
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceProbing glycosaminoglycan spectral signatures in live cells and their conditioned media by Raman microspectroscopy†
S.
Brézillon‡
*ab,
V.
Untereiner
acd,
H. T.
Mohamed§
e,
J.
Hodin§
abc,
A.
Chatron-Colliet
ab,
F.-X.
Maquart
abf and
G. D.
Sockalingum‡
 *ac
*ac
aCNRS UMR7369, Matrice Extracellulaire et Dynamique Cellulaire (MEDyC), Reims, France. E-mail: stephane.brezillon@univ-reims.fr; ganesh.sockalingum@univ-reims.fr
bUniversité de Reims Champagne-Ardenne, Laboratoire de Biochimie médicale et de Biologie Moléculaire, UFR de Médecine, Reims, France
cUniversité de Reims Champagne-Ardenne, MéDIAN-Biophotonique et Technologies pour la Santé, UFR de Pharmacie, Reims, France
dUniversité de Reims Champagne-Ardenne, Plateforme d'imagerie cellulaire et tissulaire (PICT), Reims, France
eDepartment of Zoology, Faculty of Science, Cairo University, Giza, Egypt
fLaboratoire Central de Biochimie, CHU de Reims, Reims, France
First published on 20th March 2017
Abstract
Spectroscopic markers characteristic of reference glycosaminoglycan molecules were identified previously based on their vibrational signatures. Infrared spectral signatures of glycosaminoglycans in fixed cells were also recently demonstrated but probing live cells still remains challenging. Raman microspectroscopy is potentially interesting to perform studies under physiological conditions. The aim of the present work was to identify the Raman spectral signatures of GAGs in fixed and live cells and in their conditioned media. Biochemical and Raman analyses were performed on five cell types: chondrocytes, dermal fibroblasts, melanoma (SK-MEL-28), wild type CHO, and glycosaminoglycan-defective mutant CHO-745 cells. The biochemical assay of sulfated GAGs in conditioned media was only possible for chondrocytes, dermal fibroblasts, and wild type CHO due to the detection limit of the test. In contrast, Raman microspectroscopy allowed probing total glycosaminoglycan content in conditioned media, fixed and live cells and the data were analysed by principal component analysis. Our results showed that the Raman technique is sensitive enough to identify spectral markers of glycosaminoglycans that were useful to characterise the conditioned media of the five cell types. The results were confirmed at the single cell level on both live and fixed cells with a good differentiation between the cell types. Furthermore, the principal component loadings revealed prominent glycosaminoglycan-related spectral information. Raman microspectroscopy allows monitoring of the glycosaminoglycan profiles of single live cells and could therefore be developed for cell screening purposes and holds promise for identifying glycosaminoglycan signatures as a marker of cancer progression in tissues.
1. Introduction
Proteoglycans are ubiquitous major macromolecules of extracellular matrices, cell surfaces, and some intracellular granules.1 Proteoglycans are composed of a protein core to which one or several sulfated glycosaminoglycan (GAG) chains are attached by a covalent linkage. They exhibit an architectural role and maintain tissue integrity. Due to their high structural microheterogeneities, GAGs critically modulate a large array of cell functions. During physiological processes, like embryogenesis, GAGs were described to regulate cell migration2 but also cell differentiation.3,4 Under pathophysiological conditions, GAGs were demonstrated to play key regulatory roles in proliferation,5 adhesion and migration,6 angiogenesis,7,8 extracellular matrix homeostasis,9 and tumor progression.10–13Physicochemical and biochemical analyses of GAGs from complex biological systems like tissues and cells have been reported. Analysis from tissue sections was first reported for hyaluronan and chondroitin sulfate,14,15 for chondroitin sulfate and dermatan sulfate,16 and for keratan sulfate.17,18 These studies involved ion pairing chromatography and mass spectrometry applied to enzyme digest solutions. The chemical analysis of such preparations is therefore a tedious task accomplished by enzymatic depolymerisation of the chain with specific bacterial enzymes followed by disaccharide analysis by high-performance liquid chromatography, capillary electrophoresis, or fluorophore assisted carbohydrate electrophoresis.19–22 After invasive preparation such as protein digestion of the samples and GAGs precipitation, the latter can be analyzed on cellulose acetate.23 Cell surface proteoglycans play an important part in the functional and metabolic behaviour of cells, like cancer cells6 or leucocytes.24 These methods are nevertheless time-consuming and require specific reagents and enzymes. Therefore, novel and alternative methods of GAG analysis have been developed. A non-invasive and non-destructive approach based on vibrational spectroscopy appears to be promising because it gives a global molecular fingerprint that can be used for GAG characterisation. Vibrational spectroscopy includes both infrared (IR) and Raman techniques capable of probing bond vibrations in molecular systems.25 In terms of structural and compositional analysis, vibrational spectroscopy is able to give a complete “molecular fingerprint” of the studied sample and can be both quantitative and qualitative. Furthermore, vibrational spectroscopy is rapid, non-contact and does not require external labelling. The first Raman spectra of GAGs were published nearly four decades ago.26 The association of spectroscopy with powerful data analytical methods gives more insight into the interpretation of the spectral information and molecular-level phenomena. When coupled with a microscope, vibrational spectroscopy becomes a highly sensitive method capable of probing at the micron level, thus necessitating only small amounts of the sample. Raman and Raman optical activity of GAGs was described.27 Spectroscopic markers characteristic of reference GAG molecules (hyaluronan, dermatan sulfate, keratan sulfate, chondroitin 4-sulfate, chondroitin 6-sulfate, heparin, heparan sulfate) have been identified previously based on their molecular structures by Raman and IR microspectroscopies.28,29 Other vibrational spectroscopy studies involved heparin related sulfation sites,30 hyaluronic acid complexes with DNA,31 chondroitin sulfate proteoglycans from glial cells32 and various GAG/collagen mixtures.33
