
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTrinuclear (aminonitrone)ZnII complexes as key intermediates in zinc(II)-mediated generation of 1,2,4-oxadiazoles from amidoximes and nitriles†
Dmitrii S.
Bolotin
 a,
Mikhail V.
Il'in
a,
Alexander S.
Novikov
a,
Nadezhda A.
Bokach
a,
Vitalii V.
Suslonov
b and
Vadim Yu.
Kukushkin
*a
a,
Mikhail V.
Il'in
a,
Alexander S.
Novikov
a,
Nadezhda A.
Bokach
a,
Vitalii V.
Suslonov
b and
Vadim Yu.
Kukushkin
*a
aInstitute of Chemistry, Saint Petersburg State University, Universitetskaya Nab., 7/9, Saint Petersburg, Russian Federation. E-mail: v.kukushkin@spbu.ru
bCenter for X-ray Diffraction Studies, Saint Petersburg State University, Universitetskii Pr., 26, Saint Petersburg, Russian Federation
First published on 16th January 2017
Abstract
Aliphatic and aromatic amidoximes RC(NH2)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NOH (R = Et, tBu, Ph, o-ClC6H4) react with Zn(OAc)2·2H2O in Me2CO giving [Zn(OAc)2{RC(NH2)NOH}2] complexes bearing N-bound amidoximes, which are involved in a moderate strength (7.3–11.9 kcal mol−1 by the DFT calculations) intramolecular resonance-assisted hydrogen bonding between the oxime HO group and the oxo group of the acetate ligand. The complexes [Zn(OAc)2{RC(NH2)NOH}2] react with excess Zn(OTf)2 in acetone accomplishing trinuclear species [Zn3(μ2-OAc)2{μ2-RC(NH2)N(H)O}4(H2O)6](OTf)4 featuring both O-ligated amidoximes—stabilized in the aminonitrone tautomeric form—and bridging acetate ligands. The aminonitrone trinuclear species were also prepared directly via the reaction of the amidoximes with Zn(OTf)2 in EtOAc; ethyl acetate in this reaction plays the role of the acetate donor and OAc− is generated in situ via ZnII-mediated hydrolysis of EtOAc. Although [Zn(OAc)2{RC(NH2)NOH}2] are inactive toward dimethylcyanamide, the [Zn3(μ2-OAc)2{μ2-RC(NH2)N(H)O}4(H2O)6](OTf)4 complexes readily react with Me2NCN giving, as a result of ZnII-mediated amidoxime–cyanamide coupling, the O-carbamidine amidoxime complexes [Zn(OTf)2{RC(NH2)NOC(NMe2)NH}2]. All synthesized compounds were characterized by HRESI-MS, FTIR, 1H-, CP-MAS TOSS 13C{1H}-, and 13C{1H} NMR, and additionally by single-crystal X-ray diffraction for eight species. Different types of non-covalent interactions in the obtained solid-state structures were studied by DFT calculations (M06-2X/6-311+G(d,p) level of theory) and topological analysis of the electron density distribution within the formalism of Bader's theory (QTAIM method).
NOH (R = Et, tBu, Ph, o-ClC6H4) react with Zn(OAc)2·2H2O in Me2CO giving [Zn(OAc)2{RC(NH2)NOH}2] complexes bearing N-bound amidoximes, which are involved in a moderate strength (7.3–11.9 kcal mol−1 by the DFT calculations) intramolecular resonance-assisted hydrogen bonding between the oxime HO group and the oxo group of the acetate ligand. The complexes [Zn(OAc)2{RC(NH2)NOH}2] react with excess Zn(OTf)2 in acetone accomplishing trinuclear species [Zn3(μ2-OAc)2{μ2-RC(NH2)N(H)O}4(H2O)6](OTf)4 featuring both O-ligated amidoximes—stabilized in the aminonitrone tautomeric form—and bridging acetate ligands. The aminonitrone trinuclear species were also prepared directly via the reaction of the amidoximes with Zn(OTf)2 in EtOAc; ethyl acetate in this reaction plays the role of the acetate donor and OAc− is generated in situ via ZnII-mediated hydrolysis of EtOAc. Although [Zn(OAc)2{RC(NH2)NOH}2] are inactive toward dimethylcyanamide, the [Zn3(μ2-OAc)2{μ2-RC(NH2)N(H)O}4(H2O)6](OTf)4 complexes readily react with Me2NCN giving, as a result of ZnII-mediated amidoxime–cyanamide coupling, the O-carbamidine amidoxime complexes [Zn(OTf)2{RC(NH2)NOC(NMe2)NH}2]. All synthesized compounds were characterized by HRESI-MS, FTIR, 1H-, CP-MAS TOSS 13C{1H}-, and 13C{1H} NMR, and additionally by single-crystal X-ray diffraction for eight species. Different types of non-covalent interactions in the obtained solid-state structures were studied by DFT calculations (M06-2X/6-311+G(d,p) level of theory) and topological analysis of the electron density distribution within the formalism of Bader's theory (QTAIM method).
Introduction
The zinc(II)-mediated reaction between amidoximes and nitriles1 is among the most facile and expedient routes for the synthesis of 1,2,4-oxadiazoles – a class of heterocycles that exhibit a wide range of biomedical2 (e.g., antibacterial,3 antibiotic,4 antimalarial,5 anticancer,6 anticonvulsant,7 immunomodulating8 and antihistaminic9) properties. These ring systems also act as active antihypertensive agents,10 selective inhibitors of 11β-hydroxysteroid dehydrogenase,11 dopamine D3 receptor ligands,12 glycogen phosphorylase inhibitors,13 and elective agonists of free fatty acid receptor,14 and serve as useful precursors for materials science for mastering liquid crystals,15 nanoporous networks,16 and insensitive energetic materials.17Recent synthetic works1a,18 helped to obtain some data partially explaining the mechanism of heterocyclization. In particular, intermediates A and B (Scheme 1) in the reaction between R2NCN and R′(NH2)CNOH in the presence of zinc(II) in undried EtOAc were trapped and identified. These data allowed the assumption that the overall reaction starts from the ZnII-mediated amidoxime–nitrile coupling. However, black-boxed intermediate zinc(II) complexes featuring coordinated amidoximes and/or nitriles were neither isolated/characterized nor identified in situ by physicochemical methods.
 | ||
| Scheme 1 Simplified mechanism of zinc(II)-mediated generation of 1,2,4-oxadiazoles. | ||
Being interested in understanding the detailed mechanism of ZnII-mediated generation of 1,2,4-oxadiazoles from amidoximes and nitriles1a,18 and in further development of coordination chemistry of amidoximes (⇌aminonitrones) (for our review on this subject see ref. 19), we synthesized novel zinc(II) species featuring amidoximes and studied their reactivity toward Me2NCN as a representative of cyanamides. The main idea of this work was to obtain experimental data fully explaining the mechanism of zinc(II)-mediated generation of 1,2,4-oxadiazoles from amidoximes and nitriles to provide a solid background for rational choice of reactants and reaction conditions.
The scenario of this study was as follows. Firstly, we synthesized zinc(II) amidoxime species and determined ligand coordination patterns (amidoxime and aminonitrone forms) and verified the structures of these complexes both experimentally and theoretically. Secondly, we studied the reactivity of the (amidoxime)ZnII and (aminonitrone)ZnII complexes toward Me2NCN as a model cyanamide substrate. Based on all these data we, thirdly, described the mechanism of zinc(II)-mediated generation of 1,2,4-oxadiazoles from amidoximes and nitriles. All the obtained data along with the corresponding discussions are consistently disclosed in sections that follow.
Results and discussion
Generation of zinc(II) amidoxime and aminonitrone complexes
As starting materials for the generation of zinc(II) amidoxime and aminonitrone complexes we used the aromatic and aliphatic amidoximes RC(NH2)NOH (R = Ph 1a, o-ClC6H41b, Et 1c, tBu 1d) and, as sources of zinc(II), the salts Zn(OTf)2 and Zn(OAc)2·2H2O. We also applied Me2CO and EtOAc as solvents in our synthetic experiments to understand the mysterious effect of ethyl acetate on the facilitation of the ZnII-mediated reaction.1a
We started our experiments under conditions, which were applied for synthetically optimized ZnII-mediated amidoxime–cyanamide coupling.1a When undried EtOAc was employed as a solvent in the reaction between 1a–d and Zn(OTf)2 (Scheme 2, c2), we observed the generation of trinuclear complexes [3a–d](OTf)4. We varied molar ratios between 1a–d and Zn(OTf)2 from 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to 6:1, including 2:1, and found that the trinuclear complexes are formed in all these combinations. The highest yields of [3a–d](OTf)4 (70–87%) were achieved when the molar ratio between 1a–d and Zn(OTf)2 was 4:3 and the reaction mixture was refluxed for 30 min in EtOAc (C2). We believe that in [3a–d](OTf)4, two bridging OAc− ligands are derived from ZnII-mediated hydrolysis of EtOAc; a relevant metal-mediated hydrolysis of ethyl acetate has previously been repeatedly reported (see, for example, ref. 20).
:1 to 6:1, including 2:1, and found that the trinuclear complexes are formed in all these combinations. The highest yields of [3a–d](OTf)4 (70–87%) were achieved when the molar ratio between 1a–d and Zn(OTf)2 was 4:3 and the reaction mixture was refluxed for 30 min in EtOAc (C2). We believe that in [3a–d](OTf)4, two bridging OAc− ligands are derived from ZnII-mediated hydrolysis of EtOAc; a relevant metal-mediated hydrolysis of ethyl acetate has previously been repeatedly reported (see, for example, ref. 20).
 | ||
| Scheme 2 The studied reactions. | ||
Concurrently, treatment of 1a–d with Zn(OTf)2 in a molar ratio 2:1 in acetone either at RT or 50 °C for 1 day gives a broad spectrum of products that we failed to separate. After keeping the reaction mixture for 1 week at 20–25 °C we observed the formation of [1a–dH](OTf) in the solution along with some amount of zinc-containing precipitate.
We assumed that the presence of acetate favors the generation of the trinuclear complexes. Accordingly, 1a–d were treated with Zn(OAc)2·2H2O in a molar ratio 2:1 in acetone at 50 °C and we observed the fast (5 min) formation of 2a–d that were isolated in 75–82% yields (Scheme 2, a2). Complexes 2a–d were easily transformed into the corresponding [3a–d](OTf)4 when 2a–d reacted with 2 equiv. of Zn(OTf)2 and 2 equiv. of 1a–d in undried acetone for 5 min at 50 °C (b2). The reverse process—which includes the transformation of [3a–d](OTf)4 to 2a–d—was not realized and the addition of the corresponding amidoxime (2 equiv.) and NaOAc (4 equiv.) to [3a–d](OTf)4 in acetone (5 min, 50 °C or 1 d, RT) led to amorphous zinc hydroxides and [1a–dH](OAc).
