant (i.e., turn over number, Kcat) for the complexes, the Lineweaver–Burk graph of 1/rate against 1/[substrate] is plotted (Fig. 10). Corresponding plots is linear and parameters are calculated from the following equation, 1/rate = {KM/Vmax} × {1/[substrate]} + 1/Vmax, and Kcat are evaluated by applying the equation Kcat = (Vmax/[cat.]). Some of the kinetic parameters are tabulated in Table 4. The values of Kcat are 14.72 h−1 and 26.39 h−1 for 1 and 2, respectively. Thus, solvent effect could be responsible for the differences observed in the catecholase activities. Among the cobalt complexes produced as mimics for catechol oxidase, the number of Co(II) complexes studied as models for catechol oxidase is relatively low. The Kcat values of complexes 1 and 2 and other reported Co(II) complexes are shown in Table 5. The exciting thing is that Salamo-type complexes as the catechol oxidase model complexes in our work exhibit good properties compared with other types of complexes reported.
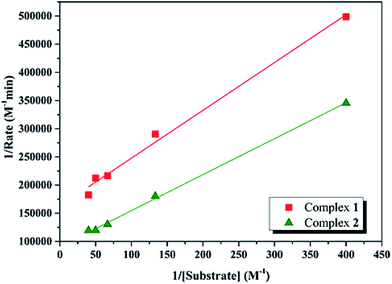 |
| | Fig. 10 Lineweaver–Burk oxidation reaction plot for 1 and 2 in MeCN. | |
Table 4 Kinetic parameters for the oxidation of 3,5-DTBC catalyzed by 1 and 2
| Complex |
Vmax (M min−1) |
KM (M) |
Kcat (h−1) |
| 1 |
6.14 × 10−6 |
5.20 × 10−3 |
14.7 |
| 2 |
1.10 × 10−5 |
7.09 × 10−3 |
26.4 |
Table 5 Kcat values of complexes 1, 2 and other reported Co(II) complexes
| Complexa |
Solvent |
Kcat (h−1) |
Ref. |
| Structures of ligands are shown in Scheme 3. Substrate: 3,5-di-tert-butylcatechol. Substrate: catechol. |
| [Co2(L2)(H2O)2(OAc)2]+b |
MeOH |
45.9 |
32 |
| [Co2(L3)(H2O)2(OAc)2]+b |
MeOH |
42.9 |
32 |
| [Co2(L1H)(H2O)2(OAc)2]2+b |
MeOH |
447 |
32 |
| [Co2(EGTB)Cl2]2+c |
MeOH–Tris–HCl buffer, pH 8.0 |
7.02 |
33 |
| MeOH–Tris–HCl buffer, pH 7.6 |
1.14 |
33 |
| MeOH–Tris–HCl buffer, pH 7.3 |
0.9 |
33 |
| 1b |
MeCN |
14.72 |
This work |
| 2b |
MeCN |
26.39 |
This work |
The catecholase model complexes have been reported to relate to several factors such as metal–metal distance, lability of exogenous ligands, coordination configuration and electrochemical properties.31 A probable explanation for the catecholase catalytic reactivity of two complexes were mainly related to its coordination environment. The five-coordinated cobalt(II) atom has an unsaturated vacancy, and it could combine with the catechol substrates. Meanwhile, the presence of acetate which may be displaced by the catechol substrate has been shown to be important. An acetate ligand would contribute a negatively charged moiety capable of facilitating the catalytic process.
Conclusions
As a result of the introduction of different solvent molecules, two new homotrinuclear Co(II) complexes 1 and 2 with a bis(Salamo)-type tetraoxime ligand H4L, were designed and synthesized. Methanol as coordinating solvent generates the coordination of methanol molecules. In the presence of i-PrOH/H2O, water molecules coordinate to the central Co(II) rather than isopropanol. 1 and 2 differ only by the solvent molecules coordinated to central Co(II) ions. Two terminal Co(II) ions, located at the N2O2 coordination spheres of Salamo moieties, are both in a distorted trigonal-bipyramidal geometries, whereas the one in the central O4 cavity shows an octahedral geometry with the two solvent molecules. As far as we know, the present work is the first to report the catecholase activities of Salamo-type complexes. The catalytic reaction follows Michaelis–Menten enzymatic reaction kinetics with turnover number 14.72 h−1 and 26.39 h−1 for 1 and 2, respectively.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (21361015), which is gratefully acknowledged.
