
Tetraphenylfuran: aggregation-induced emission or aggregation-caused quenching?†
Han
Nie‡
a,
Kun
Hu‡
a,
Yuanjing
Cai
a,
Qian
Peng
b,
Zujin
Zhao
a,
Rongrong
Hu
a,
Junwu
Chen
a,
Shi-Jian
Su
a,
Anjun
Qin
 *a and
Ben Zhong
Tang
*ac
*a and
Ben Zhong
Tang
*ac
aGuangdong Innovative Research Team, State Key Laboratory of Luminescent Materials and Devices, South China University of Technology, Guangzhou 510640, China. E-mail: msqinaj@scut.edu.cn
bKey Laboratory of Organic Solids, Beijing National Laboratory for Molecular Science, Institute of Chemistry, Chinese Academy of Sciences, Beijing 100190, China
cDepartment of Chemistry, Hong Kong Branch of Chinese National Engineering Research Center for Tissue Restoration and Reconstruction, The Hong Kong University of Science & Technology, Clear Water Bay, Kowloon, Hong Kong, China. E-mail: tangbenz@ust.hk
First published on 28th December 2016
Abstract
Tetraphenylfuran (TPF) and its control molecule tetraphenylthiophene (TPT), which are structurally similar to the aggregation-induced emission (AIE) active 2,3,4,5-tetraphenylsilole, were synthesized. Surprisingly, investigation of its photo-physical properties showed that TPF exhibits the aggregation-caused quenching effect instead of AIE characteristics, whereas TPT exhibits a quite weak AIE effect. Combining experimental results and theoretical calculations, this phenomenon was concluded to be co-caused by the restriction of intramolecular rotation (RIR), the mechanism of AIE, and the conjugation effect. Thus, this work provides an insight into RIR, which will greatly promote the development of AIE.
Introduction
The immense interest in organic fluorophores is driven mostly by their tremendous utility in the fields of organic light-emitting diodes, organic light-emitting transistors, organic lasers, chemosensors and bioprobes, etc.1 Because these fluorophores are practically used in their condensed phases, such as aggregates or solid films, it is the properties of the molecular aggregates that ultimately dominate the performance of devices in which they are used as active layers. However, most conventional organic fluorophores are highly emissive in dilute solutions, but become weakly luminescent or even non-emissive in the aggregate state due to the aggregation-caused quenching (ACQ) effect.2 This ACQ problem has limited the practical application of organic fluorophores to some extent.Delightfully, some novel luminogens display the opposite effect: they are non- or weakly emissive in dilute solutions but exhibit bright luminescence upon aggregation.3 This unique phenomenon is coined as aggregation-induced emission (AIE).4 During the past decade, systematic studies have proposed that the AIE effect is mainly caused by restriction of intramolecular motion (RIM), including rotation (RIR), vibration (RIV), etc.5 In the aggregate state, the intramolecular rotation and/or vibration in a luminogen is greatly restricted by natural physical constraints or interactions from the surrounding molecules, which suppresses the non-radiative decay channels and results in high luminescence. The AIE effect has become a very effective strategy to address the ACQ problem, and to construct efficient solid-state emitters for applications in high performance optoelectronic devices, and excellent chemo- and bio-sensors.6 Because of its great significance in fundamental research and technological applications, the study of AIE is a frontier research area in the field of chemistry and materials science nowadays.7
Attracted by the prospects of AIE, researchers worldwide are enthusiastically engaged in this area and are working on the development of new AIE luminogens (AIEgens), deciphering the underlying mechanism and exploring their practical applications. Based on the RIR, a series of AIEgens have been developed, among which, representative examples are (hetero)cyclopentadiene derivatives, such as 1,2,3,4-tetraphenyl-1,3-cyclopentadiene (TPC),8 1,1-dimethyl-2,3,4,5-tetraphenylsilole (TPS),9 1,2,3,4,5-pentaphenylpyrrole (PPP),10 1,2,3,4,5-pentaphenylphosphole oxide (PPPO),11 and tetraphenylthiophene (TPT)12 (Chart 1).
