DOI:
10.1039/C7RA00746A
(Review Article)
RSC Adv., 2017,
7, 13198-13211
N-Propargylic β-enaminocarbonyls: powerful and versatile building blocks in organic synthesis
Received
17th January 2017
, Accepted 14th February 2017
First published on 27th February 2017
Abstract
Nitrogen-containing heterocyclic compounds are not only prevalent in an extensive number of natural products and synthetic pharmaceuticals but are also used as building blocks in organic synthesis. The efficient preparation of highly functionalized N-heterocycles from cheap and easily available starting materials has therefore become of central interest for synthetic organic chemists. This review gives an overview of new developments in synthesis of highly substituted N-heterocycles, including pyrroles, pyridines, pyrrolines, piperidines, azepines, and related compounds, from N-propargylic β-enaminocarbonyls (N-propargylic β-enaminones and β-enaminoesters) in recent years. Mechanistic aspects of the reactions are considered and discussed in detail.
 Sattar Arshadi | Sattar Arshadi was born in Miandoab, West Azarbayjan province, Iran, in 1973. He received his B.Sc. degree in chemistry in the University of Kermanshah (1997) and his M.Sc. (2000) under supervision of Professor Issa Yavari and Ph.D. (2004) in organic chemistry under supervision of Professor M.Z. Kassaee Both in Tarbiat Modarres University, Tehran, Iran. Then, he went to the University of Payame Noor as an assistant professor (2005). His main research interest is computational chemistry (especially on rearrangements and interactions in nano systems), organic synthesis and spectral studies of organic compounds. |
 Esmail Vessally | Esmail Vessally was born in Sharabiyan, Sarab, Iran, in 1973. He received his B.S. degree in pure chemistry from University of Tabriz, Tabriz, Iran, and his M.S. degree in organic chemistry from Tehran University, Tehran, Iran, in 1999 under the supervision of Prof. H. Pirelahi. He completed his Ph.D. degree in 2005 under the supervision of Prof. M.Z. Kassaee. Now he is working at Payame Noor University as associate professor. His research interests include theoretical organic chemistry, new methodologies in organic synthesis and spectral studies of organic compounds. |
 Ladan Edjlali | Ladan Edjlali was born in Tabriz, Iran, in 1960. She received her B.S. degree in Applied Chemistry from University of Tabriz, Iran, and her M.S. degree in organic chemistry from University of Tabriz, Tabriz, Iran, in 1993 under the supervision of Prof. Y. Mirzaei. She completed his Ph.D. degree in 2000 under the supervision of Prof. Y. Mirzaei and Prof. S. M. Golabi. Now she is working at Islamic Azad University, Tabriz Branch as Associate Professor. Her research interests include organic synthesis and new methodologies in organic synthesis. |
 Ebrahim Ghorbani-Kalhor | Ebrahim Ghorbani Kalhor is an associate professor of analytical chemistry in the department of chemistry at the university of Islamic Azad University, Tabriz Branch, where he has been a faculty member since 2009. He was born in Ahar, Iran in 1972. He received his B.S. degree in applied chemistry from Bu Ali Sina University, Hamedan, Iran in 1995 and his M.S. degree in analytical chemistry from Urmia University, Urmia, Iran, under the supervision of Prof. M. A. Farajzadeh in 1999. Kalhor completed his Ph.D. at Tabriz University, Tabriz, Iran, in 2005 under the supervision of Prof. M. H. Sorouraddin and Prof. M.R. Rashidi. His research interests include chemometrics, nano particles, nano composites, separation science, solid phase micro extraction, sample pretreatment, molecular spectroscopy, modelling and experimental design. |
 Rahim Hosseinzadeh-Khanmiri | Rahim Hosseinzadeh-Khanmiri was born in Azarshahr, Iran, in 1979. He received his B.S. degree in Pure Chemistry from University of Tabriz, Tabriz, Iran, and his M.S. degree in organic chemistry from Kharazmi University, Tehran, Iran, in 2007 under the supervision of Prof. M. A. Bigdeli. He completed his Ph.D. degree in 2014 under the supervision of Prof. S. A. Moghimi and prof. Ahmad Shaabani in Shahid Beheshti University, Tehran, Iran. Now he is working at Tabriz Branch, Islamic Azad University, Tabriz, Iran as Assistant Professor. His research interests include organic synthesis, catalyst, nano, organic and green chemistry. |
1. Introduction
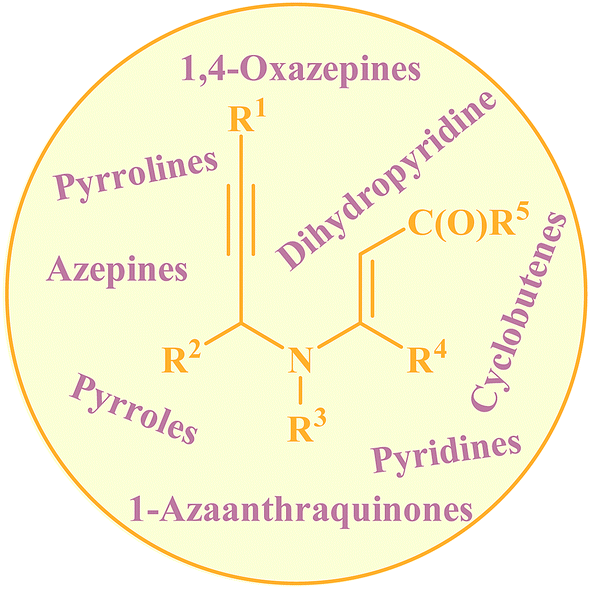
Needless to say, nitrogen-based heterocycles, including pyrroles, pyridines, pyrrolines, piperidines, azepines, and related compounds, are key structural units for a large array of drugs and natural products.1 Therefore, the development of practical and convenient methodologies that benefit from cheap, readily available, and relatively simple substrates for the construction of these N-heterocyclic compounds is highly desirable. N-propargylamines represent one of the low-cost, readily accessible, and versatile building blocks in organic synthesis for generation of N-heterocyclic molecules.2 N-propargylic β-enaminocarbonyls are one of the novel and specific class of these compounds having diverse reaction patterns and applied in the assembly of various heterocycles (Fig. 1). Since a large number of developments in the use of N-propargylic β-enaminocarbonyls in heterocyclic synthesis have occurred from 2006 to present, a comprehensive review on this interesting field seems to be timely. In this review, we have classified these reactions based on the desired products. Mechanistic aspects of the reactions are considered and discussed in detail.