References
-
(a) Y. X. Sun, S. T. Zhang, Z. L. Ren, X. Y. Dong and L. Wang, Synth. React. Inorg., Met.-Org., Nano-Met. Chem., 2013, 43, 995–1000 CrossRef CAS;
(b) H. L. Wu, G. L. Pan, Y. C. Bai, H. Wang, J. Kong, F. R. Shi, Y. H. Zhang and X. L. Wang, J. Chem. Res., 2014, 38, 211–217 CrossRef CAS;
(c) H. L. Wu, G. L. Pan, Y. C. Bai, H. Wang, J. Kong, F. R. Shi, Y. H. Zhang and X. L. Wang, Res. Chem. Intermed., 2015, 41, 3375–3388 CrossRef CAS;
(d) H. L. Wu, G. L. Pan, Y. C. Bai, H. Wang, J. Kong, F. R. Shi, Y. H. Zhang and X. L. Wang, J. Coord. Chem., 2013, 66, 2634–2646 CrossRef CAS;
(e) W. K. Dong, J. C. Ma, L. C. Zhu and Y. Zhang, New J. Chem., 2016, 40, 6998–7010 RSC.
-
(a) P. Wang and L. Zhao, Spectrochim. Acta, Part A, 2015, 135, 342–350 CrossRef CAS PubMed;
(b) P. Wang and L. Zhao, Synth. React. Inorg., Met.-Org., Nano-Met. Chem., 2016, 46, 1095–1101 CrossRef CAS;
(c) H. L. Wu, Y. C. Bai, Y. H. Zhang, G. L. Pan, J. Kong, F. R. Shi and X. L. Wang, Z. Anorg. Allg. Chem., 2014, 640, 2062–2071 CrossRef CAS;
(d) H. L. Wu, C. P. Wang, F. Wang, H. P. Peng, H. Zhang and Y. C. Bai, J. Chin. Chem. Soc., 2015, 62, 1028–1034 CrossRef CAS;
(e) L. Xu, L. C. Zhu, J. C. Ma, Y. Zhang, J. Zhang and W. K. Dong, Z. Anorg. Allg. Chem., 2015, 641, 2520–2524 CrossRef CAS;
(f) L. Q. Chai, K. Y. Zhang, L. J. Tang, J. Y. Zhang and H. S. Zhang, Polyhedron, 2017, 130, 100–107 CrossRef CAS;
(g) L. Q. Chai, L. J. Tang, L. C. Chen and J. J. Huang, Polyhedron, 2017, 122, 228–240 CrossRef CAS;
(h) W. K. Dong, P. F. Lan, W. M. Zhou and Y. Zhang, J. Coord. Chem., 2016, 7, 1–22 Search PubMed;
(i) P. P. Liu, C. Y. Wang, M. Zhang and X. Q. Song, Polyhedron, 2017, 129, 133–140 CrossRef CAS.