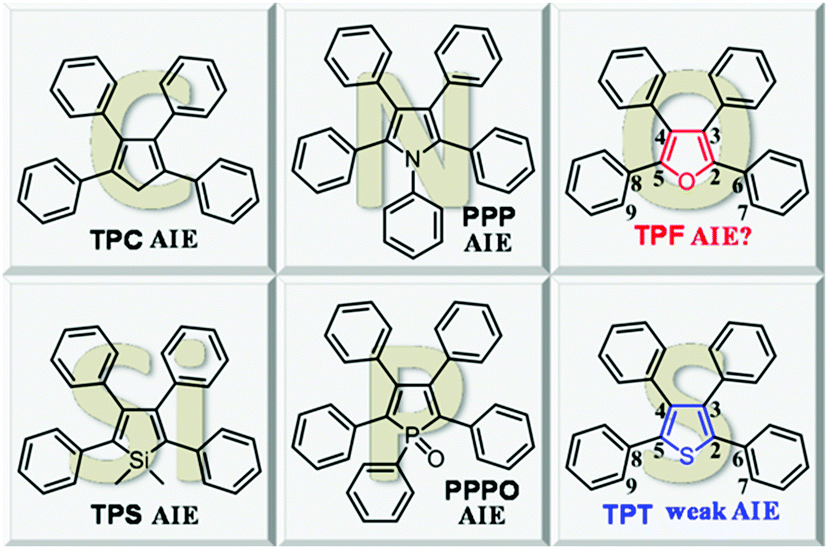 | ||
| Chart 1 Chemical structures of (hetero)cyclopentadiene derivatives. | ||
By the detailed survey of the aforementioned propeller-shaped structures, we could find that the central (hetero)cyclic rings are connected with at least four freely rotary phenyl rings. Moreover, besides the carbon atom at the 1-position of TPC, the Si, P, N, and S-containing compounds are also AIE-active although TPT displays a weak AIE effect. Given the similar structures with the aforementioned AIEgens, we envisioned that if the carbon atom of TPC were replaced with O, the resultant compound, i.e. tetraphenylfuran (TPF) would also be AIE-active. With this hypothesis in mind, in this work, we studied the photo-physical properties of TPF, and conducted a comparative study with its analogue of TPT. The unexpected ACQ effect of TPF confused us, since its structure was found to be very similar to that of TPT. Detailed theoretical calculations revealed that the RIR and molecular conjugation play a collective role in determining this interesting photo-physical behavior, which provides a deep insight into the RIR mechanism of AIE.
Results and discussion
Synthesis and photo-physical properties
TPF was synthesized by Pd-catalyzed cyclization of diphenylacetylene in the presence of oxygen,13 and, for comparison, TPT was also prepared by the Suzuki coupling of tetrabromothiophene and phenylboronic acid (Scheme S1, see the ESI† for detailed synthetic processes). Their chemical structures were characterized by 1H and 13C NMR, elemental analysis, mass spectroscopy and single-crystal X-ray diffraction analysis, and satisfactory results corresponding to their structures were obtained (Fig. S1–S5, ESI†).After confirming their structures, we investigated their photo-physical properties in their solution and aggregate states. Firstly, we measured their UV-vis spectra (Fig. S6A, ESI†). The results showed that TPF in THF solution exhibits a redder maximum absorption (327 nm) and a higher molar absorptivity (ε, 2.18 × 104 M−1 cm−1) than TPT solution (313 nm, 1.28 × 104 M−1 cm−1), which demonstrates that TPF possesses better conjugation than TPT. In addition, as shown in Fig. S6 (ESI†) the Stokes shift value of TPF in solution (4471 cm−1) is significantly smaller than that of TPT (7135 cm−1), indicative of the greater rigidity of TPF.14
The photoluminescence (PL) measurement, however, showed unexpected phenomena. TPF is highly emissive in THF solution at 383 nm (Fig. S6B, ESI†) with a fluorescence quantum yield (ΦF) of 39.9% (Fig. 1). However, this value was greatly decreased to 0.6% in a THF/water mixture with a water fraction (fw) of 99%, displaying a typical ACQ effect. TPT behaved differently. Its emission peaked at 403 nm in both THF solution and the aggregate state, though the emission was quite weak with a ΦF value of ca. 3%, and a slight increase in the ΦF value was recorded in THF/water mixtures with fw values of 80 and 85%, suggesting that TPT is a weak AIEgen (Fig. 1 and Fig. S7, ESI†).12 This discrepancy piqued our interest and made us wonder whether the RIR mechanism is still operative in TPF, and what is the cause of the observed ACQ effect and the high degree of dissimilarity between TPF and TPT. We thus performed theoretical calculations to gain insight into this “abnormal” phenomenon.