Zinc(II)-mediated amidoxime–cyanamide coupling
In order to study the reactivity of the zinc(II)-bound oximes, we reacted complexes 2a–d and [3a–d](OTf)4 with dimethylcyanamide as a model nitrile substrate. When complexes [3a–d](OTf)4 were treated with 4 equiv. of Me2NCN (EtOAc, RT, 3 h), the reaction yielded either imino complexes [4a–c(OTf)2] (R = Ph, o-ClC6H4, Et; Scheme 2, d2), or iminium salt [5d](OTf) (R = tBu; d2 and f2) in excellent (93–98%) yields. Complex [4d(OTf)2] was not obtained due to the steric hindrance of the bulky tBu group, which precluded the formation of the chelated complex and gave only uncomplexed [5d](OTf). Imino complexes [4a–c(OTf)2] and salt [5d](OTf) were also generated by the direct reaction of 1a–d with Me2NCN and Zn(OTf)2 (EtOAc, RT, 3 h; e2). The yields of [4a–c(OTf)2] and [5d](OTf) obtained via route e2 are comparable with the corresponding overall yields determined for routes c2 and d2 (approx. 70–80%). Complexes 2a–d do not react with Me2NCN at RT even for 24 h and a mixture of yet unidentified products was formed when the reaction was attempted at 50 °C for 3 h.In our previous study, we demonstrated that substituted cyanamides and nitriles RCN (R = Alk, Ar, NR2) are involved in the coupling with amidoximes in the presence of ZnCl2 achieving the complexes [ZnCl2{N(H)C(R)ONC(NH2)R′}] and this reaction is metal-mediated.1a Herein, we demonstrate that aminonitrone trinuclear species [3a–d](OTf)4 can also be involved in the zinc(II)-mediated coupling with Me2NCN and (aminonitrone)ZnII complexes, viz. [3a–d](OTf)4, serve as key intermediates of the coupling.1a
Our observations provide a background for a deeper understanding of the mechanism of ZnII-mediated amidoxime–cyanamide coupling. Insofar as 2a–d and [3a–d](OTf)4 demonstrated different reactivities toward Me2NCN, one can assume a strong effect of the coordination pattern of the amidoxime on the reaction. Thus, in 2a–d, the N-coordinated amidoxime—which is involved in the formation of intramolecular resonance-assisted hydrogen bonding (RAHB; for recent works on this type of hydrogen bonding see ref. 21) between the oxime HO group and the oxo group of the acetate ligand—does not react with Me2NCN. In [3a–d](OTf)4, O-coordinated amidoxime in its aminonitrone form reacted with Me2NCN under mild conditions (RT, 3 h). The latter observation is in agreement with the theoretical calculation data, which demonstrated that oxime–nitrile coupling proceeds via the aminonitrone form of the nucleophile.22
Analytical and spectroscopy data
Complexes 2a–d, [3a–d](OTf)4, [4a–c(OTf)2], and salts [5a–d](OTf) gave satisfactory C, H, and N elemental analysis results for the proposed formulas, and these species were also characterized by high-resolution ESI-MS, FTIR, 1H-, and CP-MAS TOSS 13C{1H} (for poorly soluble 2a–d, [3a–d](OTf)4, and [5d](OTf)) or 13C{1H} NMR spectroscopy (for [4a–c(OTf)2] and [5a–c](OTf) exhibiting sufficient solubility) and additionally by single-crystal X-ray diffraction for eight species (2a–c, [3a](OTf)4, [4b(OTf)(EtOH)](OTf), [4c(OTf)2], [5a](OTf), and [5d](OTf)).For a detailed description of the characterization see the ESI.† Briefly, the IR spectra of 2a–d, [3a–d](OTf)4, and [4a–c(OTf)2], exhibit one ν(CN) band in the range 1678–1638 cm−1, which is specific to amidoximes.19 The spectra of 2a–d and [3a–d](OTf)4 also display three strong to very strong bands in the region 1608–1340 cm−1 from the ν(CO) of the ligated acetate group. A characteristic feature of the 1H NMR spectra of 2a–d and [3a–d](OTf)4 recorded in (CD3)2CO is the absence of the OH and ONH signals due to a fast exchange with water protons. Another feature is the availability of broad singlets attributed to the NH2 resonances at 6.40–5.29 (for 2a–d) and 8.48–7.93 ppm (for [3a–d](OTf)4). The low-field shift of the signal in the spectra of [3a–d](OTf)4 is probably due to the positive charge on the HNCNH2 moiety provided by the stabilization of 1a–d ligands in the aminonitrone form.
X-ray structure determination, QTAIM and NBO analysis of the bonding
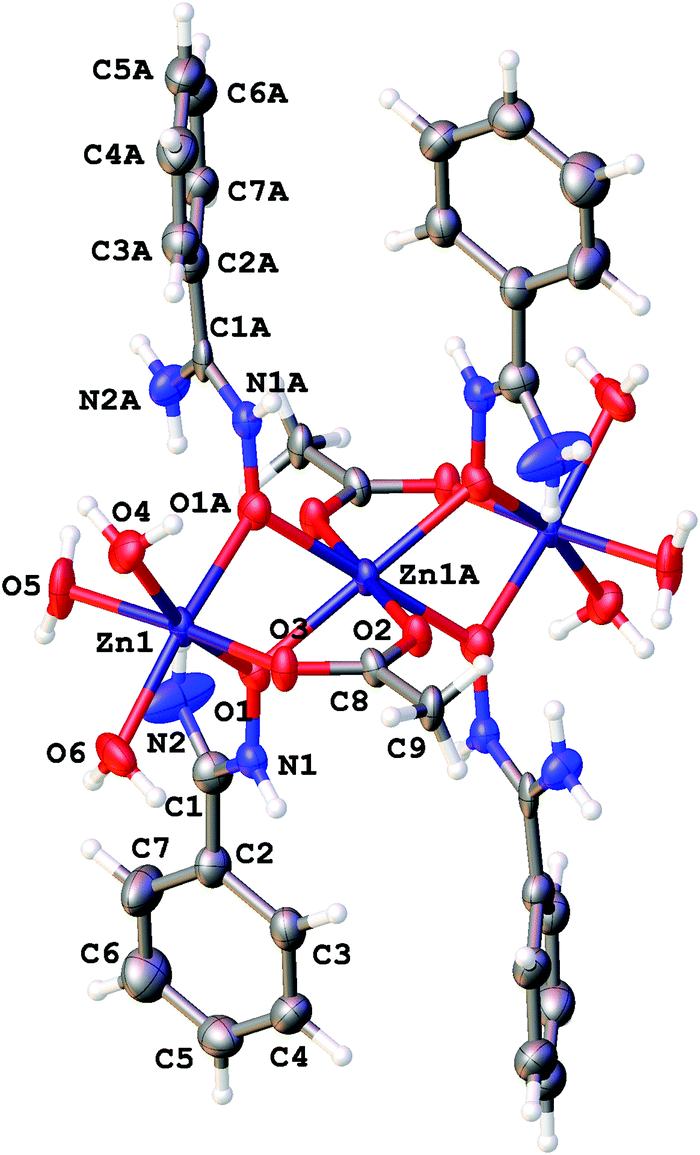 | ||
| Fig. 1 The structure of [3a]4+ showing the atomic numbering scheme. Thermal ellipsoids are given at the 50% probability level. | ||
Amidoximes and their deprotonated forms possess three nucleophilic centers, viz. two N- and one O atoms, therefore several types of coordination modes for these ligands can be realized. In our recent review,19 we analyzed the existing data on amidoxime coordination, verified nine patterns, and led to the conclusion that N-binding (as in 2a–d) is the conventional coordination pattern. The binding in the aminonitrone form, viz. [M]{ON(H)C(NR2)R′} or [M]2{μ2-ON(H)C(NR2)R′} (Fig. 2) less common.
 | ||
| Fig. 2 O-Coordinated amidoxime moiety. | ||
If the former type (A) of coordination was documented for UVI,25 PuIV,26 GeIV,19 SnIV,19 and FeIII19,27 species, the latter type (B)—which we observed in [3a–d](OTf)4—is substantially less abundant and it was previously observed only for a molybdenum(VI) center.19,28 The difference in bonding pattern can be rationalized by the application of Pearson's HSAB principle29 and charge considerations. Accordingly, the aminonitrone form coordinates to the harder and highly charged {Zn3(OAc)2}4+ moiety via a “hard” and negatively charged oxygen center, whereas neutral amidoximes tend to ligate to the neutral Zn(OAc)2 species, as in 2a–d, via a relatively “soft” nitrogen center. An additional stabilization of 2a–d by resonance-assisted hydrogen bonding (see below) should be taken into account.
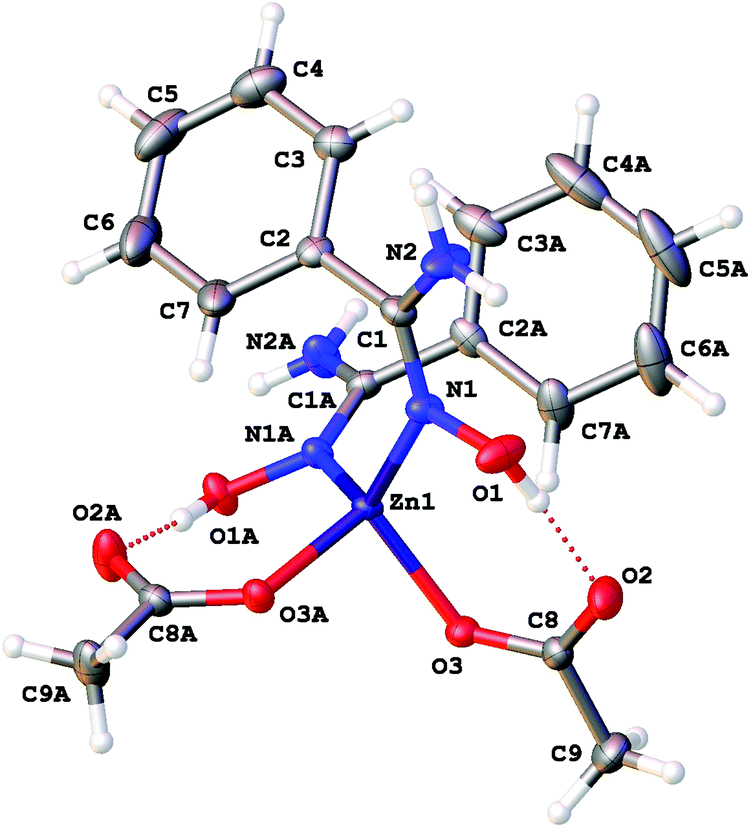 | ||
| Fig. 3 The molecular structure of 2a showing the atomic numbering scheme. Thermal ellipsoids are given at the 50% probability level. | ||
Intramolecular resonance-assisted hydrogen bonding (RAHB) between the H atom of the HO-group and the O atom of the acetate (O(1)⋯O(2) 2.600–2.696 Å; O(1)–H⋯O(2) 158.16–173.44°) was identified and this hydrogen bonding can be responsible for additional stabilization of the structure with the monodentately coordinated amidoxime. Similar RAHB stabilized structures were reported for (AcO)MII (M = Mn,31 Zn,30b,31 Ni32) complexes bearing bidentate N,N′-coordinated 2-pyridine amidoxime.
Zinc(II) complexes with terminal N-coordinated amidoximes, are known,19 while the previously reported complexes comprise bidentately coordinated ligands in which at least one coordinating group is an N-coordinated amidoxime moiety (Fig. 4, C).1a,19,30b,c,31,33
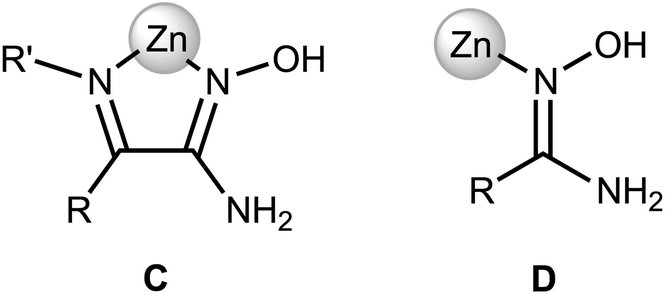 | ||
| Fig. 4 N-Coordinated amidoxime moiety. | ||
Our complexes 2a–d are the first examples of ZnII-complexes bearing monodentately N-coordinated amidoxime, but one should take into account that this binding mode is rather specific as it is supported by RAHB (Fig. 4, D).
The topological analysis of the electron density distribution within the formalism of Bader's theory (QTAIM method)34 for 2a–c (this approach has already been successfully used by us in studies of the non-covalent interactions and properties of coordination bonds in various transition metal complexes22,35) demonstrates the presence of appropriate bond critical points (BCPs) (3, −1) for intramolecular RAHB O–H⋯O in 2a–c (Fig. 5). The low magnitude of the electron density, positive values of the Laplacian, and zero or close to zero positive energy density in these BCPs are typical for hydrogen bonding (Table S3, ESI†). The balance between the Lagrangian kinetic energy G(r) and potential energy density V(r) at these BCPs (−G(r)/V(r) ≥ 1) reveals the purely non-covalent nature of these interactions. The strength of O–H⋯O contacts (7.3–11.9 kcal mol−1) corresponds to moderate hydrogen bonds following the classification of Jeffrey (“weak” HBs: <4 kcal mol−1, “moderate” HBs: 15–4 kcal mol−1, “strong” HBs: 40–15 kcal mol−1).36 The negligible values of the Wiberg bond indices for these contacts (0.01–0.04) computed by using the natural bond orbital (NBO) partitioning scheme additionally confirm the electrostatic nature of these non-covalent interactions. The electron-density delocalization in RAHB-involved 7-membered quasi-heterocyclic fragments in 2a–c has been estimated by the analysis of Wiberg bond indices (WI) for appropriate contacts (Table S4, ESI†). The WI for O–C and CO contacts in 2a–c (1.29–1.55) indicate that these bonds are significantly involved in the conjugation. For more detailed information about our DFT calculations see the ESI.†
 | ||
| Fig. 5 Contour line diagram of the Laplacian distribution ∇2ρ(r), bond paths and selected zero-flux surfaces for RAHB O–H⋯O in 2a. Bond critical points (3, −1) are shown in blue and nuclear critical points (3, −3) in pale brown, length unit – Å. | ||
 | ||
| Fig. 6 Molecular structure of [4b(OTf)(EtOH)](OTf) showing the atomic numbering scheme. Thermal ellipsoids are given at the 50% probability level. | ||
 | ||
| Fig. 7 Molecular structure of [4c(OTf)2] showing the atomic numbering scheme. Thermal ellipsoids are given at the 50% probability level. | ||
In [4c(OTf)2], the QTAIM analysis reveals the presence of two BCPs for Zn–O and four BCPs for Zn–N contacts (Fig. 8). The properties of electron density in BCPs for Zn–N contacts are typical for ordinary coordination bonds (the ρ(r) and ∇2ρ(r) values are positive and relatively high; the Hb values are significantly negative; the −G(r)/V(r) ≪ 1; Wiberg bond indices for these contacts are noticeable), whereas Zn–O contacts can be classified as non-covalent close shell interactions with some contribution of the covalent component (for more detailed information see the ESI†).