-
(a) L. Wang, J. C. Ma, W. K. Dong, L. C. Zhu and Y. Zhang, Z. Anorg. Allg. Chem., 2016, 642, 834–839 CrossRef CAS;
(b) J. C. Ma, X. Y. Dong, W. K. Dong, Y. Zhang, L. C. Zhu and J. T. Zhang, J. Coord. Chem., 2016, 69, 149–159 CrossRef CAS;
(c) X. Y. Dong, S. F. Akogun, W. M. Zhou and W. K. Dong, J. Chin. Chem. Soc., 2017, 64, 412–419 CrossRef CAS;
(d) C. H. Tao, J. C. Ma, L. C. Zhu, Y. Zhang and W. K. Dong, Polyhedron, 2017, 128, 38–45 CrossRef CAS;
(e) Y. J. Dong, X. Y. Dong, W. K. Dong, Y. Zhang and L. S. Zhang, Polyhedron, 2017, 123, 305–315 CrossRef CAS;
(f) Y. J. Dong, J. C. Ma, L. C. Zhu, W. K. Dong and Y. Zhang, J. Coord. Chem., 2017, 70, 103–115 CrossRef CAS;
(g) X. Q. Song, G. Q. Cheng and Y. A. Liu, Inorg. Chim. Acta, 2016, 450, 386–394 CrossRef CAS;
(h) X. Q. Song, P. P. Liu, Y. A. Liu, J. J. Zhou and X. L. Wang, Dalton Trans., 2016, 45, 8154–8163 RSC;
(i) X. Q. Song, Y. Q. Peng, G. Q. Cheng, X. R. Wang, P. P. Liu and W. Y. Xu, Inorg. Chim. Acta, 2015, 427, 13–21 CrossRef CAS.
-
(a) C. Y. Chen, J. W. Zhang, Y. H. Zhang, Z. H. Yang, H. L. Wu, G. L. Pan and Y. C. Bai, J. Coord. Chem., 2015, 68, 1054–1071 CrossRef CAS;
(b) H. L. Wu, Y. C. Bai, Y. H. Zhang, Z. Li, M. C. Wu, C. Y. Chen and J. W. Zhang, J. Coord. Chem., 2014, 67, 3054–3066 CrossRef CAS;
(c) H. L. Wu, H. Wang, X. L. Wang, G. L. Pan, F. R. Shi, Y. H. Zhang, Y. C. Bai and J. Kong, New J. Chem., 2014, 38, 1052–1061 RSC;
(d) H. L. Wu, G. L. Pan, Y. C. Bai, Y. H. Zhang, H. Wang, F. R. Shi, X. L. Wang and J. Kong, J. Photochem. Photobiol., B, 2014, 135, 33–43 CrossRef CAS PubMed.
- L. H. Li, W. K. Dong, Y. Zhang, S. F. Akogun and L. Xu, Appl. Organomet. Chem. DOI:10.1002/aoc.3818.
-
(a) P. P. Liu, L. Sheng, X. Q. Song, W. Y. Xu and Y. A. Liu, Inorg. Chim. Acta, 2015, 434, 252–257 CrossRef CAS;
(b) X. Q. Song, P. P. Liu, Z. R. Xiao, X. Li and Y. A. Liu, Inorg. Chim. Acta, 2015, 438, 232–244 CrossRef CAS;
(c) Y. A. Liu, C. Y. Wang, M. Zhang and X. Q. Song, Polyhedron, 2017, 127, 278–286 CrossRef CAS.
-
(a) W. K. Dong, J. C. Ma, L. C. Zhu, Y. Zhang and X. L. Li, Inorg. Chim. Acta, 2016, 445, 140–148 CrossRef CAS;
(b) W. K. Dong, P. F. Lan, W. M. Zhou and Y. Zhang, J. Coord. Chem., 2016, 65, 1272–1283 CrossRef.
-
(a) B. J. Wang, W. K. Dong, Y. Zhang and S. F. Akogun, Sens. Actuators, B, 2017, 247, 254–264 CrossRef CAS;
(b) Y. J. Dong, X. L. Li, Y. Zhang and W. K. Dong, Supramol. Chem., 2017, 29, 518–527 CrossRef CAS.
-
(a) W. K. Dong, J. C. Ma, L. C. Zhu, Y. X. Sun, S. F. Akogun and Y. Zhang, Cryst. Growth Des., 2016, 16, 6903–6914 CrossRef CAS;
(b) X. Y. Dong, Y. X. Sun, L. Wang and L. Li, J. Chem. Res., 2012, 36, 387–390 CrossRef CAS;
(c) X. Y. Dong, Q. P. Kang, B. X. Jin and W. K. Dong, Z. Naturforsch., 2017, 72, 415–420 CAS;
(d) S. Akine, T. Taniguchi and T. Nabeshima, Chem. Lett., 2001, 30, 682–683 CrossRef.