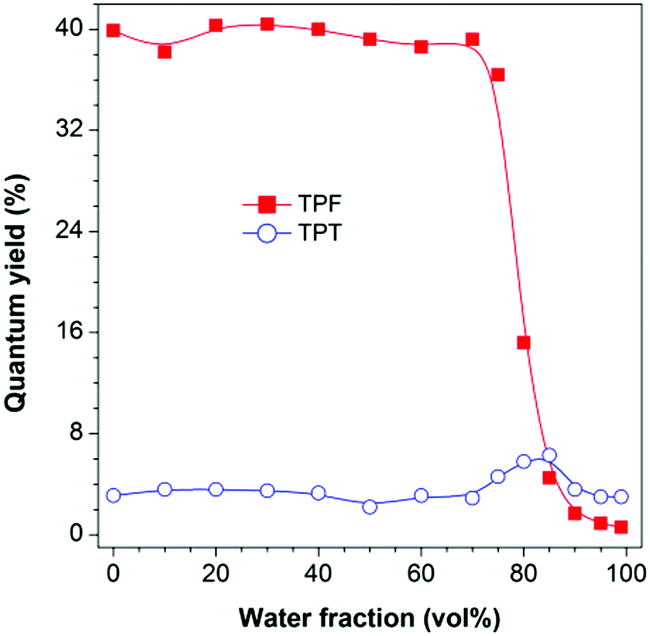 | ||
| Fig. 1 Plots of PL quantum yield versus water fraction in THF/water mixtures. Concentration: 10 μM; excitation wavelengths: 327 nm (TPF) and 320 nm (TPT). | ||
Electronic and geometric structures
The calculated absorption and emission peaks of TPF and TPT by means of the time-dependent density functional theory (TDDFT) method and the B3LYP/6-31G(d,p) level agree well with the experimental data (Table S1, ESI†), suggesting the reliability of the adopted calculation method. According to the results, the first singlet excited state (S1) is dominated by the transition between the HOMO and the LUMO for both TPF and TPT, which represents the transition of π to π* (Fig. 2). Generally, spin–orbit coupling (SOC) is very small for the π → π* electron transition, so the intersystem crossing (ISC) process from S1 to the first triplet excited state (T1) can be reasonably neglected in these two molecules.15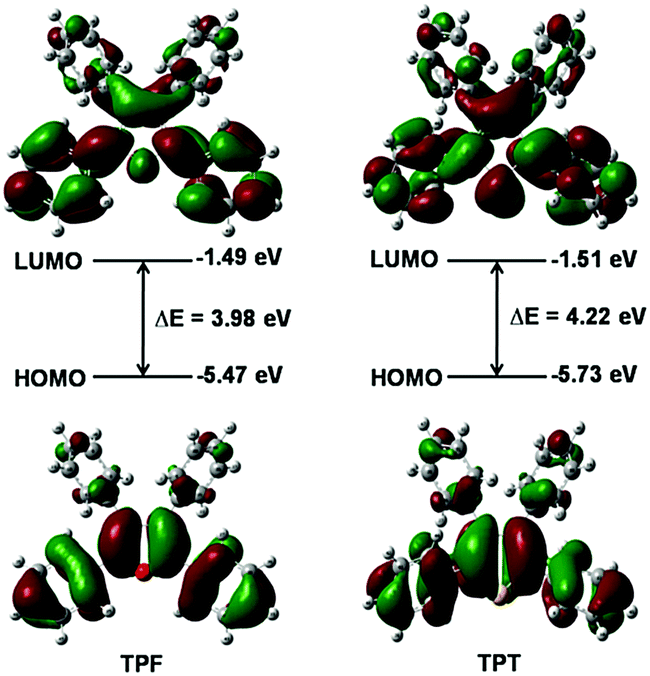 | ||
| Fig. 2 Calculated electronic density contours and energy levels of HOMOs and LUMOs for TPF and TPT. | ||
Moreover, it could be seen that the HOMOs and LUMOs are mainly localized on the central heterocyclic rings and the phenyl rings at the 2,5-positions of TPF and TPT with HOMO energy levels of −5.47 and −5.73 eV, respectively (Fig. 2), indicating that TPF shows better conjugation than TPT.16,17 Hence, it is clear that the central heterocyclic rings and the two phenyls will affect their photo-physical properties.
To unveil the underlying mechanism, we conducted a detailed survey on the optimized molecular structures. The relevant structural parameters of TPF and TPT are listed in Table 1. The bond lengths of O–C2, O–C5, C2–C6, and C5–C8 in TPF are much shorter than the corresponding ones in TPT both in the ground (S0) and excited (S1) states. Furthermore, the bond angles of the furan ring are more homogenized than those in the thiophene rings. In comparison with the relevant dihedral angles of TPT, the dihedral angles O–C2–C6–C7 and O–C5–C8–C9 of TPF are significantly smaller both in S0 and S1. It is worth noting that the geometrical modifications of the two dihedral angles (X–C2–C6–C7 and X–C5–C8–C9) between S0 and S1 are both 20.2° in TPT, while the corresponding values are less than 13.0° in TPF. These results suggest that TPF is more rigid due to its better conjugation and hence the rotation of the phenyl rings becomes more difficult than in TPT, which makes TPF highly emissive in solution.