Vibrational spectroscopy has an important advantage over physicochemical and biochemical analyses since intact cells and tissues can be directly analysed without any extraction or purification procedures. IR spectral signatures of GAG in complex biological systems like cells were also recently demonstrated.34 This study was performed on fixed cells and IR microspectroscopy of live cells remains challenging. In order to study live cells, Raman microspectroscopy is potentially interesting to perform studies under physiological conditions. It is highly sensitive to the structure, composition, and environment of the molecules constituting the studied specimen.35 Raman spectral imaging of single living cancer cells was previously described.36–38 A preliminary study of single cell confocal Raman spectroscopy of human osteoarthritic chondrocytes was recently reported.39 Surface-enhanced Raman scattering was used to map glycan expression for the identification of cancerous cells.40
Raman spectroscopy was shown to be a powerful non-invasive analytical tool for melanoma research.41 Raman spectroscopy has been developed to make real time in vivo cancer diagnosis42–45 and to differentiate melanoma and non-melanoma skin cancers.46–51 It can distinguish between melanocytes and melanoma cells, analyse a specific type of cell death in melanoma cells, and predict the susceptibility of melanoma cells to anticancer drugs.41 A characterisation of the vibrational spectral signatures for a series of melanoma sublines has been described.52
The aim of the present work is to demonstrate the potential of Raman microspectroscopy to characterize GAG signatures in cells and in their respective conditioned media, the latter being enriched in secreted protein and GAGs released by cells after a period of time as opposed to fresh culture medium containing serum. These conditioned media are commonly used to measure the concentration of secreted proteins and GAGs from cells. GAGs are synthesised in the endoplasmic reticulum and, via the Golgi apparatus, assembled, maturated (sulfation, epimerisation) and secreted. Except for hyaluronan, GAG can be covalently linked to a core protein forming thus a proteoglycan. With the exception of serglycin which remains in the cytoplasm, proteoglycans can be either secreted in the extracellular medium, fully integrated into the plasma membrane or anchored to the membrane by a glycosylphosphatidylinositol residue.53
Five cell types were investigated including chondrocytes, dermal fibroblasts, SK-MEL-28 melanoma cells, wild type CHO (CHO-WT), and GAG-defective mutant CHO-745 cells. These cells were chosen for their capacity to synthesise different levels of GAGs. Chondrocytes are the most abundant cells in cartilage which plays an important role as a shock absorber and space holder in living organisms.54 These physical properties are closely connected with their molecular structure and intermolecular interactions between polymeric components, such as GAGs. Chondrocytes are responsible for producing and maintaining the integrity of the cartilaginous matrix, which consists mainly of collagen, hyaluronan, and proteoglycans such as aggrecan with chondroitin sulfate and keratan sulfate chains. Hyaluronan and chondroitin sulfate are the most abundant in cartilage.54–56 Since they are rich in chondroitin synthase and sulfotransferase, chondrocytes are the cells with the highest capacity for GAG synthesis.57 GAGs constitute 10% of the dry weight of the cartilage. In comparison, fibroblasts secrete 90% of collagen and express only 1% of GAGs. In contrast, the CHO-WT cells being epithelial cells express less GAGs than dermal fibroblasts which are mesenchymal cells. The SK-MEL-28 human melanoma cells known to express less GAGs than dermal fibroblasts were used to evaluate the sensitivity of vibrational spectroscopy to detect GAGs. GAG-defective mutant CHO-745 cells lacking xylosyltransferase were used as a control since they express a very low level of GAGs. The cells were analysed individually under both fixed and live conditions. Raman microspectroscopy data were complemented by biochemical GAG assays.
2. Materials and methods
2.1. Cell culture
In this study, five cell cultures were carried out: three cell lines, CHO-WT (CHO-K1, ATCC® CCL-61™), CHO mutated cells lacking xylosyltransferase I (CHO-745, ATCC® CRL-2242™), SK-MEL-28 human melanoma cell line (SK-MEL-28 ATCC® HTB-72™), and two primary cultures of human chondrocytes and human dermal fibroblasts. The CHO-745 cell line, lacking xylosyltransferase, is the only known mutant deficient in the synthesis of chondroitin sulfate and heparan sulfate58,59 which results in a decreased amount of GAGs in these cells, compared to the wild-type CHO (CHO-WT). Chondrocytes and CHO cells were grown in DMEM/F-12 medium (Dulbecco's Modified Eagle Medium: Nutrient Mixture F-12, Gibco) supplemented with an antifungal agent (amphotericin B, Gibco) with 10% FBS and 1% penicillin/streptomycin. Dermal fibroblasts were grown in DMEM 1 g L−1 glucose (Gibco) supplemented with 10% FBS and 1% penicillin/streptomycin. The SK-MEL-28 melanoma cell line was grown in DMEM 1 g L−1 glucose supplemented with 10% FBS and 1% penicillin/streptomycin. Cultures were maintained at 37 °C under a humidified atmosphere containing 5% (v/v) CO2. The cells were detached at 80% of confluence with 0.5% trypsin/EDTA (Gibco). The enzymatic reaction was stopped by adding full medium. The cells in suspension were centrifuged at 420g for 3 min and then cell pellets were resuspended. Cell viability was greater than 80% as assessed by the trypan blue assay.The protocols for biochemical and Raman spectral analyses of GAGs obtained from conditioned media of CHO-WT, CHO-745, SK-MEL-28, dermal fibroblasts and chondrocytes are shown in Fig. 1a. The figure also depicts the preparation steps for the Raman analysis of these cells measured at the single cell level, under fixed and live conditions (Fig. 1b).
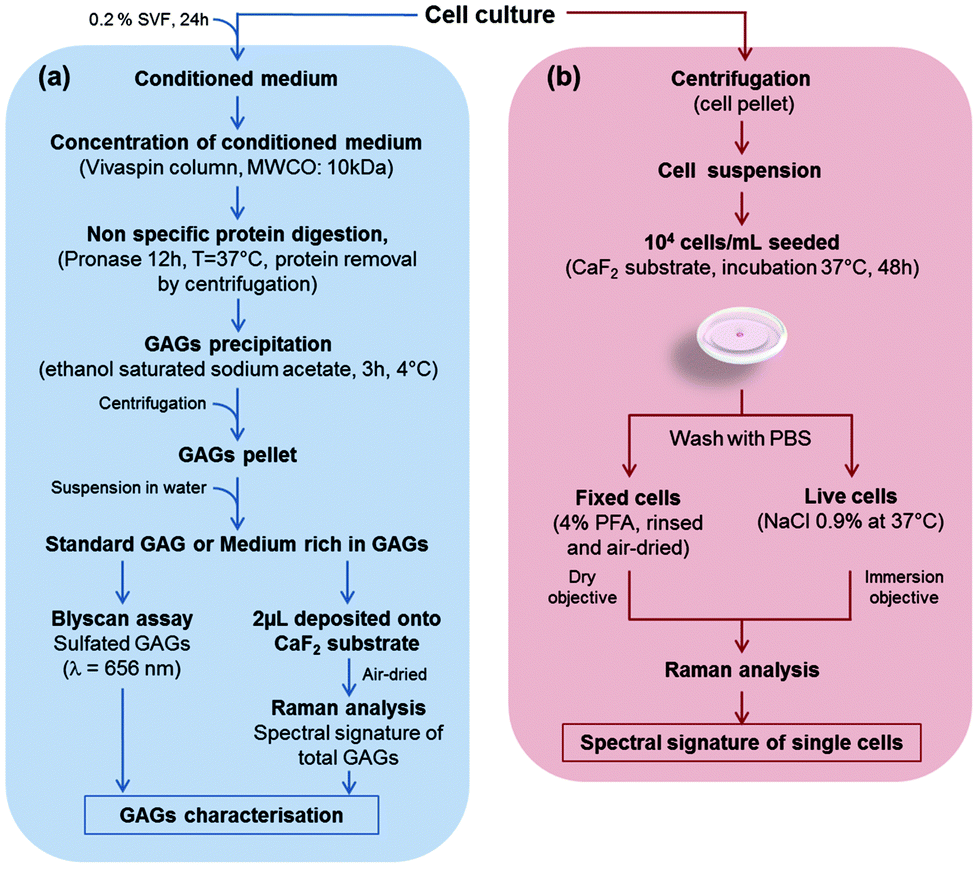 | ||
| Fig. 1 Biochemical and spectral analysis of GAGs from CHO-WT, CHO-745, SK-MEL-28, dermal fibroblasts and chondrocytes. (a) Workflow showing preparation, biochemical and Raman analyses of GAGs from conditioned media, (b) workflow showing preparation for Raman analysis of single fixed and live cells. | ||