 | ||
| Fig. 8 Contour line diagram of the Laplacian distribution ∇2ρ(r), bond paths and selected zero-flux surfaces for contacts Zn–O and Zn–N in [4c(OTf)2]. Bond critical points (3, −1) are shown in blue, nuclear critical points (3, −3) in pale brown, and ring critical points (3, +1) in orange, length unit – Å. | ||
Zinc(II)-mediated generation of 1,2,4-oxadiazoles from amidoximes and nitriles
In this work, we succeeded in isolating and identifying zinc(II) complexes featuring amidoximes and aminonitrones and studied the reaction of these (amidoxime/aminonitrone)ZnII complexes with Me2NCN. All accumulated data combined with those from our previous study18 on metal-free heterocyclization of amidinium salts [5]+ allowed the formulation of the detailed mechanism of zinc(II)-mediated generation of 1,2,4-oxadiazoles from amidoximes and nitriles that efficiently proceeds in undried ethyl acetate.Firstly, complexes 2a–d are formed via coordination of both an amidoxime and AcO− to the zinc(II) center (Scheme 3, b3 and d3). Acetate is generated via the zinc(II)-mediated hydrolysis of EtOAc and this explains why the reaction occurs so easily namely in ethyl acetate (c3). Amidoxime complexes 2a–d are inactive toward dimethylcyanamide, whereas aminonitrone complexes [3a–d]4+—which are formed from 2a–d with excess zinc(II) (e3 and f3)—react with Me2NCN giving imino species [4a–c]2+ (g3 and h3). This observation agrees well with the theoretical calculation data indicating that the nucleophilic addition of amidoximes to electrophilically activated C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N22 and CC37 bonds proceeds specifically via the aminonitrone form.
N22 and CC37 bonds proceeds specifically via the aminonitrone form.
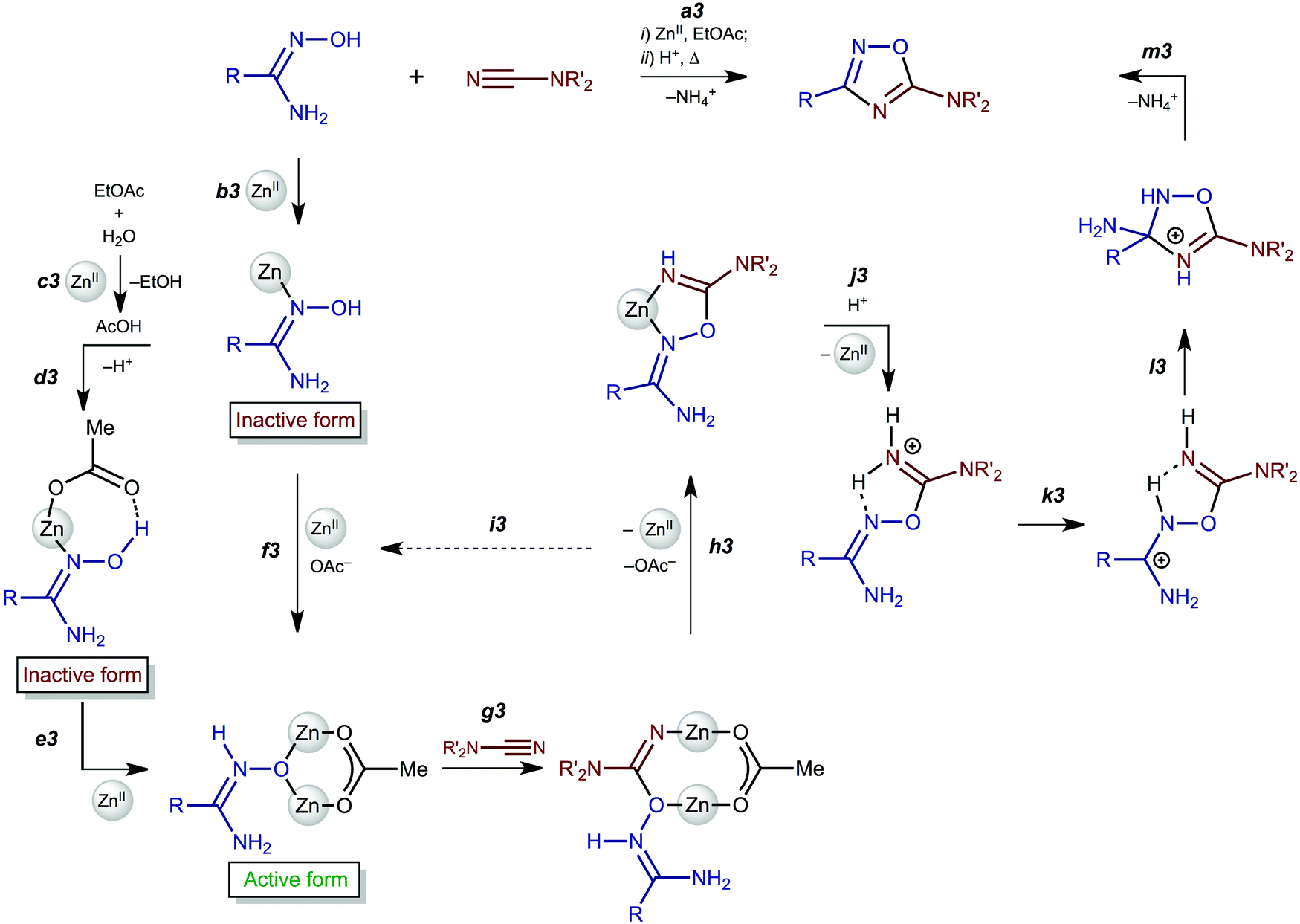 | ||
| Scheme 3 Mechanism of the zinc(II)-mediated generation of 1,2,4-oxadiazoles from amidoximes and nitriles. | ||
Addition of a strong acid (TfOH or p-TolSO3H) leads to the decoordination and liberation of amidinium salts [5a–d]+ (j3). In the case of the tBuC(NH2)NOH derivative, whose N-coordination to the zinc(II) (h3) is sterically hindered, liberation of [5d]+ (j3) occurs even in the absence of an acid. Amidinium salts [5a–d]+ undergo prototropic tautomerization to O-iminoacyl amidinium species (k3). For [RC(NH2)NOC(R′′)NH2]+, this step is accelerated by donor R and acceptor R′′ substituents. Accordingly, derivatives of conventional nitriles (R′′ = Alk or Ar) are significantly less stable than dialkylcyanamide derivatives (R′′ = NR2′ with strong +M effect). Tautomerization (k3) followed by the intramolecular nucleophilic attack of the imine N atom on the electrophilically activated C atom leads to 3-amino-1,2,4-oxadiazoline (l3). The latter compound is subject to aromatization accompanied by the elimination of NH4+ giving 1,2,4-oxadiazoles (m3).
Concluding remarks
In this work, we succeeded in generating, isolating, and fully characterizing the trinuclear zinc(II) species [3a–d](OTf)4, which most likely serve as key intermediates in the ZnII-mediated generation of 1,2,4-oxadiazoles from amidoximes and nitriles in EtOAc.1a,18 The amidoxime ligands in [3a–d](OTf)4 are stabilized in their aminonitrone form, RC(NH2)N+(H)O−, and they act as μ2-ligands being coordinated through the O atom. Before our experiments, this type of coordination was unknown for amidoxime zinc(II) complexes and this ligation pattern was observed only at a molybdenum(VI) center.19,28
All accumulated reactivity data, combined with those from our previous study18 on metal-free heterocyclization of amidinium salts [5]+, allowed the formulation of the detailed mechanism of the zinc(II)-mediated generation of 1,2,4-oxadiazoles from amidoximes and nitriles and the explanation of the effect of undried ethyl acetate on facilitation of the heterocyclization (Scheme 3). It is now clear that the overall reaction consists of four main steps namely the acetate-promoted formation of the trinuclear complexes, amidoxime–nitrile coupling, acid driven decoordination, and, eventually, the heterocyclization. All these data allowed the rational choice of the reactants and further tuning of the reaction conditions. It is noteworthy that although for a while we are satisfied with improving the synthesis of 1,2,4-oxadiazoles with stoichiometric amounts of zinc(II), our distant goal is to find out a catalytic system for the preparation of 1,2,4-oxadiazoles and we are currently testing appropriate catalytic systems.
One more issue requires additional attention. Zinc(II) centers belong to the category of kinetically labile metal centers when ambidentate or, in general, ligands with multiple donor centers are subject to facile linkage isomerization giving various coordination pattern species. In this respect, the different reactivity of 2a–d and [3a–d](OTf)4 toward the coupling with Me2NCN is surprising. The observed high reactivity of the trinuclear complexes can be further explored in at least two directions: (i) nitrones, as allyl anion type dipoles, are involved in cycloaddition reactions with various dipolarophiles.38 Although amidoximes exist in tautomeric equilibrium with their aminonitrone form RC(NH2)N+(H)O−, aminonitrone reactivity (e.g. toward cycloaddition) has never been observed in the past. The obtained aminonitrone trinuclear complexes represent available reactants for the inverse electron demand cycloaddition (type III in Sustman's classification39), when the reaction involves complexed dipole and uncoordinated dipolarophile; (ii) many organic reactions involving polynuclear metal species proceed via simultaneous coordination of two reactants to the adjacent metal centers, which, via the so-called “two-metal-mechanism”,40 couples intramolecularly. One can assume that substrates with donor centers could split the [ZnII]2{μ2-ON(H)C(NR2)R′} bridge by their ligation to a zinc(II) center followed by intramolecular coupling with aminonitrones leading to novel organic species. All works in these two directions are underway in our group.
Experimental section
Materials and instrumentation
Solvents were obtained from commercial sources and used as received. All of the syntheses were conducted in an air atmosphere. Amidoximes 1a–d were synthesized according to the literature methods.1a Melting points were measured on a Stuart SMP30 apparatus in capillaries and were not corrected. Microanalyses (C, H, N) were carried out on a Euro EA3028-HT instrument. Electrospray ionization mass-spectra were obtained on a Bruker micrOTOF spectrometer equipped with an electrospray ionization (ESI) source. The instrument was operated both in negative and positive ion modes in the m/z range 50–3000. The nebulizer gas flow was 0.4 bar and the drying gas flow was 4.0 L min−1. For HRESI, complexes were dissolved in MeOH. In the isotopic pattern, the most intensive peak is reported. Infrared spectra (4000–400 cm−1) were recorded on a Shimadzu IR Prestige-21 instrument in KBr pellets. 1H and 13C{1H} NMR spectra were measured on a Bruker Avance 400 at ambient temperature; residual solvent signals were used as the internal standard. Solid state CP-MAS TOSS 13C{1H} NMR spectra were measured on Bruker Avance III WB 400 with the magic angle spinning at 6 kHz frequency.X-ray structure determination
The single-crystal X-ray diffraction experiment was carried out using Agilent Technologies “Xcalibur” and “Supernova” diffractometers with monochromated MoKα or CuKα radiation, respectively. The crystal was fixed on a micro mount, placed on the diffractometer and measured at a temperature of 100 K. The unit cell parameters and other summarized data are represented in Tables 1S and 2S (ESI†). The structure has been solved by using the Superflip41 structure solution program using charge flipping and refined using the ShelXL42 refinement incorporated in the OLEX2 program package.43 The hydrogen atoms were placed in calculated positions and were included in the refinement in the ‘riding’ model approximation. Empirical absorption correction was applied in the CrysAlisPro44 program complex using spherical harmonics, implemented in SCALE3 ABSPACK scaling algorithm. CCDC 1503999–1504006.Computational details
The single point calculations based on the experimental X-ray geometries (quasi-solid-state approach) have been carried out at the DFT level of theory using the M06-2X functional34 (this functional was specifically developed to describe weak dispersion forces and non-covalent interactions) with the help of Gaussian-0945 program package. The standard 6-311+G(d,p) basis sets have been used for all atoms. The topological analysis of the electron density distribution with the help of the atoms-in-molecules (QTAIM) method developed by Bader34 has been performed by using the Multiwfn program (version 3.3.7).46 The Wiberg bond indices (WI) were computed by using the natural bond orbital (NBO) partitioning scheme.47 The Cartesian atomic coordinates of model structures are presented in ESI,† Table S5.Syntheses and characterization
 NOH–OAc]+, calcd 259.0056), 335.0469 ([M–2OAc–H]+, calcd 335.0481), 382.9346 ([M + Zn–PhC(NH2)NOH–H]+, calcd 382.9372), 458.9771 ([M + Zn–OAc–2H]+, calcd 458.9798), 518.9991 ([M + Zn–H]+, calcd 519.0001), 579.0156 ([M + Zn + OAc]+, calcd 579.0222). HRESI−-MS (m/z): 240.9730 ([Zn(OAc)3]−, calcd 240.9685), 424.9288 ([Zn2(OAc)5]−, calcd 424.9213). IR (KBr, selected bands, cm−1): 3464(s), 3318(s), 3178(m) ν(N–H); 3060 (m), 2876(w–m) ν(C–H); 1662(vs) ν(CN); 1564(vs), 1420(vs), 1340(s) ν(CO). 1H NMR ((CD3)2CO, δ): 7.64 (d, br, 2H, o-CH), 7.46–7.38 (m, 3H, p-CH and m-CH), 5.92 (s, br, 2H, NH2), 1.91 (s, 3H, CH3). CP-MAS TOSS 13C{1H} NMR (δ): 180.23, 178.18 (CH3CO2); 158.59 (CN); 132.73, 130.56, 128.23, 126.98, 126.15 (Ar); 24.03, 22.03 (CH3). Crystals of 2a suitable for X-ray diffraction were obtained by slow evaporation of Me2CO solution at RT in air.