| TPF | TPT | |||
|---|---|---|---|---|
| S0 | S1 | S0 | S1 | |
| TPF: X = O; TPT: X = S. | ||||
| X–C2 | 1.369 | 1.386 | 1.746 | 1.782 |
| X–C5 | 1.369 | 1.384 | 1.746 | 1.782 |
| C2–C6 | 1.463 | 1.431 | 1.476 | 1.433 |
| C5–C8 | 1.463 | 1.418 | 1.476 | 1.433 |
| C2–C3 | 1.379 | 1.409 | 1.384 | 1.431 |
| C4–C5 | 1.379 | 1.436 | 1.384 | 1.431 |
| C3–C4 | 1.447 | 1.418 | 1.448 | 1.413 |
| C5–X–C2 | 108.8 | 107.9 | 92.6 | 91.1 |
| X–C2–C3 | 109.2 | 109.3 | 110.7 | 111.1 |
| C2–C3–C4 | 106.4 | 107.5 | 112.9 | 113.2 |
| X–C2–C6–C7 | 22.8 | 9.8 | 42.8 | 22.6 |
| X–C5–C8–C9 | 22.8 | 15.0 | 42.8 | 22.6 |
Radiative and nonradiative decay channels
It is well known that PL is directly related to the radiative and nonradiative decay of the excited state of a molecule. Their rates (kr and knr) are expressed in eqn (1) and (2):15| ΦF = kr/(kr + knr) | (1) |
| 〈τ〉 = 1/(kr + knr) | (2) |
As aforementioned, the ISC process from S1 to T1 could be neglected in TPF and TPT; thus, the internal conversion (IC) process from S1 to S0 is regarded as the main nonradiative decay pathway in their THF solutions. According to the approximate form of the IC rate (kIC), the adiabatic excitation energy (ΔEad) and the reorganization energy (λ) are two important factors to determine the kIC.17 The ΔEad values of TPF and TPT were calculated to be 3.41 and 3.36 eV, respectively. Since these values are quite close, λ is the key factor that dominates the kIC. A larger λ will lead to a higher kIC value and consequently a smaller ΦF.17
On the basis of the normal mode method using the DUSHIN program,17 the total reorganization energy of S1 for TPT was calculated to be 4107 cm−1, whereas the value of only 2599 cm−1 was obtained for TPF, which agrees well with the trend of their Stokes shifts in experiments. This result proves that the reorganization energies of TPF and TPT greatly affect their ΦF values in solution. From the plots of the reorganization energies versus normal mode frequencies of TPT and TPF (Fig. 3), we could find that the smaller total reorganization energy of the TPF molecule is largely caused by the restriction of low frequency motion modes, which is assigned to the rotations of the peripheral phenyl rings. To clearly demonstrate the relationship between the IC process and the molecular structure, we projected the reorganization energy into the geometry relaxation in the internal coordinates. As illustrated in the inset of Fig. 3, the contribution of the rotations of the phenyl rings at the 2,5-positions of TPF to the total reorganization energy (607 cm−1) is remarkably reduced compared to that of TPT (2012 cm−1), although other motion modes for TPF and TPT are quite similar. So we could conclude that the RIR of the phenyl rings at the 2,5-positions of TPF mainly accounts for the higher ΦF in solution.