2.2. Preparation of conditioned media
As depicted in Fig. 1a, when cell growth reached 80% confluence, all cells were serum starved for 24 h. Conditioned media of all cell types were harvested, concentrated after centrifugation (3220g, 20 min, 4 °C) on a Vivaspin™ column (10 kDa MWCO) and subjected to pronase (Sigma-Aldrich) digestion for 12 h at 37 °C to digest unspecifically proteins. The digested proteins–pronase mix was centrifuged (1700g, 10 min, 4 °C). From the supernatant, GAGs were precipitated with ethanol saturated with sodium acetate for 3 h at 4 °C. GAG pellets were suspended in sterile water. These GAG solutions were studied by both biochemical analysis and vibrational spectroscopy. For Raman analysis of conditioned media, one drop (2 μL each) per cell type was deposited on the calcium fluoride (CaF2) substrate and air-dried at room temperature.2.3. Glycosaminoglycan quantification
Sulfated glycosaminoglycan content was measured using a Blyscan™ assay (Biocolor Ltd, Westbury, NY, USA) according to the manufacturer's instructions. The addition of Blyscan™ dye reagent is used to precipitate the sulfated GAG–dye complex. A sulfated GAG standard (chondroitin 4-sulfate purified from bovine trachea) and the blank reagent (0 μg) were used to produce a calibration curve. More precisely, 12 μL of each conditioned medium obtained from the 5 cell cultures and the sulfated GAG standard (100 μg ml−1) from the Blyscan™ kit were used as follows: conditioned media and sulfated GAG standard were prepared by adding 12 μL of sample to 3 μL of 50 mM Tris-HCl buffer pH 7.5 (Fig. 1a). The sulfated GAG standard was diluted at 10, 20, 30, 40, and 50 μg mL−1. In all tubes, 500 μL of Blyscan™ dye reagent were added and the samples were mixed every 5 min for 30 min at RT. During this time period, a sulfated GAG–dye complex was formed and precipitated out from the soluble unbound dye. Then, the samples were centrifuged (10 min, 420g, RT) to pellet the precipitated sulfated GAGs linked to the dye reagent. The supernatant was discarded and 500 μL of dissociation agent were added. After strong shaking, the dye reagent was dissociated from sulfated GAGs. Then, 200 μL of each sample were withdrawn and loaded in duplicate on a 96-well microplate and ODs were measured at 656 nm on a microplate reader. The staining was proportional to the concentration of sulfated GAGs. The concentrations of the sulfated GAGs were calculated from the calibration curve. Three independent experiments were performed.2.4. Sample preparation for Raman microspectroscopy
The five different cell types were plated on a CaF2 substrate at 104 cells per mL and allowed to adhere (Fig. 1b). The CaF2 substrates used here were from the same batch and are specific for Raman measurements. For each experiment, ten spectra of the substrate were recorded under the same conditions as the sample. These spectra were stored in a file containing calibration spectra (silicon, neon lamp, NIST, optics, dark current). They were then used for preprocessing of raw Raman cell spectra as we reported previously.36 This procedure was repeated for each measurement. Since we use a modeling based on a report by Lasch,60 batch to batch variations were also taken into account.After 24 h of culture, two different protocols were applied for fixed and live cells. For fixed cell preparation, the cells seeded on CaF2 substrates were rinsed five times with Dulbecco's phosphate buffer saline (DPBS, Gibco). Cell fixation was performed using 4% paraformaldehyde (PFA, Thermo Fisher Scientific) for 30 min at room temperature. The cells were then rinsed with DPBS and distilled water to remove PFA, and then air-dried. The integrity of cell morphology was checked under a microscope before and after Raman measurement.
For live cell preparation, the cells were rinsed with 0.9% sterilized NaCl three times to remove the culture medium. Live cells were then immersed in 0.9% sterilized NaCl at 37 °C. Under such conditions, the integrity of cells is not compromised for an hour. Besides verifying that there were no morphological modifications, a trypan blue exclusion assay was also performed for testing cell viability.37,60,61
2.5. Raman data acquisition and analysis
Conditioned media and fixed cells were analysed with a LabRam Raman micro-spectrometer (Horiba Jobin Yvon, Villeneuve d'Ascq, France) equipped with a laser excitation at 660 nm, 950 lines per mm grating and using a confocal hole of 300 μm. The objective used on the microscope (Olympus BX40) was a 100× short working distance (NA: 0.9) giving a laser spot size of ∼1 μm.Live cells were analysed with an ARAMIS Raman micro-spectrometer (Horiba Jobin Yvon, Villeneuve d'Ascq, France) equipped with a laser excitation at 532 nm, 1200 lines per mm grating and using a confocal hole of 300 μm. The objective used on the microscope (Olympus BX41) was a 100× water immersion objective (NA: 1.0) giving a laser spot size of ∼1 μm.
For all measurements, the laser power at the sample was 28 mW.
The acquisition was performed in point mode in the spectral range of 600 to 1750 cm−1 with 30 s for conditioned media and 45 s for cells with a number of accumulation of 3 for fixed cells and 1 for live cells. A total of 35 spectra were recorded from each dried drop of conditioned media (Fig. ESC 1†). For fixed and live cells, four Raman spectra from their cytoplasms were recorded of nine different cells as shown in Fig. ESC 2.† Raman spectra were analysed using an in-house routine built in the Matlab software (MathWorks, Natick, MA, USA). First, spectra were smoothed using a Savitzky–Golay function (second polynomial order for live cells and third polynomial order for fixed cells and conditioned media) and corrected for instrument response. The instrument response and correction have been performed as previously described in detail.36 The baseline was corrected using a polynomial function (order 4) and vector normalised in the 600–1750 cm−1 spectral range. A principal component analysis (PCA) was performed using the spectral range 600–1350 cm−1 to focus on the spectral window containing carbohydrate information and to limit the importance of other components.
PCA is represented as a 2D scatterplot and the difference in spectral data can be observed in scores of the principal components (PCs) explaining the variance in the data.