NOH–OAc]+, calcd 259.0056), 335.0469 ([M–2OAc–H]+, calcd 335.0481), 382.9346 ([M + Zn–PhC(NH2)NOH–H]+, calcd 382.9372), 458.9771 ([M + Zn–OAc–2H]+, calcd 458.9798), 518.9991 ([M + Zn–H]+, calcd 519.0001), 579.0156 ([M + Zn + OAc]+, calcd 579.0222). HRESI−-MS (m/z): 240.9730 ([Zn(OAc)3]−, calcd 240.9685), 424.9288 ([Zn2(OAc)5]−, calcd 424.9213). IR (KBr, selected bands, cm−1): 3464(s), 3318(s), 3178(m) ν(N–H); 3060 (m), 2876(w–m) ν(C–H); 1662(vs) ν(CN); 1564(vs), 1420(vs), 1340(s) ν(CO). 1H NMR ((CD3)2CO, δ): 7.64 (d, br, 2H, o-CH), 7.46–7.38 (m, 3H, p-CH and m-CH), 5.92 (s, br, 2H, NH2), 1.91 (s, 3H, CH3). CP-MAS TOSS 13C{1H} NMR (δ): 180.23, 178.18 (CH3CO2); 158.59 (CN); 132.73, 130.56, 128.23, 126.98, 126.15 (Ar); 24.03, 22.03 (CH3). Crystals of 2a suitable for X-ray diffraction were obtained by slow evaporation of Me2CO solution at RT in air.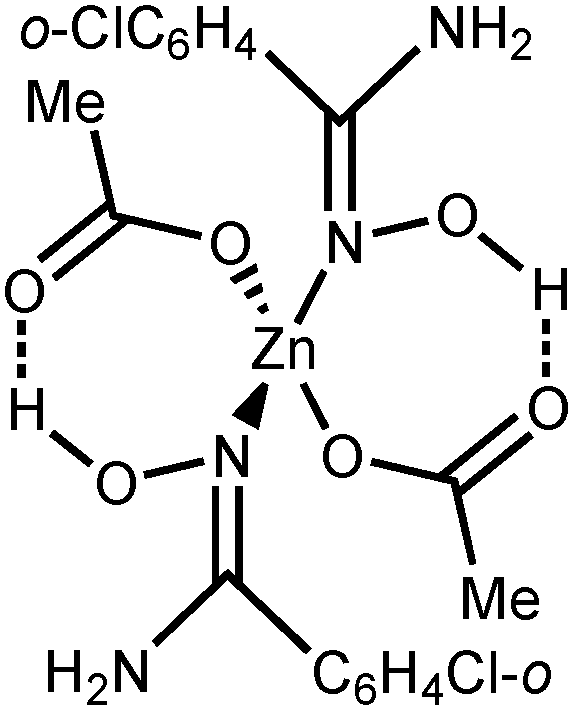 NOH–OAc]+, calcd 292.9666), 404.9651 ([M–2OAc–H]+, calcd 404.9672), 528.8963 ([M + Zn–OAc–2H]+, calcd 528.8997), 588.9170 ([M + Zn–H]+, calcd 588.9209), 648.9371 ([M + Zn + OAc]+, calcd 648.9421). HRESI−-MS (m/z): 240.9745 ([Zn(OAc)3]−, calcd 240.9685), 424.9309 ([Zn2(OAc)5]−, calcd 424.9213). IR (KBr, selected bands, cm−1): 3498(s), 3406(s), 3362(s), 3202(m–s) ν(N–H); 3078(m–s), 2832(s) ν(C–H); 1662(vs) ν(CN); 1574(vs), 1402(vs), 1340(s) ν(CO). 1H NMR ((CD3)2CO, δ): 7.50–7.43 (m, 3H, CH), 7.40–7.36 (m, 1H, CH), 6.27 (s, br, 2H, NH2), 1.78 (s, 3H, CH3). CP-MAS TOSS 13C NMR (δ): 176.94, 175.69 (CH3CO2); 155.53, 153.57 (CN); 135.27, 132.56, 130.67, 127.51, 126.48 (Ar); 22.94, 20.36 (CH3). Crystals of 2b suitable for X-ray diffraction were obtained by slow evaporation of Me2CO solution at RT in air.
NOH–OAc]+, calcd 292.9666), 404.9651 ([M–2OAc–H]+, calcd 404.9672), 528.8963 ([M + Zn–OAc–2H]+, calcd 528.8997), 588.9170 ([M + Zn–H]+, calcd 588.9209), 648.9371 ([M + Zn + OAc]+, calcd 648.9421). HRESI−-MS (m/z): 240.9745 ([Zn(OAc)3]−, calcd 240.9685), 424.9309 ([Zn2(OAc)5]−, calcd 424.9213). IR (KBr, selected bands, cm−1): 3498(s), 3406(s), 3362(s), 3202(m–s) ν(N–H); 3078(m–s), 2832(s) ν(C–H); 1662(vs) ν(CN); 1574(vs), 1402(vs), 1340(s) ν(CO). 1H NMR ((CD3)2CO, δ): 7.50–7.43 (m, 3H, CH), 7.40–7.36 (m, 1H, CH), 6.27 (s, br, 2H, NH2), 1.78 (s, 3H, CH3). CP-MAS TOSS 13C NMR (δ): 176.94, 175.69 (CH3CO2); 155.53, 153.57 (CN); 135.27, 132.56, 130.67, 127.51, 126.48 (Ar); 22.94, 20.36 (CH3). Crystals of 2b suitable for X-ray diffraction were obtained by slow evaporation of Me2CO solution at RT in air. NOH–H]+, calcd 334.9372), 362.9748 ([M + Zn–OAc–2H]+, calcd 362.9797), 422.9979 ([M + Zn–H]+, calcd 423.0009), 483.0190 ([M + Zn + OAc]+, calcd 483.0221). HRESI−-MS (m/z): 240.9729 ([Zn(OAc)3]−, calcd 240.9685), 424.9282 ([Zn2(OAc)5]−, calcd 424.9213). IR (KBr, selected bands, cm−1): 3484(s), 3428(s), 3344(s–vs), 3218(s) ν(N–H); 3088(m), 2990(s), 2880(m–s), 2772(m) ν(C–H); 1672(vs) ν(CN); 1562(vs), 1412(vs), 1346(vs) ν(CO). 1H NMR ((CD3)2CO, δ): 6.40 (s, br, 2H, NH2), 2.30 (q, 2H, CH2), 1.92 (s, 3H, CH3), 1.16 (t, 3H, CH3). CP-MAS TOSS 13C{1H} NMR (δ): 182.47, 178.12 (CH3CO2); 160.93 (CN); 25.41 (CH2CH3); 22.98, 21.56 (CH3CO2); 12.85, 11.43 (CH2CH3). Crystals of 2c suitable for X-ray diffraction were obtained by slow evaporation of Me2CO solution at RT in air.
NOH–H]+, calcd 334.9372), 362.9748 ([M + Zn–OAc–2H]+, calcd 362.9797), 422.9979 ([M + Zn–H]+, calcd 423.0009), 483.0190 ([M + Zn + OAc]+, calcd 483.0221). HRESI−-MS (m/z): 240.9729 ([Zn(OAc)3]−, calcd 240.9685), 424.9282 ([Zn2(OAc)5]−, calcd 424.9213). IR (KBr, selected bands, cm−1): 3484(s), 3428(s), 3344(s–vs), 3218(s) ν(N–H); 3088(m), 2990(s), 2880(m–s), 2772(m) ν(C–H); 1672(vs) ν(CN); 1562(vs), 1412(vs), 1346(vs) ν(CO). 1H NMR ((CD3)2CO, δ): 6.40 (s, br, 2H, NH2), 2.30 (q, 2H, CH2), 1.92 (s, 3H, CH3), 1.16 (t, 3H, CH3). CP-MAS TOSS 13C{1H} NMR (δ): 182.47, 178.12 (CH3CO2); 160.93 (CN); 25.41 (CH2CH3); 22.98, 21.56 (CH3CO2); 12.85, 11.43 (CH2CH3). Crystals of 2c suitable for X-ray diffraction were obtained by slow evaporation of Me2CO solution at RT in air.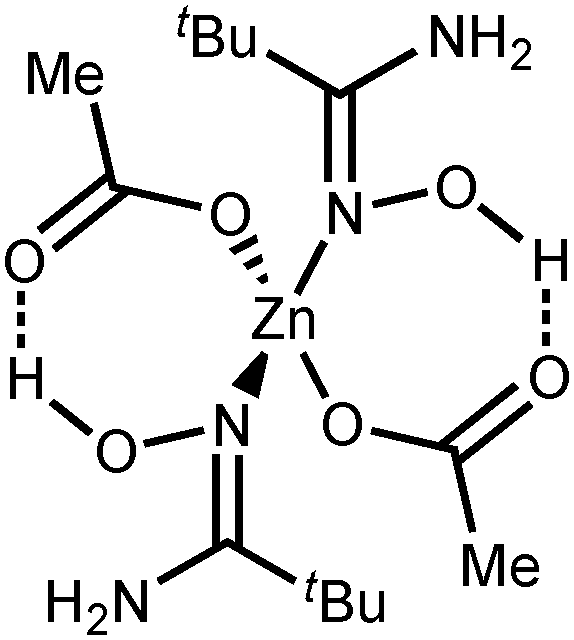 NOH–OAc]+, calcd 239.0369), 295.1099 ([M–2OAc–H]+, calcd 295.1107), 419.0406 ([M + Zn–OAc–2H]+, calcd 419.0423), 479.0717 ([M + Zn–H]+, calcd 479.0831), 539.0819 ([M + Zn + OAc]+, calcd 539.0847). HRESI−-MS (m/z): 240.9727 ([Zn(OAc)3]−, calcd 240.9685), 424.9351 ([Zn2(OAc)5]−, calcd 424.9213). IR (KBr, selected bands, cm−1): 3434(s), 3302(s), 3164(m–s) ν(N–H); 2976(s) ν(C–H); 1668(vs) ν(CN); 1608(vs), 1498(s), 1396(vs) ν(CO). 1H NMR ((CD3)2CO, δ): 5.29 (s, br, 2H, NH2), 1.97 (s, 3H, CH3), 1.19 (s, 9H, CH3). CP-MAS TOSS 13C{1H} NMR (δ): 176.91; 176.40 (CH3CO2); 160.93 (CN); 33.87, 33.50 (C(CH3)3); 27.27, 27.00 (C(CH3)3); 23.47, 22.89 (CH3CO2).
NOH–OAc]+, calcd 239.0369), 295.1099 ([M–2OAc–H]+, calcd 295.1107), 419.0406 ([M + Zn–OAc–2H]+, calcd 419.0423), 479.0717 ([M + Zn–H]+, calcd 479.0831), 539.0819 ([M + Zn + OAc]+, calcd 539.0847). HRESI−-MS (m/z): 240.9727 ([Zn(OAc)3]−, calcd 240.9685), 424.9351 ([Zn2(OAc)5]−, calcd 424.9213). IR (KBr, selected bands, cm−1): 3434(s), 3302(s), 3164(m–s) ν(N–H); 2976(s) ν(C–H); 1668(vs) ν(CN); 1608(vs), 1498(s), 1396(vs) ν(CO). 1H NMR ((CD3)2CO, δ): 5.29 (s, br, 2H, NH2), 1.97 (s, 3H, CH3), 1.19 (s, 9H, CH3). CP-MAS TOSS 13C{1H} NMR (δ): 176.91; 176.40 (CH3CO2); 160.93 (CN); 33.87, 33.50 (C(CH3)3); 27.27, 27.00 (C(CH3)3); 23.47, 22.89 (CH3CO2).