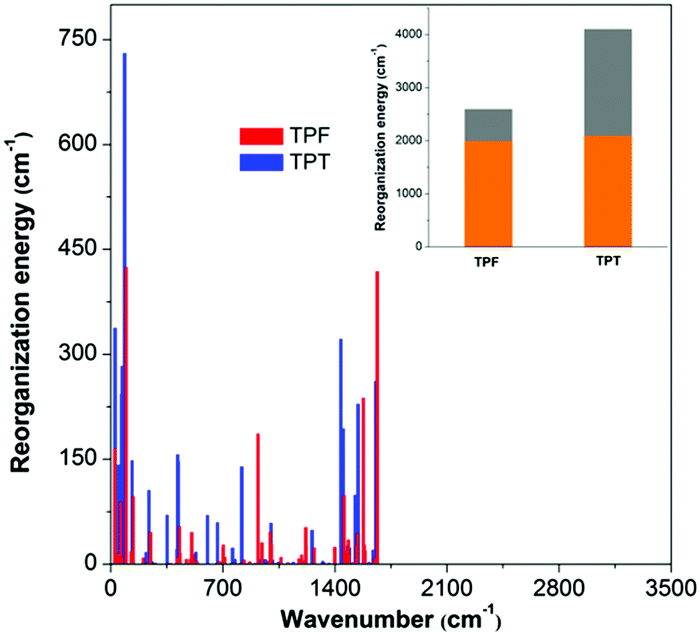 | ||
| Fig. 3 The calculated reorganization energies versus the normal mode frequencies for isolated TPT and TPF molecules. Inset: The gray bar represents the contribution of the rotation motions of the phenyl groups at the 2,5-positions to the total reorganization energy; the orange one represents the contribution of the other motion modes. | ||
Single crystal packing
The single crystals of TPF (CCDC: 1494293) and TPT (CCDC: 1494294) were successfully obtained during our experiments, which enabled us to directly investigate the effect of molecular packing on their emission in the aggregate/solid states (Fig. 4). The packing mode of TPF in its crystal showed that the distance of the two adjacent oxygen atoms is between 4.155 Å and 4.413 Å. Textbook knowledge tells us that the oxygen atom in the furan ring has two lone pairs, one is perpendicular to the furan ring and the other is coplanar with the furan ring. According to a report, the length of the perpendicular lone pair is 1.804 Å.18 So there are probably intensive intermolecular n → π* interactions in TPF aggregates, which possibly could suppress the radiative process, prompt the nonradiative channel, and efficiently quench the emission in its aggregate state (Table S3, ESI†).19 In contrast, the TPT crystal displays a relatively loose packing with weak intermolecular interactions and the phenyl rings could still rotate similarly to that in the solution state. Thus, the radiative and nonradiative decay rates are slightly changed, which leads to the similar small ΦF in both the solution and aggregate states.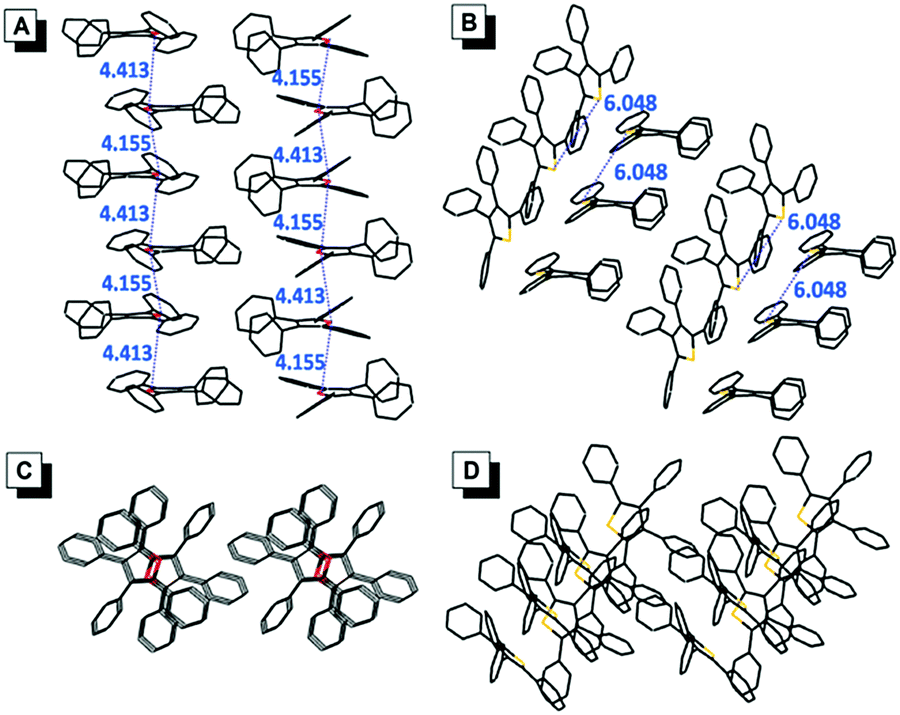 | ||
| Fig. 4 Side views of the packing arrangements in crystals of (A) TPF and (B) TPT. Top views of the packing arrangements in crystals of (C) TPF and (D) TPT. The distances are in the unit of Å. | ||
Conclusion
In summary, according to the structural similarity with silole and the RIR mechanism of AIE, we synthesized a fluorophore of TPF. For comparison, TPT was also prepared. Surprisingly, the photo-physical investigation showed that TPF is an ACQ fluorophore instead of an AIEgen, whereas TPT exhibited weak AIE-activity with low ΦF values both in the solution and aggregate states. Based on theoretical calculations and single crystal investigation, we conclude that the better conjugation in TPF readily enhances its radiative decay rate and blocks its nonradiative channel via the RIR of the phenyl rings at its 2,5-postions, which provides TPF with a much higher ΦF in solution. However, the conjugated planar skeleton of TPF easily forms intermolecular interactions in the aggregate state through n → π* interactions, conversely quenching its emission. Thus, this work provides direct evidence to support the RIR mechanism of AIE. At the same time, it suggests that the photo-physical properties can be fine-tuned dramatically by simply changing the molecular structure.3Acknowledgements