3. Results and discussion
3.1. Quantification of sulfated GAGs in conditioned media
In order to assess the total amount of sulfated GAG synthesised by each cell type, a Blyscan™ assay was performed on the respective conditioned media as described in the Materials and methods section. Table 1 shows the concentration of sulfated GAGs for the five different cell conditioned media. The data show that chondrocytes, dermal fibroblasts and CHO-WT were capable of synthesising detectable levels of sulfated GAGs (3.39 ± 0.03 μg mL−1, 2.81 ± 0.02 μg mL−1, 0.96 ± 0.01 μg mL−1, respectively). For a better comparison, these values were normalised to the number of cells and ×107: 161.38 ± 1.38 μg per mL per cell for chondrocytes, 80.26 ± 0.60 μg per mL per cell for dermal fibroblasts and 7.99 ± 0.01 μg per mL per cell for CHO-WT. In contrast, the concentrations of sulfated GAGs in CHO-745 and SK-MEL-28 cells were below the detection limit (<0.25 μg mL−1) of the Blyscan™ assay and are not reported here. This assay shows that the chondrocytes secrete approximately 2-fold and 20-fold higher values of GAGs compared to dermal fibroblasts and CHO-WT, respectively.| Cell type | Mean ± SD (μg mL−1) | Mean ± SD (μg per mL per cell) × 107 |
|---|---|---|
| The results of the final concentration of total sulfated GAGs in conditioned media are expressed as mean ± SD (n = 3). nd: not determined (value below the detection limit). | ||
| CHO-745 | Nd | — |
| CHO-WT | 0.96 ± 0.01 | 7.99 ± 0.01 |
| Dermal fibroblasts | 2.81 ± 0.02 | 80.26 ± 0.60 |
| Chondrocytes | 3.39 ± 0.03 | 161.38 ± 1.38 |
| SK-MEL-28 | Nd | — |
3.2. Raman analysis of conditioned media of different cell types
The biochemical assay shows a difference in the synthesis of GAGs between the different cell types. However, this information only relates to sulfated GAGs. In order to have more molecular information on secreted GAGs, each conditioned medium was analysed by Raman spectroscopy.Fig. 2a displays the mean of 35 spectra recorded on dried drops of conditioned media obtained from different cell cultures: CHO-745 (green curve), CHO-WT (pink curve), SK-MEL-28 (black curve), dermal fibroblasts (blue curve), and chondrocytes (red curve). The spectra show very similar profiles and low variability between the different spectra of conditioned media (Fig. ESC 1†). Vibrational bands were attributed to polysaccharides and GAG-specific signatures. Some characteristic vibrational bands can be associated with GAGs like keratan sulfate, chondroitin sulfate, dermatan sulfate and hyaluronan. These spectra also show vibrations (1004, 1320, 1665 cm−1) of proteins due to a partial pronase digestion. To avoid some protein bands, the spectral range has been reduced in the following spectral analysis. Raman spectra were then analysed by PCA (Fig. 2b) in the spectral range 600–1350 cm−1 including sulfate and glucosidic related information. The PCA score plot using the first two PCs (81% of explained variance) is displayed in Fig. 2b for CHO-745 (green dots), CHO-WT (pink dots), SK-MEL-28 (black squares), dermal fibroblasts (blue triangles) and chondrocytes (red crosses). The first two PCs showed low intra-group variability and high inter-group variability enabling a good discrimination of the five cell types allowing a good separation between the conditioned media based on the spectral information from polysaccharides and GAG molecular vibrations. This information is associated with sulfate vibrational modes of GAGs at 853, 1001 and 1248 cm−1. Other vibrations at 860, 941, 1043, and 1137 cm−1 correspond to C–O–C of polysaccharides and C–OH of GAG respectively as shown by the loadings of PC1 and PC2 (Fig. 2c).
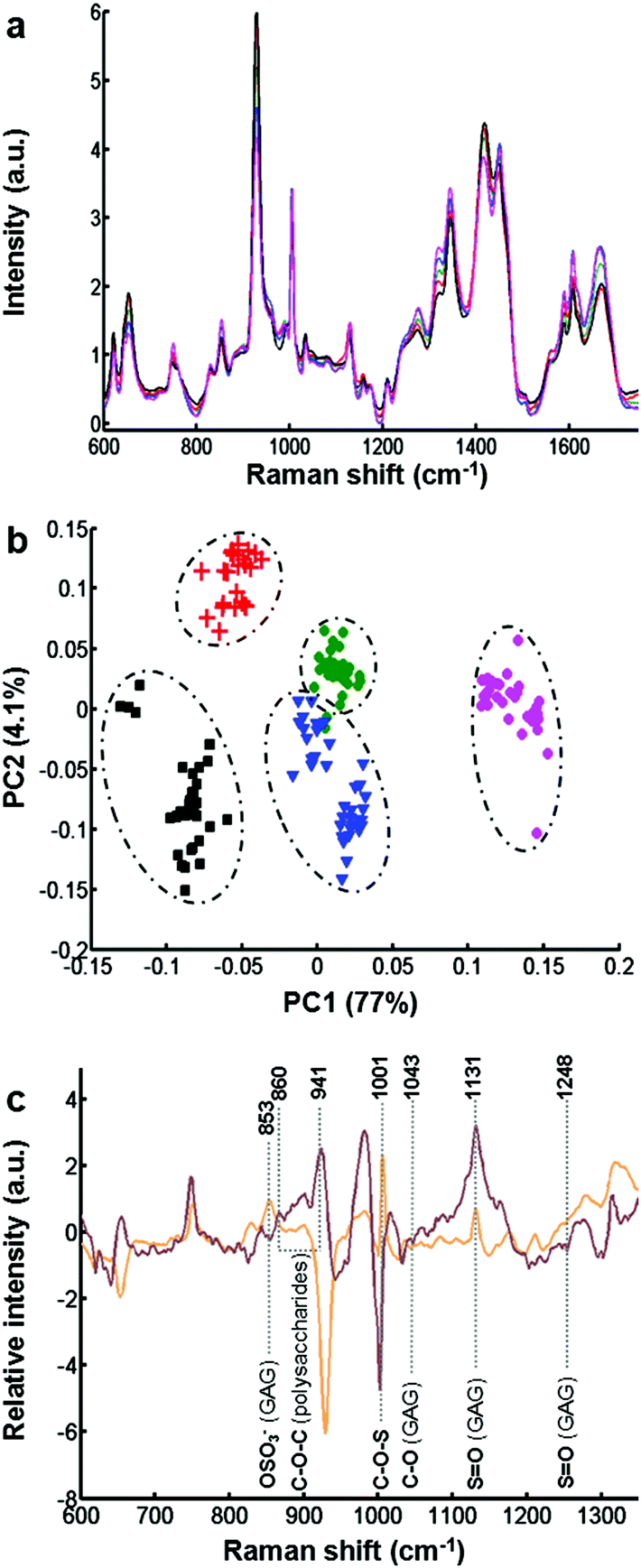 | ||