 |
| | Fig. 1 Some important synthetic compounds derived from N-propargylic β-enaminocarbonyls. | |
2. Pyrroles
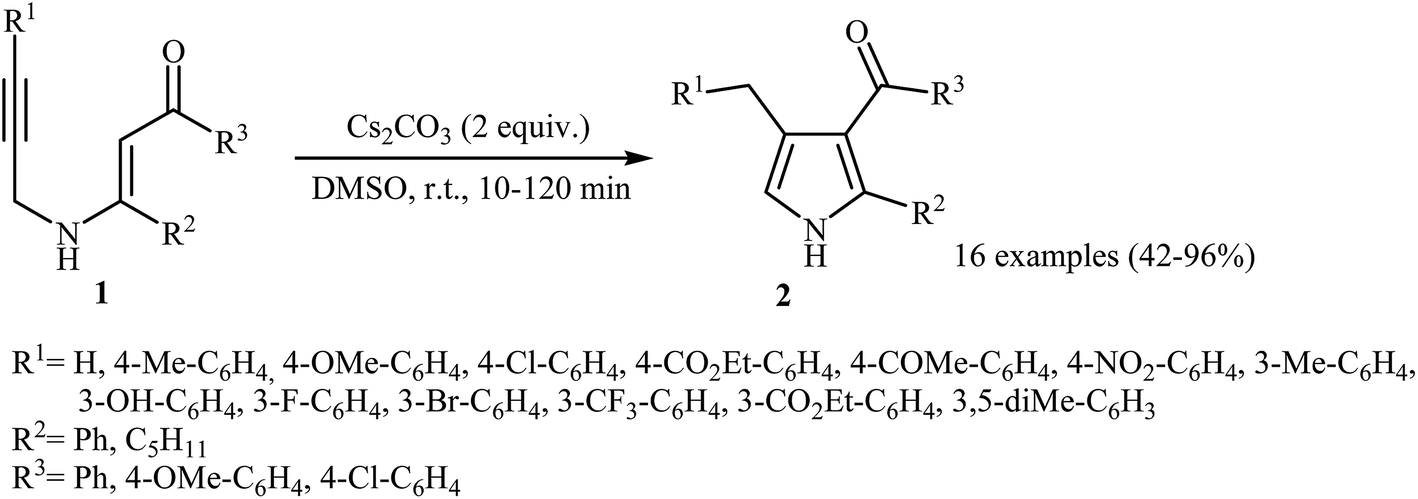
Pioneering works devoted to the synthesis of functionalized pyrroles 2 through intramolecular cyclization of N-propargylic β-enaminones 1 have been reported by Cacchi et al., who used Cs2CO3 as catalyst. This reaction was run in anhydrous DMSO at room temperature and tolerated the presence of a variety of sensitive functional groups, such as hydroxyl, nitro, methoxy, acetyl, ester, bromo, and chloro groups (Scheme 1). This made possible the further derivatization of the products. According to the author proposed mechanism, this transformation proceeded via a 5-exo-dig cyclization/protonation/isomerization sequential process.3
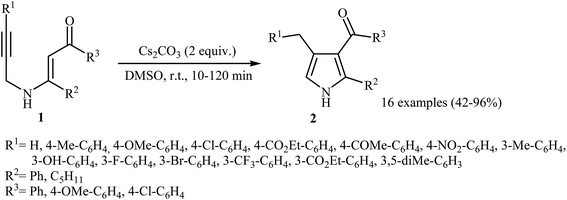 |
| | Scheme 1 Base-catalyzed synthesis of pyrroles 2 from N-propargylic β-enaminones 1. | |
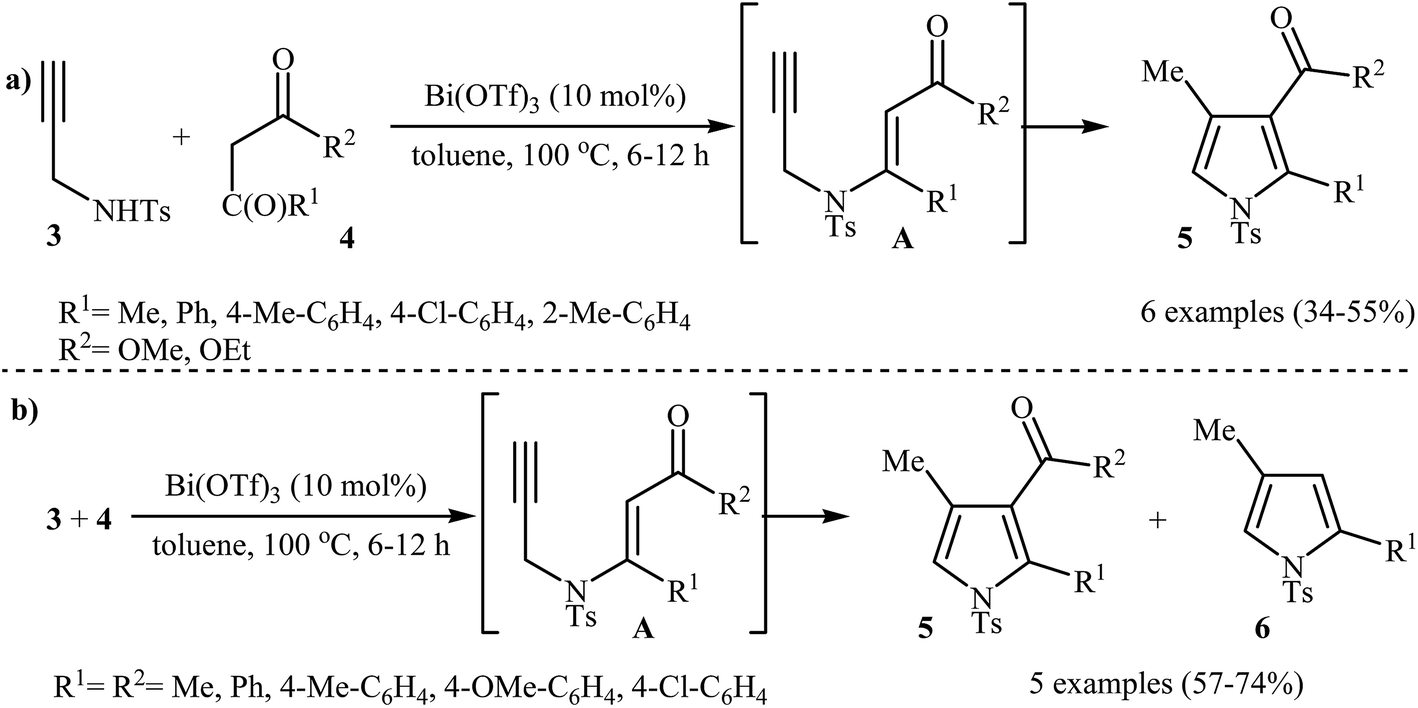
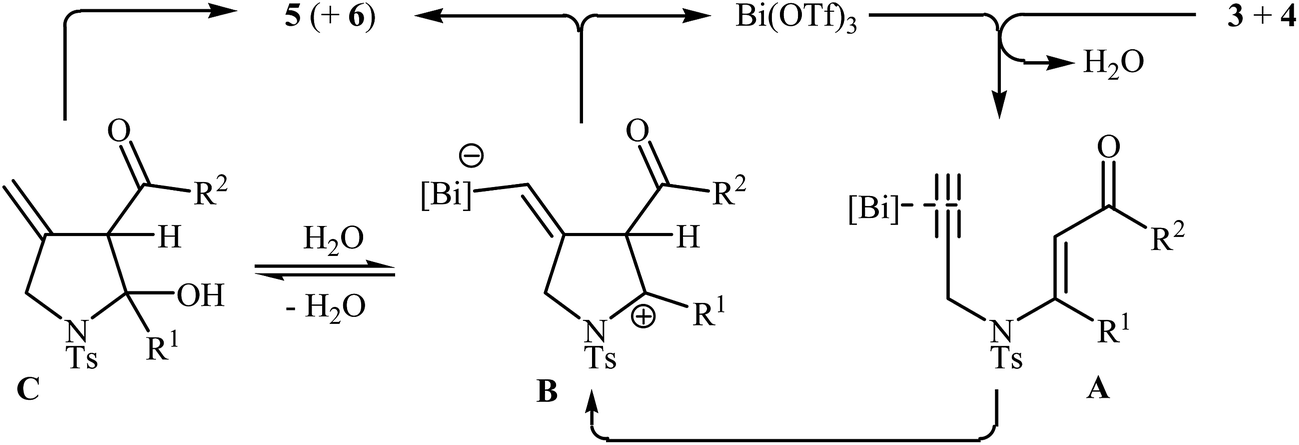
Subsequently, the Komeyama laboratory reported the synthesis of 3-carboxylated pyrroles 5 through a bismuth catalyzed intramolecular cyclization of N-propargylic β-enaminoesters A (generated in situ from N-propargyl amine 3 and β-keto esters 4) in refluxing toluene (Scheme 2a). They showed that the use of 1,3-diketones (instead of β-keto esters) as starting materials gave mixtures of pyrroles 5 and 6 (Scheme 2b). These observations would be explained by the instability of +C(O)OR. Thus, in the case of β-keto esters, deprotonate is preferred to decarboxylation. But in the case of 1,3-diketones, competition between the deprotonation and deacylation gave a mixture of products (Scheme 3).4
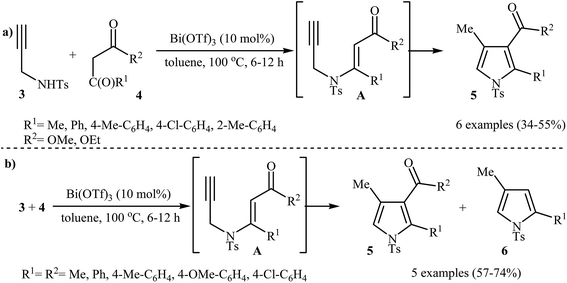 |
| | Scheme 2 (a) One-pot synthesis of pyrroles 5 from N-propargyl amine 3 and β-keto esters 4; (b) Synthesis of pyrroles 5 and 6 from N-propargyl amine 3 and 1,3-diketones 4. | |
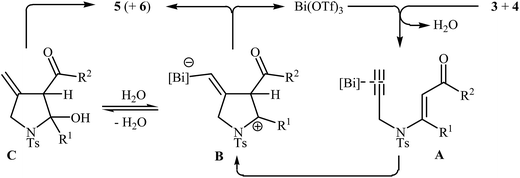 |
| | Scheme 3 Possible route for the formation of 5 and 6. | |
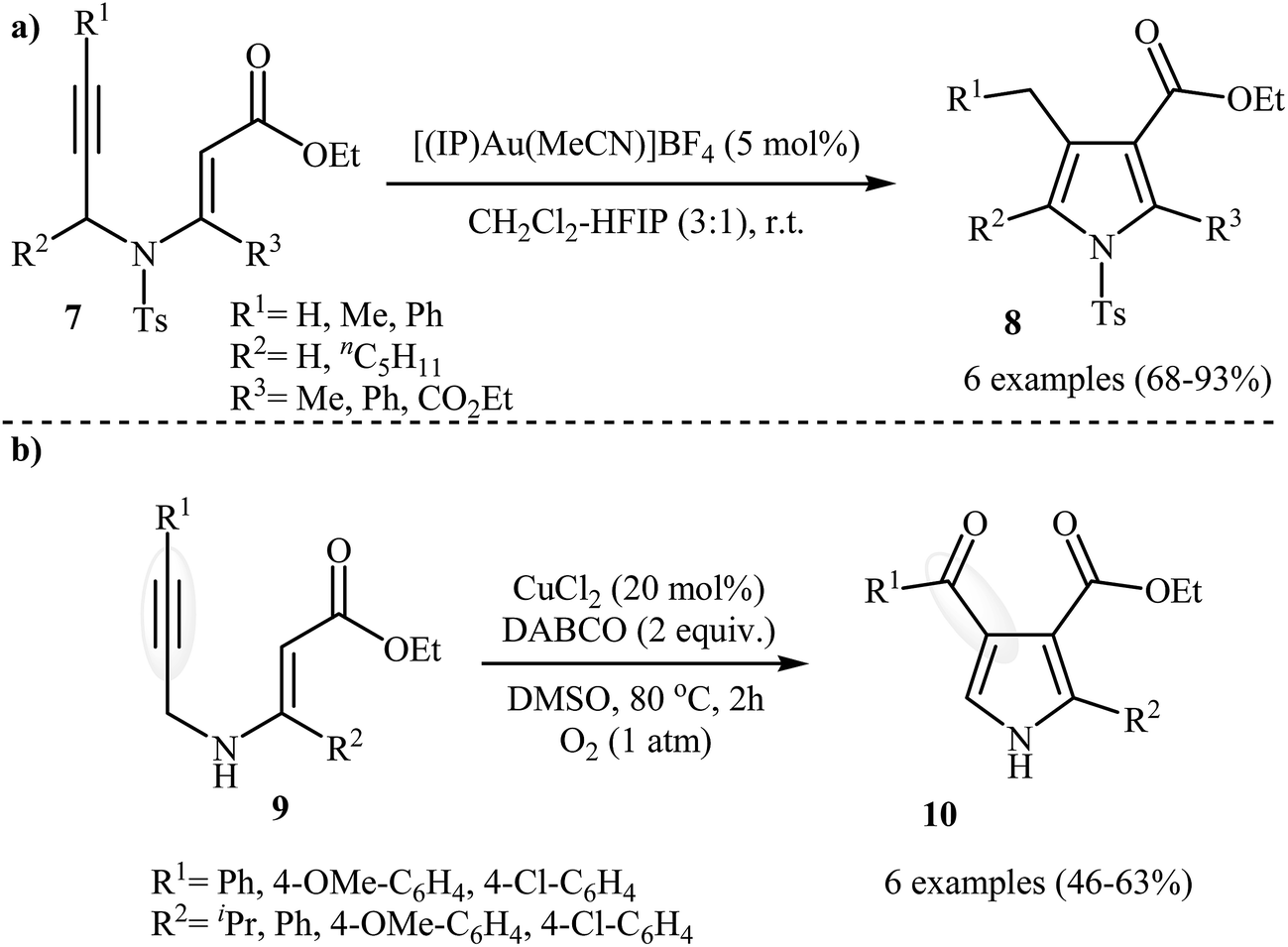
Following these works, Saito, Konishi, and Hanzawa were able to demonstrate that a series of 3-carboxylated pyrroles 8 could be obtained from corresponding N-propargylic β-enaminoesters 7 through a silver catalyzed amino-Claisen rearrangement/heterocyclization sequential process. This [(IP)Au(MeCN)]BF4 catalyzed reaction is equally efficient for both the internal and external alkynes. The reaction was run in CH2Cl2–HFIP (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) at room temperature and generally provided the highly substituted pyrroles 8 in good to high yields (Scheme 4a).5 Shortly afterward, Chiba et al. developed a new methodology for the construction of 4-carbonylpyrroles 10 by the copper-mediated aerobic carbooxygenation of N-propargylic β-enaminoesters 9 under oxygen atmosphere (Scheme 4b). Among the various copper catalysts like CuBr˖SMe2, CuCl2, CuCl, CuBr2, Cu(OAc)2; CuCl2 was the most efficient for the transformation. DABCO (1,4-diazabicyclo[2.2.2]octane) is required as additive and the DMSO as the solvent for this method and this protocol is applicable for the both electron reach and electron poor substrates.6
:1) at room temperature and generally provided the highly substituted pyrroles 8 in good to high yields (Scheme 4a).5 Shortly afterward, Chiba et al. developed a new methodology for the construction of 4-carbonylpyrroles 10 by the copper-mediated aerobic carbooxygenation of N-propargylic β-enaminoesters 9 under oxygen atmosphere (Scheme 4b). Among the various copper catalysts like CuBr˖SMe2, CuCl2, CuCl, CuBr2, Cu(OAc)2; CuCl2 was the most efficient for the transformation. DABCO (1,4-diazabicyclo[2.2.2]octane) is required as additive and the DMSO as the solvent for this method and this protocol is applicable for the both electron reach and electron poor substrates.6
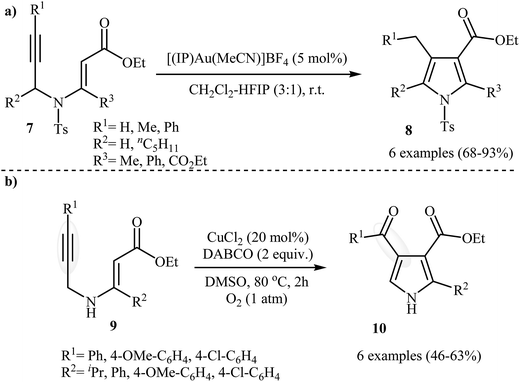 |
| | Scheme 4 (a) Synthesis of highly substituted pyrroles 8 through Au-catalyzed cyclization of N-propargylic β-enaminoesters 7; (b) Synthesis of 4-carbonylpyrroles 10 via copper-mediated aerobic carbooxygenation of N-propargylic β-enaminoesters 9. | |
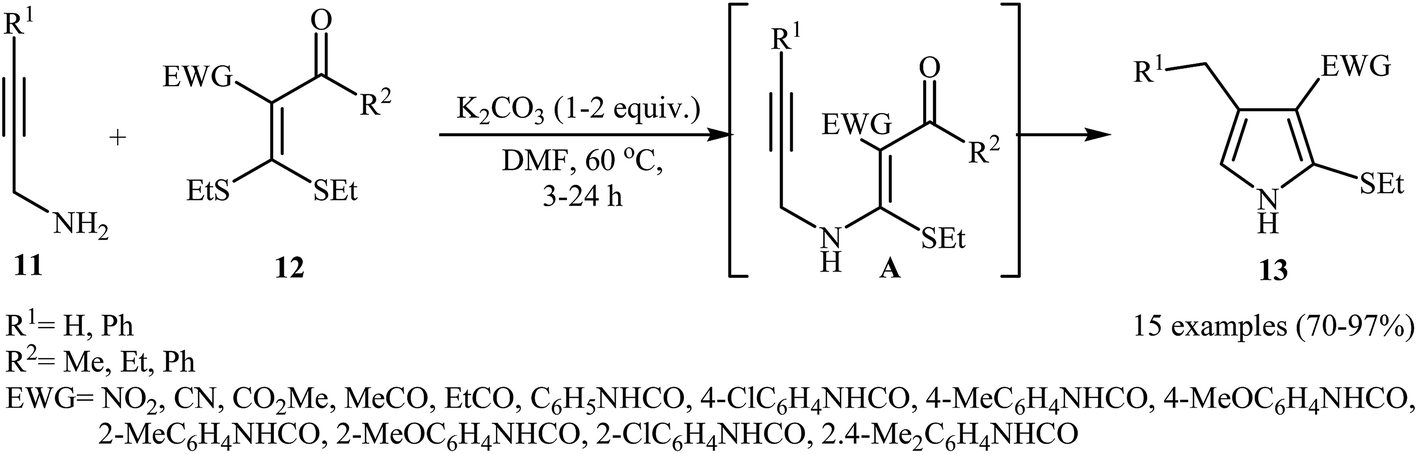
In 2012, Zhao and co-workers published an efficient protocol for the synthesis 2-ethylthio-pyrroles 13 via base catalyzed 5-exo-dig cyclization of N-propargylic β-enaminoes A were prepared in situ by intermolecular Michael addition of N-propargylamines 11 to α-acylketene dithioacetals 12 (Scheme 5). The authors determined that the use of K2CO3/DMF system provided the optimal conditions for this reaction. The most striking feature of this protocol is excellent functional group tolerance to produce highly functionalized pyrroles 13 from simple starting materials. It is noted that when external N-propargylamines (R1 = H) were used as substrates, the reaction was performed in the absence of an external base and the corresponding products were obtained in high to excellent yields.7