This work was financially supported by the key project of the Ministry of Science and Technology of China (2013CB834702), the National Natural Science Foundation of China (21525417); the Natural Science Foundation of Guangdong Province (2016A030312002); The National Program for Support of Top-Notch Young Professionals; the Fundamental Research Funds for the Central Universities (2015ZY013) and the Innovation and Technology Commission of Hong Kong (ITC-CNERC14S01). A. J. Q. and B. Z. T. are thankful for the support from Guangdong Innovative Research Team Program (201101C0105067115).Notes and references
- (a) X. Y. Yan, T. R. Cook, P. Wang, F. H. Huang and P. J. Stang, Nat. Chem., 2015, 7, 342 CrossRef CAS PubMed; (b) J.-H. Jou, S. Kumar, A. Agrawal, T.-H. Li and S. Sahoo, J. Mater. Chem. C, 2015, 3, 2974 RSC; (c) J. Liu, H. Zhang, H. Dong, L. Meng, L. Jiang, L. Jiang, Y. Wang, J. Yu, Y. Sun, W. Hu and A. J. Heeger, Nat. Commun., 2015, 6, 10032 CrossRef CAS PubMed; (d) I. D. W. Samuel and G. A. Turnbull, Chem. Rev., 2007, 107, 1272 CrossRef CAS PubMed; (e) Y. Yang, Q. Zhao, W. Feng and F. Li, Chem. Rev., 2013, 113, 192 CrossRef CAS PubMed; (f) T. D. Ashton, K. A. Jolliffeb and F. M. Pfeffer, Chem. Soc. Rev., 2015, 44, 4547 RSC; (g) K. Li and B. Liu, Chem. Soc. Rev., 2014, 43, 6570 RSC; (h) M. Shimizu and T. Hiyama, Chem. – Asian J., 2010, 5, 1516 CrossRef CAS PubMed.
- (a) J. B. Birks, Photophysics of Aromatic Molecules, Wiley, London, 1970 Search PubMed; (b) S. A. Jenekhe and J. A. Osaheni, Science, 1994, 265, 765 CAS.
- (a) D. Oelkrug, A. Tompert, H.-J. Egelhaaf, M. Hanack, E. Steinhuber, M. Hohloch, H. Meier and U. Stalmach, Synth. Met., 1996, 83, 231 CrossRef CAS; (b) D. Oelkrug, A. Tompert, J. Gierschner, H.-J. Egelhaaf, M. Hanack, M. Hohloch and E. Steinhuber, J. Phys. Chem. B, 1998, 102, 1902 CrossRef CAS.
- (a) J. Luo, Z. Xie, J. W. Y. Lam, L. Cheng, H. Chen, C. Qiu, H. S. Kwok, X. Zhan, Y. Liu, D. Zhu and B. Z. Tang, Chem. Commun., 2001, 1740 RSC; (b) Y. Dong, J. W. Y. Lam, A. Qin, J. Liu, Z. Li, B. Z. Tang, J. Sun and H. S. Kwok, Appl. Phys. Lett., 2007, 91, 011111 CrossRef; (c) J. He, B. Xu, F. Chen, H. Xia, K. Li, L. Ye and W. Tian, J. Phys. Chem. C, 2009, 113, 9892 CrossRef CAS; (d) N. L. Leung, N. Xie, W. Yuan, Y. Liu, Q. Wu, Q. Peng, Q. Miao, J. W. Y. Lam and B. Z. Tang, Chem. – Eur. J., 2014, 20, 15349 CrossRef CAS PubMed; (e) F. Bu, R. Duan, Y. Xie, Y. Yi, Q. Peng, R. Hu, A. Qin, Z. Zhao and B. Z. Tang, Angew. Chem., Int. Ed., 2015, 54, 14492 CrossRef CAS PubMed; (f) M. Chen, L. Li, H. Nie, J. Tong, L. Yan, B. Xu, J. Z. Sun, W. Tian, Z. Zhao, A. Qin and B. Z. Tang, Chem. Sci., 2015, 6, 1932 RSC; (g) Z. T. Wang, Y. Fang, J. Z. Sun, A. J. Qin and B. Z. Tang, Sci. China: Chem., 2013, 56, 1187 CrossRef CAS.