 NOH)]+, calcd 259.0056), 335.0433 ([Zn(PhC(NH2)NOH)–H]+, calcd 335.0481), 382.9308 ([Zn2(OAc)2(PhC(NH2)NOH)–H]+, calcd 382.9372), 458.9737 ([Zn2(OAc)(PhC(NH2)NOH)2–2H]+, calcd 458.9798), 518.9933 ([Zn2(OAc)2(PhC(NH2)NOH)2–H]+, calcd 519.0001), 579.0156 ([Zn2(OAc)3(PhC(NH2)NOH)2]+, calcd 579.0222). HRESI−-MS (m/z): 646.8494 ([Zn(OTf)3(PhC(NH2)NOH)]−, calcd 646.8483), 862.7197 ([Zn2(OTf)4(PhC(NH2)NOH)–H]−, calcd 862.7167). IR (KBr, selected bands, cm−1): 3412(s), 3216(s) ν(O–H) and ν(N–H); 2968(w), 2868(w) ν(C–H); 1658(vs) ν(CN); 1570(vs), 1448(s), 1404(s) ν(CO); 1246(vs), 1174(vs) ν(SO). 1H NMR ((CD3)2CO, δ): 8.48 (s, br, 2H, NH2), 7.83 (m, br, 2H, o-CH), 7.73 (t, br, 1H, p-CH), 7.62 (t, br, 2H, m-CH), 1.95 (s, 3H, CH3). CP-MAS TOSS 13C{1H} NMR (δ): 178.42 (CH3CO2); 157.59 (CN); 133.77, 133.09, 131.54, 127.15, 124.37 (Ar); 119.12 (CF3); 26.10 (CH3). Crystals of [3a](OTf)4 suitable for X-ray diffraction were obtained by slow evaporation of Me2CO solution at RT in air.
NOH)]+, calcd 259.0056), 335.0433 ([Zn(PhC(NH2)NOH)–H]+, calcd 335.0481), 382.9308 ([Zn2(OAc)2(PhC(NH2)NOH)–H]+, calcd 382.9372), 458.9737 ([Zn2(OAc)(PhC(NH2)NOH)2–2H]+, calcd 458.9798), 518.9933 ([Zn2(OAc)2(PhC(NH2)NOH)2–H]+, calcd 519.0001), 579.0156 ([Zn2(OAc)3(PhC(NH2)NOH)2]+, calcd 579.0222). HRESI−-MS (m/z): 646.8494 ([Zn(OTf)3(PhC(NH2)NOH)]−, calcd 646.8483), 862.7197 ([Zn2(OTf)4(PhC(NH2)NOH)–H]−, calcd 862.7167). IR (KBr, selected bands, cm−1): 3412(s), 3216(s) ν(O–H) and ν(N–H); 2968(w), 2868(w) ν(C–H); 1658(vs) ν(CN); 1570(vs), 1448(s), 1404(s) ν(CO); 1246(vs), 1174(vs) ν(SO). 1H NMR ((CD3)2CO, δ): 8.48 (s, br, 2H, NH2), 7.83 (m, br, 2H, o-CH), 7.73 (t, br, 1H, p-CH), 7.62 (t, br, 2H, m-CH), 1.95 (s, 3H, CH3). CP-MAS TOSS 13C{1H} NMR (δ): 178.42 (CH3CO2); 157.59 (CN); 133.77, 133.09, 131.54, 127.15, 124.37 (Ar); 119.12 (CF3); 26.10 (CH3). Crystals of [3a](OTf)4 suitable for X-ray diffraction were obtained by slow evaporation of Me2CO solution at RT in air. NOH)]+, calcd 292.9666), 404.9625 ([Zn(ClC6H4C(NH2)NOH)2–H]+, calcd 404.9672), 416.8932 ([Zn2(OAc)2(ClC6H4C(NH2)NOH)–H]+, calcd 416.8982), 528.8937 ([Zn2(OAc)(ClC6H4C(NH2)NOH)2–2H]+, calcd 528.8997), 588.9144 ([Zn2(OAc)2(ClC6H4C(NH2)NOH)2–H]+, calcd 588.9209), 648.9352 ([Zn2(OAc)3(ClC6H4C(NH2)NOH)2]+, calcd 648.9421). HRESI−-MS (m/z): 682.7972 ([Zn(OTf)3(ClC6H4C(NH2)NOH)]−, calcd 682.8064), 896.6801 ([Zn2(OTf)4(ClC6H4C(NH2)NOH)–H]−, calcd 896.6775). IR (KBr, selected bands, cm−1): 3422(s), 3314(w–m), 3234(s) ν(O–H) and ν(N–H); 2994(sh), 2898(w) ν(C–H); 1670(s) ν(CN); 1574(vs), 1438(s), 1406(s), 1350(m) ν(CO); 1250(vs), 1174(vs) ν(SO). 1H NMR ((CD3)2CO, δ): 7.93 (s, br, 2H, NH2), 7.68 (m, 1H, CH), 7.62 (m, 1H, CH), 7.52 (m, 2H, CH), 1.95 (s, 3H, CH3). CP-MAS TOSS 13C{1H} NMR (δ): 177.82 (CH3CO2); 152.40 (CN); 130.61, 128.63, 124.60 (Ar); 119.31, 116.28 (CF3); 23.48, 22.31, 21.36 (CH3).
NOH)]+, calcd 292.9666), 404.9625 ([Zn(ClC6H4C(NH2)NOH)2–H]+, calcd 404.9672), 416.8932 ([Zn2(OAc)2(ClC6H4C(NH2)NOH)–H]+, calcd 416.8982), 528.8937 ([Zn2(OAc)(ClC6H4C(NH2)NOH)2–2H]+, calcd 528.8997), 588.9144 ([Zn2(OAc)2(ClC6H4C(NH2)NOH)2–H]+, calcd 588.9209), 648.9352 ([Zn2(OAc)3(ClC6H4C(NH2)NOH)2]+, calcd 648.9421). HRESI−-MS (m/z): 682.7972 ([Zn(OTf)3(ClC6H4C(NH2)NOH)]−, calcd 682.8064), 896.6801 ([Zn2(OTf)4(ClC6H4C(NH2)NOH)–H]−, calcd 896.6775). IR (KBr, selected bands, cm−1): 3422(s), 3314(w–m), 3234(s) ν(O–H) and ν(N–H); 2994(sh), 2898(w) ν(C–H); 1670(s) ν(CN); 1574(vs), 1438(s), 1406(s), 1350(m) ν(CO); 1250(vs), 1174(vs) ν(SO). 1H NMR ((CD3)2CO, δ): 7.93 (s, br, 2H, NH2), 7.68 (m, 1H, CH), 7.62 (m, 1H, CH), 7.52 (m, 2H, CH), 1.95 (s, 3H, CH3). CP-MAS TOSS 13C{1H} NMR (δ): 177.82 (CH3CO2); 152.40 (CN); 130.61, 128.63, 124.60 (Ar); 119.31, 116.28 (CF3); 23.48, 22.31, 21.36 (CH3). NOH)]+, calcd 211.0056), 239.0471 ([Zn(EtC(NH2)NOH)2–H]+, calcd 239.0481), 334.9362 ([Zn2(OAc)2(EtC(NH2)NOH)–H]+, calcd 334.9372), 362.9773 ([Zn2(OAc)(EtC(NH2)NOH)–2H]+, calcd 362.9797), 422.9981 ([Zn2(OAc)2(EtC(NH2)NOH)2–H]+, calcd 423.0009), 483.0202 ([Zn2(OAc)3(EtC(NH2)NOH)2]+, calcd 483.0221). HRESI−-MS (m/z): 598.8526 ([Zn(OTf)3(EtC(NH2)NOH)]−, calcd 598.8483). IR (KBr, selected bands, cm−1): 3454(s), 3364(s), 3232(s), 3126(m) ν(O–H) and ν(N–H); 2994(m), 2894(w–m) ν(C–H); 1676(vs) ν(CN); 1572(vs), 1444(s), 1404(s) ν(CO); 1252(vs), 1170(vs) ν(SO). 1H NMR ((CD3)2CO, δ): 8.44 (s, br, 2H, NH2), 2.68, 2.66 (q, 2H, CH2), 2.02 (s, 3H, CH3), 1.35 (t, 3H, CH3). CP-MAS TOSS 13C{1H} NMR (δ): 177.12 (CH3CO2); 159.71 (CN), 117.79 (CF3); 24.93, 20.12 (CH2 and CH3CO2); 6.70, 5.89, 5.14 (CH2CH3).
NOH)]+, calcd 211.0056), 239.0471 ([Zn(EtC(NH2)NOH)2–H]+, calcd 239.0481), 334.9362 ([Zn2(OAc)2(EtC(NH2)NOH)–H]+, calcd 334.9372), 362.9773 ([Zn2(OAc)(EtC(NH2)NOH)–2H]+, calcd 362.9797), 422.9981 ([Zn2(OAc)2(EtC(NH2)NOH)2–H]+, calcd 423.0009), 483.0202 ([Zn2(OAc)3(EtC(NH2)NOH)2]+, calcd 483.0221). HRESI−-MS (m/z): 598.8526 ([Zn(OTf)3(EtC(NH2)NOH)]−, calcd 598.8483). IR (KBr, selected bands, cm−1): 3454(s), 3364(s), 3232(s), 3126(m) ν(O–H) and ν(N–H); 2994(m), 2894(w–m) ν(C–H); 1676(vs) ν(CN); 1572(vs), 1444(s), 1404(s) ν(CO); 1252(vs), 1170(vs) ν(SO). 1H NMR ((CD3)2CO, δ): 8.44 (s, br, 2H, NH2), 2.68, 2.66 (q, 2H, CH2), 2.02 (s, 3H, CH3), 1.35 (t, 3H, CH3). CP-MAS TOSS 13C{1H} NMR (δ): 177.12 (CH3CO2); 159.71 (CN), 117.79 (CF3); 24.93, 20.12 (CH2 and CH3CO2); 6.70, 5.89, 5.14 (CH2CH3). NOH)]+, calcd 239.0369), 295.1095 ([Zn(tBuC(NH2)NOH)2–H]+, calcd 295.1107), 419.0386 ([Zn2(OAc)(tBuC(NH2)NOH)2–2H]+, calcd 419.0423), 479.0626 ([Zn2(OAc)2(tBuC(NH2)NOH)2–H]+, calcd 479.0635), 539.0819 ([Zn2(OAc)3(tBuC(NH2)NOH)3]+, calcd 539.0847). HRESI−-MS (m/z): 626.8803 ([Zn(OTf)3(tBuC(NH2)NOH)]−, calcd 626.8796), 840.7540 ([Zn2(OTf)4(tBuC(NH2)NOH)–H]−, calcd 840.7499). IR (KBr, selected bands, cm−1): 3446(vs), 3314(vs), 3238(vs) ν(O–H) and ν(N–H); 2980(s), 2884(m) ν(C–H); 1668(vs) ν(CN); 1564(s–vs), 1440(s), 1376(s) ν(CO); 1250(vs), 1178(vs) ν(SO). 1H NMR ((CD3)2CO, δ): 8.37 (s, br, 2H, NH2), 2.01 (s, 3H, CH3), 1.45 (s, 9H, CH3). CP-MAS TOSS 13C{1H} NMR (δ): 179.94 (CH3CO2); 164.47 (CN); 120.62, 118.24 (CF3); 34.45, 33.30 (C(CH3)3); 26.38, 25.77, 25.57, 23.30 (CH3CO2 and C(CH3)3).
NOH)]+, calcd 239.0369), 295.1095 ([Zn(tBuC(NH2)NOH)2–H]+, calcd 295.1107), 419.0386 ([Zn2(OAc)(tBuC(NH2)NOH)2–2H]+, calcd 419.0423), 479.0626 ([Zn2(OAc)2(tBuC(NH2)NOH)2–H]+, calcd 479.0635), 539.0819 ([Zn2(OAc)3(tBuC(NH2)NOH)3]+, calcd 539.0847). HRESI−-MS (m/z): 626.8803 ([Zn(OTf)3(tBuC(NH2)NOH)]−, calcd 626.8796), 840.7540 ([Zn2(OTf)4(tBuC(NH2)NOH)–H]−, calcd 840.7499). IR (KBr, selected bands, cm−1): 3446(vs), 3314(vs), 3238(vs) ν(O–H) and ν(N–H); 2980(s), 2884(m) ν(C–H); 1668(vs) ν(CN); 1564(s–vs), 1440(s), 1376(s) ν(CO); 1250(vs), 1178(vs) ν(SO). 1H NMR ((CD3)2CO, δ): 8.37 (s, br, 2H, NH2), 2.01 (s, 3H, CH3), 1.45 (s, 9H, CH3). CP-MAS TOSS 13C{1H} NMR (δ): 179.94 (CH3CO2); 164.47 (CN); 120.62, 118.24 (CF3); 34.45, 33.30 (C(CH3)3); 26.38, 25.77, 25.57, 23.30 (CH3CO2 and C(CH3)3).