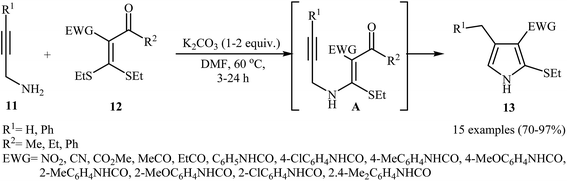 |
| | Scheme 5 Synthesis 2-ethylthio-pyrroles 13 developed by Zhao. | |
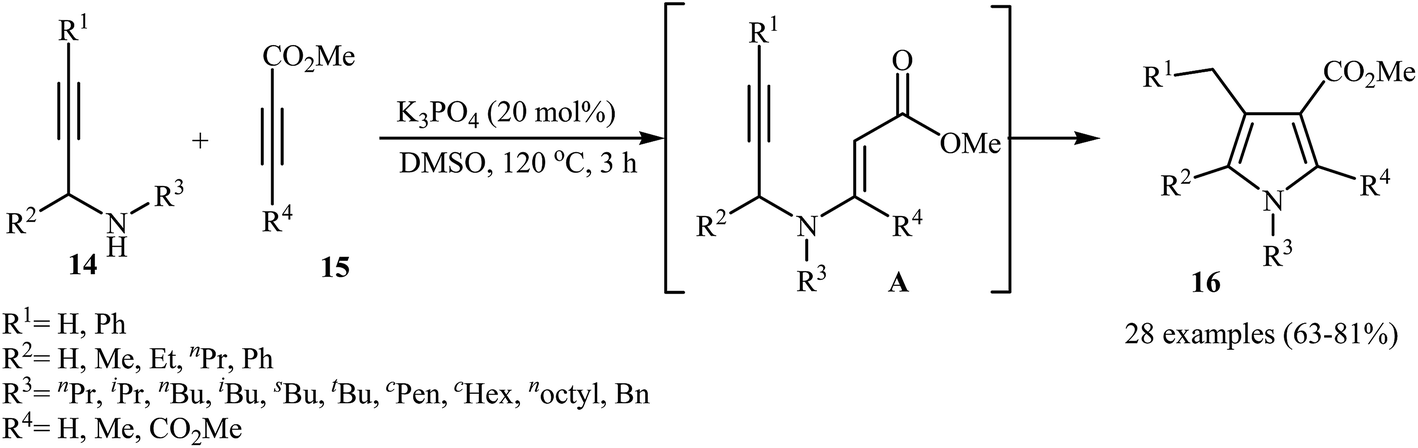
Recently, Jin and co-workers demonstrated that polysubstituted pyrroles 16 can be readily prepared in the reaction of N-propargylamines 14 with activated alkynes 15 in the presence of 20 mol% of K3PO4 as catalyst in DMSO at 120 °C (Scheme 6) The authors suggested that the reaction proceeds via the formation of N-propargylic β-enaminoester intermediates A through Michael addition of N-propargylamines 14 to alkynes 15, which after a 5-exo-dig cyclization process delivers the 3-carboxylated pyrroles 16 in good yields.8
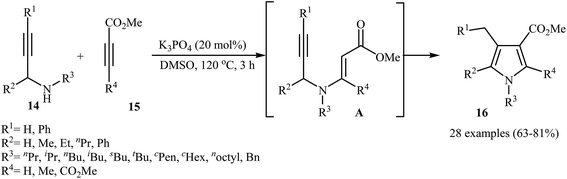 |
| | Scheme 6 Synthesis of polysubstituted pyrroles 16 from N-propargylamines 14 and activated alkynes 15. | |
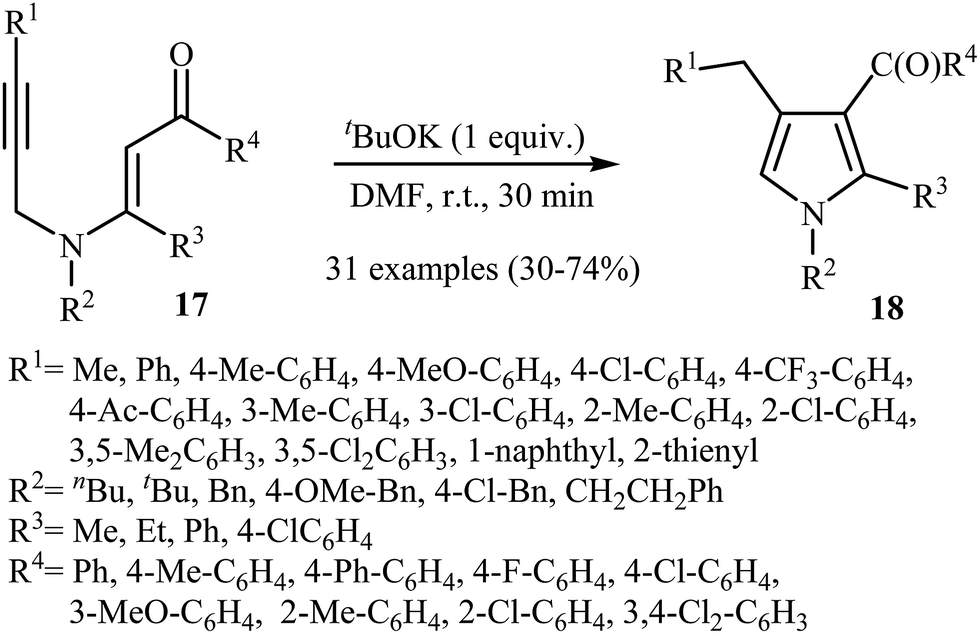
Later, Zhang's research team reported a direct synthesis of polysubstituted pyrrole derivatives 18 via base-catalyzed intramolecular cyclization of N-propargylic β-enaminones 17 (Scheme 7). Thus, the careful analysis of the optimized reactions revealed that the optimum condition for this transformation was the addition of 1 equiv. of tBuOK, at room temperature, to a solution of N-propargylic β-enaminones 17 in DMF. The reaction conditions allowed the use of substrates bearing a variety of sensitive functional groups including flouro, chloro, methoxy, and acetyl functionalities. According to the author proposed mechanism, the reaction proceeded via a propargyl−allenyl isomerization/5-exo-dig cyclization/proton transfer sequential process.9
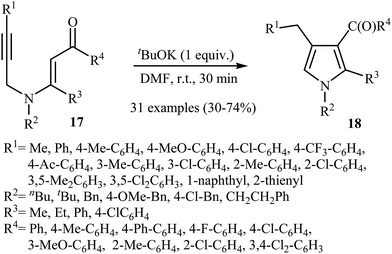 |
| | Scheme 7 Base-catalyzed synthesis of pyrrole derivatives 18 described by Zhang. | |
A notable contribution to this field was reported by Wan and co-workers in 2012. They found that β-(arylsulfonyl)methyl pyrroles 20 could be prepared from N-propargylic β-enaminoesters 19 using 10 mol% of Cs2CO3 as base in DMF at 80 °C. According to a plausible mechanistic pathway, this reaction proceeds through an aza-Claisen rearrangment/cyclization/sulfonyl group migration/aromatization sequential process (Scheme 8a). The authors claimed their reaction was the first example of sulfonyl group migration in pyrrole synthesis. The results demonstrated that the electronic characters of substrates had little effect on the rate of the reaction. Under optimized conditions, the reaction tolerates both electron-donating and electron-withdrawing substituents and gave the corresponding highly substituted N–H free pyrroles 20 in high to excellent yields. Interestingly, when the reaction was carried out in DMF at 140 °C under catalyst-free conditions, α-(arylsulfonyl)methyl pyrroles 21 were isolated instead of β-(arylsulfonyl)methyl pyrroles 20. The mechanism proposed by the authors to explain this transformation is outlined in Scheme 8b.10 Recently, the same authors applied this methodology to synthesis of a series of 2-trifluoromethyl-5-(arylsulfonyl)methyl pyrroles and 2-trifluoromethyl-4-(arylsulfonyl)methyl pyrroles from corresponding trifluoromethyl-substituted N-propargylic β-enaminoesters.11
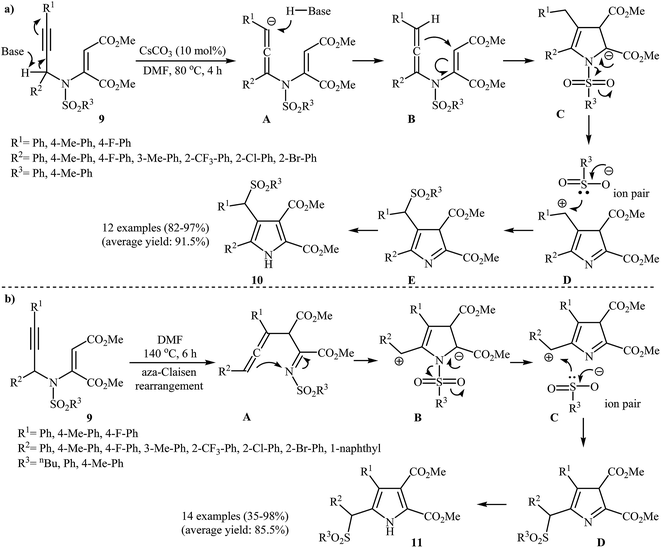 |
| | Scheme 8 (a) Base-catalyzed synthesis of β-(arylsulfonyl)methyl pyrroles 20 developed by Wan; (b) thermal cyclization of 19 for the synthesis of α-(arylsulfonyl)methyl pyrroles 21 reported by Wan. | |
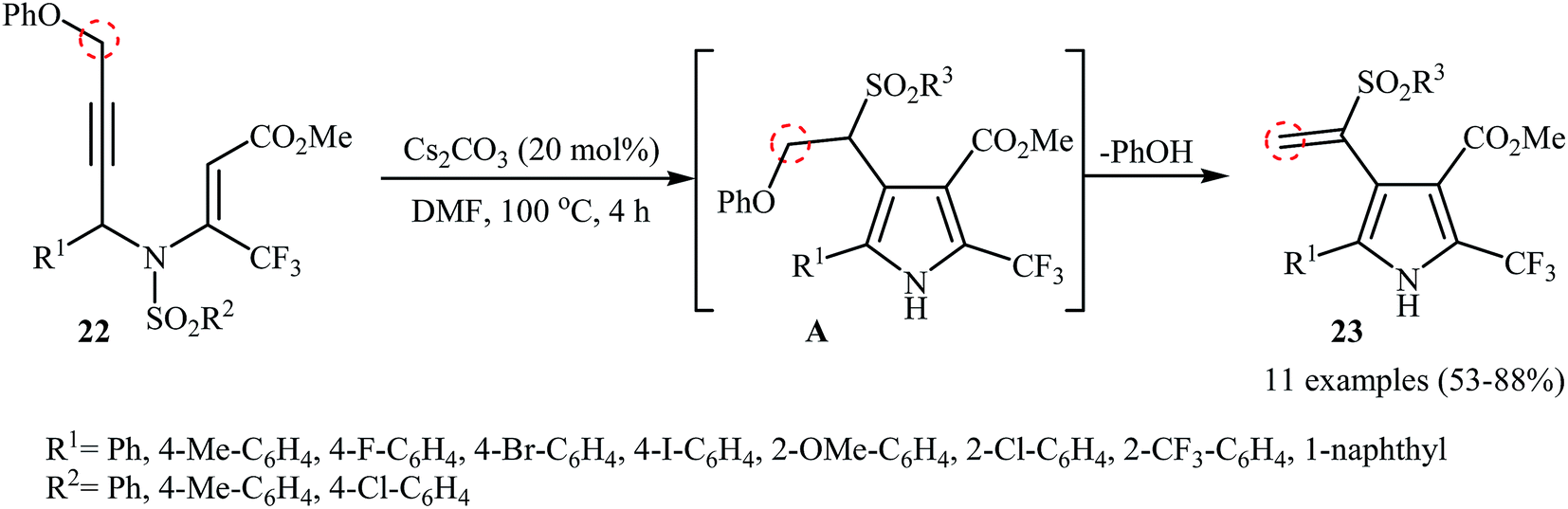
The groups of Wan also were able to demonstrate that a series of sulfonyl vinyl-substituted pyrroles 23 could be obtained from the corresponding phenoxysubstituted N-propargylic β-enaminoesters 22 employing 20 mol% of Cs2CO3 as catalyst in anhydrous DMF (Scheme 9). The reaction proceeds along the similar mechanistic pathway that described in Scheme 4a to afford intermediate A. This intermediate undergoes spontaneous phenol elimination to produce the observed products 23.12
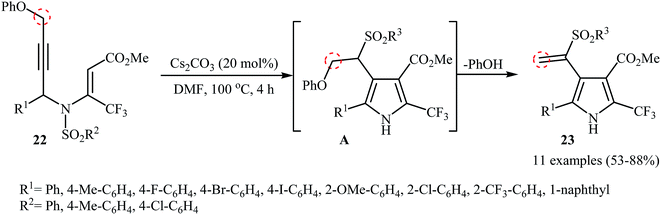 |
| | Scheme 9 Wan's synthesis of sulfonyl vinyl-substituted pyrroles 23. | |
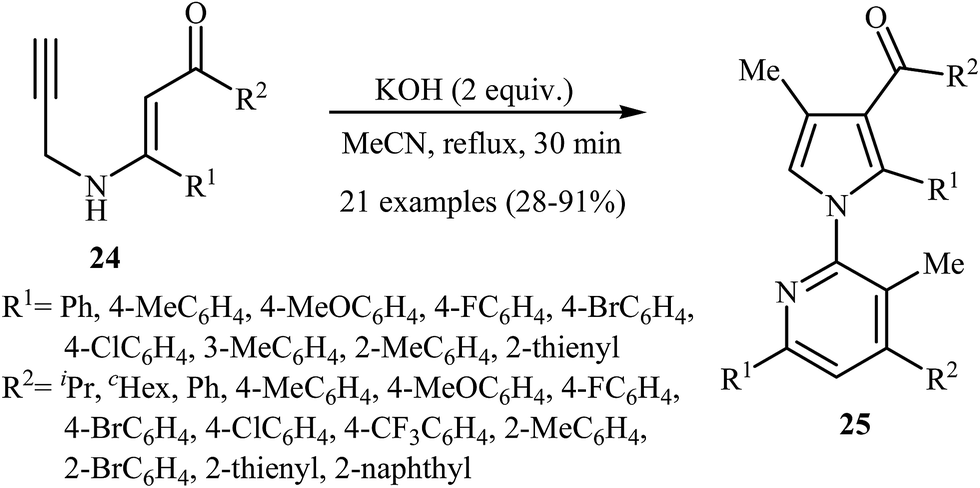
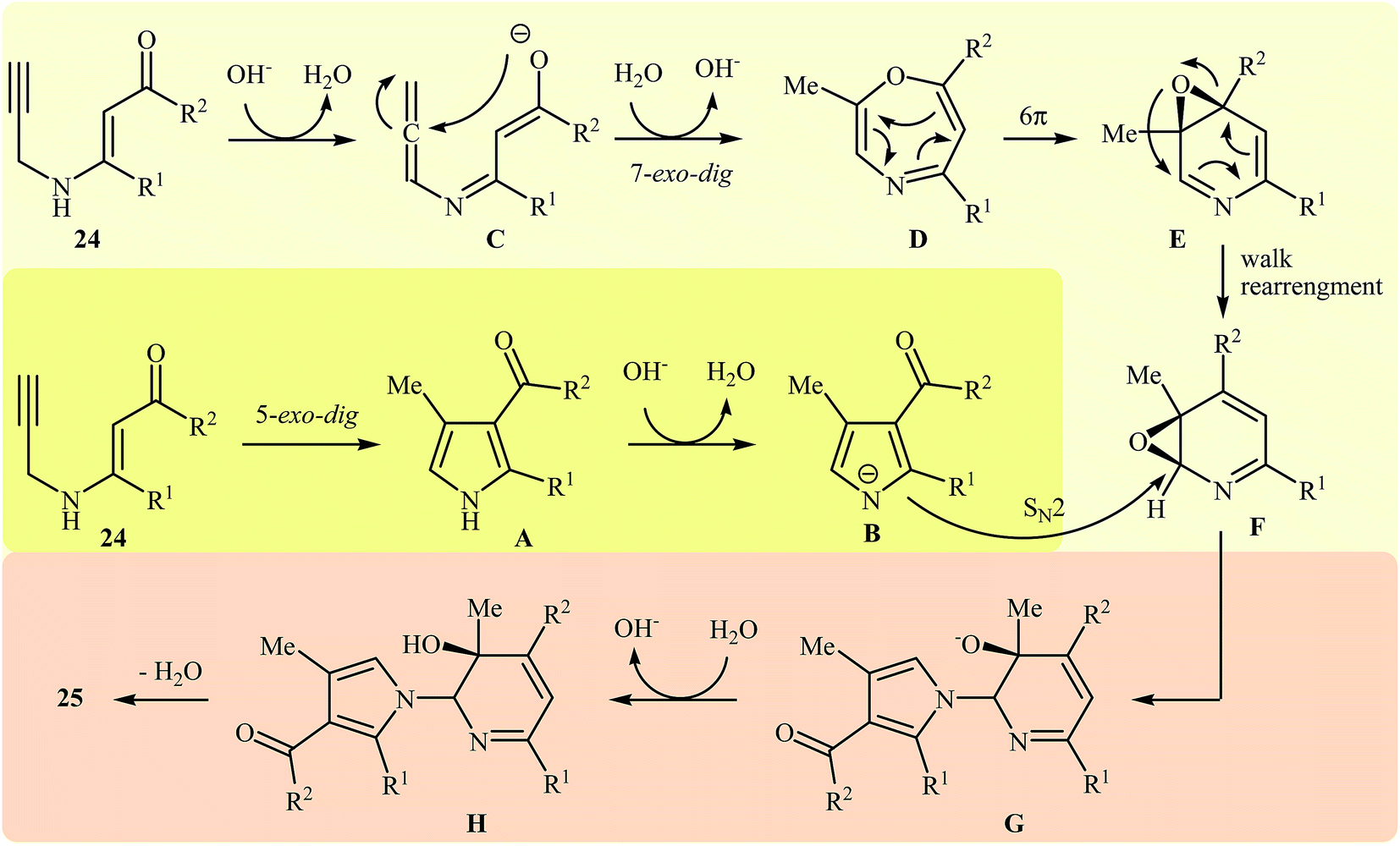
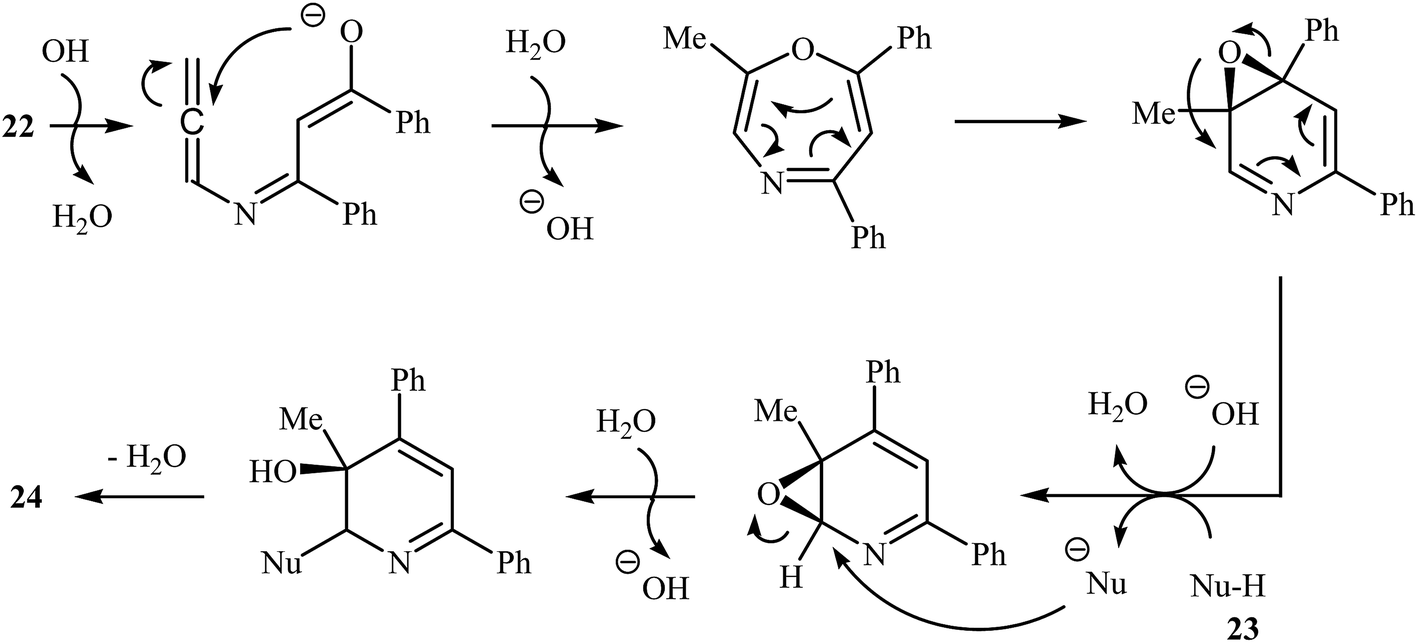
More recently, Cui and co-workers described a general and efficient synthesis of a diverse collection of biologically important highly substituted N-(2-pyridyl)pyrroles 25 via the KOH-promoted regiospecific cyclization of the corresponding N-propargylic β-enaminones 24 under reflux conditions in acetonitrile (Scheme 10). This procedure simultaneously facilitated the construction of a pyridine scaffold and generation of a pyrrole ring from simple starting materials and the only by-product is one equivalent of water. The proposed mechanism for this reaction involves the following steps (Scheme 11): (i) formation of a pyrrole anion intermediate B through intramolecular 5-exo-dig cyclization of N-propargylic β-enaminone 24; (ii) formation of a 7-oxa-5-aza-bicyclo[4.1.0]hepta-2,4-diene intermediate F from another molecule of N-propargylic β-enaminone 24 via propargyl−allenyl isomerization/intramolecular 7-exo-dig cyclization/6π-electrocyclization/walk rearrangement sequential process; (iii) SN2 attack of intermediate B to dihydropyridine F to generate trans-2,3- dihydropyridine intermediate G; and (iv) protonation and aromatization of G to produce final product 25.13