| Fig. 2 Raman analysis of conditioned media obtained from different cell types. (a) Raman spectra were recorded in the spectral range 600–1750 cm−1 on dried drops of conditioned media obtained from different cell types: CHO-745 (green curve), CHO-WT (pink curve), SK-MEL-28 (black curve), dermal fibroblasts (blue curve), and chondrocytes (red curve). (b) PCA score plot (PC1 vs. PC2) of conditioned media calculated in the 600–1350 cm−1 spectral range. CHO-745 (green circles), CHO-WT (pink circles), SK-MEL-28 (black squares), dermal fibroblasts (blue triangles), and chondrocytes (red crosses). (c) Loadings of PC1 (orange curve) and PC2 (brown curve) in the spectral range 600–1350 cm−1 showing specific GAG signatures (vertical grey lines). | ||
3.3 Raman microspectroscopy of fixed and live single cells
Following the results obtained from the conditioned media, further analysis was performed on single fixed and live cells to determine the capacity of these cell lines to synthesise GAGs. Raman analysis was performed on the cytoplasm of the different cells (n = 35 spectra for each cell line). For each cell line, the mean and standard deviation of Raman spectra of live cells are illustrated in Fig. ESC 2.† PCA was carried out on normalised spectra in the 600–1350 cm−1 spectral range. Fig. 3 shows the PCA score plot (PC1 vs. PC2) of the GAG spectra recorded from cytoplasms of fixed (Fig. 3a) and live (Fig. 3c) cell types: CHO-745 (green circles), CHO-WT (pink circles), SK-MEL-28 (black squares), dermal fibroblasts (blue triangles) and chondrocytes (red crosses). The first two PCs carry 66% and 71% of the total variance for fixed and live cells, respectively. PCA scores showed low intra-group variability and high inter-group variability sufficient to distinguish between different cell types. However, it can be noticed that CHO-745 (green circles) and CHO-WT (pink circles) spectra exhibited some overlapping in fixed cells (Fig. 3a) while in the case of live cells these two cell lines were well separated (Fig. 3c). Fig. 3b and d show the loadings of PC1 (orange curve) and PC2 (brown curve) corresponding to the PCA score plot of fixed (Fig. 3a) and live cells (Fig. 3b), respectively. Shaded regions of the loading plots (Fig. 3b and d) indicate the anti-correlations in the polysaccharides and GAG Raman signals. The separation was based on the differences observed in di- and polysaccharide vibrations (840–860, 941, 1090–1150 cm−1) and sulfate group vibrations (822, 1004, 1248 cm−1). Since Raman spectra were recorded from cell cytoplasms and PCA analysis was performed in the spectral range 600–1350 cm−1, the loadings show a weak correlation with lipids and nucleic acids.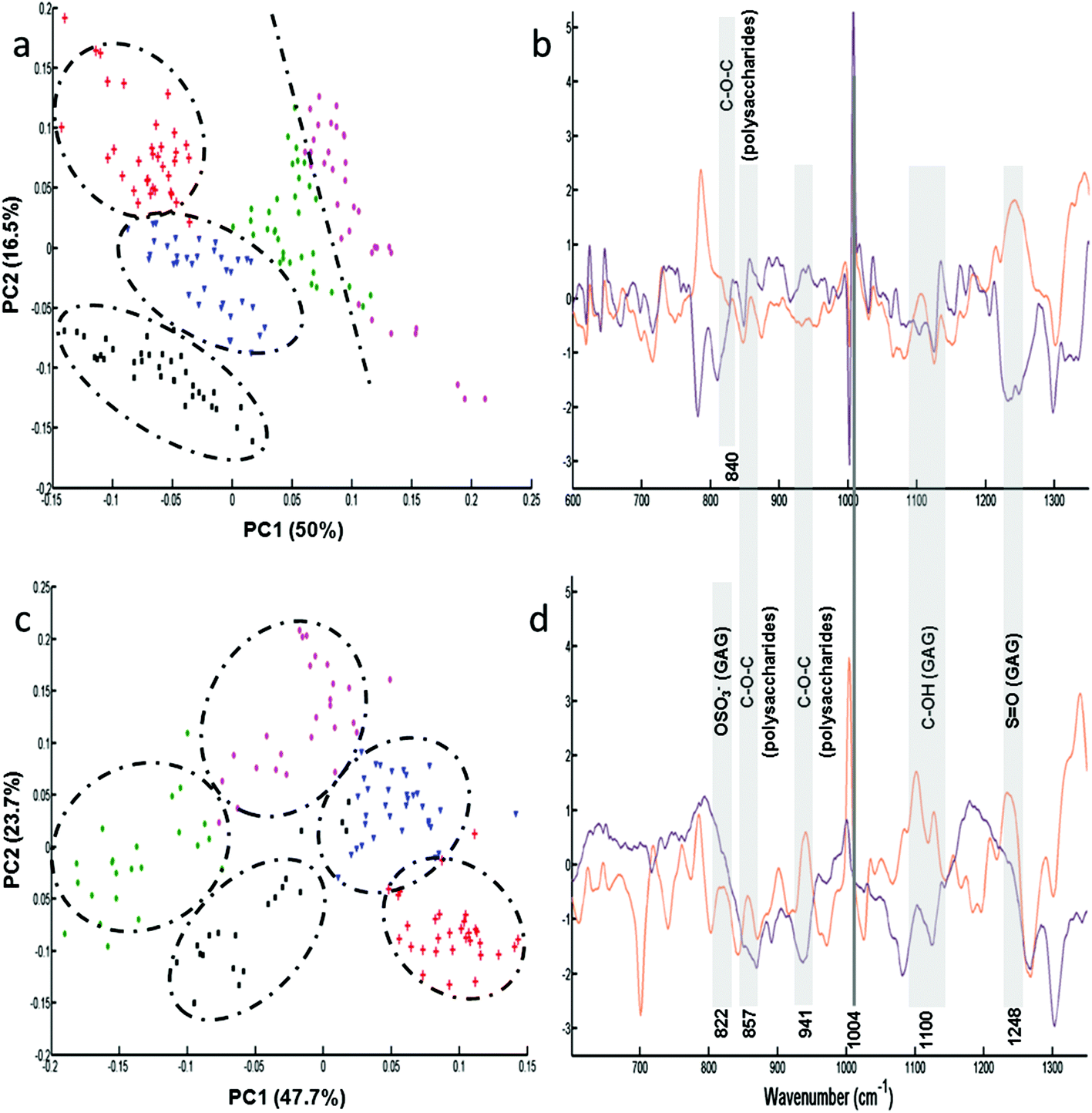 | ||
| Fig. 3 Raman analysis of fixed and live single cells. Micro-Raman spectra recorded from cytoplasms of fixed and live cells were analysed by PCA in the spectral range 600–1350 cm−1. (a) and (c) PCA score plots (PC1 vs. PC2) of fixed and live cells, respectively: CHO-745 (green circles), CHO-WT (pink circles), SK-MEL-28 (black squares), dermal fibroblasts (blue triangles) and chondrocytes (red crosses). (b) Loadings of PC1 (orange curve) and PC2 (brown curve) corresponding to PCA score plot (a); (d) loadings of PC1 (orange curve) and PC2 (brown curve) corresponding to PCA score plot (c). Vertical grey lines of the loading plots (c and d) highlight polysaccharides and GAG Raman signals. | ||
In order to further assign the spectroscopic signatures from live chondrocytes and CHO-745 cells and their corresponding conditioned media, their Raman signatures were compared in Fig. 4 with three specific standard GAGs (chondroitin sulfate, heparan sulfate, and hyaluronan). Chondroitin sulfate and heparan sulfate are known to be highly expressed in chondrocytes. Heparan sulfate was selected because it is not expressed in CHO-745 cells. In addition, chondroitin sulfate and heparan sulfate are highly sulfated GAGs in contrast to hyaluronan which is not sulfated. In the 820–1000 cm−1 region, the main difference between the two conditioned media is the additional peak appearing as a