- (a) J. Mei, Y. Hong, J. W. Y. Lam, A. Qin, Y. Tang and B. Z. Tang, Adv. Mater., 2014, 26, 5429 CrossRef CAS PubMed; (b) J. Chen, C. C. W. Law, J. W. Y. Lam, Y. Dong, S. M. F. Lo, I. D. Williams, D. Zhu and B. Z. Tang, Chem. Mater., 2003, 15, 1535 CrossRef CAS; (c) S. L. Deng, T. L. Chen, W. L. Chien and J. L. Hong, J. Mater. Chem. C, 2014, 2, 651 RSC; (d) Y. J. Jin, H. Kim, J. J. Kim, N. H. Heo, J. W. Shin, M. Teraguchi, T. Kaneko, T. Aoki and G. Kwak, Cryst. Growth Des., 2016, 16, 2804 CrossRef CAS; (e) Z. Y. Yang, W. Qin, N. L. C. Leung, M. Arseneault, J. W. Y. Lam, G. D. Liang, H. H. Y. Sung, I. D. Williams and B. Z. Tang, J. Mater. Chem. C, 2016, 4, 99 RSC; (f) G. F. Zhang, Z. Q. Chen, M. P. Aldred, Z. Hu, T. Chen, Z. L. Huang, X. G. Meng and M. Q. Zhu, Chem. Commun., 2014, 50, 12058 RSC; (g) A. J. Qin, J. W. Y. Lam, F. Mahtab, C. K. W. Jim, L. Tang, J. Z. Sun, H. H. Y. Sung, I. D. Williams and B. Z. Tang, Appl. Phys. Lett., 2009, 94, 253308 CrossRef; (h) B.-K. An, S.-K. Kwon, S.-D. Jung and S. Y. Park, J. Am. Chem. Soc., 2002, 124, 14410 CrossRef CAS PubMed.
- (a) Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361 RSC; (b) J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718 CrossRef CAS PubMed; (c) X. Z. Yan, H. Z. Wang, C. E. Hauke, T. R. Cook, M. Wang, M. L. Saha, Z. X. Zhou, M. M. Zhang, X. P. Li, F. H. Huang and P. J. Stang, J. Am. Chem. Soc., 2015, 137, 15276 CrossRef CAS PubMed; (d) X. Z. Yan, M. Wang, T. R. Cook, M. M. Zhang, M. L. Saha, Z. X. Zhou, X. P. Li, F. H. Huang and P. J. Stang, J. Am. Chem. Soc., 2016, 138, 4580 CrossRef CAS PubMed; (e) A. D. Shao, Y. S. Xie, S. J. Zhu, Z. Q. Guo, S. Q. Zhu, J. Guo, P. Shi, T. D. James, H. Tian and W. H. Zhu, Angew. Chem., Int. Ed., 2015, 54, 7275 CrossRef CAS PubMed; (f) C. Wang, B. J. Xu, M. S. Li, Z. G. Chi, Y. J. Xie, Q. Q. Li and Z. Li, Mater. Horiz., 2016, 3, 220 RSC; (g) Z. L. Xie, C. J. Chen, S. D. Xu, J. Li, Y. Zhang, S. W. Liu, J. R. Xu and Z. G. Chi, Angew. Chem., Int. Ed., 2015, 54, 7181 CrossRef CAS PubMed; (h) Y. T. Gao, G. X. Feng, T. Jiang, C. C. Goh, L. G. Ng, B. Liu, B. Li, L. Yang, J. L. Hua and H. Tian, Adv. Funct. Mater., 2015, 25, 2857 CrossRef CAS; (i) W. Dong, T. Fei, A. Palma-Cando and U. Scherf, Polym. Chem., 2014, 5, 4048 RSC.
- (a) T. Butler, W. A. Morris, J. Samonina-Kosicka and C. L. Fraser, ACS Appl. Mater. Interfaces, 2016, 8, 1242 CrossRef CAS PubMed; (b) Z. S. Wang, B. S. Gelfand, P. C. Dong, S. Trudel and T. Baumgartner, J. Mater. Chem. C, 2016, 4, 2936 RSC; (c) O. Shynkaruk, G. He, R. McDonald, M. J. Ferguson and E. Rivard, Chem. – Eur. J., 2016, 22, 248 CrossRef CAS PubMed; (d) M. T. Gabr and F. C. Pigge, RSC Adv., 2015, 5, 90226 RSC; (e) W. A. Morris, T. D. Liu and C. L. Fraser, J. Mater. Chem. C, 2015, 3, 352 RSC; (f) K. R. Ghosh, S. K. Saha and Z. Y. Wang, Polym. Chem., 2014, 5, 5638 RSC; (g) N. B. Shustova, A. F. Cozzolino, S. Reineke, M. Baldo and M. Dinca, J. Am. Chem. Soc., 2013, 135, 13326 CrossRef CAS PubMed; (h) F. Hu, G. X. Zhang, C. Zhan, W. Zhang, Y. L. Yan, Y. S. Zhao, H. B. Fu and D. Q. Zhang, Small, 2015, 11, 1335 CrossRef CAS PubMed; (i) C. Wang, H. Zhang, L. Tian, W. Zhu, Y. Lan, J. Li, H. Wang, G. X. Zhang, D. Q. Zhang, S. L. Yuan and G. T. Li, Sci. China: Chem., 2016, 59, 89 CrossRef CAS.