 NOC(NMe2)NH − OTf]+, calcd 418.9974), 625.1183 ([M − OTf]+, calcd 625.1141). HRESI−-MS (m/z): 505.0375 ([PhC(NH2)NOC(N(CH3)2)NH2 + 2OTf]−, calcd 505.0281). IR (KBr, selected bands, cm−1): 3344(m–s), 3202(m–s) ν(N–H); 3074(w), 2948(w–m), 2826(w) ν(C–H); 1642(vs) ν(CN); 1286(vs), 1246(vs), 1168(vs) ν(SO). 1H NMR ((CD3)2SO, δ): 7.95 (s, br, 1H, NH2), 7.64–7.61 (m, br, 3H, o-CH and NH2), 7.54–7.47 (m, br, 4H, p-CH, m-CH and NH2), 5.18 (s, 1H, NH), 2.83 (s, br, 6H, NMe2). 13C{1H} NMR ((CD3)2SO, δ): 160.30, 159.28 (PhC(N)–NH2 and OC(N)NMe2); 131.85, 130.67, 128.91, 128.76 (Ph); 121.16 (q, CF3); 37.44 (CH3).
NOC(NMe2)NH − OTf]+, calcd 418.9974), 625.1183 ([M − OTf]+, calcd 625.1141). HRESI−-MS (m/z): 505.0375 ([PhC(NH2)NOC(N(CH3)2)NH2 + 2OTf]−, calcd 505.0281). IR (KBr, selected bands, cm−1): 3344(m–s), 3202(m–s) ν(N–H); 3074(w), 2948(w–m), 2826(w) ν(C–H); 1642(vs) ν(CN); 1286(vs), 1246(vs), 1168(vs) ν(SO). 1H NMR ((CD3)2SO, δ): 7.95 (s, br, 1H, NH2), 7.64–7.61 (m, br, 3H, o-CH and NH2), 7.54–7.47 (m, br, 4H, p-CH, m-CH and NH2), 5.18 (s, 1H, NH), 2.83 (s, br, 6H, NMe2). 13C{1H} NMR ((CD3)2SO, δ): 160.30, 159.28 (PhC(N)–NH2 and OC(N)NMe2); 131.85, 130.67, 128.91, 128.76 (Ph); 121.16 (q, CF3); 37.44 (CH3).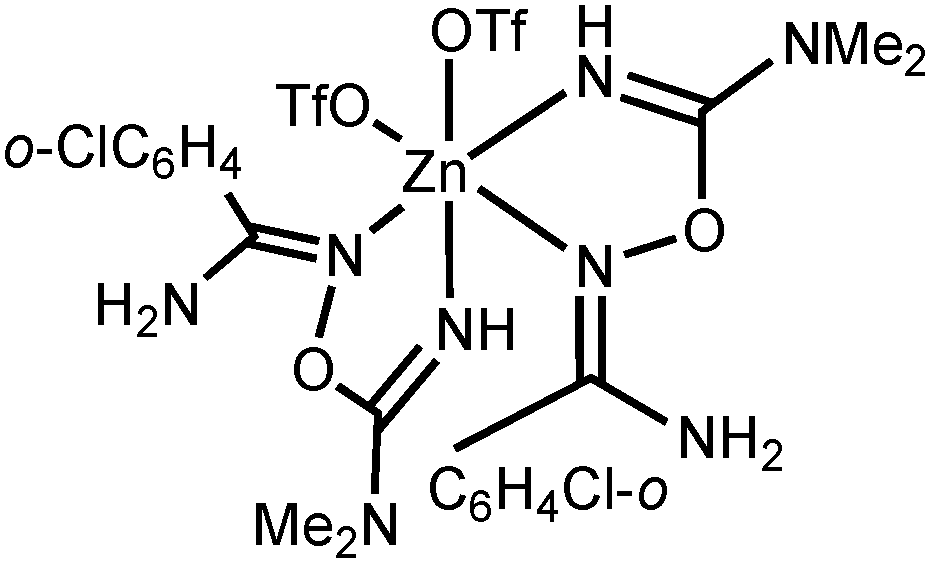 NOC(N(CH3)2)NH2 + 2OTf]−, calcd 538.9891). IR (KBr, selected bands, cm−1): 3350(s), 3206(m–s) ν(N–H); 2944(w–m) ν(C–H); 1638(vs) ν(CN); 1280(vs), 1248(vs), 1160(vs) ν(SO). 1H NMR ((CD3)2SO, δ): 7.98 (s, br, 1H, NH2), 7.60 (m, br, 2H, CH and NH2), 7.47 (m, br, 4H, CH and NH2), 4.78 (s, 1H, NH), 2.80 (s, br, 3H, NMe), 2.64 (s, br, 3H, NMe). 13C{1H} NMR ((CD3)2SO, δ): 159.52, 155.68 (o-ClC6H4C(N)NH2 and OC(N)NMe2); 132.72, 132.38, 131.52, 130.33, 129.89, 127.36 (Ar); 121.16 (q, CF3); 37.32 (CH3). Crystals of [4b(OTf)(EtOH)](OTf) suitable for X-ray diffraction were obtained by slow evaporation of undried EtOAc solution of the complex at RT in air.
NOC(N(CH3)2)NH2 + 2OTf]−, calcd 538.9891). IR (KBr, selected bands, cm−1): 3350(s), 3206(m–s) ν(N–H); 2944(w–m) ν(C–H); 1638(vs) ν(CN); 1280(vs), 1248(vs), 1160(vs) ν(SO). 1H NMR ((CD3)2SO, δ): 7.98 (s, br, 1H, NH2), 7.60 (m, br, 2H, CH and NH2), 7.47 (m, br, 4H, CH and NH2), 4.78 (s, 1H, NH), 2.80 (s, br, 3H, NMe), 2.64 (s, br, 3H, NMe). 13C{1H} NMR ((CD3)2SO, δ): 159.52, 155.68 (o-ClC6H4C(N)NH2 and OC(N)NMe2); 132.72, 132.38, 131.52, 130.33, 129.89, 127.36 (Ar); 121.16 (q, CF3); 37.32 (CH3). Crystals of [4b(OTf)(EtOH)](OTf) suitable for X-ray diffraction were obtained by slow evaporation of undried EtOAc solution of the complex at RT in air.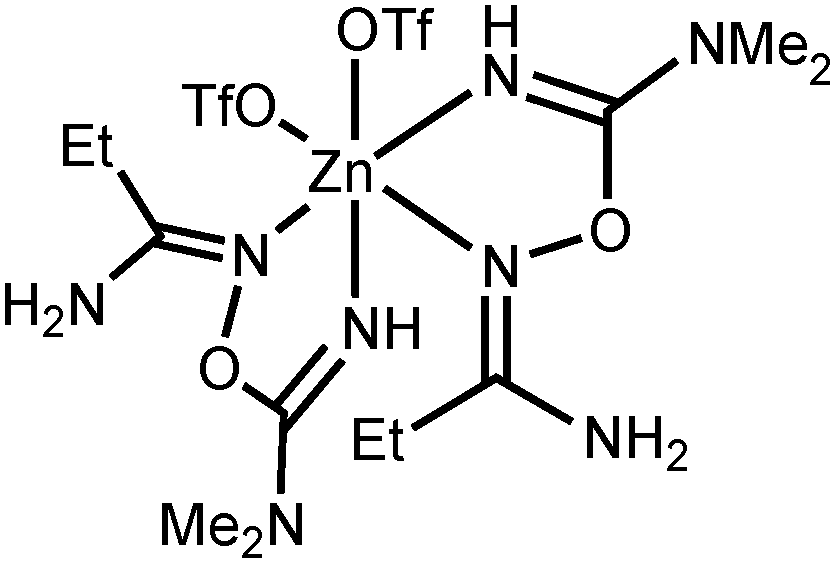 NOC(NMe2)NH − OTf]+, calcd 370.9974), 529.1189 ([M − OTf]+, calcd 529.1141). HRESI−-MS (m/z): 457.0401 ([EtC(NH2)NOC(N(CH3)2)NH2 + OTf]−, calcd 457.0290). IR (KBr, selected bands, cm−1): 3460(m–s), 3374(s), 3224(m) ν(N–H); 2982(m), 2946(m), 2890(w–m) ν(C–H); 1642(vs) ν(CN); 1290(vs), 1244(vs), 1162(vs) ν(SO). 1H NMR ((CD3)2SO, δ): 7.57 (s, br, 1H, NH2), 7.12 (s, br, 1H, NH2), 6.21 (s, 1H, NH), 3.02 (s, br, 6H, NMe2), 2.20 (q, 2H, CH2), 1.19 (t, 3H, CH3). 13C{1H} NMR ((CD3)2SO, δ): 161.23, 160.85 (CH2C(N)NH2 and OC(N)NMe2); 121.15 (q, CF3); 37.61 (NMe2); 24.61 (CH2); 12.13 (CH2CH3). Crystals of [4c(OTf)2] suitable for X-ray diffraction were obtained by slow evaporation of wet EtOAc solution of the complex at RT in air.
NOC(NMe2)NH − OTf]+, calcd 370.9974), 529.1189 ([M − OTf]+, calcd 529.1141). HRESI−-MS (m/z): 457.0401 ([EtC(NH2)NOC(N(CH3)2)NH2 + OTf]−, calcd 457.0290). IR (KBr, selected bands, cm−1): 3460(m–s), 3374(s), 3224(m) ν(N–H); 2982(m), 2946(m), 2890(w–m) ν(C–H); 1642(vs) ν(CN); 1290(vs), 1244(vs), 1162(vs) ν(SO). 1H NMR ((CD3)2SO, δ): 7.57 (s, br, 1H, NH2), 7.12 (s, br, 1H, NH2), 6.21 (s, 1H, NH), 3.02 (s, br, 6H, NMe2), 2.20 (q, 2H, CH2), 1.19 (t, 3H, CH3). 13C{1H} NMR ((CD3)2SO, δ): 161.23, 160.85 (CH2C(N)NH2 and OC(N)NMe2); 121.15 (q, CF3); 37.61 (NMe2); 24.61 (CH2); 12.13 (CH2CH3). Crystals of [4c(OTf)2] suitable for X-ray diffraction were obtained by slow evaporation of wet EtOAc solution of the complex at RT in air. N)oxime; 1652(vs) ν(CN)imine; 1264(vs), 1162(vs) ν(SO). 1H NMR ((CD3)2SO, δ): 8.51 (s, br, 1H, NH2), 7.92 (s, br, 1H, NH2), 6.63 (s, 2H, NH2), 3.05 (s, 6H, NMe2), 1.20 (s, 9H, CH3). CP-MAS TOSS 13C{1H} NMR (δ): 162.95, 156.86 (CC(N)NH2 and OC(N)NMe2); 116.01 (CF3); 35.20 (NMe2); 32.96 (CMe3); 24.28 (Me). Crystals of [5d](OTf) suitable for X-ray diffraction were obtained by slow evaporation of Me2CO solution at RT in air.
N)oxime; 1652(vs) ν(CN)imine; 1264(vs), 1162(vs) ν(SO). 1H NMR ((CD3)2SO, δ): 8.51 (s, br, 1H, NH2), 7.92 (s, br, 1H, NH2), 6.63 (s, 2H, NH2), 3.05 (s, 6H, NMe2), 1.20 (s, 9H, CH3). CP-MAS TOSS 13C{1H} NMR (δ): 162.95, 156.86 (CC(N)NH2 and OC(N)NMe2); 116.01 (CF3); 35.20 (NMe2); 32.96 (CMe3); 24.28 (Me). Crystals of [5d](OTf) suitable for X-ray diffraction were obtained by slow evaporation of Me2CO solution at RT in air.