shoulder at 933 cm−1 in chondrocytes. Interestingly, this peak is also detected in the spectrum of chondroitin sulfate standard and can be assigned to skeletal C–O–C linkage vibrations.26 The 958 cm−1 peak which is common to both conditioned media is mainly assigned to C–O–C modes. The 999 cm−1 peak, also common to both, is found in chondroitin sulfate and heparan sulfate, and can be assigned to C–O–S modes. This mode is more active in Raman than in infrared. This peak is not present in hyaluronan as it is a non-sulfated GAG. Coincidently, a peak also appears at this frequency in the spectra of cells. However, in cells it can be easily identified as the phenylalanine peak due to its characteristic sharpness. In the 1000–1200 cm−1 region, the peak at 1065 cm−1 assigned to the OSO3− symmetric stretch is stronger in chondrocyte conditioned media. This peak is present in chondroitin sulfate and heparan sulfate standards and absent in hyaluronan. This mode is more active in Raman spectra while its asymmetric counterpart is more active in infrared spectra and observed around 1248 cm−1.28 The 1000–1200 cm−1 region is also characteristic of C–OH modes, clearly visible in the hyaluronan spectrum but overlaps with the sulfate modes in sulfated GAGs. Above 1200 cm−1, a peak at 1376 cm−1, characteristic of CH3 symmetric deformation, is observed in standard GAGs but not in the other spectra. Another interesting peak characteristic of GAGs is the symmetric COO− stretch of the glucuronate residue at 1411 cm−1. Its presence is important in chondrocyte conditioned media and standard GAGs while it is not visible in cell spectra. The latter show characteristic CH2 deformation peaks at 1452 cm−1 (lipids and proteins) as well as protein peaks corresponding to amide I and amide III at 1654 and 1230–1300 cm−1, respectively. The weak amide I peak also observed in standard GAGs and conditioned media could be due to the residual protein. Bansil and collaborators reported amide I and amide II signatures originating from aqueous solutions of GAGs.26
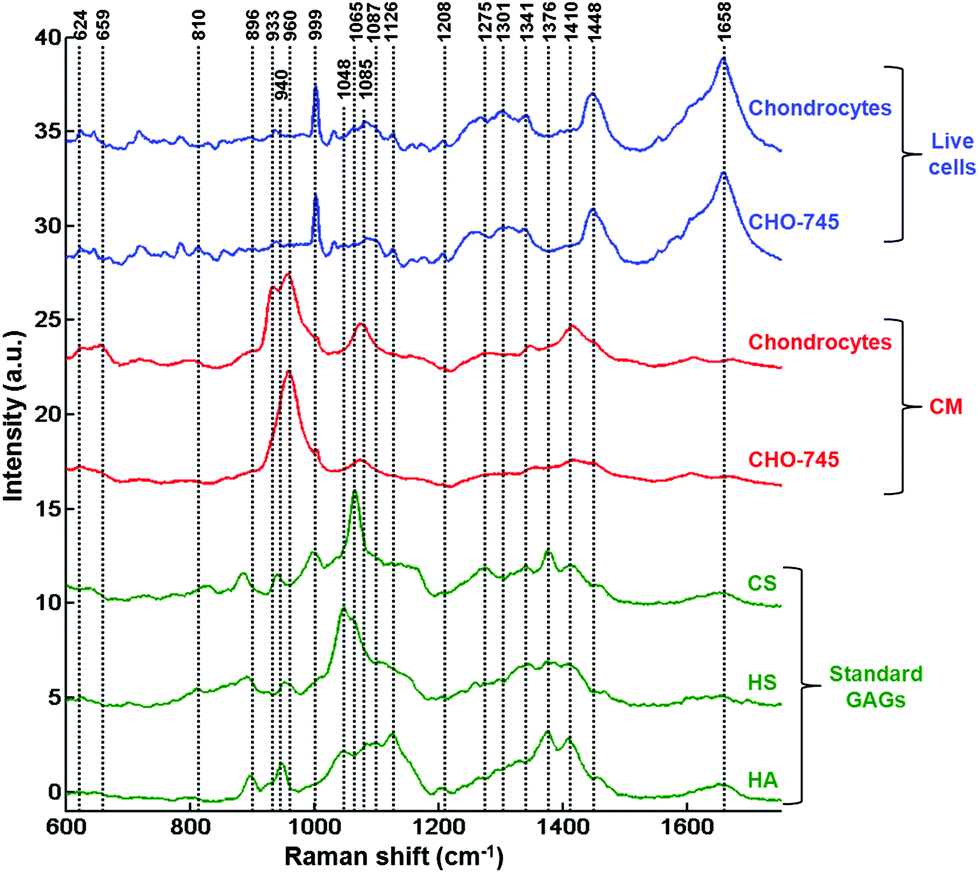 | ||
| Fig. 4 Comparison of Raman microspectroscopy spectra recorded from cytoplasms of single live cells, their corresponding conditioned media and standard GAGs. Mean Raman spectra of chondrocytes and CHO-745 cells (blue curves), conditioned media obtained from chondrocytes and CHO-745 cell cultures (red curves) and standard GAGs CS, HS, and HA (green curves). CS: chondroitin sulfate, HS: heparan sulfate, HA: hyaluronic acid. | ||
Our analysis shows that Raman microspectroscopy analysis of conditioned media can reveal characteristic GAG features directly from their spectra. However, due to the complex nature of the cell spectra, the GAG features are less visible in a direct manner from Raman spectra and therefore necessitate a multivariate approach like PCA where the PCs can reveal such information. It can be envisaged to better characterise the GAG features after isotopic labelling as it has been described previously with deuterium labelling of standard GAGs, disaccharide and tetrasaccharide fragments.62 However, even if the original structure was substantially retained, high performance liquid chromatography/size exclusion chromatography data indicated some depolymerisation of heparin and dermatan sulfate in the N-deacetylation step of the labelling reactions. Raman spectroscopy being a label-free approach, the structures of GAGs are not altered by the addition of extrinsic molecules and the technique represents a real advantage for GAG characterisation at the live cell level. A more interesting approach to reveal GAG spectral signatures without labelling could be to compare the expression of GAGs in non-stimulated and stimulated cells as it has been described for chondrocytes using TGFβ for inducing specific proteoglycan synthesis and sulfation.63 This second approach is being undertaken in an ongoing study.