- Q. Zeng, Z. Li, Y. Dong, C. Di, A. Qin, Y. Hong, L. Ji, Z. Zhu, C. K. W. Jim, G. Yu, Q. Li, Z. Li, Y. Liu, A. J. Qin and B. Z. Tang, Chem. Commun., 2007, 70 RSC.
- G. Yu, S. Yin, Y. Liu, J. Chen, X. Xu, X. Sun, D. Ma, X. Zhan, Q. Peng, Z. Shuai, B. Z. Tang, D. Zhu, W. Fang and Y. Luo, J. Am. Chem. Soc., 2005, 127, 6335 CrossRef CAS PubMed.
- (a) X. Feng, B. Tong, J. Shen, J. B. Shi, T. Han, L. Chen, J. Zhi, P. Lu, Y. Ma and Y. Dong, J. Phys. Chem. B, 2010, 114, 16731 CrossRef CAS PubMed; (b) Y. X. Guo, X. Feng, T. Y. Han, S. Wang, Z. G. Lin, Y. P. Dong and B. Wang, J. Am. Chem. Soc., 2014, 136, 15485 CrossRef CAS PubMed.
- F. Bu, E. Wang, Q. Peng, R. Hu, A. Qin, Z. Zhao and B. Z. Tang, Chem. – Eur. J., 2015, 21, 4440 CrossRef CAS PubMed.
- C. T. Lai and J. L. Hong, J. Phys. Chem. B, 2010, 114, 10302 CrossRef CAS PubMed.
- Y. M. Wen, S. F. Zhu, H. F. Jiang, A. Z. Wang and Z. W. Chen, Synlett, 2011, 1023 CAS.
- O. Gidron and M. Bendikov, Angew. Chem., Int. Ed., 2014, 53, 2546 CrossRef CAS PubMed.
- (a) B. Valeur, Molecular Fluorescence: Principles and Application, Wiley-VCH, Weinheim, 2002, p. 46 Search PubMed; (b) Q. Wu, Q. Peng, Y. Niu, X. Gao and Z. Shuai, J. Phys. Chem. A, 2012, 116, 3881 CrossRef CAS PubMed.
- H. Nie, B. Chen, C. Y. Quan, J. Zhou, H. Y. Qiu, R. R. Hu, S. J. Su, A. J. Qin, Z. J. Zhao and B. Z. Tang, Chem. – Eur. J., 2015, 21, 8137 CrossRef CAS PubMed.
- (a) Q. Wu, C. Deng, Q. Peng, Y. Niu and Z. Shuai, J. Comput. Chem., 2012, 33, 1862 CrossRef CAS PubMed; (b) T. Zhang, Y. Jiang, Y. Niu, D. Wang, Q. Peng and Z. Shuai, J. Phys. Chem. A, 2014, 118, 9094 CrossRef CAS PubMed; (c) Y. Xie, T. Zhang, Z. Li, Q. Peng, Y. Yi and Z. Shuai, Chem. – Asian J., 2015, 10, 2154 CrossRef CAS PubMed.
- A. Kumar, S. R. Gadre, N. Mohan and C. H. Suresh, J. Phys. Chem. A, 2014, 118, 526 CrossRef CAS PubMed.
- (a) S. K. Singh, K. K. Mishra, N. Sharma and A. Das, Angew. Chem., Int. Ed., 2016, 55, 7801 CrossRef CAS PubMed; (b) Y. Huang, J. J. Ferrie, X. Chen, Y. Zhang, D. M. Szantai-Kis, D. M. Chenoweth and E. J. Petersson, Chem. Commun., 2016, 52, 7798 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental and computational details, 1H and 13C NMR spectra, UV and PL spectra, X-ray structure refinement data, coordinates and energies of stationary points of TPF and TPT. CCDC 1494293 and 1494294. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6qm00343e |
| ‡ These authors contributed equally. |
| This journal is © the Partner Organisations 2017 |