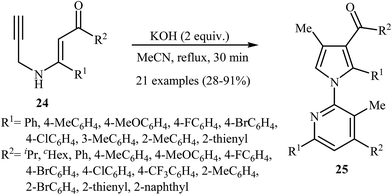 |
| | Scheme 10 Base-mediated cascade synthesis of N-(2-pyridyl)pyrroles 25 from N-propargylic β-enaminones 24 developed by Cui. | |
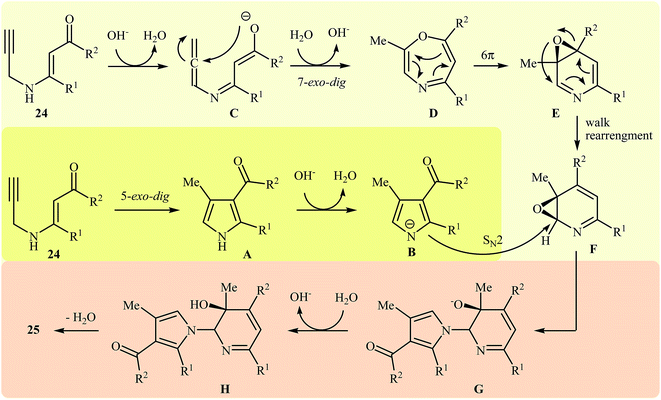 |
| | Scheme 11 Mechanism that accounts for the formation of 25. | |
3. Pyrrolines
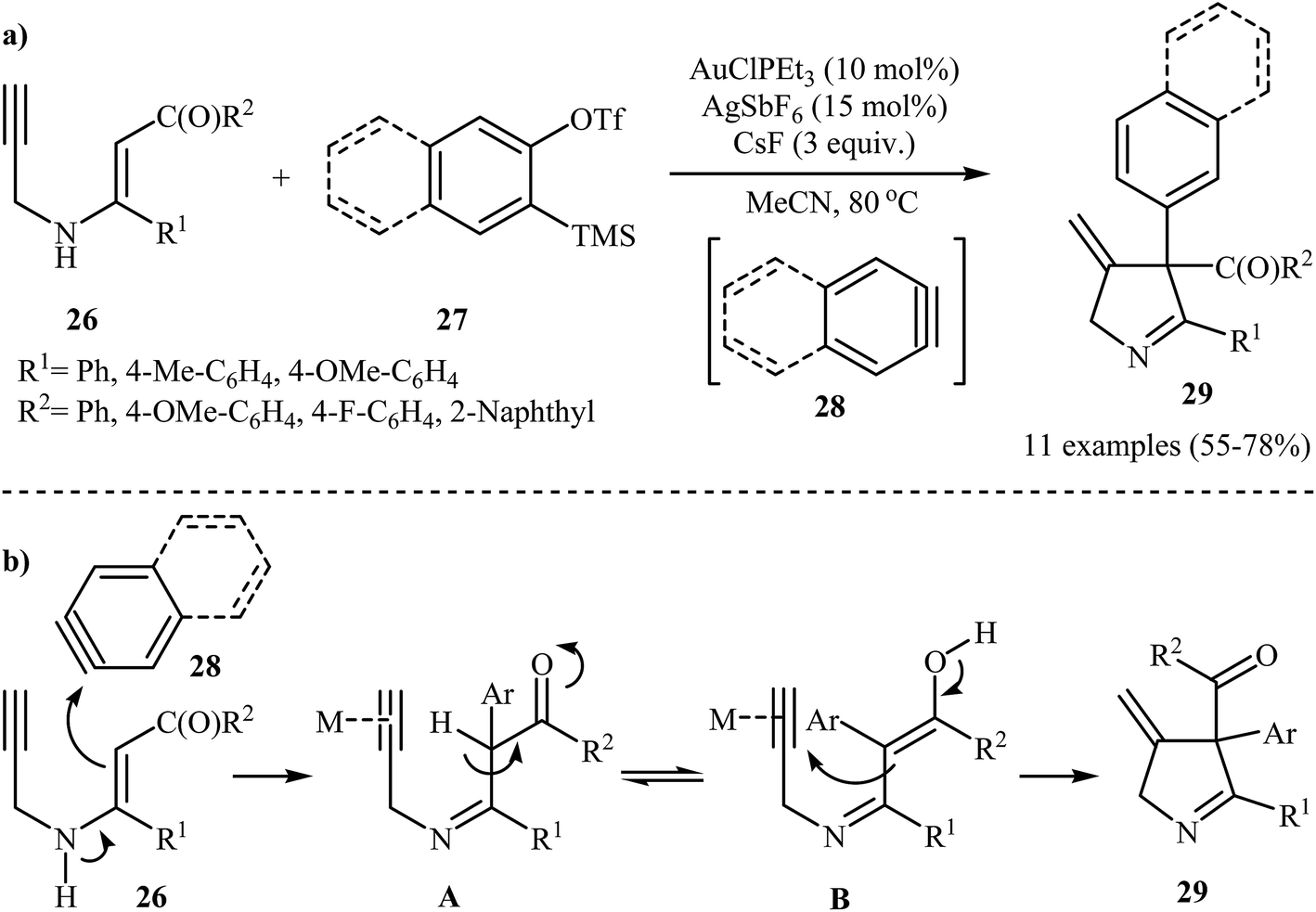
In 2014, the Karunakar group have developed a new and straightforward approach for the synthesis of substituted 1-pyrrolines 29 via a one-pot, Au(I)-catalyzed reaction between N-propargylic β-enaminones 26 and arynes 28 (generated in situ from 2-(trimethylsilyl)aryl trifluoromethanesulfonates 27 and CsF) (Scheme 12a). AuClPEt3/AgSbF6 was the best catalytic system and acetonitrile was the best solvent for this reaction. The results demonstrated that the enaminones bearing electron donating groups, like Me and OMe, gave higher yields than those having an electron withdrawing group, like F group. The author proposed mechanism for this transformation is depicted in Scheme 12b.14
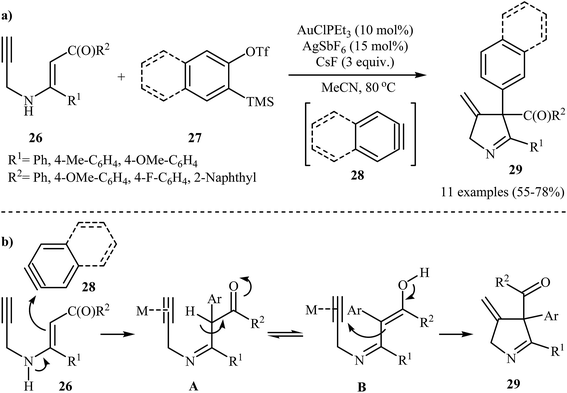 |
| | Scheme 12 (a) Synthesis of pyrrolines 29 developed by Karunakar; (b) plausible mechanism for the formation of 29. | |
4. Pyridines
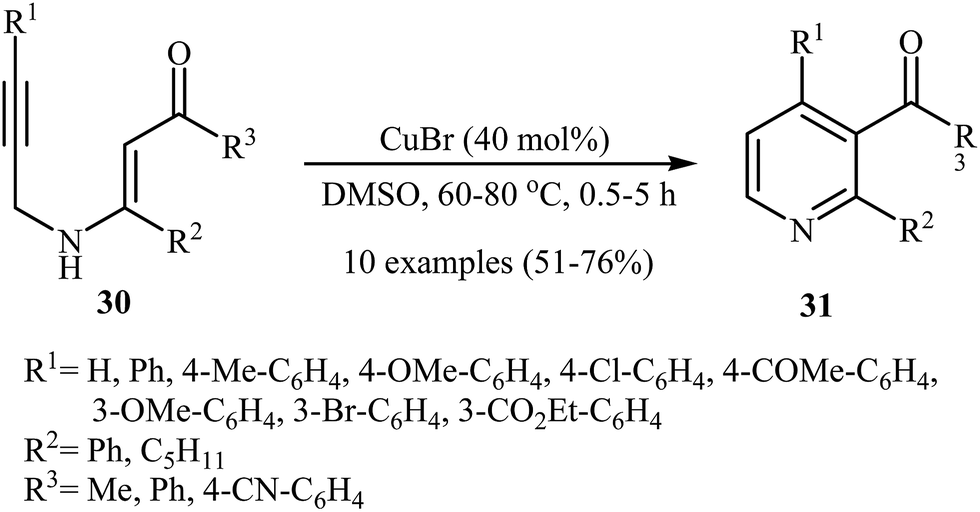
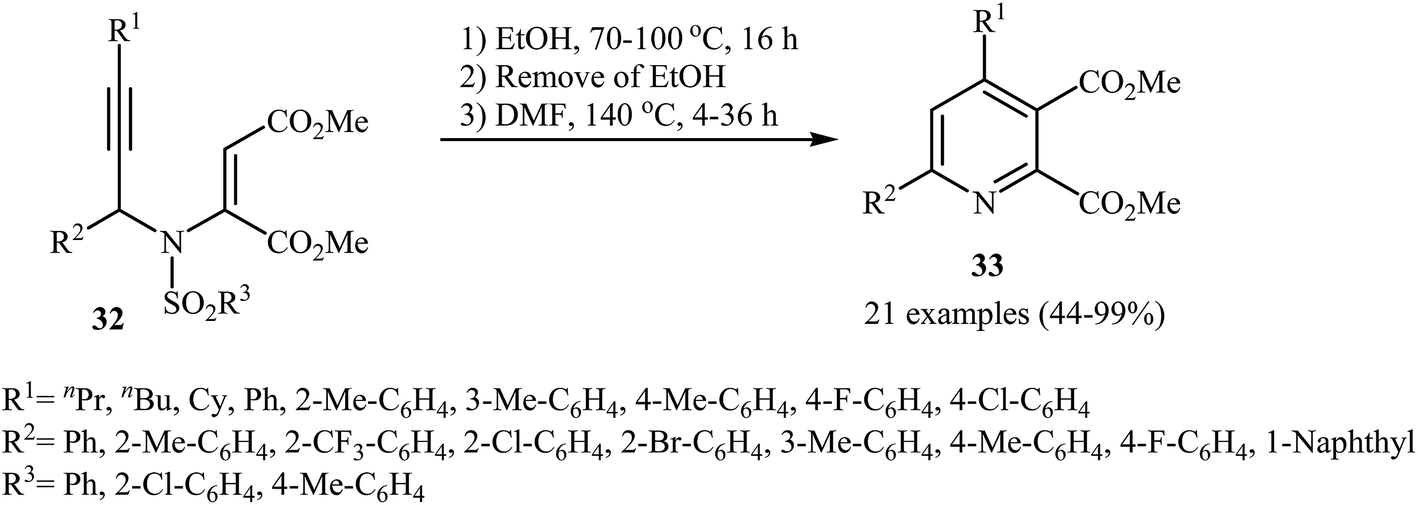
After the pioneering work of Cacchi and co-workers in 2008, who showed that highly substituted pyridines 31 could be readily synthesized in one step from easy available N-propargylic β-enaminones 30 through Cu-catalyzed 6-endo-dig cyclization strategy (Scheme 13),3 this methodology has developed into a general powerful tool for the synthesis of a wide range of pyridine derivatives, including natural products.15 In 2013, Wan's group developed a convenient one-pot three step methodology for the synthesis of 2,3,4,6-tetrasubstituted pyridines 33 from the N-sufonylated N-propargylic β-enaminoesters 32 through an intramolecular cyclization/sulfinyl acids elimination cascade process (Scheme 14). Thus, in the first step a solution of 32 (0.2 mmol) in ethanol (2 mL) was heated at 70-100 °C for 16 h under argon atmosphere. In the next step the ethanol solvent was removed in vacuo. Finally, to the residue was added DMF (1 mL) and the mixture was heated at 140 °C for 4-36 h under argon atmosphere.16
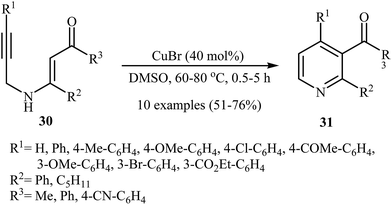 |
| | Scheme 13 Cu(I)-catalyzed synthesis of pyridines 31 from N-propargylic β-enaminones 30. | |
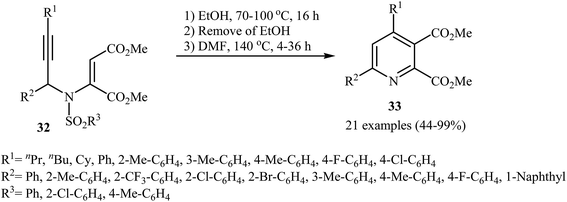 |
| | Scheme 14 Synthesis of 2,3,4,6-tetrasubstituted pyridines 33 described by Cacchi. | |
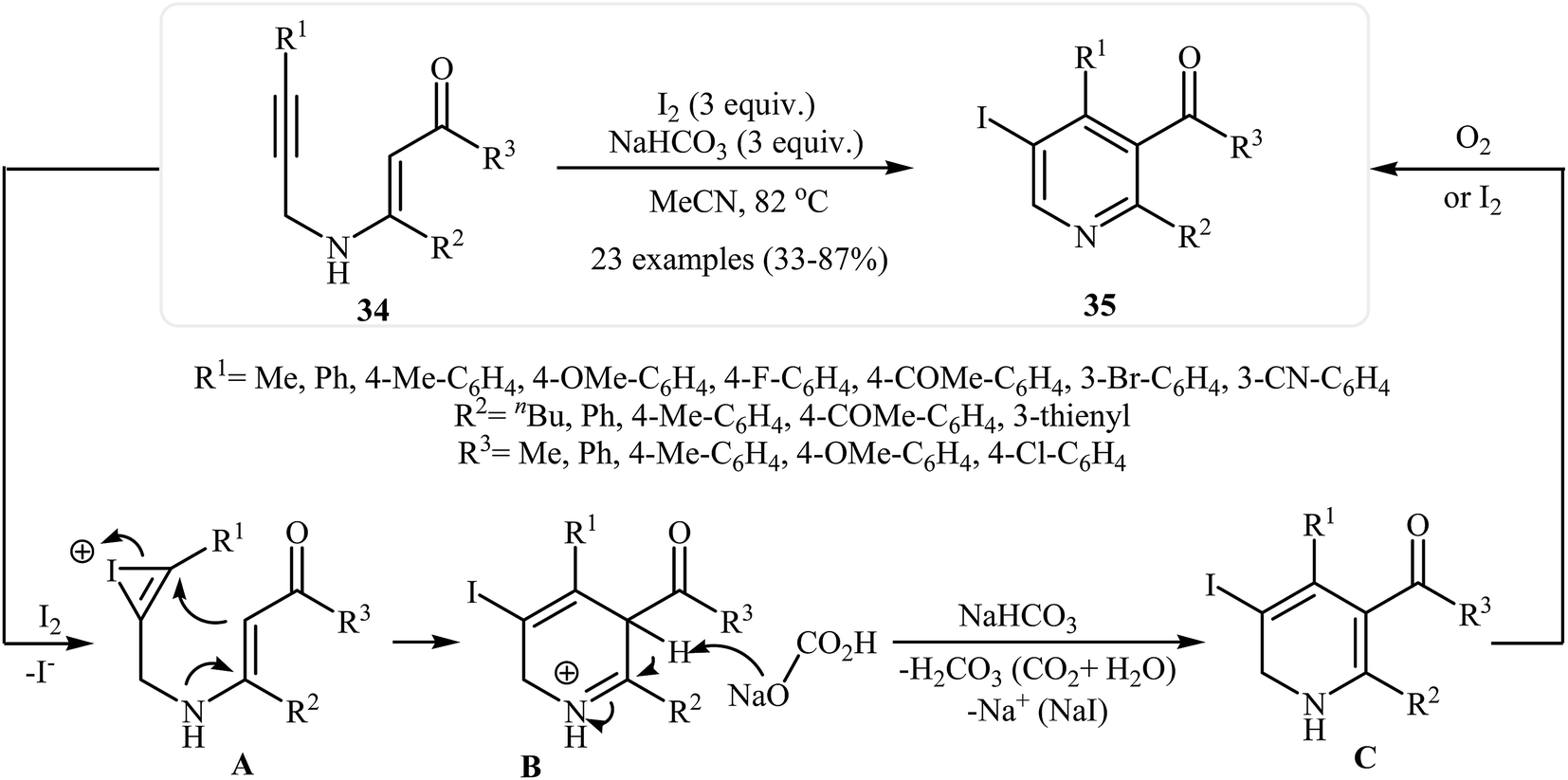
Recently an alternative intramolecular electrophilic cyclization of N-propargylic β-enaminones was reported by Zora and coworkers. In this study, 5-iodopyridines 35 were obtained in good yields by reaction of N-propargylic β-enaminones 34 with 3.0 equiv of I2 in MeCN at 82 °C in the presence of NaHCO3 as base. The reaction proceeded via a 6-endo-dig process, giving only the six-membered rings. The authors proposed a mechanism initiated by the reaction of the alkyne moiety of starting β-enaminone 34 and iodine followed by an electrophilic cyclization with subsequent deprotonation/oxidation to explain the cyclized products obtained (Scheme 15).17 Subsequently, the above cyclization products, applied by the same authors to the synthesis of more functionalized pyridines via transition metal-catalyzed processes, such as Sonogashira and Suzuki–Miyaura cross-coupling reactions.18,19
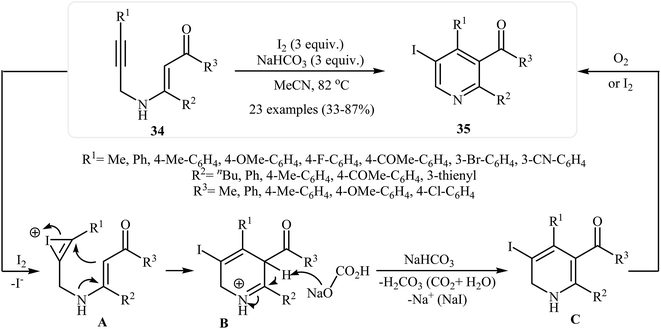 |
| | Scheme 15 Synthesis of 5-iodopyridines 35 from N-propargylic β-enaminones 34 via iodinemediated electrophilic cyclization. | |
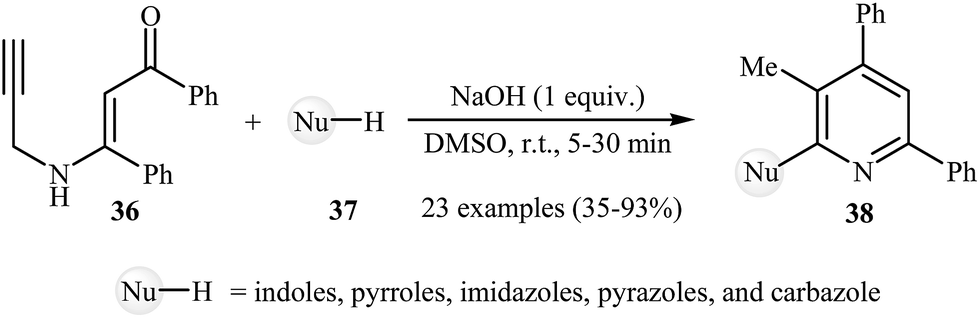
Recently, Cheng et al. developed a simple and efficient methodology for the synthesis of 2-(1-heteroaryl) pyridines 38 by the reaction between 1,3-diphenyl-3-(prop-2-ynylamino)prop-2-en-1-one 36 and N-heteroarenes 37 (Scheme 16).20 NaOH was the most efficient catalyst among the bases such as LiOtBu, NaOtBu, K2CO3, KOH, Et3N etc for this reaction. This NaOH-catalyzed reaction is applicable for a diverse set of nitrogen nucleophilic source, such as indole, pyrrole, imidazole, pyrazole, carbazole, and benzimidazole. However, the reaction of benzimidazole and 36 gave the desired product in unsatisfactory yield and aniline, benzylamine, succinimide, acetanilide, and benzyl thiol failed to participate in the reaction. According to the suggested reaction mechanism (Scheme 17), the reaction proceeds via a propargyl−allenyl isomerization/enolization/7-exo-dig cyclization/6π-electrocyclization/walk rearrangement/epoxide ring opening/dehydrative aromatization sequential process.