- S. Akine, T. Taniguchi and T. Nabeshima, Inorg. Chem., 2008, 47, 3255–3264 CrossRef CAS PubMed.
-
(a) Y. X. Sun and X. H. Gao, Synth. React. Inorg., Met.-Org., Nano-Met. Chem., 2011, 41, 973–978 CrossRef CAS;
(b) Y. X. Sun, L. Xu, T. H. Zhao, S. H. Liu, G. H. Liu and X. T. Dong, Synth. React. Inorg., Met.-Org., Nano-Met. Chem., 2013, 43, 509–513 CrossRef CAS;
(c) L. Q. Chai, G. Wang, Y. X. Sun, W. K. Dong, L. Zhao and X. H. Gao, J. Coord. Chem., 2012, 65, 1621–1631 CrossRef CAS.
-
(a) Z. F. Li, S. J. Wu, H. Ding, H. Lu, J. Y. Liu, Q. S. Huo, J. Q. Guan and Q. B. Kan, New J. Chem., 2013, 37, 4220–4229 RSC;
(b) P. Yu, A. L. Li, H. Liang and J. Lu, J. Polym. Sci., 2007, 45, 3739–3746 CrossRef CAS.
- C. Gerdemann, C. Eicken and B. Krebs, Acc. Chem. Res., 2002, 35, 183–191 CrossRef CAS PubMed.
- A. Banerjee, S. Sarkar, D. Chopra, E. Colacio and K. K. Rajak, Inorg. Chem., 2008, 47, 4023–4031 CrossRef CAS PubMed.
- J. Anekwe, A. Hammerschmidt, A. Rompel and B. Krebs, Z. Anorg. Allg. Chem., 2006, 632, 1057–1066 CrossRef CAS.
- P. Chakraborty, J. Adhikary, B. Ghosh, R. Sanyal, S. K. Chattopadhyay, A. Bauzá, A. Frontera, E. Zangrando and D. Das, Inorg. Chem., 2014, 53, 8257–8269 CrossRef CAS PubMed.
- S. Torelli, C. Belle, I. Gautier-Luneau, J. L. Pierre, E. Saint-Aman, J. M. Latour, L. Le Pape and D. Luneau, Inorg. Chem., 2000, 39, 3526–3536 CrossRef CAS PubMed.
-
(a) M. J. Gajewska, W. M. Ching, Y. S. Wen and C. H. Hung, Dalton Trans., 2014, 43, 14726–14736 RSC;
(b) P. Kar, Y. Ida, T. Kanetomo, M. G. Drew, T. Ishida and A. Ghosh, Dalton Trans., 2015, 44, 9795–9804 RSC.
- S. Akine, T. Taniguchi, W. K. Dong, S. Masubuchi and T. Nabeshima, J. Org. Chem., 2005, 70, 1704–1711 CrossRef CAS PubMed.
- H. A. Tran, J. Collins and P. E. Georghiou, New J. Chem., 2008, 32, 1175–1182 RSC.
- G. M. Sheldrick, SHELXS 97, Program for crystal structure solution, University of Göttingen, Göttingen, Germany, 1997 Search PubMed.
- G. M. Sheldrick, SHELXL 97, Program for crystal structure refinement, University of Göttingen, Göttingen, Germany, 1997 Search PubMed.
- M. Tümer, H. Köksal, M. K. Sener and S. Serin, Transition Met. Chem., 1999, 24, 414–420 CrossRef.
- Y. Yang, P. F. Yan, P. Gao, T. Gao, G. F. Hou and G. M. Li, J. Inorg. Organomet. Polym., 2013, 23, 1211–1218 CrossRef CAS.