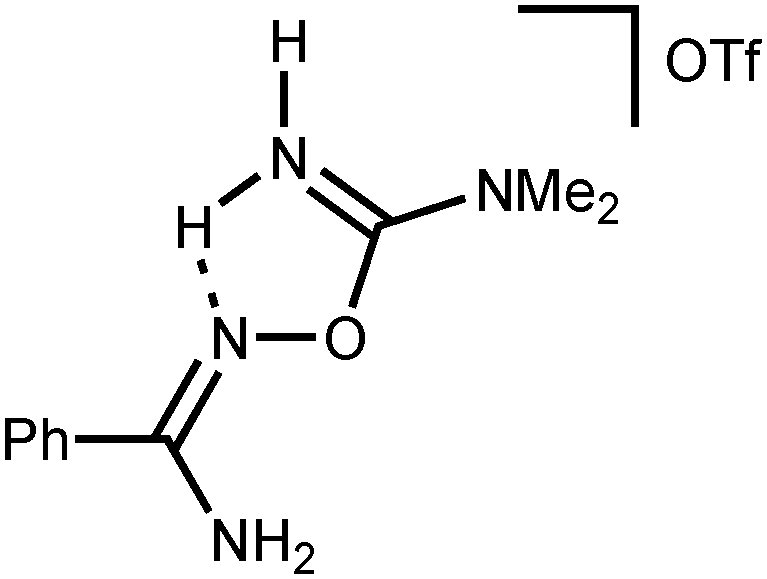 N)oxime; 1654(vs) ν(CN)imine; 1258(vs), 1168(vs) ν(SO). 1H NMR ((CD3)2SO, δ): 8.68 (s, br, 2H, NH2), 7.87–7.85 (m, 2H, o-CH), 7.60–7.56 (m, 1H, p-CH), 7.53–7.49 (m, 2H, m-CH) 7.34 (s, br, 2H, NH2), 3.14 (s, br, 6H, NMe2). 13C{1H} NMR ((CD3)2SO, δ): 159.96, 158.16 (PhC(N)NH2 and OC(N)NMe2); 131.73, 130.50, 128.93, 127.86 (Ph); 121.17 (q, CF3); 38.35, 37.27 (CH3). Crystals of [5a](OTf) suitable for X-ray diffraction were obtained by slow evaporation of Me2CO solution at RT in air.
N)oxime; 1654(vs) ν(CN)imine; 1258(vs), 1168(vs) ν(SO). 1H NMR ((CD3)2SO, δ): 8.68 (s, br, 2H, NH2), 7.87–7.85 (m, 2H, o-CH), 7.60–7.56 (m, 1H, p-CH), 7.53–7.49 (m, 2H, m-CH) 7.34 (s, br, 2H, NH2), 3.14 (s, br, 6H, NMe2). 13C{1H} NMR ((CD3)2SO, δ): 159.96, 158.16 (PhC(N)NH2 and OC(N)NMe2); 131.73, 130.50, 128.93, 127.86 (Ph); 121.17 (q, CF3); 38.35, 37.27 (CH3). Crystals of [5a](OTf) suitable for X-ray diffraction were obtained by slow evaporation of Me2CO solution at RT in air. N)oxime; 1650(vs) ν(CN)imine; 1254(vs), 1172(vs) ν(SO). 1H NMR ((CD3)2SO, δ): 8.74 (s, br, 1H, NH2), 8.48 (s, br, 1H, NH2), 7.62–7.54 (m, 5H, m-, p-CH and NH2), 7.49–7.45 (m, 2H, o-CH and NH2), 3.12 (s, br, 6H, NMe2). 13C{1H} NMR ((CD3)2SO, δ): 159.75, 157.34 (ClC6H4C(N)NH2 and OC(N)NMe2); 132.82, 132.43, 131.86, 130.68, 130.17, 127.66 (Ar); 121.16 (q, CF3); 38.29, 37.28 (CH3).
N)oxime; 1650(vs) ν(CN)imine; 1254(vs), 1172(vs) ν(SO). 1H NMR ((CD3)2SO, δ): 8.74 (s, br, 1H, NH2), 8.48 (s, br, 1H, NH2), 7.62–7.54 (m, 5H, m-, p-CH and NH2), 7.49–7.45 (m, 2H, o-CH and NH2), 3.12 (s, br, 6H, NMe2). 13C{1H} NMR ((CD3)2SO, δ): 159.75, 157.34 (ClC6H4C(N)NH2 and OC(N)NMe2); 132.82, 132.43, 131.86, 130.68, 130.17, 127.66 (Ar); 121.16 (q, CF3); 38.29, 37.28 (CH3).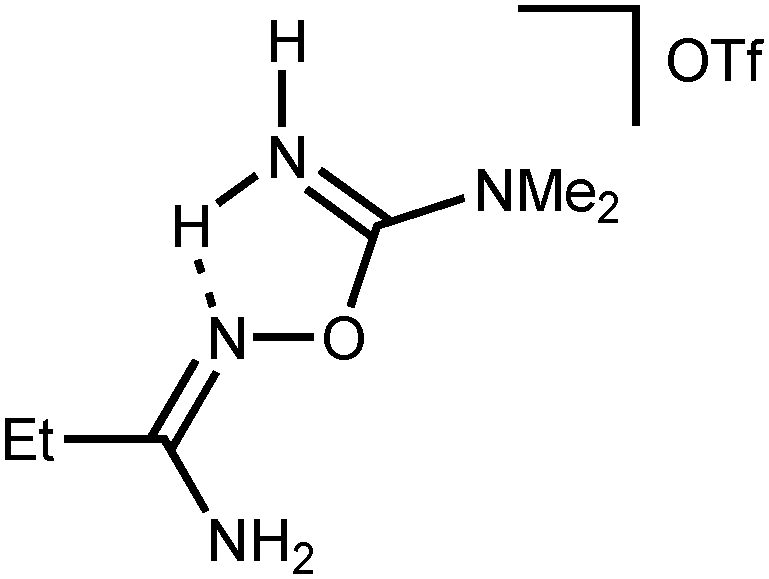 N)oxime; 1666(vs) ν(CN)imine; 1274(vs), 1160(vs) ν(SO). 1H NMR ((CD3)2SO, δ): 8.63 (s, br, 1H, NH2), 8.37 (s, br, 1H, NH2), 6.97 (s, br, 1H, NH), 6.78 (s, br 1H, NH), 3.06 (s, br, 6H, NMe2), 2.16 (q, 2H, CH2), 1.15 (t, 3H, CH3). 13C{1H} NMR ((CD3)2SO, δ): 161.89, 159.88 (CH2C(N)NH2 and OC(N)NMe2); 121.14 (q, CF3); 38.17, 37.09 (NMe2); 24.06 (CH2); 11.91 (CH3).
N)oxime; 1666(vs) ν(CN)imine; 1274(vs), 1160(vs) ν(SO). 1H NMR ((CD3)2SO, δ): 8.63 (s, br, 1H, NH2), 8.37 (s, br, 1H, NH2), 6.97 (s, br, 1H, NH), 6.78 (s, br 1H, NH), 3.06 (s, br, 6H, NMe2), 2.16 (q, 2H, CH2), 1.15 (t, 3H, CH3). 13C{1H} NMR ((CD3)2SO, δ): 161.89, 159.88 (CH2C(N)NH2 and OC(N)NMe2); 121.14 (q, CF3); 38.17, 37.09 (NMe2); 24.06 (CH2); 11.91 (CH3).
Acknowledgements
The work was supported by the Russian Science Foundation (grant 14-13-00060). The authors thank the Center for X-ray Diffraction Studies, Center for Magnetic Resonance, Center for Chemical Analysis and Materials Research, and Chemistry Educational Centre (all belong to Saint Petersburg State University) for the physicochemical measurements.References
- (a) D. S. Bolotin, K. I. Kulish, N. A. Bokach, G. L. Starova, V. V. Gurzhiy and V. Y. Kukushkin, Inorg. Chem., 2014, 53, 10312–10324 CrossRef CAS PubMed; (b) J. K. Augustine, V. Akabote, S. G. Hegde and P. Alagarsamy, J. Org. Chem., 2009, 74, 5640–5643 CrossRef CAS PubMed; (c) V. N. Yarovenko, V. K. Taralashvili, I. V. Zavarzin and M. M. Krayushkin, Tetrahedron, 1990, 46, 3941–3952 CrossRef CAS; (d) V. N. Yarovenko, B. I. Ugrak, M. M. Krayushkin, V. Z. Shirinyan and I. V. Zavarzin, Russ. Chem. Bull., 1994, 43, 627–629 CrossRef; (e) V. N. Yarovenko, I. V. Zavarzin and M. M. Krayushkin, Bull. Acad. Sci. USSR, Div. Chem. Sci., 1986, 35, 1106 CrossRef.
- O. B. Rajesh, D. Bashir, P. Vidya and F. Mazahar, Mini-Rev. Med. Chem., 2014, 14, 355–369 CrossRef.
- C. G. Fortuna, R. Berardozzi, C. Bonaccorso, G. Caltabiano, L. Di Bari, L. Goracci, A. Guarcello, A. Pace, A. Palumbo Piccionello, G. Pescitelli, P. Pierro, E. Lonati, A. Bulbarelli, C. E. A. Cocuzza, G. Musumarra and R. Musumeci, Bioorg. Med. Chem., 2014, 22, 6814–6825 CrossRef CAS PubMed.
- M. Chang, Sh. Mobashery, E. Spink, D. Ding, S. Testero, E. Leemans and M. Bouderau, WO2016049586A2, 2016.
- J. M. dos Santos Filho, D. M. A. de Queiroz e Silva, T. S. Macedo, H. M. P. Teixeira, D. R. M. Moreira, S. Challal, J.-L. Wolfender, E. F. Queiroz and M. B. P. Soares, Bioorg. Med. Chem., 2016, 24, 5693–5701 CrossRef CAS PubMed.
- (a) C. V. Maftei, E. Fodor, P. G. Jones, C. G. Daniliuc, M. H. Franz, G. Kelter, H.-H. Fiebig, M. Tamm and I. Neda, Tetrahedron, 2016, 72, 1185–1199 CrossRef CAS; (b) K. Challa, M. V. Bhargavi and G. L. D. Krupadanam, J. Asian Nat. Prod. Res., 2016, 18, 1158–1168 CrossRef CAS PubMed; (c) K. N. Nandeesh, H. A. Swarup, N. C. Sandhya, C. D. Mohan, C. S. Pavan Kumar, M. N. Kumara, K. Mantelingu, S. Ananda and K. S. Rangappa, New J. Chem., 2016, 40, 2823–2828 RSC.
- M. Mohammadi-Khanaposhtani, M. Shabani, M. Faizi, I. Aghaei, R. Jahani, Z. Sharafi, N. Shamsaei Zafarghandi, M. Mahdavi, T. Akbarzadeh, S. Emami, A. Shafiee and A. Foroumadi, Eur. J. Med. Chem., 2016, 112, 91–98 CrossRef CAS PubMed.
- (a) P. G. N. Sasikumar, M. Ramachandra, A. Prasad and S. S. S. Naremaddepalli, WO2016142886A2, 2016; (b) P. G. N. Sasikumar, M. Ramachandra and S. S. S. Naremaddepalli, WO2016142833A1, 2016.
- S. K. Jain and J. Nandan, World J. Pharm. Pharm. Sci., 2014, 3, 762–773 Search PubMed , 712 p.
- W. Zhu, X. Bao, H. Ren, P. Liao, W. Zhu, Y. Yan, L. Wang and Z. Chen, Clin. Exp. Hypertens., 2016, 38, 435–442 CrossRef CAS PubMed.
- D. V. Ziolkovskiy, V. V. Lipson, A. D. Nikitina and V. A. Chebanov, Lett. Drug Des. Discovery, 2016, 13, 226–233 CrossRef CAS.
- Y. Cao, C. Min, S. Acharya, K.-M. Kim and S. H. Cheon, Bioorg. Med. Chem., 2016, 24, 191–200 CrossRef CAS PubMed.
- M. Donnier-Marechal, D. Goyard, V. Folliard, T. Docsa, P. Gergely, J.-P. Praly and S. Vidal, Beilstein J. Org. Chem., 2015, 11, 499–503 CrossRef CAS PubMed.
- (a) I. Zahanich, I. Kondratov, V. Naumchyk, Y. Kheylik, M. Platonov, S. Zozulya and M. Krasavin, Bioorg. Med. Chem. Lett., 2015, 25, 3105–3111 CrossRef CAS PubMed; (b) A. Tolmachev, A. V. Bogolubsky, S. E. Pipko, A. V. Grishchenko, D. V. Ushakov, A. V. Zhemera, O. O. Viniychuk, A. I. Konovets, O. A. Zaporozhets, P. K. Mykhailiuk and Y. S. Moroz, ACS Comb. Sci., 2016, 18, 616–624 CrossRef CAS PubMed.
- (a) E. Girotto, I. H. Bechtold and H. Gallardo, Liq. Cryst., 2016, 43, 1768–1777 CrossRef CAS; (b) I. H. R. Tomi, M. M. Abdul Razzaq Al-Obaidy and A. H. R. Al-Daraji, Liq. Cryst., 2016 DOI:10.1080/02678292.2016.1225837; (c) M. Subrao, D. M. Potukuchi, G. S. Ramachandra, P. Bhagavath, S. G. Bhat and S. Maddasani, Beilstein J. Org. Chem., 2015, 11, 233–241 CrossRef PubMed , 239 p.
- D. Ko, H. A. Patel and C. T. Yavuz, Chem. Commun., 2015, 51, 2915–2917 RSC.
- V. Thottempudi, J. Zhang, C. He and J. M. Shreeve, RSC Adv., 2014, 4, 50361–50364 RSC.