 |
| | Scheme 16 Synthesis of 2-(1-heteroaryl) pyridines 38 developed by Cheng. | |
 |
| | Scheme 17 Mechanistic proposal for the reaction in Scheme 15. | |
5. Dihydropyridine
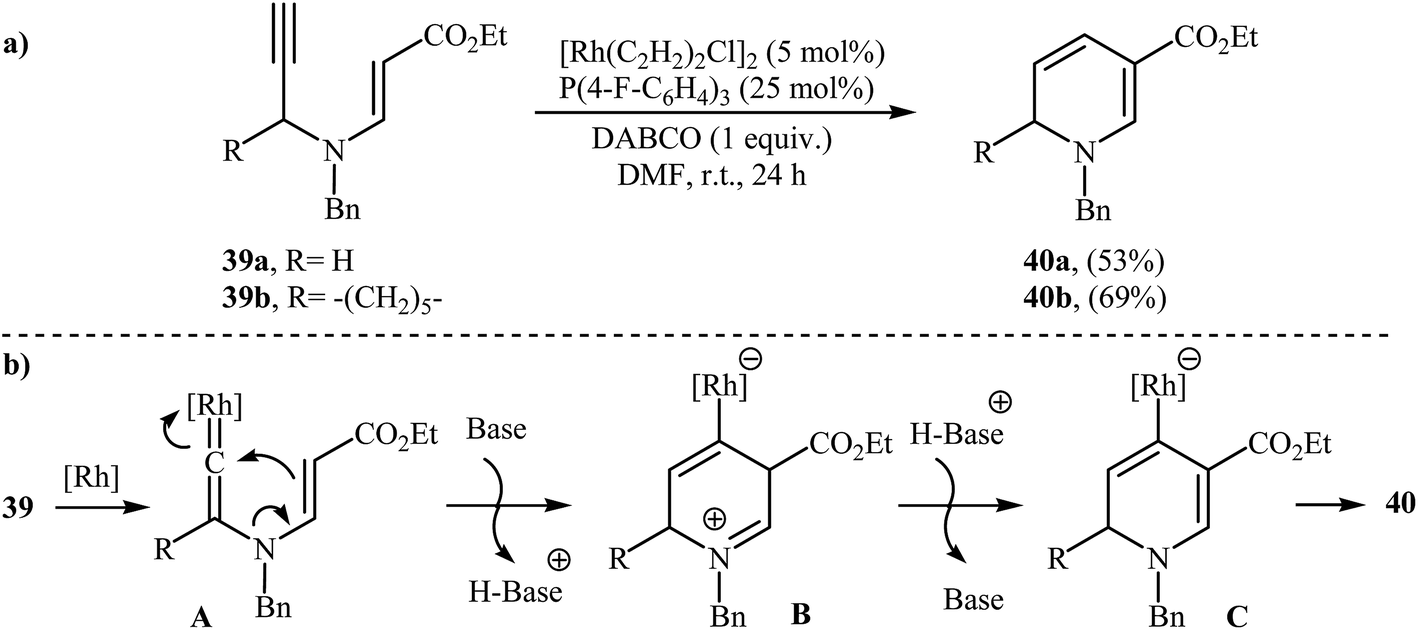
N-propargylic β-enaminoesters 39 were found by Kim and Lee to undergo intramolecular cyclization in the presence of catalytic amount of [Rh(C2H2)2Cl]2, using P(4 F-C6H4)3 as ligand and DABCO as base in DMF at room temperature, to afford 1,6-dihydropyridine-3-carboxylates 40 in moderate yields (Scheme 18a). The mechanistic course of this reaction sequence is shown in Scheme 18b, and involves the initial formation of the rhodium vinylidene complex A from the reaction of alkyne moiety of 39 with rhodium. Intramolecular cyclization of A yields intermediate B, which undergoes deprotonation to furnish intermediate C. Finally, protodemetalation of intermediate C affords the observed dihydropyridines 40.21
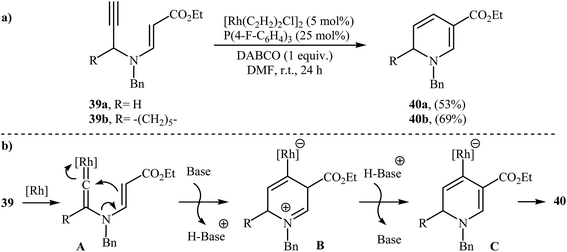 |
| | Scheme 18 (a) Rh-catalyzed synthesis of 1,6-dihydropyridine-3-carboxylates 40 reported by Kim and Lee; (b) Mechanism that accounts for the formation of 40. | |
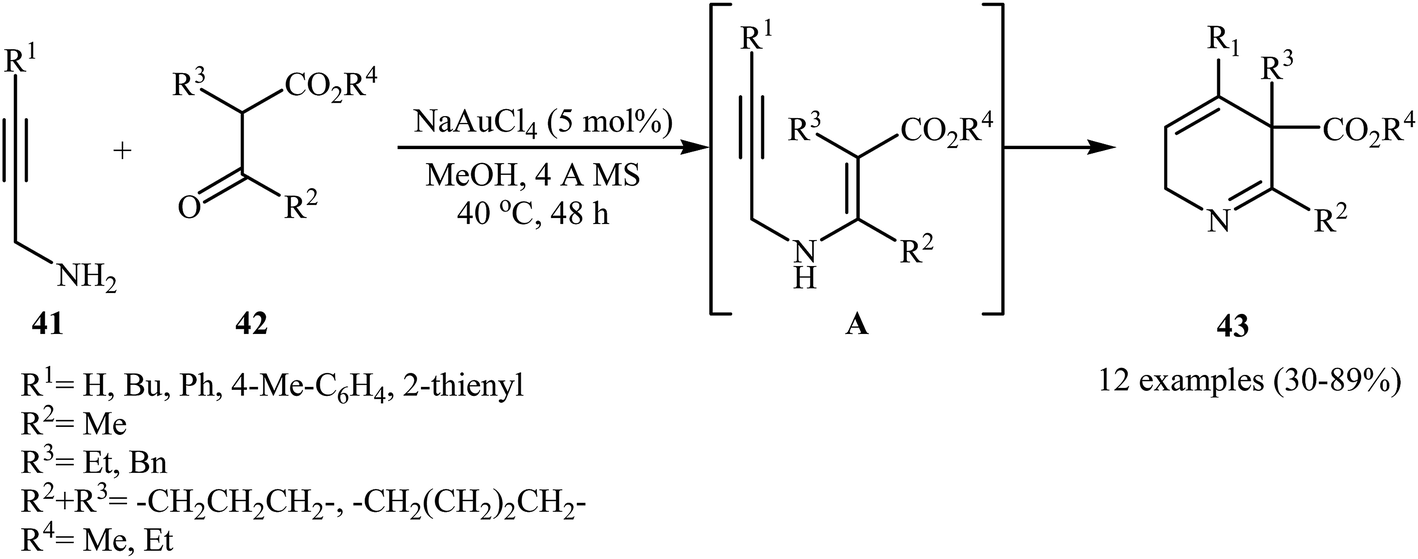
The preparation of 2,5-dihydropyridines 43 can be achieved through a 6-endo-dig cyclization of N-propargylic β-enaminesters A (generated in situ from condensation of N-propargyl amines 41 and β-keto esters 42) (Scheme 19). The best condition used for this transformation was 5 mol% of NaAuCl4 and MeOH at 40 °C.22
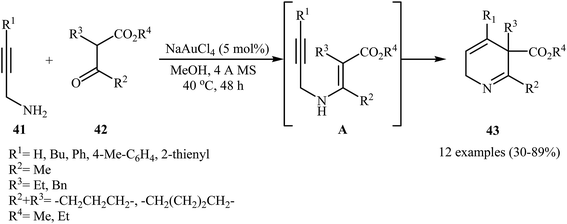 |
| | Scheme 19 Au-catalyzed synthesis of highly substituted 2,5-dihydropyridines 43 from N-propargyl amines 41 and β-keto esters 42 through a N-propargylic β-enaminoester intermediate A. | |
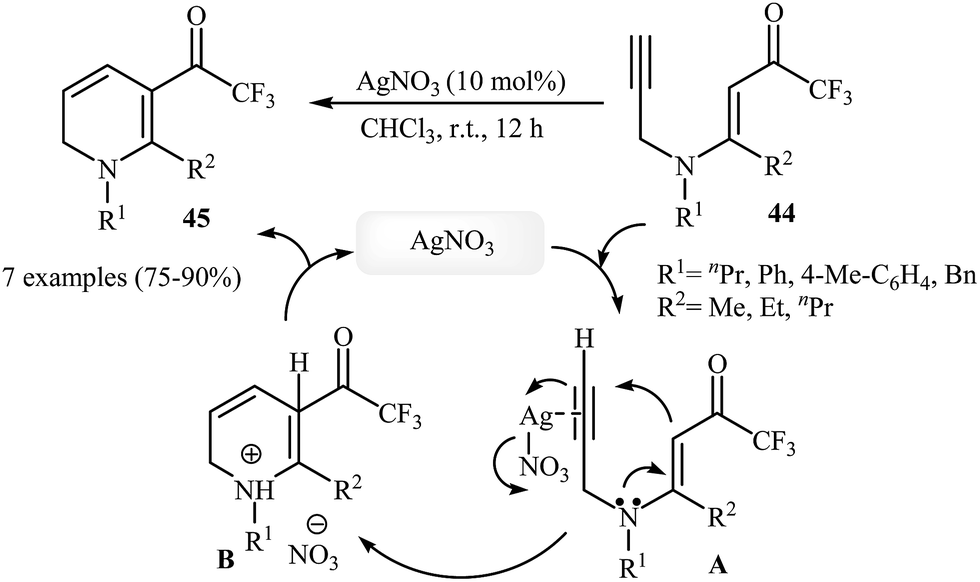
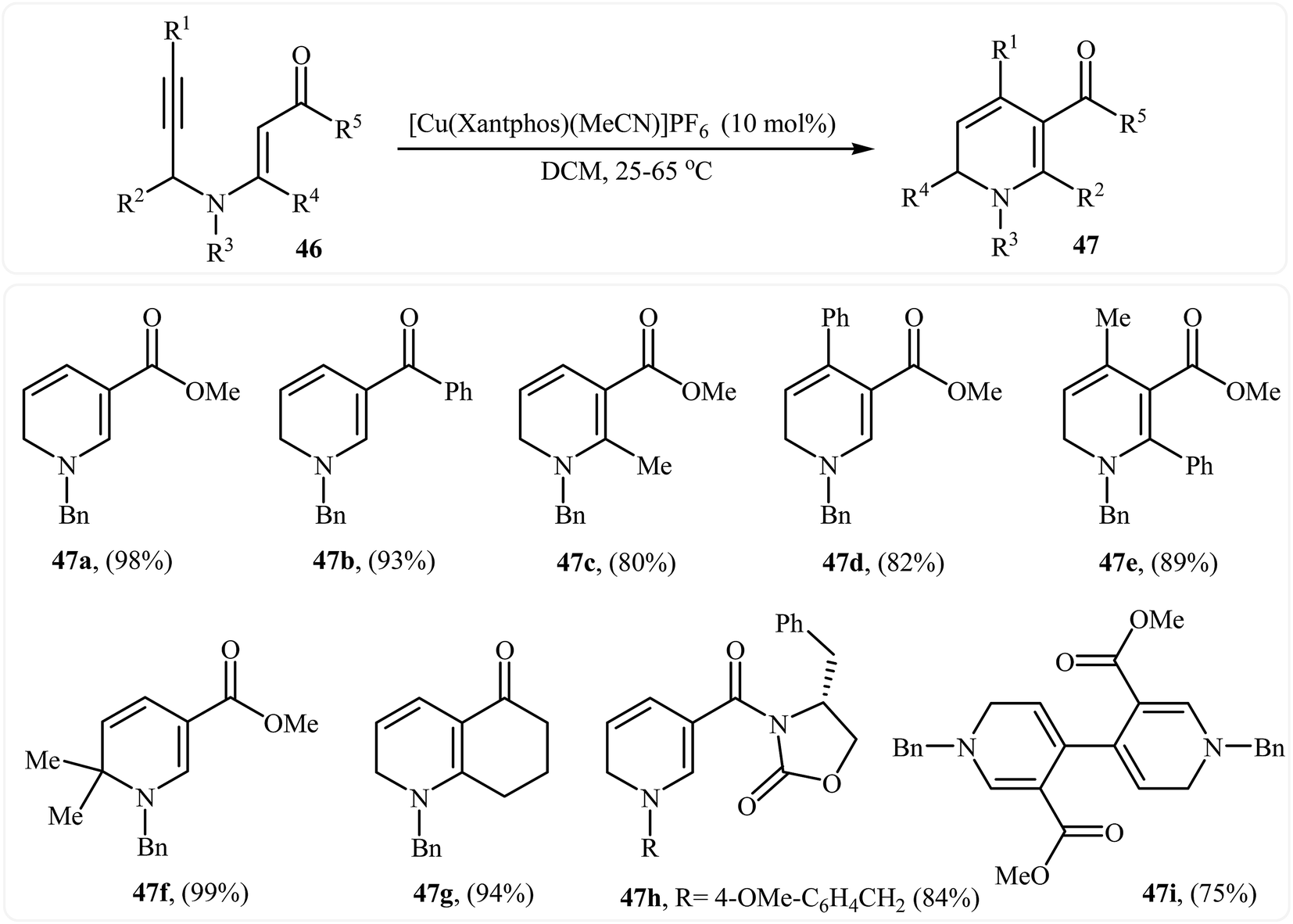
In 2013, Martins and co-workers disclosed a mild and practical access to trifluoromethylated 1,2-dihydropyridines 45 through Au(I)-catalyzed 6-endo-dig cyclization of corresponding N-propargylic β-enaminones 44 (Scheme 20). A number of 1,2-dihydropyridines containing aromatic or aliphatic motifs were readily afforded in high yields. Mechanistically, the triple bond is activated by coordination with silver to form a π-alkyne complex A, which then undergoes a 6-endo-dig cyclization to produce cationic intermediate B. Subsequently, an intramolecular proto-demetalation leads to the product 44.23 In a closely related investigation, the Oguri group found that N-propargylic β-enaminocarbonyls 46 were converted to the corresponding substituted 1,2-dihydropyridines 47, via 6-endo-dig cyclization using [Cu(Xantphos)(MeCN)]PF6 as catalyst in DCM at room temperature (Scheme 21).24
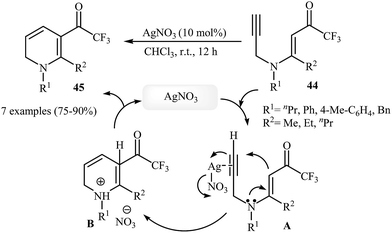 |
| | Scheme 20 Construction of trifluoromethylated 1,2-dihydropyridines 45 through Au(I)-catalyzed 6-endo-dig cyclization of N-propargylic β-enaminones 44. | |
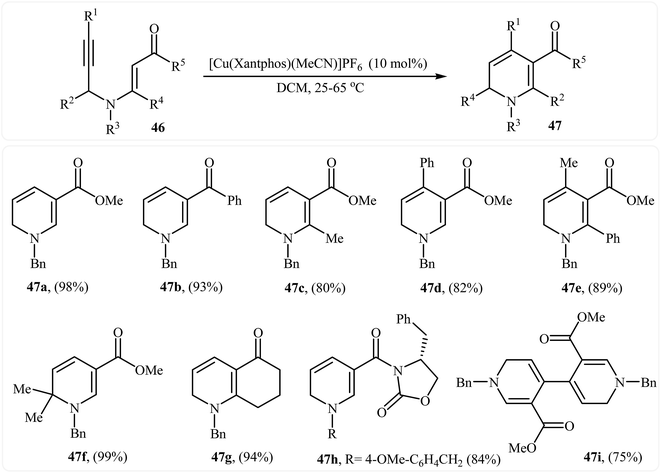 |
| | Scheme 21 Synthesis of highly substituted 1,2-dihydropyridines 47 through Cu(I)-catalyzed 6-endo cyclization of N-propargylic β-enaminocarbonyls 46. | |
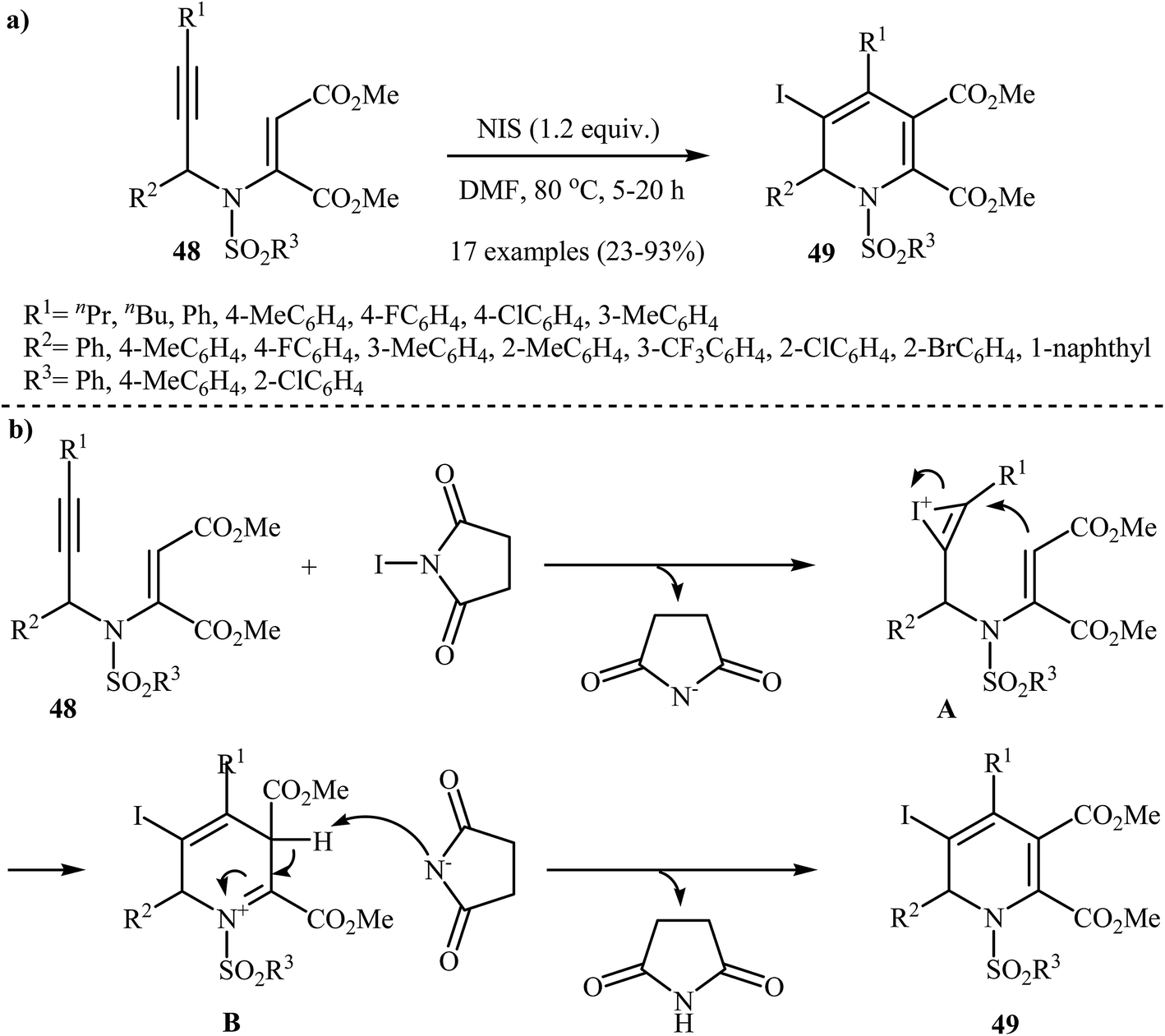
Recently, Wan and co-workers performed the iodocyclization of N-propargylic β-enaminoesters 48 with N-iodosuccinimide as electrophilic source and developed a route to obtain 3-iodo-1,2 dihydropyridines 49 in good yields (Scheme 22a). A plausible mechanism for these electrophilic cyclizations is shown in Scheme 22b, where intermediate A, generated by the attack of iodonium ion to the alkyne moiety, undergoes 6-endo-dig cyclization, and then deprotonation, to afford 3-iodo-1,2-dihydropyridine product 49.25
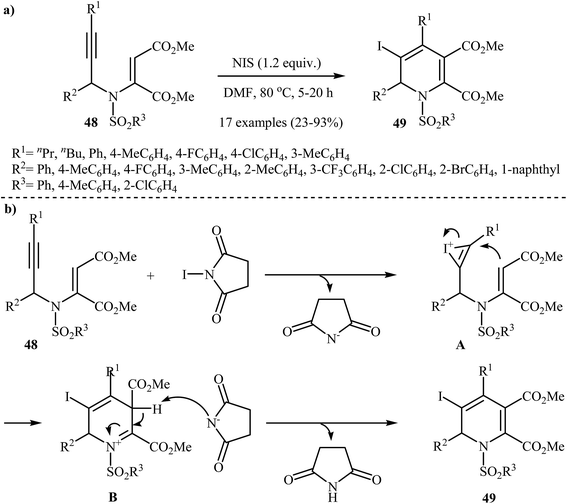 |
| | Scheme 22 (a) N-iodosuccinimide-induced electrophilic iodocyclization of N-propargylic β-enaminoesters 48 to 3-iodo-1,2-dihydropyridine product 49; (b) Proposed mechanism for formation of 49. | |
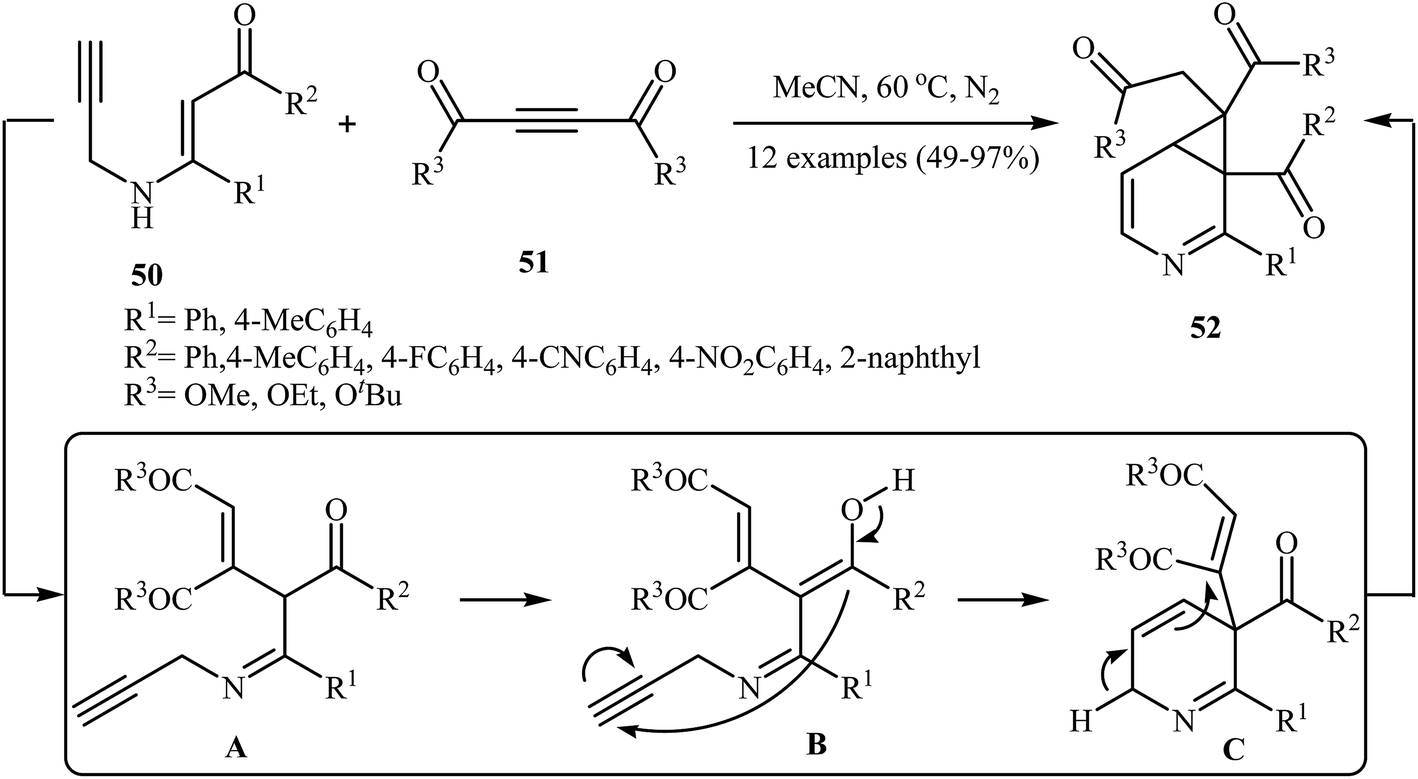
In 2014, Karunakar et al. synthesized 3-azabicyclo[4.1.0]hepta-2,4-dienes 52, a cyclopropane fused dihydropyridine system, by the catalyst- and base-free cascade reaction between internal N-propargylic β-enaminones 50 and acetylenedicarboxylates 51 (Scheme 23). Among the various solvents like DMF, THF, DCM, EtOH, EtOAc, CHCl3, MeCN, toluene, 1,4-dioxane, H2O; MeCN was the most efficient for this reaction. The results demonstrated that N-propargylic β-enaminones bearing electron-donating groups afforded higher yields in comparison to those bearing electron- withdrawing groups. Following is the decreasing order of reactivity of acetylenedicarboxylates in this reaction: diethyl but-2-ynedioate > di-tert-butyl but-2-ynedioate > dimethyl but-2-ynedioate.26
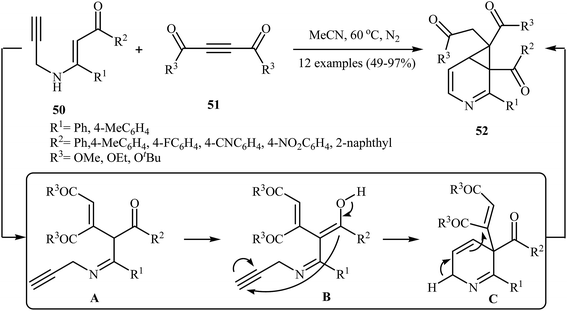 |
| | Scheme 23 Catalyst-free synthesis of 3-azabicyclo[4.1.0]hepta-2,4-dienes 52 reported by Karunakar. | |
6. 1-Azaanthraquinones
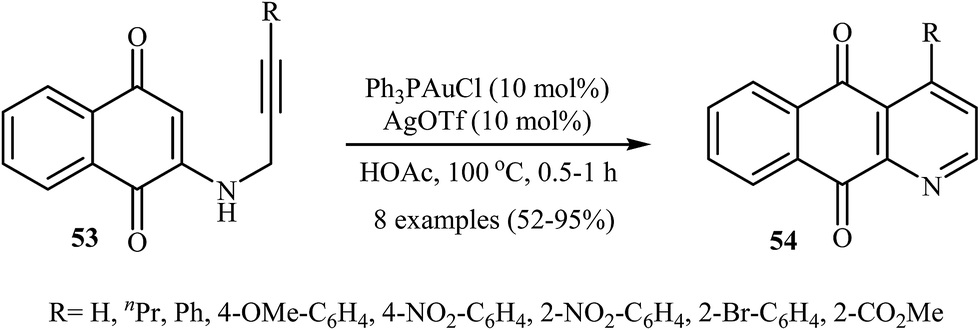
In 2010, the groups of Wang published an elegant and novel protocol for the synthesis of substituted azaanthraquinones using Au(I)-catalyzed 6-endo-dig cycloisomerization of N-propargylamino quinones 53.27 Thus, the optimized reactions revealed that the optimum condition for this cyclization reaction was the combination of Ph3PAuCl (10 mol%) and AgOTf (10 mol%) as catalytic system using acetic acid as the solvent, at 100 °C. Under optimized conditions, the reaction tolerates both terminal and internal alkynes and gave the products 54 in good yields (Scheme 24). This reaction proceeds through 6-endo-dig cyclization followed by a sequential tautomerization/aromatization. It is noted that the protocol was successfully applied for the synthesis of alkaloid cleistopholine and its analogues.