- W. K. Dong, Y. X. Sun, C. Y. Zhao, X. Y. Dong and L. Xu, Polyhedron, 2010, 29, 2087–2097 CrossRef CAS.
- T. Konno, K. Tokuda, J. Sakurai and K. I. Okamoto, Bull. Chem. Soc. Jpn., 2000, 73, 2767–2773 CrossRef CAS.
-
(a) Y. X. Sun, C. Y. Li, C. J. Yang, Y. Y. Zhao, J. Q. Guo and B. Yu, Chin. J. Inorg. Chem., 2016, 32, 327–335 CAS;
(b) Y. X. Sun, L. Wang, X. Y. Dong, Z. L. Ren and W. S. Meng, Synth. React. Inorg., Met.-Org., Nano-Met. Chem., 2013, 43, 599–603 CrossRef CAS;
(c) Y. X. Sun, R. E. Lu, X. R. Li, Y. Y. Zhao and C. Y. Li, Chin. J. Inorg. Chem., 2015, 31, 1055–1062 CAS;
(d) L. Q. Chai, Y. X. Li, L. C. Chen, J. Y. Zhang and J. J. Huang, Inorg. Chim. Acta, 2016, 444, 193–201 CrossRef CAS;
(e) L. Zhao, X. T. Dang, Q. Chen, J. X. Zhao and L. Wang, Synth. React. Inorg., Met.-Org., Nano-Met. Chem., 2013, 43, 1241–1246 CrossRef CAS;
(f) L. Zhao, L. Wang, Y. X. Sun, W. K. Dong, X. L. Tang and X. H. Gao, Synth. React. Inorg., Met.-Org., Nano-Met. Chem., 2012, 42, 1303–1308 CrossRef CAS.
-
(a) X. Q. Song, G. Q. Cheng, X. R. Wang, W. Y. Xu and P. P. Liu, Inorg. Chim. Acta, 2015, 425, 145–153 CrossRef CAS;
(b) Y. X. Sun, Y. Y. Zhao, C. Y. Li, B. Yu, J. Q. Guo and J. Li, Chin. J. Inorg. Chem., 2016, 32, 913–920 CAS;
(c) L. Q. Chai, K. H. Mao, J. Y. Zhang, K. Y. Zhang and H. S. Zhang, Inorg. Chim. Acta, 2017, 457, 34–40 CrossRef CAS;
(d) L. Q. Chai, J. J. Huang, J. Y. Zhang and Y. X. Li, J. Coord. Chem., 2015, 68, 1224–1237 CrossRef CAS.
- P. Seth, L. K. Das, M. G. B. Drew and A. Ghosh, Eur. J. Inorg. Chem., 2012, 13, 2232–2242 CrossRef.
- F. Zippel, F. Ahlers, R. Werner, W. Haase, H. F. Nolting and B. Krebs, Inorg. Chem., 1996, 35, 3409–3419 CrossRef CAS PubMed.
-
(a) S. J. Smith, C. J. Noble, R. C. Palmer, G. R. Hanson, G. Schenk, L. R. Gahan and M. J. Riley, J. Biol. Inorg. Chem., 2008, 13, 499–510 CrossRef CAS PubMed;
(b) S. Y. Shaban, R. Aelm, M. M. Ibrahim, M. A. Mohamed and E. R. Van, Dalton Trans., 2015, 44, 14110–14121 RSC;
(c) S. Y. Shaban, A. M. Ramadan and E. R. Van, J. Coord. Chem., 2012, 65, 2415–2431 CrossRef CAS.
- A. Banerjee, A. Guha, J. Adhikary, A. Khan, K. Manna, S. Dey, E. Zangrando and D. Das, Polyhedron, 2013, 60, 102–109 CrossRef CAS.
- J. H. Qiu, Z. R. Liao, X. G. Meng, L. Zhu, Z. M. Wang and K. B. Yu, Polyhedron, 2005, 24, 1617–1623 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1508735 and 1506120. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra06796h |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *
*