4. Conclusions
Raman microspectroscopy was applied in this study to probe GAG spectral signatures both intracellularly from the cytoplasms of single cells and extracellularly from their corresponding conditioned media using five different cell types exhibiting varying levels of GAG synthesis. Standard GAGs were used as control samples for peak assignment. In parallel, biochemical assay showed that chondrocytes secreted approximately 2-fold and 20-fold higher values of sulfated GAGs in the conditioned media compared to dermal fibroblasts and CHO-WT, respectively. While the total sulfated GAGs of only these three cell types could be assessed biochemically, Raman spectral profiles gave information on both sulfated and non-sulfated GAGs that could be synthesised and secreted by all five cell types showing the potential of this approach. Thus, after PCA analysis of the Raman data, the results showed that the Raman technique was sensitive enough to clearly distinguish the conditioned media of the five cell types. The results were confirmed at the single cell level on both fixed and live cells with a better differentiation of these cell types under the physiological conditions. While characteristic GAG features could be directly identified from the conditioned media Raman spectra, a multivariate approach like PCA was necessary to observe the GAG features in cells due to the complex nature of the cell spectra. This study shows that Raman microspectroscopy allows extraction of GAG-related information at the single live cell level, in intra- and extracellular compartments, and could therefore be developed for cell screening purposes. Since GAGs have been reported to play a key role in tumour progression, identifying GAG spectral signatures can be perspectively interesting as a cancer biomarker in tissues.Acknowledgements
The authors thank the Reims PICT-IBiSA Platform and the Service de Chirurgie Orthopédique, CHU de Reims. The financial support of the Ligue Nationale contre le Cancer (Comité de la Marne et Comité de Haute-Marne, Conférence de Coordination InterRégionale du Grand Est (CCIR-GE)), the FEDER and the Région Champagne-Ardenne (CPER 2015–2019) is gratefully acknowledged. HTM acknowledges funding from RISE for secondment visit to URCA (Project GLYCANC No. 645756).References
- R. V. Iozzo and L. Schaefer, Matrix Biol., 2015, 42, 11–55 CrossRef CAS PubMed.
- U. Häcker, K. Nybakken and N. Perrimon, Nat. Rev. Mol. Cell Biol., 2005, 6, 530–541 CrossRef PubMed.
- D. Kawamura, T. Funakoshi, S. Mizumoto, K. Sugahara and N. Iwasaki, J. Orthop. Sci., 2014, 19, 1028–1035 CrossRef CAS PubMed.
- A. Xiong, S. Kundu and K. Forsberg-Nilsson, FEBS J., 2014, 281, 4993–5008 CrossRef CAS PubMed.
- D. Nikitovic, P. Katonis, A. Tsatsakis, N. K. Karamanos and G. N. Tzanakakis, IUBMB Life, 2008, 60, 818–823 CrossRef CAS PubMed.
- H. Hassan, B. Greve, M. S. Pavao, L. Kiesel, S. A. Ibrahim and M. Götte, FEBS J., 2013, 280, 2216–2227 CrossRef CAS PubMed.
- G. Frescaline, T. Bouderlique, L. Mansoor, G. Carpentier, B. Baroukh, F. Sineriz, M. Trouillas, J.-L. Saffar, J. Courty and J.-J. Lataillade, Tissue Eng., Part A, 2013, 19, 1641–1653 CrossRef CAS PubMed.
- B. Zhang, W. Xiao, H. Qiu, F. Zhang, H. A. Moniz, A. Jaworski, E. Condac, G. Gutierrez-Sanchez, C. Heiss and R. D. Clugston, J. Clin. Invest., 2014, 124, 209 CAS.
- M. Bernfield, M. Götte, P. W. Park, O. Reizes, M. L. Fitzgerald, J. Lincecum and M. Zako, Annu. Rev. Biochem., 1999, 68, 729–777 CrossRef CAS PubMed.
- N. Afratis, C. Gialeli, D. Nikitovic, T. Tsegenidis, E. Karousou, A. D. Theocharis, M. S. Pavão, G. N. Tzanakakis and N. K. Karamanos, FEBS J., 2012, 279, 1177–1197 CrossRef CAS PubMed.
- F. H. Blackhall, C. L. Merry, E. Davies and G. C. Jayson, Br. J. Cancer, 2001, 85, 1094 CrossRef CAS PubMed.
- R. Sasisekharan, Z. Shriver, G. Venkataraman and U. Narayanasami, Nat. Rev. Cancer, 2002, 2, 521–528 CrossRef CAS PubMed.
- H. Siiskonen, M. Poukka, K. Tyynelä-Korhonen, R. Sironen and S. Pasonen-Seppänen, BMC Cancer, 2013, 13, 1–12 CrossRef PubMed.
- I. Koshiishi, E. Horikoshi and T. Imanari, Anal. Biochem., 1999, 267, 222–226 CrossRef CAS PubMed.
- I. Koshiishi, M. Takenouchi, T. Hasegawa and T. Imanari, Anal. Biochem., 1998, 265, 49–54 CrossRef CAS PubMed.
- T. Oguma, H. Toyoda, T. Toida and T. Imanari, Biomed. Chromatogr., 2001, 15, 356–362 CrossRef CAS PubMed.
- A. H. Conrad, Y. Zhang, A. R. Walker, L. A. Olberding, A. Hanzlick, A. J. Zimmer, R. Morffi and G. W. Conrad, Invest. Ophthalmol. Visual Sci., 2006, 47, 120–132 Search PubMed.
- Y. Zhang, A. H. Conrad, E. S. Tasheva, K. An, L. M. Corpuz, Y. Kariya, K. Suzuki and G. W. Conrad, Invest. Ophthalmol. Visual Sci., 2005, 46, 1604–1614 Search PubMed.
- S. B. Frazier, K. A. Roodhouse, D. E. Hourcade and L. Zhang, Open Glycosci., 2008, 1, 31–39 CrossRef CAS PubMed.
- N. K. Karamanos and A. Hjerpe, Biomed. Chromatogr., 1999, 13, 507–512 CrossRef CAS PubMed.
- F. Lamari, M. Militsopoulou, T. Mitropoulou, A. Hjerpe and N. Karamanos, Biomed. Chromatogr., 2002, 16, 95–102 CrossRef CAS PubMed.
- A. H. Plaas, L. West, R. J. Midura and V. C. Hascall, Methods Mol. Biol., 2001, 171, 117–128 CAS.
- Y. Wegrowski and F.-X. Maquart, Proteoglycan. Protocols, 2001, vol. 171, pp. 175–179 Search PubMed.
- Y. Wegrowski, A. L. Milard, G. Kotlarz, E. Toulmonde, F. X. Maquart and J. Bernard, Clin. Exp. Immunol., 2006, 144, 485–493 CrossRef CAS PubMed.
- M. J. Baker, J. Trevisan, P. Bassan, R. Bhargava, H. J. Butler, K. M. Dorling, P. R. Fielden, S. W. Fogarty, N. J. Fullwood and K. A. Heys, Nat. Protoc., 2014, 9, 1771–1791 CrossRef CAS PubMed.
- R. Bansil, I. Yannas and H. Stanley, Biochim. Biophys. Acta, 1978, 541, 535–542 CrossRef CAS.
- T. R. Rudd, R. Hussain, G. Siligardi and E. A. Yates, Chem. Commun., 2010, 46, 4124–4126 RSC.
- N. Mainreck, S. Brézillon, G. D. Sockalingum, F. X. Maquart, M. Manfait and Y. Wegrowski, J. Pharm. Sci., 2011, 100, 441–450 CrossRef CAS PubMed.
- N. Mainreck, S. Brézillon, G. D. Sockalingum, F. X. Maquart, M. Manfait and Y. Wegrowski, in Proteoglycans, Springer, New-York, 2012, pp. 117–130 Search PubMed.