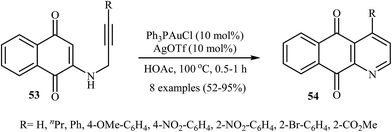 |
| | Scheme 24 Wang's synthesis of azaanthraquinones 54 from N-propargylamino quinones 53. | |
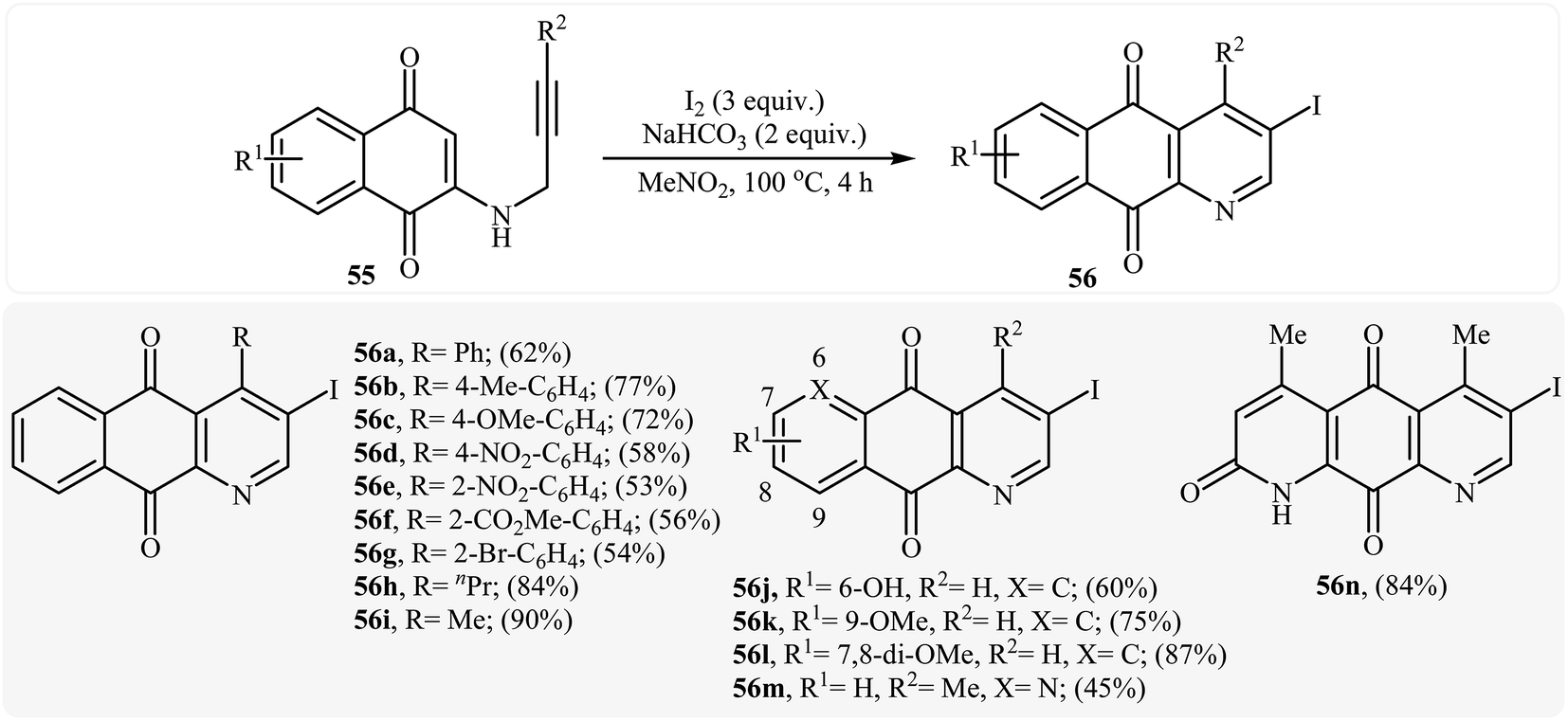
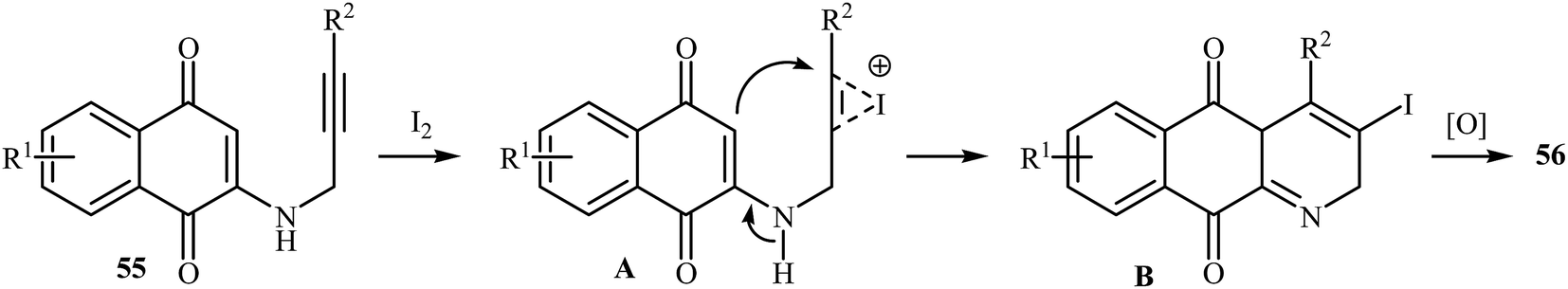
In a subsequent study, the same research team reported the preparation of a variety of 3-iodo-1-azaanthraquinones 56 in moderate to high yields via iodocyclization of N-propargylamino quinones 55 (Scheme 25). The reactions were carried out in nitromethane at 100 °C with 3 equiv of molecular iodine as the electrophilic source and NaHCO3 (2 equiv) as the base. The mechanism shown in Scheme 26 was proposed for this process. It consists of the following key steps: (1) initial formation of iodonium ion A via coordination of iodine with the C–C triple bond of alkyne; (2) nucleophilic attack by the double bond of the aminoquinone through 6-endo-dig manner to afford the intermediate B; and (3) oxidative aromatization of intermediate B to give the observed product 36.28 Soon after, the authors extended this chemistry to synthesis of 3-chloro-1-azaanthraquinones employing CuCl2 as electrophilic source.29
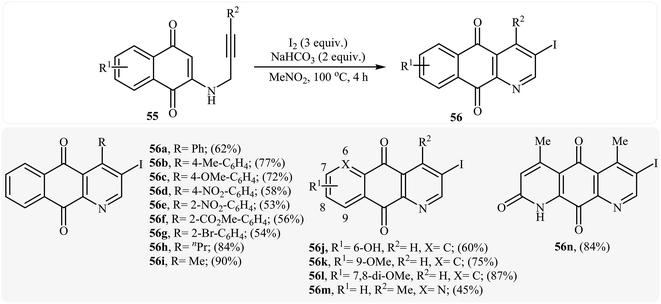 |
| | Scheme 25 Formation of 3-iodo-1-azaanthraquinones 56 by the iodocyclization of N-propargylamino quinones 55. | |
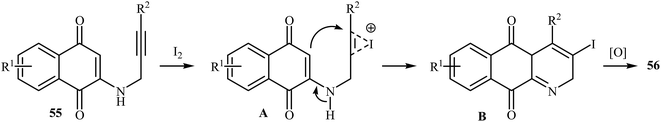 |
| | Scheme 26 Mechanism that accounts for the formation of 56. | |
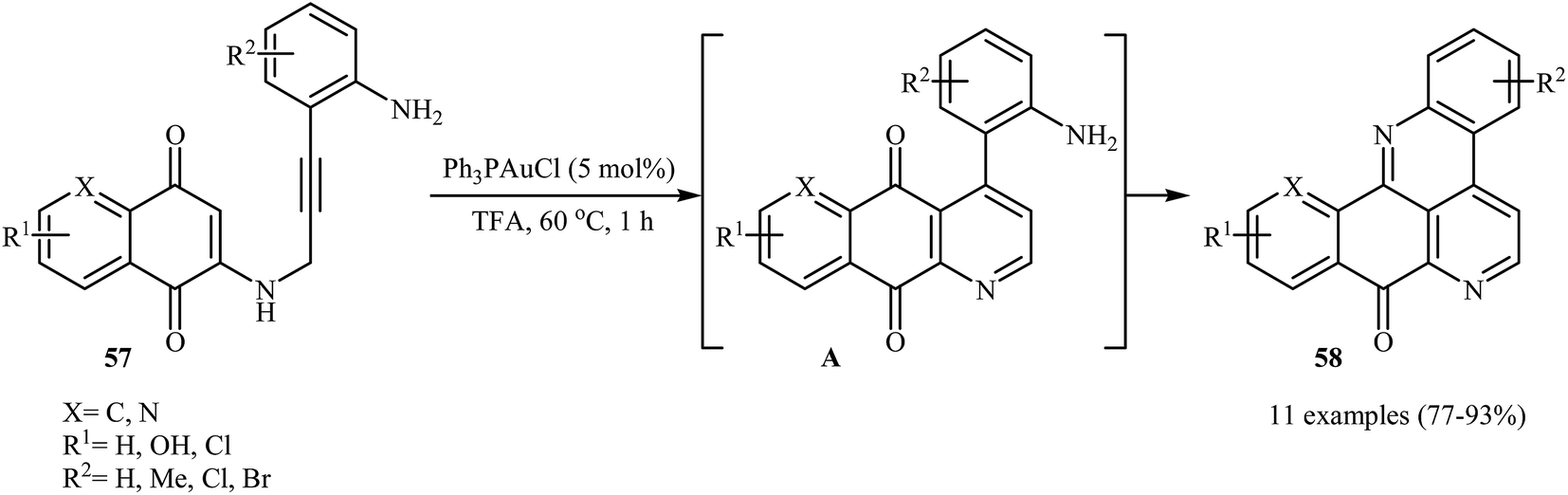
Follow these works, the authors reported the preparation of a variety of pentacyclic pyrido[4,3,2-mn]acridin-8-ones 58 in good to high yields via an Au(I)-catalyzed domino reaction of N-propargylaminoquinones 57 (Scheme 27). Among the various gold catalysts like AuCl, AuCl3, Ph3PAuCl, NaAuCl4˖H2O; Ph3PAuCl was the most efficient for the transformation. The results demonstrated that the substrates bearing electron-donating groups on the aniline ring afforded relatively higher yields in comparison to those bearing electron withdrawing groups.30
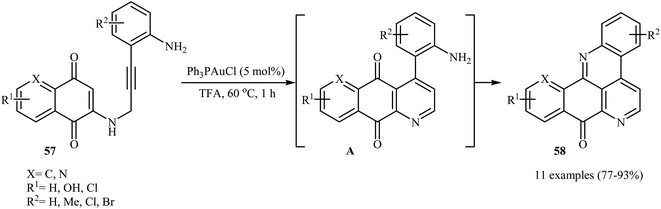 |
| | Scheme 27 Au(I)-catalyzed synthesis of pentacyclic pyrido[4,3,2-mn]acridin-8-ones 58 from N-propargylaminoquinones 57. | |
7. 1,4-Oxazepines
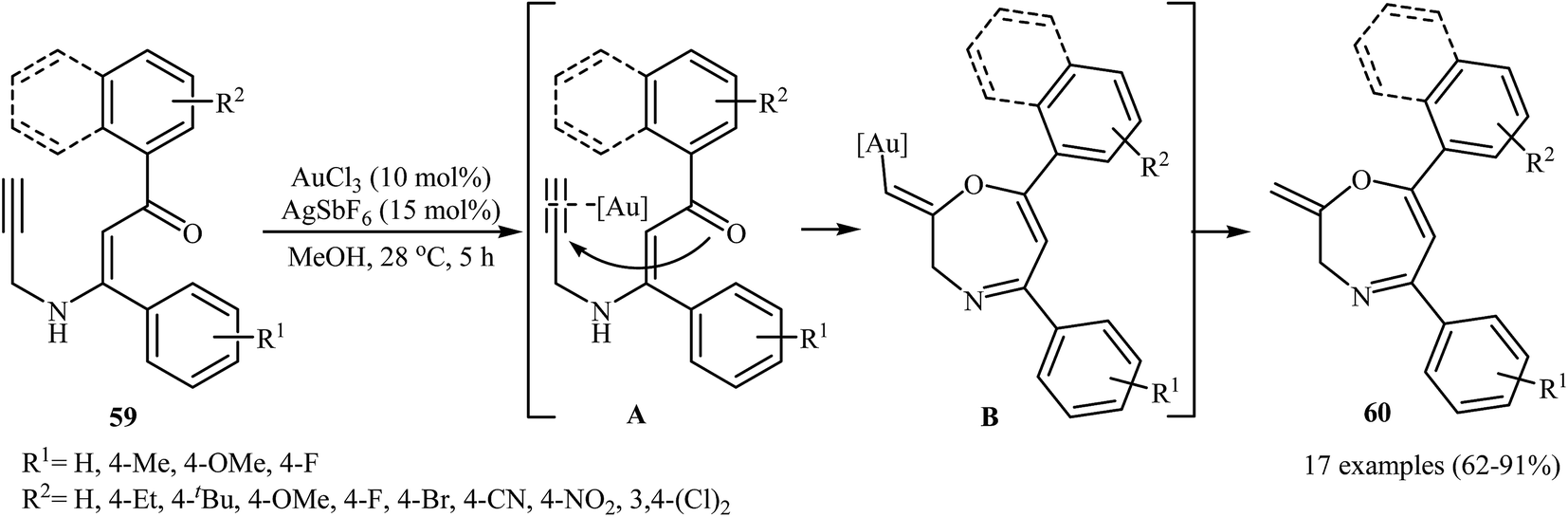
In 2015, the Karunakar group have developed a new and straightforward approach for the synthesis of 1,4-oxazepine derivatives 60 via a one-pot gold-catalyzed intramolecular cyclization of N-propargylic β-enaminones 59 (Scheme 28). Thus, the careful analysis of the optimized reactions revealed that the optimum condition for this cyclization was the addition of AuCl3 (10 mol%), and AgSbF6 (15 mol%), at 28 °C, to a solution of N-propargylic β-enaminones in MeOH. The reaction scope appears that the N-propargylic β-enaminones with electron-donating groups on the benzene rings are reactive than those with electron-poor aryl groups. According to mechanistic studies, it proceeds through the coordination of AuCl3 to the triple bond of 59, following the 7-exo-dig cyclization via nucleophilic attack of carbonyl oxygen onto the activated triple bond to give intermediate B, which undergoes protodemetalation to yield 1,4-oxazepine derivatives 60.31
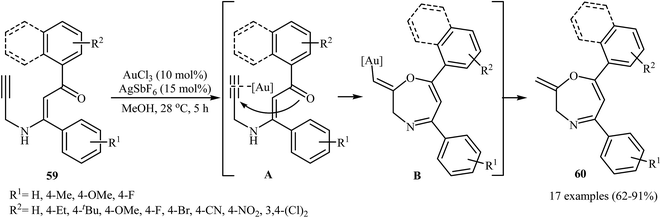 |
| | Scheme 28 Au-catalyzed intramolecular cyclization of N-propargylic β-enaminones 59 for the synthesis of 1,4-Oxazepine derivatives 60. | |
8. Azepines
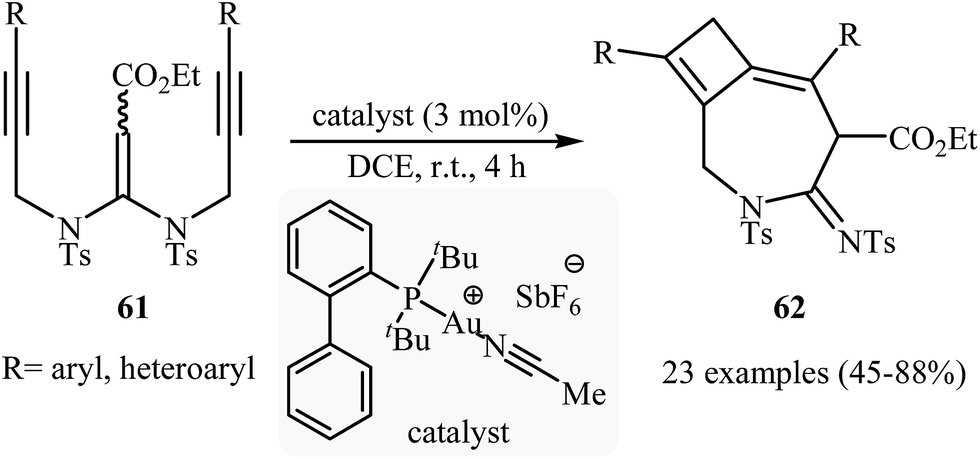
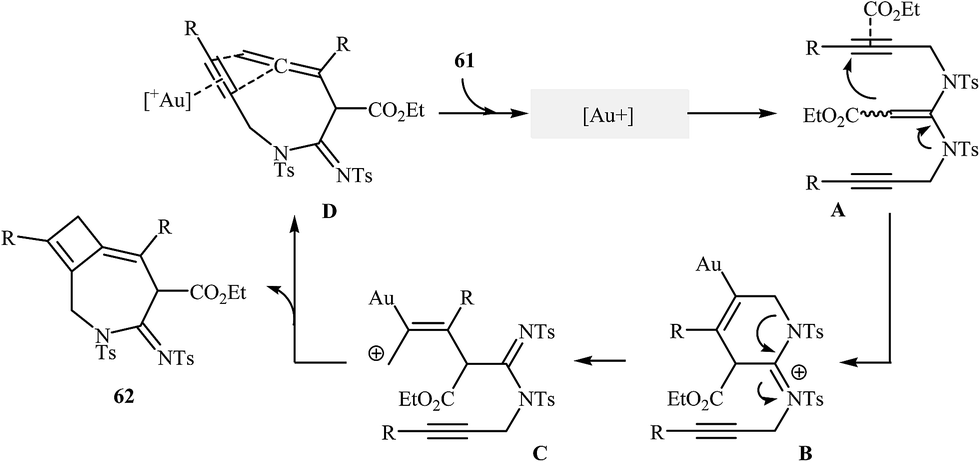
The reported methods for synthesis of azepine cores via intramolecular cyclization of N-propargylic β-enaminocarbonyls are very scarce and to the best of our awareness there is only one report for such reactions. More recently, Sahoo's research team reported a beautiful protocol for synthesis of hitherto unknown cyclobutene fused azepine heterocycles 62 by cycloisomerization of corresponding alkyne-tethered ketene N,N-acetals 61 in the presence of [JohnphosAu(I) (MeCN)]+SbF6- as catalyst (Scheme 29). This reaction was run in 1,2-dichloroethane at room temperature and tolerated various sensitive functional groups (including nitro, cyano, chloro, ketone, and ester), and in all cases provided fused azepines 2 in moderate to good yields.32 In this study, the authors also evaluated the mechanistic details of this electrophilic cyclization. To determine the advanced intermediates of the reaction, the authors monitored the reaction by 1H NMR spectroscopy. Among the data that could be obtained by 1H NMR spectroscopy, the most relevant for the proposal was those shown in Scheme 30.
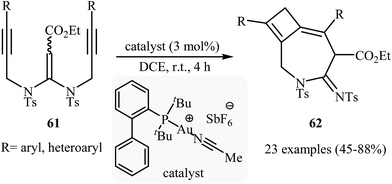 |
| | Scheme 29 Au-catalyzed intramolecular cyclization of alkyne-tethered ketene N,N-acetals 61 for the synthesis of cyclobutene fused azepine heterocycles 62. | |
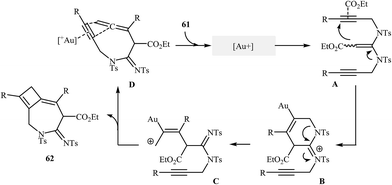 |
| | Scheme 30 Mechanistic proposal for the reactions in Scheme 18. | |
9. Summary and outlook
Synthesis of nitrogen-based heterocycles via cyclization of N-propargylic β-enaminocarbonyls, in both inter- and interamolecular manner, have witnessed rapid and comprehensive development in the past ten years. In comparison with many well-established traditional and modern methods, the preparation of N-heterocycles (such as pyrroles, pyrrolines, pyridines, piperidine as well as azaanthraquinones, and so forth) from titled compounds showed a wider tolerance to important functional groups. This made possible the further derivatization of the products. As illustrated, high atom economy and shorter synthetic routes are the key features of these reactions. Also interesting is the fact that most of the cyclization reactions covered in this review could be easily adapted to the gram-scale synthesis of N-heterocycles. The present methodologies are meaningful and particularly attractive for the fact that those N-heterocycles are the structural component of a vast number of biologically active natural and unnatural compounds. We believe that these salient features of N-propargylic β-enaminocarbonyls in the synthesis of N-heterocycles will further elicit widespread attention in the quest for more applications and utilities, serving as a powerful and versatile substrate in the synthesis of important N-heterocycles and complex natural products.
Acknowledgements
Payame Noor University is fully acknowledged for their financial support. The authors would like to thank Tabriz Branch, Islamic Azad University for the financial support of this research. The authors would like to thank Dr. Akram Hosseinian from Tehran University for their valuable comments and suggestions to improve the quality of the paper.
References
- Selected references:
(a) V. Bhardwaj, D. Gumber, V. Abbot, S. Dhiman and P. Sharma, RSC Adv., 2015, 5, 15233–15266 RSC;
(b) M. Baumann and I. R. Baxendale, Beilstein J. Org. Chem., 2013, 9, 2265–2319 CrossRef PubMed;
(c) T. Sravanthi and S. Manju, Eur. J. Pharm. Sci., 2016, 91, 1–10 CrossRef CAS PubMed;
(d) S. B. Ferreira and C. R. Kaiser, Expert Opin. Ther. Pat., 2012, 22, 1033–1051 CrossRef CAS PubMed;
(e) A. Marella, O. P. Tanwar, R. Saha, M. R. Ali, S. Srivastava, M. Akhter, M. Shaquiquzzaman and M. M. Alam, Saudi Pharm. J., 2013, 21, 1–12 CrossRef PubMed;
(f) L. Zhang, X. M. Peng, G. L. Damu, R. X. Geng and C. H. Zhou, Med. Res. Rev., 2014, 34, 340–437 CrossRef CAS PubMed;
(g) J. W. Blunt, B. R. Copp, R. A. Keyzers, M. H. Munro and M. R. Prinsep, Nat. Prod. Rep., 2014, 31, 160–258 RSC;
(h) D. O'Hagan, Nat. Prod. Rep., 2000, 17, 435–446 RSC;
(i) Z. Jin, Nat. Prod. Rep., 2016, 33, 1268–1317 RSC;
(j) M. Ishikura, T. Abe, T. Choshi and S. Hibino, Nat. Prod. Rep., 2013, 30, 694–752 RSC;
(k) G. K Gupta, V. Kumar and K. Kaur, Nat. Prod. J., 2014, 4, 73–81 Search PubMed;
(l) E. Vessally and M. Abdoli, J. Iran. Chem. Soc., 2016, 13, 1235–1256 CrossRef CAS;
(m) E. Vessally, H. Saeidian, A. Hosseinian, L. Edjlali and A. Bekhradnia, Curr. Org. Chem., 2017, 21, 249–271 CrossRef CAS;
(n) E. Vessally, A. Hassanpour, R. Hosseinzadeh-Khanmiri, M. Babazadeh and J. Abolhasani, Monatsh. Chem., 2017, 148, 321–326 CrossRef CAS;
(o) L. Dinparast, H. Valizadeh, E. Vessally and M. B. Bahadori, J. Mol. Struct., 2016, 1105, 118–127 CrossRef CAS;
(p) Z. Asadi, M. B. Asnaashariisfahani, E. Vessally and M. D. Esrafili, Spectrochim. Acta, Part A, 2015, 140, 585–599 CrossRef CAS PubMed;
(q) E. Vessally, E. Fereyduni, H. Shabrendi and M. D. Esrafili, Spectrochim. Acta, Part A, 2013, 116, 65–73 CrossRef CAS PubMed;
(r) E. Vessally, M. Ghasemisarabbadeih, Z. Ekhteyari, R. Hosseinzadeh-Khanmiri, E. Ghorbani-Kalhor and L. Ejlali, RSC Adv., 2016, 6, 106769–106777 RSC;
(s) E. Vessally, R. Hosseinzadeh-Khanmiri, M. Babazadeh, E. Ghorbani-Kalhor and L. Ejlali, Appl. Organomet. Chem., DOI:10.1002/aoc.3603;
(t) E. Vessally, R. Hosseinzadeh-Khanmiri, E. Ghorbani-Kalhor, M. Es'haghi and L. Ejlali, Appl. Organomet. Chem., DOI:10.1002/aoc.3729.
- Selected references:
(a) E. Vessally, RSC Adv., 2016, 6, 18619–18631 RSC;
(b) E. Vessally, A. Hosseinian, L. Edjlali, A. Bekhradnia and M. D. Esrafili, RSC Adv., 2016, 6, 71662–71675 RSC;
(c) E. Vessally, L. Edjlali, A. Hosseinian, A. Bekhradnia and M. D. Esrafili, RSC Adv., 2016, 6, 49730–49746 RSC;
(d) E. Vessally, A. Hosseinian, L. Edjlali, A. Bekhradnia and M. D. Esrafili, RSC Adv., 2016, 6, 99781–99793 RSC;
(e) E. Vessally, A. Hosseinian, L. Edjlali, A. Bekhradnia and M. D. Esrafili, Curr. Org. Synth., 2017, 4, 557–567 Search PubMed;
(f) E. Vessally, S. Soleimani-Amiri, A. Hosseinian, L. Edjlali and A. Bekhradnia, RSC Adv., 2017, 7, 7079–7091 RSC.
- S. Cacchi, G. Fabrizi and E. Filisti, Org. Lett., 2008, 10, 2629–2632 CrossRef CAS PubMed.
- K. Komeyama, M. Miyagi and K. Takaki, Chem. Lett., 2009, 38, 224–225 CrossRef CAS.
- A. Saito, T. Konishi and Y. Hanzawa, Org. Lett., 2010, 12, 372–374 CrossRef CAS PubMed.
- K. K. Toh, Y.-F. Wang, E. P. J. Ng and S. Chiba, J. Am. Chem. Soc., 2011, 133, 13942–13945 CrossRef CAS PubMed.
- Y. L. Zhao, C. H. Di, S. D. Liu, J. Meng and Q. Liu, Adv. Synth. Catal., 2012, 354, 3545–3550 CrossRef CAS.
- J. Weng, Y. Chen, B. Yue, M. Xu and H. Jin, Eur. J. Org. Chem., 2015, 3164–3170 CrossRef CAS.
- X. Yang, Y. Wang, F. Hu, X. Kan, C. Yang, J. Liu, P. Liu and Q. Zhang, RSC Adv., 2016, 6, 68454–68459 RSC.
- X. Xin, D. Wang, X. Li and B. Wan, Angew. Chem., Int. Ed., 2012, 51, 1693–1697 CrossRef CAS PubMed.
- Y. Zhao, H. Wang, X. Li, D. Wang, X. Xin and B. Wan, Org. Biomol. Chem., 2016, 14, 526–541 CAS.
- X. Xin, H. Wang, X. Li, D. Wang and B. Wan, Org. Lett., 2015, 17, 3944–3947 CrossRef CAS PubMed.
- J. Shen, X. Yang, F. Wang, Y. Wang, G. Cheng and X. Cui, RSC Adv., 2016, 6, 48905–48909 RSC.
- K. Goutham, N. R. Mangina, S. Suresh, P. Raghavaiah and G. V. Karunakar, Org. Biomol. Chem., 2014, 12, 2869–2873 CAS.
- H. Mizoguchi, H. Oikawa and H. Oguri, Nat. Chem., 2014, 6, 57–64 CrossRef CAS PubMed.
- X. Xin, D. Wang, X. Li and B. Wan, Tetrahedron, 2013, 69, 10245–10248 CrossRef CAS.
- S. Karabiyikoglu, Y. Kelgokmen and M. Zora, Tetrahedron, 2015, 71, 4324–4333 CrossRef CAS.
- E. Karadeniz, M. Zora and N. Z. Kılıçaslan, Tetrahedron, 2015, 71, 8943–8952 CrossRef CAS.
- Y. Kelgokmen and M. Zora, RSC Adv., 2016, 6, 4608–4621 RSC.
- G. Cheng, Y. Weng, X. Yang and X. Cui, Org. Lett., 2015, 17, 3790–3793 CrossRef CAS PubMed.
- H. Kim and C. Lee, J. Am. Chem. Soc., 2006, 128, 6336–6337 CrossRef CAS PubMed.
- F. J. Fananás, T. Arto, A. Mendoza and F. Rodriguez, Org. Lett., 2011, 13, 4184–4187 CrossRef PubMed.
- M. A. Martins, M. Rossatto, C. P. Frizzo, E. Scapin, L. Buriol, N. Zanatta and H. G. Bonacorso, Tetrahedron Lett., 2013, 54, 847–849 CrossRef CAS.
- H. Mizoguchi, R. Watanabe, S. Minami, H. Oikawa and H. Oguri, Org. Biomol. Chem., 2015, 13, 5955–5963 CAS.
- X. Xin, D. Wang, F. Wu, X. Li and B. Wan, J. Org. Chem., 2013, 78, 4065–4074 CrossRef CAS PubMed.
- K. Goutham, V. Nagaraju, S. Suresh, P. Raghavaiah and G. V. Karunakar, RSC Adv., 2014, 4, 21054–21059 RSC.
- C. Jiang, M. Xu, S. Wang, H. Wang and Z.-J. Yao, J. Org. Chem., 2010, 75, 4323–4325 CrossRef CAS PubMed.
- N. Fei, Q. Hou, S. Wang, H. Wang and Z.-J. Yao, Org. Biomol. Chem., 2010, 8, 4096–4103 CAS.
- N. Fei, H. Yin, S. Wang, H. Wang and Z.-J. Yao, Org. Lett., 2011, 13, 4208–4211 CrossRef CAS PubMed.
- H. Yin, F. Kong, S. Wang and Z.-J. Yao, Tetrahedron Lett., 2012, 53, 7078–7082 CrossRef CAS.
- K. Goutham, D. Ashok Kumar, S. Suresh, B. Sridhar, R. Narender and G. V. Karunakar, J. Org. Chem., 2015, 80, 11162–11168 CrossRef CAS PubMed.
- S. Nayak, N. Ghosh and A. K. Sahoo, Org. Lett., 2014, 16, 2996–2999 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *a,
Ladan Edjlali
*b,
Ebrahim Ghorbani-Kalhor
b and
Rahim Hosseinzadeh-Khanmiri
b
*a,
Ladan Edjlali
*b,
Ebrahim Ghorbani-Kalhor
b and
Rahim Hosseinzadeh-Khanmiri
b