- K. I. Kulish, A. S. Novikov, P. M. Tolstoy, D. S. Bolotin, N. A. Bokach, A. A. Zolotarev and V. Y. Kukushkin, J. Mol. Struct., 2016, 1111, 142–150 CrossRef CAS.
- D. S. Bolotin, N. A. Bokach and V. Y. Kukushkin, Coord. Chem. Rev., 2016, 313, 62–93 CrossRef CAS.
- (a) E. Tílvez, G. I. Cárdenas-Jirón, M. I. Menéndez and R. López, Inorg. Chem., 2015, 54, 1223–1231 CrossRef PubMed; (b) K. L. Breno, M. D. Pluth, C. W. Landorf and D. R. Tyler, Organometallics, 2004, 23, 1738–1746 CrossRef CAS.
- (a) P. E. Hansen, Molecules, 2015, 20, 2405–2424 CrossRef PubMed; (b) A. Nowroozi, S. Rahmani, A. Eshraghi and K. Shayan, Struct. Chem., 2016, 27, 829–838 CrossRef CAS; (c) M. P. Romero-Fernandez, M. Avalos, R. Babiano, P. Cintas, J. L. Jimenez and J. C. Palacios, Tetrahedron, 2016, 72, 95–104 CrossRef CAS; (d) D. Rusinska-Roszak, J. Phys. Chem. A, 2015, 119, 3674–3687 CrossRef CAS PubMed; (e) M. Vatanparast and A. R. Nekoei, Struct. Chem., 2015, 26, 1039–1048 CrossRef CAS; (f) Non-covalent Interactions in the Synthesis and Design of New Compounds, ed. A. M. Maharramov, K. T. Mahmudov, M. N. Kopylovich, and A. J. L. Pombeiro, Wiley, 2016 Search PubMed; (g) K. T. Mahmudov, M. N. Kopylovich and A. J. L. Pombeiro, Coord. Chem. Rev., 2013, 257, 1244–1281 CrossRef CAS; (h) M. N. Kopylovich, M. F. C. Guedes da Silva, L. M. D. R. S. Martins, M. L. Kouznetsov, K. T. Mahmudov and A. J. L. Pombeiro, Polyhedron, 2013, 50, 374–382 CrossRef CAS; (i) K. T. Mahmudov, M. N. Kopylovich, M. F. C. Guedes da Silva, G. S. Mahmudova, M. Sutradhar and A. J. L. Pombeiro, Polyhedron, 2013, 60, 78–84 CrossRef CAS; (j) M. N. Kopylovich, A. C. C. Nunes, K. T. Mahmudov, M. Haukka, T. C. O. Mac Leod, L. M. D. R. S. Martins, M. L. Kuznetsov and A. J. L. Pombeiro, Dalton Trans., 2011, 40, 2822–2836 RSC; (k) S. Wanninger, V. Lorenz, A. Subhan and F. T. Edelmann, Chem. Soc. Rev., 2015, 44, 4986–5002 RSC; (l) A. B. Davis, R. E. Lambert, F. R. Fronczek, P. J. Cragg and K. J. Wallace, New J. Chem., 2014, 38, 4678–4683 RSC; (m) X. Su, T. F. Robbins and I. Aprahamian, Angew. Chem., Int. Ed., 2011, 50, 1841–1844 CrossRef CAS PubMed; (n) K. T. Mahmudov, M. N. Kopylovich, M. F. C. Guedes da Silva and A. J. L. Pombeiro, ChemPlusChem, 2014, 79, 1523–1531 CrossRef CAS; (o) K. T. Mahmudov, M. F. C. Guedes da Silva, M. Sutradhar, M. N. Kopylovich, F. E. Huseynov, N. T. Shamilov, A. A. Voronina, T. M. Buslaeva and A. J. L. Pombeiro, Dalton Trans., 2015, 44, 5602–5610 RSC; (p) K. T. Mahmudov, M. N. Kopylovich, A. Sabbatini, M. G. B. Drew, L. M. D. R. S. Martins, C. Pettinari and A. J. L. Pombeiro, Inorg. Chem., 2014, 53, 9946–9958 CrossRef CAS PubMed; (q) M. N. Kopylovich, K. T. Mahmudov, A. Mizar and A. J. L. Pombeiro, Chem. Commun., 2011, 47, 7248–7250 RSC; (r) M. N. Kopylovich, A. Mizar, M. F. C. Guedes da Silva, T. C. O. Mac Leod, K. T. Mahmudov and A. J. L. Pombeiro, Chem. – Eur. J., 2013, 19, 588–600 CrossRef CAS PubMed.
- D. S. Bolotin, V. K. Burianova, A. S. Novikov, M. Y. Demakova, C. Pretorius, P. P. Mokolokolo, A. Roodt, N. A. Bokach, V. V. Suslonov, A. P. Zhdanov, K. Y. Zhizhin, N. T. Kuznetsov and V. Y. Kukushkin, Organometallics, 2016, 35, 3612–3623 CrossRef CAS.
- F. H. Allen, O. Kennard, D. G. Watson, L. Brammer and G. Orpen, J. Chem. Soc., Perkin Trans. 2, 1987, S1–S19 RSC.
- (a) L. Zhu, D. Liu, L. Wu, W. Feng, X. Zhang, J. Wu, D. Fan, X. Lü, R. Lu and Q. Shi, Inorg. Chem. Commun., 2013, 37, 182–185 CrossRef CAS; (b) D. Dey, G. Kaur, A. Ranjani, L. Gayathri, P. Chakraborty, J. Adhikary, J. Pasan, D. Dhanasekaran, A. R. Choudhury, M. A. Akbarsha, N. Kole and B. Biswas, Eur. J. Inorg. Chem., 2014, 3350–3358 CrossRef CAS; (c) R. Olejník, M. Bílek, Z. Růžičková, Z. Hoštálek, J. Merna and A. Růžička, J. Organomet. Chem., 2015, 794, 237–246 CrossRef.
- E. G. Witte, K. S. Schwochau, G. Henkel and B. Krebs, Inorg. Chim. Acta, 1984, 94, 323–331 CrossRef CAS.
- L. Xian, G. Tian, C. M. Beavers, S. J. Teat and D. K. Shuh, Angew. Chem., Int. Ed., 2016, 55, 4671–4673 CrossRef CAS PubMed.
- D. Lefevre-Groboillot, S. Dijols, J.-L. Boucher, J.-P. Mahy, R. Ricoux, A. Desbois, J.-L. Zimmermann and D. Mansuy, Biochemistry, 2001, 40, 9909–9917 CrossRef CAS PubMed.
- S.-G. Roh, A. Proust, P. Gouzerh and F. Robert, J. Chem. Soc., Chem. Commun., 1993, 836–838 RSC.
- R. G. Pearson, J. Am. Chem. Soc., 1963, 85, 3533–3539 CrossRef CAS.
- (a) A. G. Orpen, L. Brammer, F. H. Allen, O. Kennard, D. G. Watson and R. Taylor, J. Chem. Soc., Dalton Trans., 1989, 12, S1–S83 RSC; (b) K. F. Konidaris, V. Bekiari, E. Katsoulakou, C. P. Raptopoulou, V. Psycharis, S. P. Perlepes, T. C. Stamatatos and E. Manessi-Zoupa, Inorg. Chim. Acta, 2011, 376, 470–478 CrossRef CAS; (c) K. F. Konidaris, M. Giouli, C. P. Raptopoulou, V. Psycharis, I. I. Verginadis, A. Vasiliadis, A. S. Afendra, S. Karkabounas, E. Manessi-Zoupa and T. C. Stamatatos, Inorg. Chim. Acta, 2013, 396, 49–59 CrossRef CAS.
- J. Ran and Y.-P. Tong, Struct. Chem., 2011, 22, 1113–1118 CrossRef CAS.
- M. Werner, J. Berner and P. G. Jones, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1996, 52, 72–74 CrossRef.
- M. Y. Demakova, D. S. Bolotin, N. A. Bokach, G. L. Starova and V. Y. Kukushkin, Inorg. Chim. Acta, 2015, 425, 114–117 CrossRef CAS.
- R. F. W. Bader, Chem. Rev., 1991, 91, 893–928 CrossRef CAS.
- (a) A. S. Mikherdov, M. A. Kinzhalov, A. S. Novikov, V. P. Boyarskiy, I. A. Boyarskaya, D. V. Dar'in, G. L. Starova and V. Y. Kukushkin, J. Am. Chem. Soc., 2016, 138, 14129–14137 CrossRef CAS PubMed; (b) A. S. Novikov, M. L. Kuznetsov and A. J. L. Pombeiro, Chem. – Eur. J., 2013, 19, 2874–2888 CrossRef CAS PubMed; (c) V. N. Mikhaylov, V. N. Sorokoumov, K. A. Korvinson, A. S. Novikov and I. A. Balova, Organometallics, 2016, 35, 1684–1697 CrossRef CAS; (d) X. Ding, M. J. Tuikka, P. Hirva, V. Y. Kukushkin, A. S. Novikov and M. Haukka, CrystEngComm, 2016, 18, 1987–1995 RSC; (e) D. M. Ivanov, A. S. Novikov, G. L. Starova, M. Haukka and V. Y. Kukushkin, CrystEngComm, 2016, 18, 5278–5286 RSC; (f) T. V. Serebryanskaya, A. S. Novikov, P. V. Gushchin, M. Haukka, R. E. Asfin, P. M. Tolstoy and V. Y. Kukushkin, Phys. Chem. Chem. Phys., 2016, 18, 14104–14112 RSC.
- T. Steiner, Angew. Chem., Int. Ed., 2002, 41, 48–76 CrossRef CAS.
- D. Roca-Lopez, A. Daru, T. Tejero and P. Merino, RSC Adv., 2016, 6, 22161–22173 RSC.
- (a) L. L. Anderson, Asian J. Org. Chem., 2016, 5, 9–30 CrossRef CAS; (b) A. Badoiu, G. Bernardinelli, J. Mareda, E. P. Kundig and F. Viton, Chem. – Asian J., 2008, 3, 1298–1311 CrossRef CAS PubMed; (c) N. A. Bokach, M. L. Kuznetsov and V. Y. Kukushkin, Coord. Chem. Rev., 2011, 255, 2946–2967 CrossRef CAS; (d) L. Maiuolo and A. De Nino, in Targets in Heterocyclic Systems: Chemistry and Properties, ed. O. A. Attanasi, P. Merino and D. Spinelli, 2015, vol. 19, pp. 299–345 Search PubMed; (e) V. Nair and T. D. Suja, Tetrahedron, 2007, 63, 12247–12275 CrossRef CAS; (f) H. Pellissier, Tetrahedron, 2007, 63, 3235–3285 CrossRef CAS; (g) W. M. Shi, X. P. Ma, G. F. Su and D. L. Mo, Org. Chem. Front., 2016, 3, 116–130 RSC; (h) H. Suga and N. Hayashi, in Heterocyclic Supramolecules II, ed. K. Matsumoto, 2009, vol. 18, pp. 119–154 Search PubMed.
- (a) R. Sustmann, Tetrahedron Lett., 1971, 12, 2717–2720 CrossRef; (b) R. Sustmann, Pure Appl. Chem., 1974, 40, 569–593 CrossRef CAS; (c) R. Sustmann, Tetrahedron Lett., 1971, 12, 2721–2724 CrossRef.
- (a) T. A. Steitz and J. A. Steitz, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 6496 CrossRef; (b) V. Y. Kukushkin and A. J. L. Pombeiro, Inorg. Chim. Acta, 2005, 358, 1–21 CrossRef CAS.
- (a) L. Palatinus and G. Chapuis, J. Appl. Crystallogr., 2007, 40, 786–790 CrossRef CAS; (b) L. Palatinus and A. van der Lee, J. Appl. Crystallogr., 2008, 41, 975–984 CrossRef CAS; (c) L. Palatinus, S. J. Prathapa and S. van Smaalen, J. Appl. Crystallogr., 2012, 45, 575–580 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, C71, 3–8 CrossRef PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- K. Marakchi, O. K. Kabbaj and N. Komiha, J. Fluorine Chem., 2002, 114, 81–89 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision A.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- E. D. Glendening, C. R. Landis and F. Weinhold, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 1–42 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Analytical and spectroscopy data; the structures of the O-iminoacylated oximes; spectra of 2a–d, [3a–d](OTf)4, [4a–c(OTf)2], and [5a–d](OTf); crystal data for 2a–c, [3a](OTf)4, [4b(OTf)(EtOH)](OTf), [4c(OTf)2], [5a](OTf), and [5d](OTf); theoretical study of bonding situation in solid state structures of 2 and [4](OTf)2, calculated Wiberg bond indices for selected bonds in 2a–c; Cartesian atomic coordinates of model structures; crystallographic information files for 2a–c, [3a](OTf)4, [4b(OTf)(EtOH)](OTf), [4c(OTf)2], [5a](OTf), and [5d](OTf). CCDC 1503999–1504006. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6nj03508f |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2017 |