- G. J. Miller, S. U. Hansen, M. Baráth, C. Johannessen, E. W. Blanch, G. C. Jayson and J. M. Gardiner, Carbohydr. Res., 2014, 400, 44–53 CrossRef CAS PubMed.
- T. Ito, Y. Koyama and M. Otsuka, J. Pharm. Biomed. Anal., 2010, 51, 268–272 CrossRef CAS PubMed.
- T. Saxena, B. Deng, D. Stelzner, J. Hasenwinkel and J. Chaiken, J. Biomed. Opt., 2011, 16, 027003–027001 CrossRef PubMed.
- E. Kamilari, C. Kontoyannis, F. Lamari and M. Orkoula, 9th Panhellenic Scientific Chemical Engineering Congress, Athens, 2013.
- S. Brézillon, V. Untereiner, L. Lovergne, I. Tadeo, R. Noguera, F.-X. Maquart, Y. Wegrowski and G. D. Sockalingum, Anal. Bioanal. Chem., 2014, 406, 5795–5803 CrossRef PubMed.
- Z. Movasaghi, S. Rehman and I. U. Rehman, Appl. Spectrosc. Rev., 2007, 42, 493–541 CrossRef CAS.
- F. Draux, C. Gobinet, J. Sulé-Suso, A. Trussardi, M. Manfait, P. Jeannesson and G. D. Sockalingum, Anal. Bioanal. Chem., 2010, 397, 2727–2737 CrossRef CAS PubMed.
- F. Draux, P. Jeannesson, A. Beljebbar, A. Tfayli, N. Fourre, M. Manfait, J. Sulé-Suso and G. D. Sockalingum, Analyst, 2009, 134, 542–548 RSC.
- F. Draux, P. Jeannesson, C. Gobinet, J. Sule-Suso, J. Pijanka, C. Sandt, P. Dumas, M. Manfait and G. D. Sockalingum, Anal. Bioanal. Chem., 2009, 395, 2293–2301 CrossRef CAS PubMed.
- R. Kumar, G. P. Singh, K. M. Grønhaug, N. K. Afseth, C. de, L. Davies, J. O. Drogset and M. B. Lilledahl, Int. J. Mol. Sci., 2015, 16, 9341–9353 CrossRef CAS PubMed.
- D. Craig, S. McAughtrie, J. Simpson, C. McCraw, K. Faulds and D. Graham, Anal. Chem., 2014, 86, 4775–4782 CrossRef CAS PubMed.
- E. Brauchle, S. Noor, E. Holtorf, C. Garbe, K. Schenke-Layland and C. Busch, Clin. Exp. Dermatol., 2014, 39, 636–645 CrossRef CAS PubMed.
- K. Kong, C. Kendall, N. Stone and I. Notingher, Adv. Drug Delivery Rev., 2015, 89, 121–134 CrossRef CAS PubMed.
- N. Kourkoumelis, I. Balatsoukas, V. Moulia, A. Elka, G. Gaitanis and I. D. Bassukas, Int. J. Mol. Sci., 2015, 16, 14554–14570 CrossRef CAS PubMed.
- M. Sharma, E. Marple, J. Reichenberg and J. W. Tunnell, Rev. Sci. Instrum., 2014, 85, 083101 CrossRef PubMed.
- W. Wang, J. Zhao, M. Short and H. Zeng, J. Biophotonics, 2015, 8, 527–545 CrossRef PubMed.
- B. Bodanese, F. L. Silveira, R. A. Zangaro, M. T. T. Pacheco, C. A. Pasqualucci and L. Silveira Jr., Photomed. Laser Surg., 2012, 30, 381–387 CrossRef CAS PubMed.
- S. B. Cartaxo, I. D. d. A. O. Santos, R. Bitar, A. F. Oliveira, L. M. Ferreira, H. S. Martinho and A. A. Martin, Acta Cir. Bras., 2010, 25, 351–356 Search PubMed.
- L. Lim, B. Nichols, M. R. Migden, N. Rajaram, J. S. Reichenberg, M. K. Markey, M. I. Ross and J. W. Tunnell, J. Biomed. Opt., 2014, 19, 117003–117001 CrossRef PubMed.
- P. Philipsen, L. Knudsen, M. Gniadecka, M. Ravnbak and H. Wulf, Photochem. Photobiol. Sci., 2013, 12, 770–776 CAS.
- L. Silveira, F. L. Silveira, B. Bodanese, R. A. Zangaro and M. T. T. Pacheco, J. Biomed. Opt., 2012, 17, 077003–077001 CrossRef PubMed.
- H. Wang, T. H. Tsai, J. Zhao, A. Lee, B. K. K. Lo, M. Yu, H. Lui, D. I. McLean and H. Zeng, Photodermatol., Photoimmunol. Photomed., 2012, 28, 147–152 CrossRef CAS PubMed.
- C. Danciu, A. Falamas, C. Dehelean, C. Soica, H. Radeke, L. Barbu-Tudoran, F. Bojin, S. C. Pînzaru and M. F. Munteanu, Cancer Cell Int., 2013, 13, 1–12 CrossRef PubMed.
- T. Uyama, H. Kitagawa and K. Sugahara, Compr. Glycosci., 2007, 3, 79–104 CAS.
- A. Maroudas and K. E. Kuettner, Methods in carticlage research, ed. K. K. Maroudas, Academic Press, London, 1990, pp. 209–239 Search PubMed.
- M. T. Bayliss and S. Y. Ali, Biochem. J., 1978, 176, 683–693 CrossRef CAS PubMed.
- M. T. Bayliss, M. Venn, A. Maroudas and S. Y. Ali, Biochem. J., 1983, 209, 387–400 CrossRef CAS PubMed.
- C. B. Knudson and W. Knudson, Semin. Cell Dev. Biol., 2001, 12, 69–78 CrossRef CAS PubMed.
- J. D. Esko, T. E. Stewart and W. H. Taylor, Proc. Natl. Acad. Sci. U. S. A., 1985, 82, 3197–3201 CrossRef CAS.
- C. R. Franco, H. A. Rocha, E. S. Trindade, I. A. Santos, E. L. Leite, S. S. Veiga, H. B. Nader and C. P. Dietrich, Braz. J. Med. Biol. Res., 2001, 34, 971–975 CrossRef CAS PubMed.
- P. Lasch, Chemom. Intell. Lab. Syst., 2012, 117, 100–114 CrossRef CAS.
- A. Azan, V. Untereiner, C. Gobinet, G. D. Sockalingum, M. Breton, O. Piot and L. M. Mir, Sci. Rep., 2017, 7, 40448 CrossRef CAS PubMed.
- A. Naggi, B. Casu, B. Crippa, S. Magnaghi, L. Silvestro and G. Torri, Semin. Thromb. Hemostasis, 1994, 20, 168–175 CrossRef CAS PubMed.
- R. Chavez, G. Coricor, J. Perez, H.-S. Seo and R. Serra, Osteoarthr. Cartil., 2016, 30318–30311 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6an01951j |
| ‡ The authors contributed equally to this work and project supervision. |
| § The authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |