
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthetic control over intra- and intermolecular charge transfer can turn on the fluorescence emission of non-emissive coumarin†
Byung Hak
Jhun
a,
Kei
Ohkubo
bc,
Shunichi
Fukuzumi
*cd and
Youngmin
You
*a
aDivision of Chemical Engineering and Materials Science, Ewha Womans University, Seoul 03760, Korea. E-mail: odds2@ewha.ac.kr
bDepartment of Material and Life Science, Graduate School of Engineering, Osaka University, ALCA and SENTAN, Japan Science and Technology (JST), Suita, Osaka 565-0871, Japan. E-mail: fukuzumi@chem.eng.osaka-u.ac.jp
cDepartment of Chemistry and Nano Science, Ewha Womans University, Seoul 03760, Korea
dFaculty of Science and Engineering, Meijo University, ALCA and SENTAN, Japan Science and Technology Agency (JST), Nagoya, Aichi 468-0073, Japan
First published on 6th April 2016
Abstract
Conjugated molecules bearing carbonyl groups typically exhibit weak fluorescence emission due to the presence of a non-radiative n–π* transition state. Strong fluorescence emission from n–π* chromophores has been sought through the use of synthetic approaches that incorporate strong electron donors, such as amines, into the conjugated structures. As an alternative to these existing approaches, we investigated two charge-transfer strategies using a series of 1-benzopyran-2-one (coumarin) derivatives. The first strategy involved attaching chromophoric aryl moieties at the 7 position of coumarin. This molecular control produced two effects: the n–π* transition state was destabilized and an intramolecular charge-transfer (ICT) state was generated. The photoluminescence quantum yields (PLQYs) of the bichromophoric dyads increased with the π-conjugation length of the aryl groups, and a PLQY as high as 0.80 was achieved. The second strategy facilitated exciplex fluorescence in poly(N-vinylcarbazole) (PVK) films within which coumarin derivatives have been molecularly dispersed. A thermodynamic analysis based on electrochemical data indicated that exciplex generation involved electron transfer from PVK to photoexcited coumarin molecules. Exciplex fluorescence was uniquely advantageous in its ability to tune the fluorescence emission color upon addition of electron donors having oxidation potentials less positive than that of PVK. Mechanistic studies, including femtosecond laser flash photolysis, were conducted to identify the molecular parameters that governed the two fluorescence properties. A mechanistic understanding may provide useful insights into the development of electrofluorescent materials that harness triplet as well as singlet excitons.
Introduction
A variety of fluorescent compounds have been developed based on the 1-benzopyran-2-one (coumarin) platform.1,2 This platform is non-fluorescent and provides a photoluminescence quantum yield (PLQY) below 0.001 in cyclohexane.3,4 The non-emissive properties of coumarin originate from the presence of a carbonyl moiety. The photophysical mechanism by which carbonyl groups quench fluorescence emission remains an intensive research subject. Previous studies provided evidence that the n–π* transition in the carbonyl group plays a central role in quenching radiation through adiabatic relaxation,5 intersystem crossing,6 vibronic deactivation,7 and irreversible ring opening of the lactone.7 Strong emission may be achieved by modifying coumarin to incorporate electron-donating groups, such as amino and hydroxyl groups. This modification facilitates strong charge separation, generating an emissive intramolecular charge-transfer (ICT) state. Synthetic modifications involve condensation reactions that require boiling temperatures and strong acids. Although recent advances have introduced significant versatility into the syntheses of modified coumarins,2,8–18 molecular libraries have not been fully developed. Furthermore, the amino and hydroxyl groups render the coumarin fluorescence emission extremely sensitive to substitution patterns and the rigidity and the Lewis acidity of the medium.19 Therefore, the development of versatile principles for harnessing fluorescence emission from a non-emissive coumarin backbone continues to pose significant challenges.One potential approach to addressing this challenge may involve replacing the amino and hydroxyl groups with aryl units. Aromatic hydrocarbons with extended π conjugation can donate electrons to the electron-deficient coumarin upon photoexcitation, promoting an ICT transition. Depending on the oxidation potentials of the aromatic units, the ICT transition state may be located below the n–π* transition state. Aromatic units with low band gap energies may align energetically with the coumarin levels to enable this radiative process. This ICT fluorescence is advantageous over the fluorescence produced by other coumarin derivatives bearing amino or hydroxyl groups. The fluorescence transition probability may be enhanced because the ICT transition state involves an allowed π–π* transition in the aromatic component. In addition, this strategy does not rely on the use of heteroatoms with lone-pair electrons, obviating the dependence of the fluorescence emission on the solvent medium. Facilitated ICT fluorescence is enabled by introducing aromatic units with judiciously controlled oxidation potentials and π–π* transition energies.
An alternative approach to harnessing the fluorescence emission of a non-emissive coumarin backbone involves enabling exciplex formation. Exciplexes are excited-state bimolecular complexes formed during excited-state electron transfer.20 The energy level of an exciplex, which is defined by the difference between the electrochemical potentials of an electron donor and an electron acceptor, is usually located below the excitonic states of the individual molecules.21,22 Exciplex fluorescence offers a viable strategy for harnessing the excited-state energy of the non-emissive n–π* transition state in coumarin. Previous studies have identified exciplex fluorescence in binary mixtures of semiconducting polymers and electron-deficient materials, such as poly(benzothiadiazole),20,23 poly(benzobisthiazole),24 poly(quinoline),25,26 cyano-substituted poly(p-phenylenevinylene),27,28 1,4-dicyanobenzene,29 a cyclometalated Ir(III) complex,30 and derivatives of cyano-substituted stilbene,31 diaryloxadiazole,32 quinoxaline,33 phenanthroline,34 and benzanthrone.35 Recent studies suggested that exciplex formation played a decisive role in electroluminescence.36–51 We envisioned that an electron-deficient coumarin could produce exciplex when embedded in electron-rich polymers, such as poly(N-vinylcarbazole) (PVK). Indeed, a positive driving force as large as 0.66 eV was calculated for electron transfer from PVK to the lowest singlet excited state of coumarin (vide infra). However, to our surprise, few examples of exciplexes in coumarin compounds have been reported to date, and all the previous reports lacked spectroscopic studies.52–56
Herein, we report the execution of two prototypical strategies for turning on the fluorescence of coumarin derivatives (Scheme 1). Photoinduced charge transfer was exploited in bichromophoric dyads involving coumarin and aromatic hydrocarbons, as well as in PVK films molecularly doped with a series of coumarins, to obtain strong fluorescence emission. A series of coumarin compounds with aryl rings of increasing π conjugation lengths were prepared (Fig. 1), and their ICT and exciplex fluorescence behaviors were systematically investigated. A photoluminescence quantum yield as high as 0.80 was obtained using the ICT approach. The exciplex strategy enabled strong and color-tuned fluorescence emission without synthetically modifying the coumarins. Mechanistic studies, including femtosecond laser flash photolysis experiments and quantum chemical calculations based on time-dependent density functional theory, were conducted to characterize the structure–property relationships that governed the intramolecular and intermolecular charge-transfer fluorescence. Our studies provide valuable insights into the future development of electrofluorescent materials.
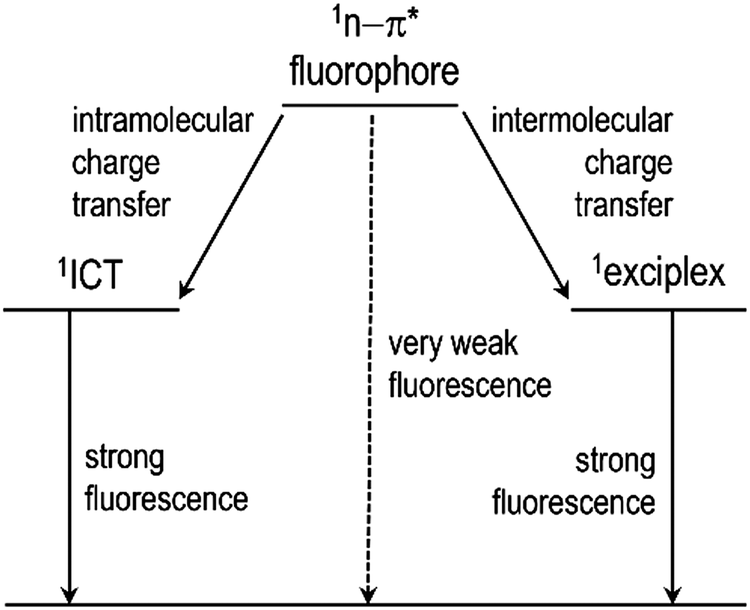 | ||
| Scheme 1 Two molecular strategies for achieving strong fluorescence emission from non-fluorescent n–π* fluorophores. | ||
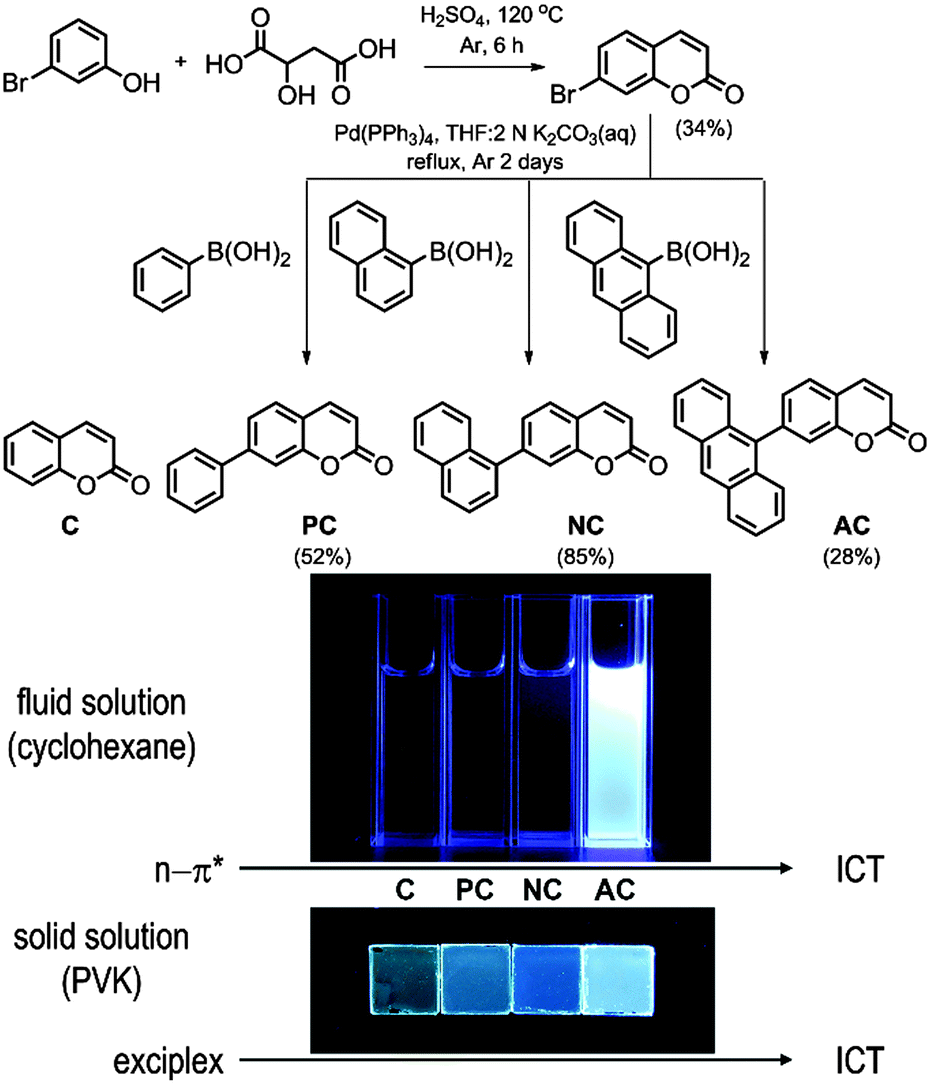 | ||
| Fig. 1 Syntheses and fluorescence emission properties of the coumarin compounds. | ||
Results and discussion
Molecular design and synthesis
Fig. 1 depicts the chemical structures of the coumarin molecules examined in this study. A series of coumarin compounds were designed to incorporate aryl rings with increasing π-conjugation lengths. Phenyl (PC), 1-naphthyl (NC), and 9-anthryl (AC) moieties were introduced at the 7 position of 1-benzopyran-2-one (C). Structural control was used to precisely tailor the electrochemical potentials of the resulting bichromophoric coumarins. Specifically, variations in the aryl groups decreased the ground-state oxidation potential (Eox) in the following order: C > PC > NC > AC, without significantly altering the ground-state reduction potential (Ered) of the coumarin derivatives. As a consequence, the ICT transition state energy, which was primarily estimated based on Eox − Ered, decreased from PC to AC. This energetic shift eventually situated the ICT transition state below the non-fluorescent n–π* transition state, thereby activating a fluorescence channel.Synthetic control over the excited states was designed based on quantum chemical calculations using time-dependent density functional theory (TD-DFT, CAM-B3LYP/6-311+G(d,p)//TD-CAM-B3LYP/6-311+G(d,p):CPCM(THF)). Twenty singlet and triplet states were modeled, and their electronic energies are depicted in Fig. 2. The lowest singlet states (S1) were predicted to predominantly possess π–π* character, except for AC, due to the use of the conductor-like polarizable continuum model (CPCM) parameterized for polar THF. The orthogonal disposition of the aryl ring and the coumarin plane disrupted conjugation across the entire molecule and localized the π–π* transition within the coumarin moiety (ESI,† Fig. S1). Interestingly, increased π conjugation in the aryl ring increased the singlet n–π* transition energies, likely due to stabilization of the non-bonding orbital on the carbonyl group. In contrast, the ICT transition energies decreased with the conjugation length. This trend persisted across the coumarin series and eventually the lowest singlet state in AC was predominantly ICT in nature. A summary of the TD-DFT calculation results for C, PC, NC, and AC is listed in the ESI,† Table S1.
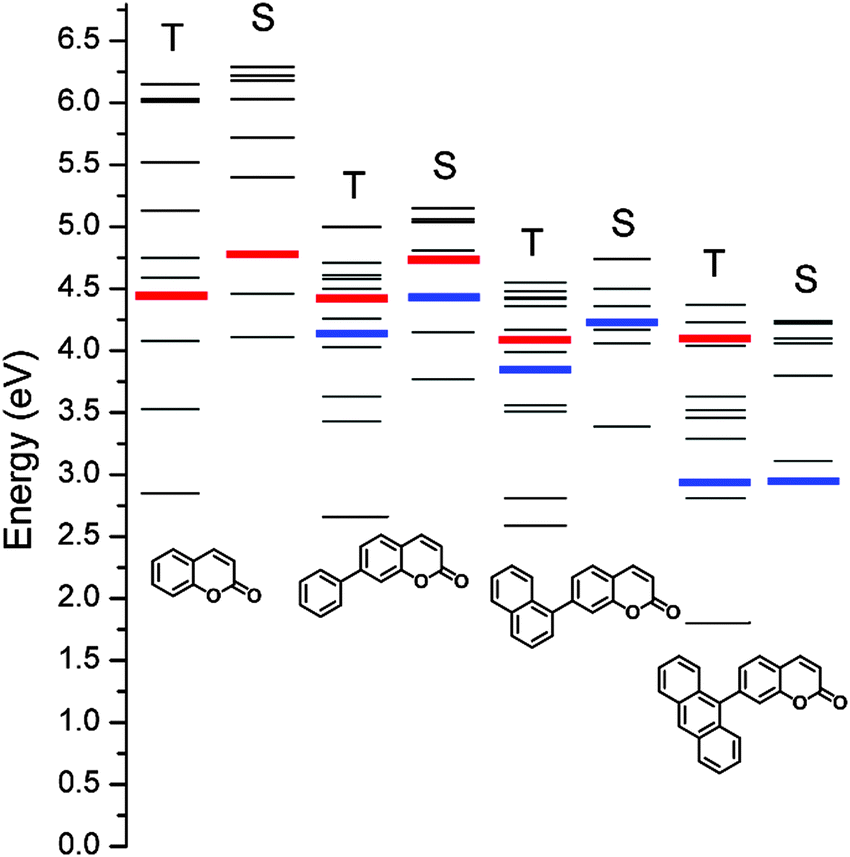 | ||
| Fig. 2 Schematic representation of the calculated (CAM-B3LYP/6-311+G(d,p)//TD-CAM-B3LYP/6-311+G(d,p):CPCM(THF)) energies of the singlet (S) and triplet (T) excited states of the coumarin compounds. Red and blue bars indicate n–π* and ICT transition states, respectively. | ||
Control over the aryl group was intimately linked to the formation of the exciplex. Because the excited-state reduction potential (Ered*) was estimated as Ered – band gap energy, the evolution of the aryl groups from phenyl (PC) to 9-anthryl (AC) led to a cathodic shift in Ered*. The free energy change for electron transfer from PVK to the excited-state coumarin compounds (ΔGeT) was calculated using ΔGeT = −e·[Eox(PVK) − Ered*(coumarin)]. Therefore, AC, which was characterized by the smallest Ered*, was expected to be least predisposed to exciplex formation, whereas C, characterized by the largest bandgap energy (i.e., the largest Ered*), favored exciplex formation.
The molecules were synthesized through a two-step procedure, which included a Pechmann condensation of m-bromophenol and malic acid, followed by Pd(0)-catalyzed Suzuki–Miyaura coupling using commercially available aryl boronic acid. The purified compounds were characterized by multi-nuclear NMR spectroscopy, high-resolution mass spectrometry, and elemental analysis. The characterization data were fully consistent with the proposed structures. The synthetic details and structural identification results are summarized in Experimental details.
Fluorescence turn-on by intramolecular charge transfer
With the exception of AC, the coumarin derivatives were minimally fluorescent in cyclohexane solutions at 298 K. This observation was consistent with the calculated predictions and previous studies, which suggested that a non-radiative n–π* transition provided the dominant channel for excited-state relaxation in non-polar solvents.3,4 Indeed, the photoluminescence quantum yield (PLQY, 9,10-diphenylanthracene standard) remained as low as 0.005 for C. The PLQY value increased in proportion to the conjugation length of the aryl ring (Table 1). An identical trend was found in the photoluminescence lifetimes (τobs) recorded at the photoluminescence peak wavelengths after picosecond pulsed photoexcitation at 377 nm (Table 1). τobs increased from 0.098 ns for C to 2.8 ns for AC. The increase was ascribed to the diminished influence of the non-emissive n–π* transition, as predicted by the TD-DFT calculations. The non-radiative rates (knr, knr = (1 − PLQY)/τobs) decreased as the number of fused phenyl rings in the aryl groups increased: C, 1.0 × 1010 s−1; PC, 8.2 × 109 s−1; NC, 2.6 × 109 s−1; and AC, 7.1 × 107 s−1.| Fluid solution | Solid solutiona | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| λ abs (nm; ε, 104 M−1 cm−1) | ΔET (eV) | E ox (V vs. SCE) | E red (V vs. SCE) | λ ems (nm) | PLQYb,f | τ obs (ns) | λ ems (nm) | PLQYh | τ avg (ns) | |
| a 10 wt% in PVK, 298 K. b 10 μM in cyclohexane solutions, 298 K. c 50 μM in 2-MeTHF, 78 K. d 50 μM in iodoethane, 78 K. e Determined by cyclic and differential pulse voltammetry using 2.0 mM CH3CN solutions containing 0.10 M TBAPF6. Pt working and counter electrodes, and an Ag/AgNO3 pseudo reference electrode were employed. Scan rate = 0.10 V s−1 (cyclic voltammetry) and 4.0 mV s−1 (differential pulse voltammetry). f Photoluminescence quantum yields were determined relative to that of a 9,10-diphenylanthracene standard (toluene, PLQY = 1.00).57 g Photoluminescence decay traces for 50 μM coumarin compounds in Ar-saturated toluene solutions or PVK films doped with 10 wt% coumarin compounds were monitored at the emission maximum after picosecond pulsed laser photoexcitation at 377 nm. The photoluminescence lifetime (τobs) values were determined through nonlinear least-squares fits to monoexponential (toluene solutions) or triexponential (PVK films) decay models. The weighted average lifetime (τavg) of triexponential decay traces was calculated using τavg = (∑ai·τi2)/(∑ai·τi) (i = 1–3), where ai and τi are the pre-exponential factor and the time constant, respectively. h Photoluminescence quantum yields determined absolutely by integrating over a sphere. The measurements were performed in triplicate. | ||||||||||
| C | 311 (0.53) | 2.73c | 2.27 | −1.75 | 388 | 0.005 | 0.098 | 458 | 0.02 ± 0.002 | 23 |
| PC | 327 (1.57) | 2.55d | 1.86 | −1.66 | 396 | 0.012 | 0.12 | 469 | 0.05 ± 0.002 | 24 |
| NC | 327 (1.50) | 2.55d | 1.60 | −1.68 | 402 | 0.081 | 0.36 | 468 | 0.04 ± 0.01 | 21 |
| AC | 385 (0.61) | N.A. | 1.24 | −1.67 | 444 | 0.80 | 2.8 | 457 | 0.18 ± 0.006 | 3.1 |
The fluorescence properties of AC differed markedly from those of the other coumarin compounds. The photoluminescence emission spectrum exhibited a large 2300 cm−1 bathochromic shift relative to the spectra of the other coumarin compounds (Fig. 3). The spectral profile of AC was broad, unlike the vibronically resolved fluorescence spectrum of 9,10-diphenylanthracene. These properties suggested that an ICT transition was responsible for the observed fluorescence of AC. Strong solvatochromism, as demonstrated by the Lippert–Mataga plot, corroborated this hypothesis (ESI,† Fig. S2). It should be noted, however, that the extent of the solvatochromic shift was not as large as the shifts observed for conventional coumarin molecules bearing strong electron donors.2 Cyclic and differential pulse voltammetry measurements revealed a cathodic shift in the oxidation potential of AC (Eox = 1.24 V vs. SCE) relative to the Eox values (1.60–2.27 V vs. SCE) of the other coumarin compounds (Table 1; see the ESI,† Fig. S3 for the cyclic and differential pulse voltammograms), further supporting the presence of ICT fluorescence. The ICT fluorescence was very bright, with a PLQY as high as 0.80. This unusually high PLQY value can be ascribed to effective suppression of non-radiative transition. Indeed, the knr value (7.1 × 107 s−1) was two orders of magnitude smaller than the values (109–1010 s−1) obtained for the other coumarins. It is likely that the non-radiative control dominated over weak radiative transition of the ICT fluorescence. Taken together, these results demonstrated that the incorporation of anthracene boosted the fluorescence intensity by activating an ICT transition.
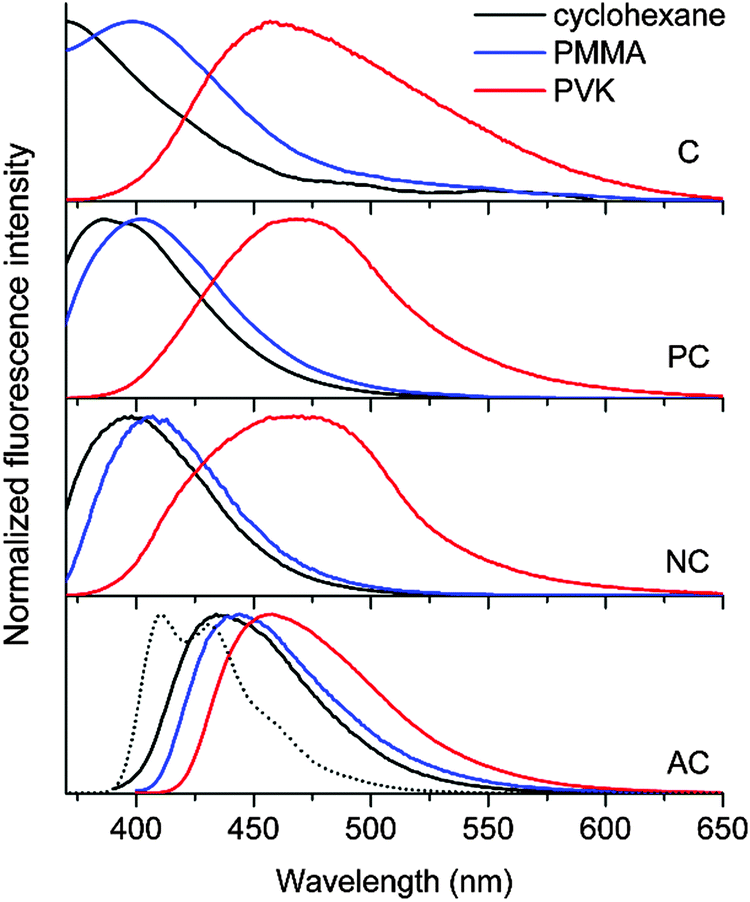 | ||
| Fig. 3 Photoluminescence spectra of the coumarin compounds in fluid (10 μM in cyclohexane (black)) or solid (10 wt% in PMMA (blue) and PVK (red)) solutions. The fluorescence spectrum of 9,10-diphenylanthracene is shown for comparison (dotted black curve). The spectra obtained in cyclohexane were truncated due to a significant overlap with the excitation beam (375 nm). | ||
Fluorescence turn-on by exciplex formation
The non-fluorescent coumarin compounds, C, PC, and NC, became emissive when they were doped into PVK at 10 wt% (Fig. 1). As shown in Fig. 3, the fluorescence peak wavelengths (λems) exhibited bathochromic shifts of 3500–3900 cm−1 relative to the solution spectra. The shifts did not result from planarization of the π frameworks in the solid state because much smaller bathochromic shifts of 300–700 cm−1 were observed in poly(methyl methacrylate) (PMMA) films doped with C, PC, or NC at identical doping concentrations (10 wt%). The PLQY values determined absolutely by integrating over a sphere were 0.02, 0.05, and 0.04 for C, PC, and NC, respectively. These values were one order of magnitude better than the PLQY values obtained in cyclohexane solutions. By contrast, the PLQY values of the PMMA films containing 10 wt% coumarin compounds were relatively low: C, 0; PC, 0.019; and NC, 0.027, suggesting that rigidification did not significantly contribute to the fluorescence observed in the PVK films.58The λems and PLQY values obtained from the PVK films were compared with the values obtained from the cyclohexane solutions and from the PMMA films, suggesting that the fluorescence emission did not originate from an excitonic state (a π–π* transition state or an ICT transition state) in the coumarin compounds. The photoluminescence peak wavelengths varied linearly with the difference between Eox(PVK) (1.18 V vs. SCE)59 and the ground-state reduction potential (Ered: C, −1.75 V; PC, −1.66 V; NC, −1.68 V vs. SCE) of the coumarin derivatives (Table 1). The UV-vis absorption spectrum of C in the 2-MeTHF solution (10 μM) was compared with the spectra obtained from the PMMA (10 wt%) and PVK (10 wt%) solid solutions to reveal the absence of ground-state charge-transfer interactions between the coumarin derivatives and PVK (Fig. 4).
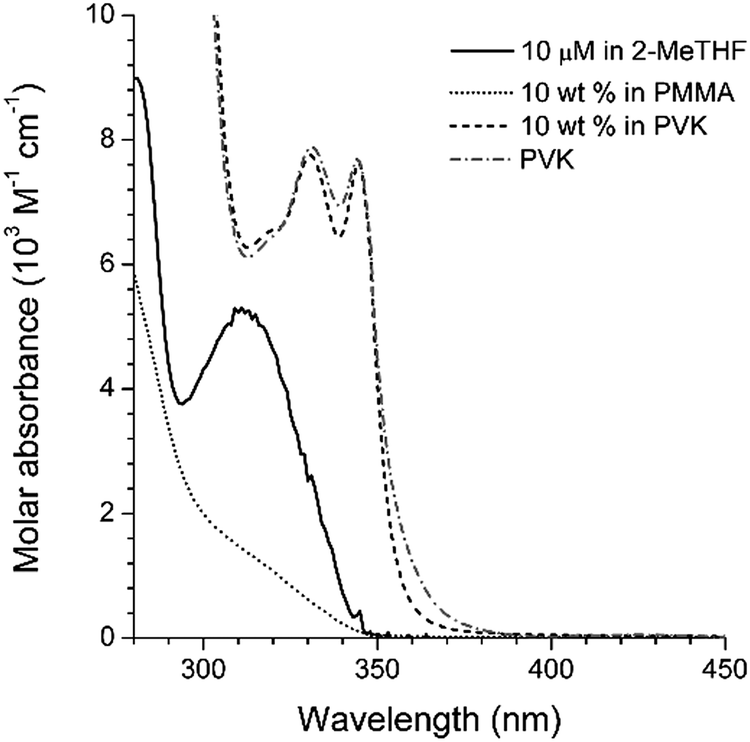 | ||
| Fig. 4 UV-vis absorption spectra of C measured in fluid (10 μM in 2-MeTHF (solid black line)) or solid (10 wt% in PMMA (dotted grey line) or PVK (dashed black line)) solutions. The absorption spectrum of PVK (dashed grey line) is included for comparison. See the ESI,† Fig. S4 for the spectra of the other compounds. | ||
To gain additional information about the nature of the fluorescence emission, photoluminescence decays were monitored for the coumarin-doped (10 wt%) PVK films at the emission peak wavelengths after picosecond pulsed laser photoexcitation at 377 nm. The decay trace of a PVK film containing NC exhibited a multi-exponential decay behavior (Fig. 5). This multiphasic decay markedly differed from those of NC in a fluid solution (50 μM in Ar-saturated toluene) and a PMMA film (10 wt%), which adhered to a monoexponential decay model with τobs values in the subnanosecond ranges. A weighted average photoluminescence lifetime of the PVK film of NC was 21 ns, being two orders of magnitude longer than those of the solution (0.36 ns; Table 1). The transient photophysical properties were consistent with the previous observations of exciplex fluorescence.23,31,60,61 The exciplex fluorescence featured several unique properties: (1) prolonged lifetimes of the emissive state, and (2) emission color which was determined by the relative position of electrochemical potentials of individual components (i.e., coumarin and PVK). Inspection of Table 1 reveals interesting trends; the τobs values of the fluid solutions increased with the π conjugation lengths of the aryl ring (C, 0.098 ns; PC, 0.12 ns; NC, 0.36 ns; and AC, 2.8 ns). This observation could be explained on the basis of the decreasing contribution of n–π* transition for the coumarin compounds with π-extended rings, as explained earlier. The influence of the fluorescence quenching n–π* transition was suppressed in PVK, because emissive exciplex species provided the lowest energy path for excited-state relaxation. In fact, such non-radiative control yielded the observed trend in the ratio of PLQY(PVK)/PLQY(cyclohexane). For instance, C with the strong quenching contribution by n–π* transition in fluid solutions showed the largest value for the ratio (12) of PLQY(PVK)/PLQY(cyclohexane). The ratio decreased with the conjugation length of the aryl rings: PC, 4.2; NC, 0.49. In the case of AC, the fluorescence properties were not affected by PVK. The emission peak wavelength of the PVK film (10 wt% doping ratio, 457 nm) did not exhibit a significant bathochromic shift from that (444 nm) of the cyclohexane solution. Similarly, the τobs value (3.1 ns) was nearly identical to that obtained in the cyclohexane solution (2.8 ns). Time-resolved photoluminescence spectra of PVK films doped with either 10 wt% PC or 10 wt% AC revealed contrasting behaviors (Fig. 6). The retention of the fluorescence properties may be indicative of the absence of the exciplex channel in AC.
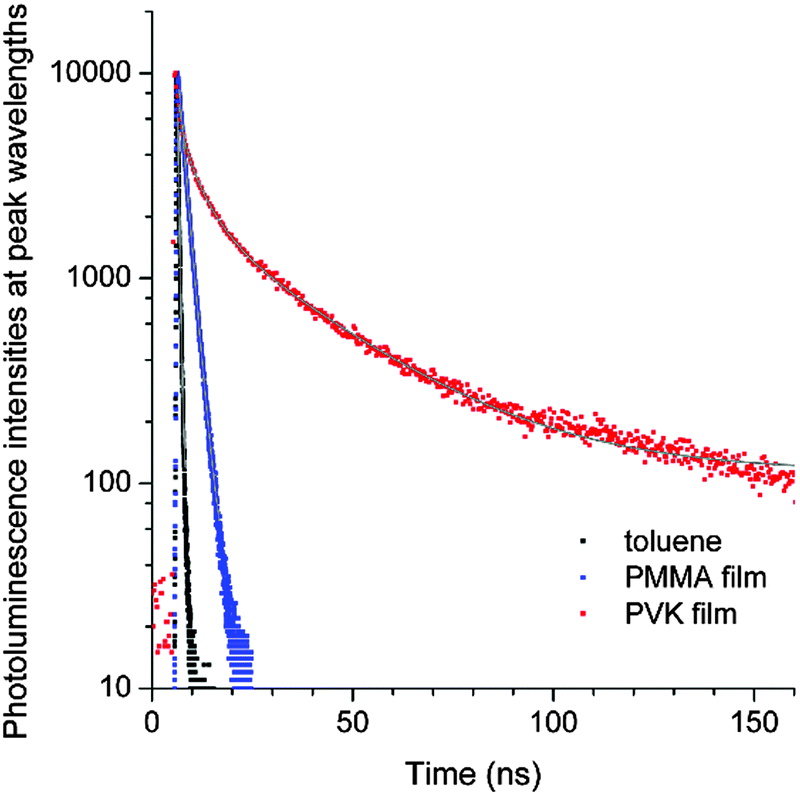 | ||
| Fig. 5 Photoluminescence decay traces of NC after picosecond pulsed laser excitation at 377 nm (pulse duration = 8 ps): black, 50 μM in an Ar-saturated toluene solution; blue, 10 wt% in a PMMA film; and red, 10 wt% in a PVK film. The grey curves are non-linear least squares fits to monoexponential (black and blue) and triexponential (red) decay models. Refer to the ESI,† Fig. S5 for the decay traces of C, PC and AC, and Table 1 for the fit results. | ||
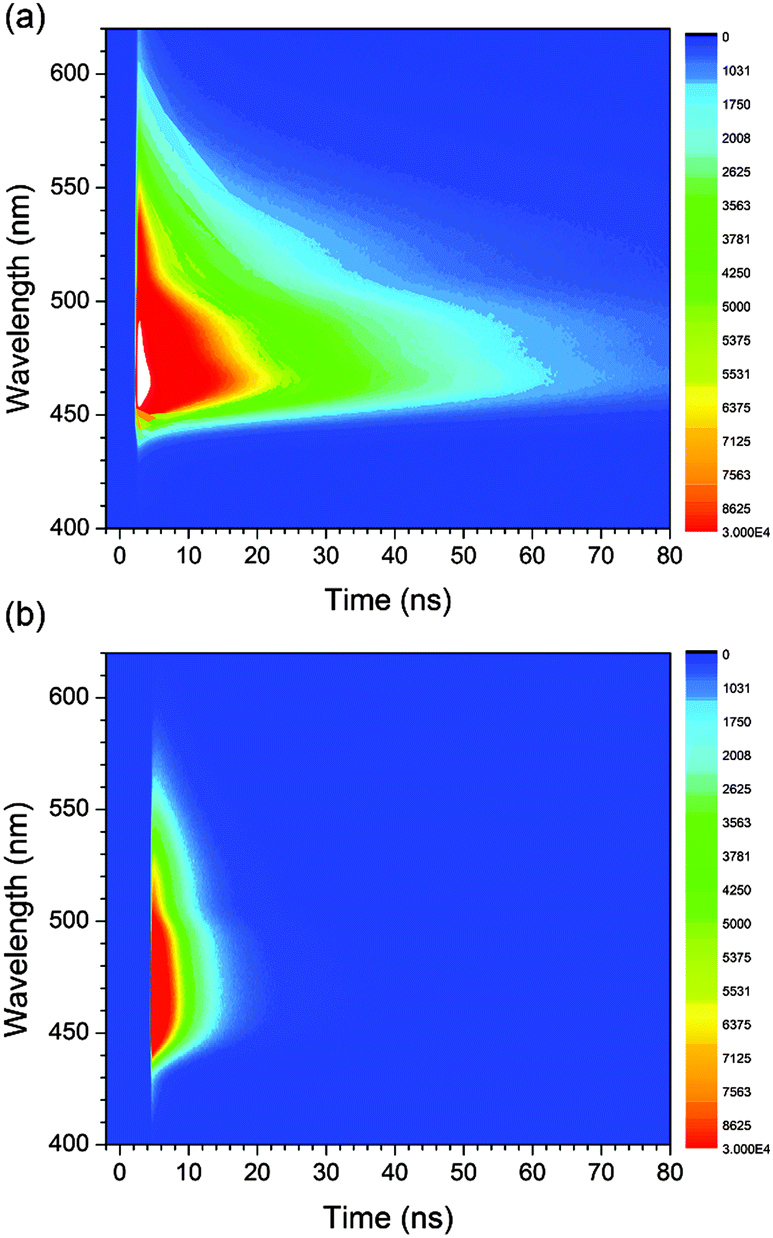 | ||
| Fig. 6 Time-resolved photoluminescence spectra of PVK films doped with 10 wt% PC (a) and 10 wt% AC (b) after picosecond pulsed laser excitation at 377 nm. | ||
Fluorescence utility of the exciplex of the coumarin compounds was demonstrated by emission color tuning. PVK films containing 10 wt% C were additionally doped with several electron donors (30 wt%) having the Eox values smaller than that of PVK: 1,2-dimethylindole (DMI), 1.06 V; N,N-dimethylaniline (DMA), 0.77 V; and N,N-dimethyl-p-toluidine (DMT), 0.72 V (all potentials are vs. SCE).62 As shown in Fig. 7a, the exciplex fluorescence emission of the ternary mixtures shifted bathochromically with decreasing Eox of the electron donors. Indeed, the fluorescence peak energies exhibited linear proportionality to the values of Eox(donor) − Ered(C) (Fig. 7b). The photoluminescence decay traces monitored at the emission peak wavelengths followed a triexponential decay model, as seen for the binary mixture of C and PVK. The weighted average values of the photoluminescence decay were as long as 8.3 ns, 13.8 ns, and 9.2 ns for 1,2-dimethylindole, N,N-dimethylaniline, and N,N-dimethyl-p-toluidine, respectively (ESI,† Fig. S7). These results unambiguously demonstrated that the exciplex is the origin of fluorescence emission, and provided a versatile strategy to tailor fluorescence emission.
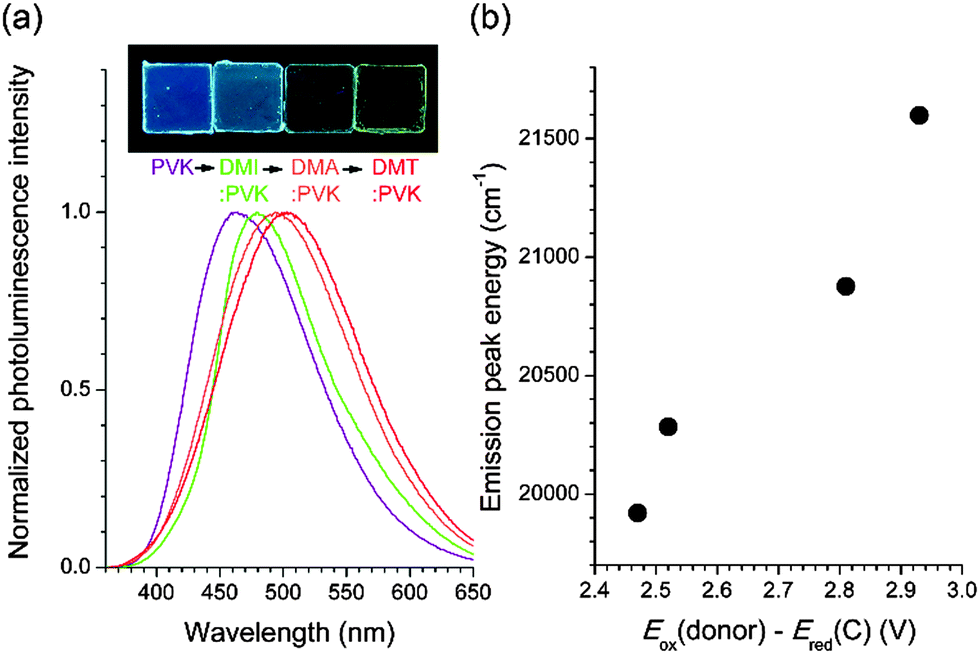 | ||
| Fig. 7 (a) Photoluminescence spectra of PVK films doped with 10 wt% C and a 30 wt% electron donor. Electron donors: DMI, 1,2-dimethylindole; DMA, N,N-dimethylaniline; and DMT, N,N-dimethyl-p-toluidine. The inset photograph shows the fluorescence emission of the PVK films of 10 wt% C in the absence (far left) and presence of DMI, DMA, and DMT (from the second left). (b) Plot of the fluorescence peak wavelength energies and the difference between the ground-state oxidation potentials of the electron donors (Eox(donor)) and the ground-state reduction potentials of C (Ered(C)). | ||
Photophysical monitoring of the excited-state processes
Having demonstrated the ICT and exciplex emission, we sought to investigate the photophysical processes. Scheme 2 depicts the plausible mechanisms for the photophysical processes in AC (i.e., ICT fluorescence) and PVK films containing PC (i.e., exciplex fluorescence). In both cases, the charge transfer provides fluorescence channels that can harvest the excited-state energy of the n–π*transition state. It should be noted that, without the new channels mediated by charge transfer, the n–π* transition state relaxes to the ground state non-radiatively.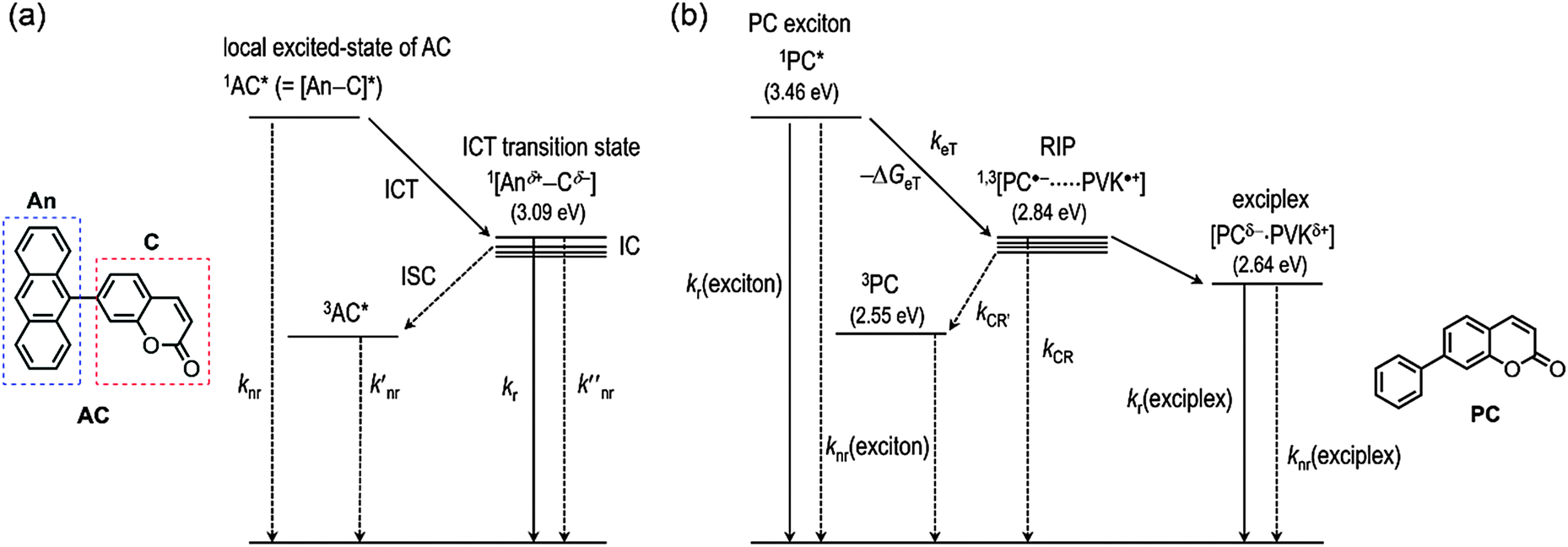 | ||
| Scheme 2 Plausible mechanisms for (a) the ICT fluorescence of AC and (b) generation and relaxation of the exciplex in PVK films doped with PC upon photoexcitation: ISC, intersystem crossing; IC, internal conversion; RIP, radical ion pair; knr, the rate constant for non-radiative decay of the singlet local excited state of AC; k′nr, the rate constant for non-radiative decay of the triplet local excited state of AC; kr, the rate constant for radiative decay of the ICT transition state of AC; k′′nr, the rate constant for non-radiative decay of the ICT transition state of AC; kr(exciton), the rate constant for radiative decay of PC; knr(exciton), the rate constant for non-radiative decay of PC; keT, the rate constant for photoinduced electron transfer from PVK to PC; kCR′, the rate constant for charge recombination to the triplet state of PC; kCR, the rate constant for charge recombination to the ground state of PC; kr(exciplex), the rate constant for radiative decay of the exciplex; and knr(exciplex), the rate constant for non-radiative decay of the exciplex. | ||
The steady-state and transient photoluminescence results revealed that the ICT transition state is responsible for the fluorescence emission of AC. To directly probe the ICT process, femtosecond laser flash photolysis experiments were performed. The transient absorption spectra of toluene solutions of AC (O.D. = 0.2 at 350 nm) were acquired after femtosecond pulsed laser photoexcitation at 350 nm. As shown in Fig. 8a, a strong absorption band at 620 nm appeared at a rise time of 0.78 ps, and decayed with a time constant of 1.2 ns. The 620 nm band was due to the radical cation of the coumarin moiety,63 while the anthracene radical cation was shown at 720 nm as a shoulder band.63–67 The rise (0.78 ps) and decay (1.2 ns) times may correspond to the time required for relaxation to the lowest ICT transition and ground states, respectively. The latter time constant was indistinguishable from the photoluminescence lifetime of AC recorded in toluene (2.8 ns), further supporting this notion. By contrast, the transient absorption spectra of the other coumarin compounds differed from that of AC, and included a transient absorption band at 550 nm. The 550 nm absorption bands were long-lived and did not decay appreciably until 3000 ps (ESI,† Fig. S8).
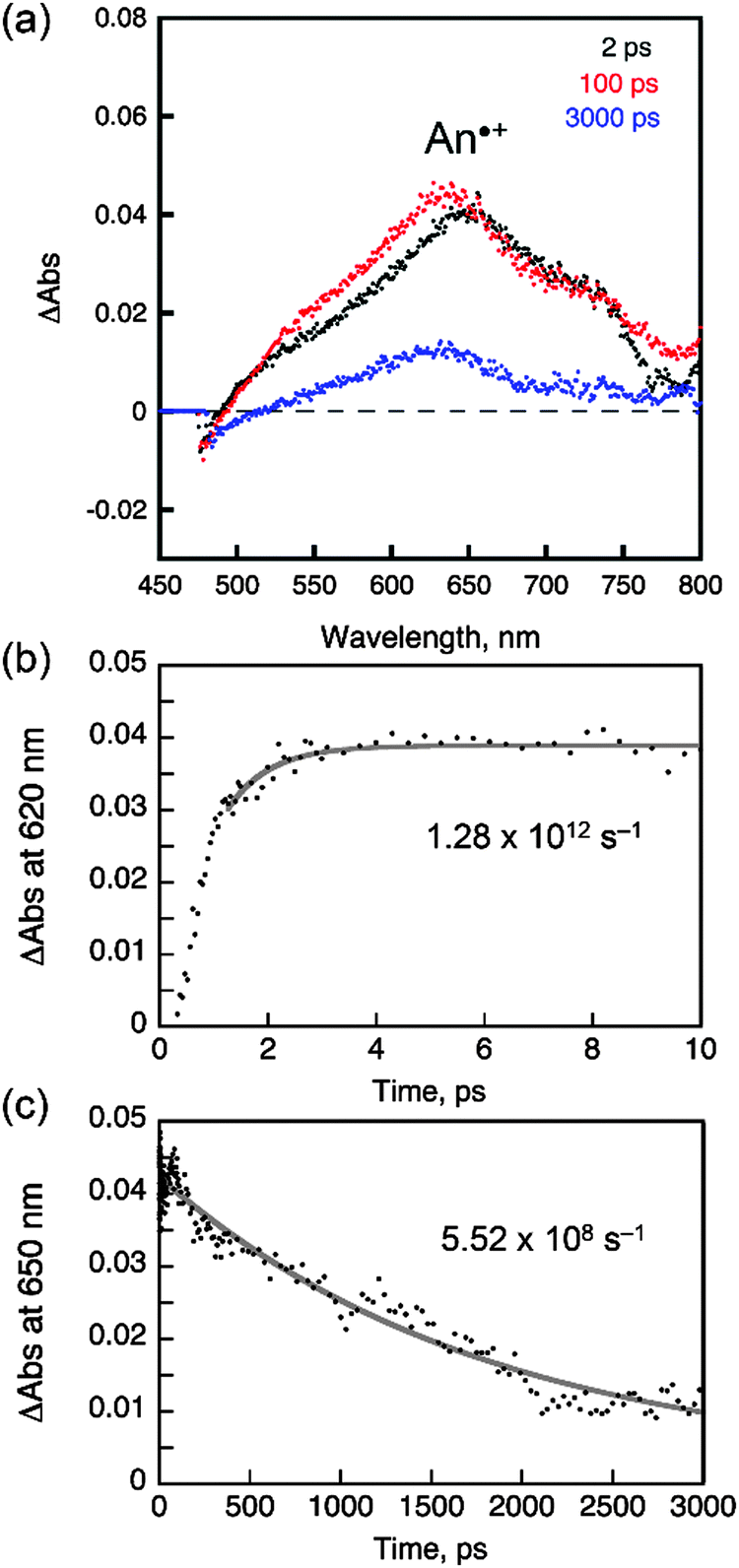 | ||
| Fig. 8 Femtosecond laser flash photolysis of a toluene solution of AC (O.D. = 0.2 at 350 nm, 2 mm pathlength). (a) Transient absorption spectra recorded at 2, 100 and 3000 ps after photoexcitation at 350 nm. (b) Rising traces of the transient absorption at 620 nm and the nonlinear least squares fit. (c) Decay traces of the transient absorption at 650 nm and the nonlinear least squares fit. See the ESI,† Fig. S8 for the laser flash photolysis results obtained from C, PC and NC. | ||
Photoexcitation under UV irradiation (365 nm) produced excited states in both PC (PC*) and PVK (PVK*); however, electron transfer was only allowed from the ground state of PVK to PC* due to a positive driving force (i.e., −ΔGeT) of 0.62 eV, which was determined by the difference between the Ered* of PC and Eox of PVK. Other coumarin derivatives also displayed a positive driving force for the photoinduced reductive electron transfer from PVK: 0.66 eV (C), 0.57 eV (NC), and 0.24 eV (AC). By contrast, electron transfer from the coumarin derivative to the excited state of PVK (PVK*) was forbidden due to the negative driving force (−ΔGeT = −0.10 eV to −0.19 eV). The photoinduced electron transfer produced a radical ion pair ([PC˙−PVK˙+]) consisting of a one-electron reduced PC species (PC˙−) and a one-electron oxidized PVK species (PVK˙+). We attempted to determine the rate constant for intermolecular electron transfer using transient photoluminescence techniques by adding N-ethylcarbazole into the coumarin solutions; however, the experiments were hampered by a significant overlap between the fluorescence spectra of N-ethylcarbazole and the coumarin derivatives. Femtosecond laser flash photolysis revealed an ultrafast increase in the PVK˙+ signal within the temporal resolution of the instrument (vide infra), indicating the presence of forward electron transfer. Once the [PC˙−PVK˙+] has been generated, it underwent charge recombination to produce ground states (CR in Scheme 2b) in PC and PVK or a triplet excited state (CR′ in Scheme 2b) in PC. An alternative pathway may involve the formation of a fluorescent exciplex. Although the limited spectral resolution impeded discrimination among these three processes, we detected the transient absorption band of PVK˙+ at 770 nm in PVK films composed of 10 wt% PC after femtosecond pulsed laser photoexcitation at 350 nm (Fig. 9a). A high-energy absorption band at 490 nm corresponded to a triplet transition (T1 → Tn) in PC.7 These assignments were consistent with those reported previously.62,68–70 The observation of the PVK˙+ species enabled us to follow the decay of [PC˙−PVK˙+]. As shown in Fig. 9b, the transient absorption of PVK˙+ underwent biexponential decay with a weighted average rate (kdecay(RIP)) of 2.6 × 109 s−1. The kdecay(RIP) corresponded to a sum of rate constants for charge recombination to the ground state (kCR in Scheme 2b) and the triplet state (kCR′ in Scheme 2b) and conversion to the exciplex. The decay rate exceeded the radiative (kr(exciplex), 2.1 × 106 s−1) and non-radiative (knr(exciplex), 4.0 × 107 s−1) decay rates of the exciplex of PC by three and two orders of magnitudes, respectively (Table 2), indicating that charge recombination was faster than the relaxation of the exciplex. Similarly, the kdecay(RIP) values of C and NC were as large as 9.8 × 109 s−1 and 3.8 × 109 s−1, respectively (Table 2). Unlike the transient absorption spectra of C, PC, and NC compounds, the transient absorption spectrum of PVK films of AC lacked the PVK˙+ band (ESI,† Fig. S9). This observation indicated that intermolecular electron transfer between PVK and AC was suppressed, despite the positive driving force (0.24 eV), most likely due to the presence of ICT transition.
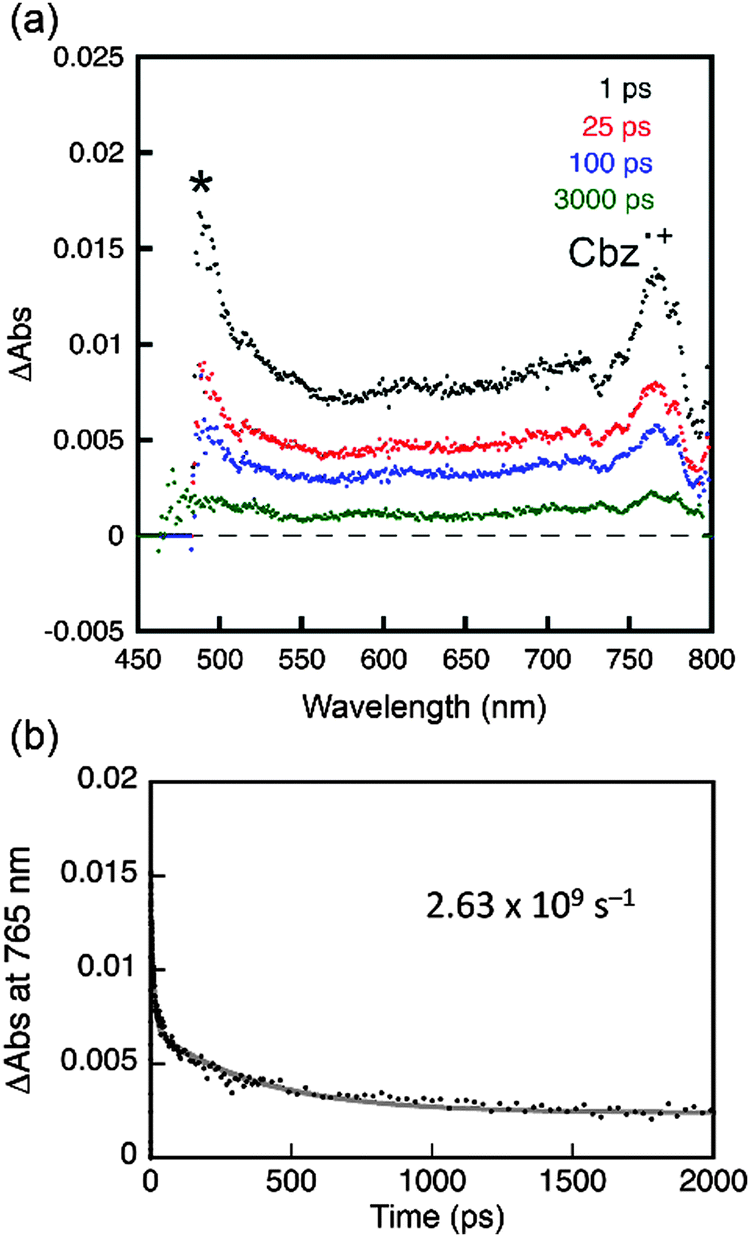 | ||
| Fig. 9 Femtosecond laser flash photolysis of a PVK film doped with 10 wt% PC. (a) Transient absorption spectra recorded at 1, 25, 100, and 1000 ps after photoexcitation at 350 nm. The absorption band indicated by an asterisk (*) corresponds to the T1 → Tn transition in PC. (b) Decay traces of the transient absorption at 765 nm and a nonlinear least squares fit to a biexponential decay model. See the ESI,† Fig. S9 for the laser flash photolysis results obtained from C, NC, and AC. | ||
| Excitona | Exciplexb | k decay(RIP)c (109 s−1) | |||
|---|---|---|---|---|---|
| k r (107 s−1) | k nr (107 s−1) | k r (107 s−1) | k nr (107 s−1) | ||
| a 10 wt% in PMMA films, 298 K. b 10 wt% in PVK films, 298 K. c Decay rate of the radical ion pair of PVK and coumarin compound species, determined by laser flash photolysis after femtosecond laser photoexcitation of the PVK films containing 10 wt% coumarin compounds at 330 nm (C) and 350 nm (PC, NC, and AC). d Radiative rate constant, kr = PLQY/τobs: PLQY = 0 (C), 0.019 ± 0.0008 (PC), 0.27 ± 0.007 (NC), and 0.41 ± 0.002 (AC); τobs = 7.4 ns (C), 8.4 ns (PC), 1.9 ns (NC), and 1.7 ns (AC). e Non-radiative rate constant, knr = (1 − PLQY)/τobs. f Radiative rate constant, kr = PLQY/τobs: see Table 1 for the PLQY and τobs values. g Non-radiative rate constant, knr = (1 − PLQY)/τobs. | |||||
| C | 0 | 14 | 0.26 | 4.1 | 9.8 |
| PC | 0.23 | 12 | 0.21 | 4.0 | 2.6 |
| NC | 1.4 | 38 | 0.16 | 4.6 | 3.8 |
| AC | 24 | 35 | N.A. | N.A. | N.A. |
It should be noted that the decay rates were proportional to the free energy change for back electron transfer within the radical ion pair to yield the ground states (ΔGBeT(G): C, −2.93 eV; PC, −2.84 eV; NC, −2.86 eV). The ΔGBeT(G) values were calculated using the relationship, ΔGBeT(G) = −e·[Ered(coumarin) − Eox(PVK)], where e is the elementary charge of an electron. The free energy change for charge recombination to the triplet state of coumarin (ΔGBeT(T): C, −0.20 eV; PC, −0.29 eV; NC, −0.31 eV) was calculated from the difference between −ΔGBeT(G) and the triplet state energies (ΔET) of the coumarin compounds. The ΔET values were determined from the low temperature (78 K) phosphorescence spectra collected from solutions containing 50 μM C in 2-MeTHF or 50 μM PC or NC in iodoethane (ESI,† Fig. S5). As shown in Fig. 10, the kdecay(RIP) varied linearly with ΔGBeT(G) but not with ΔGBeT(T). Taken together, these results suggest that the favored relaxation process for the photo-produced radical ion pair involved non-radiative charge recombination to the ground state, which occurred in the Marcus-normal region of electron transfer.
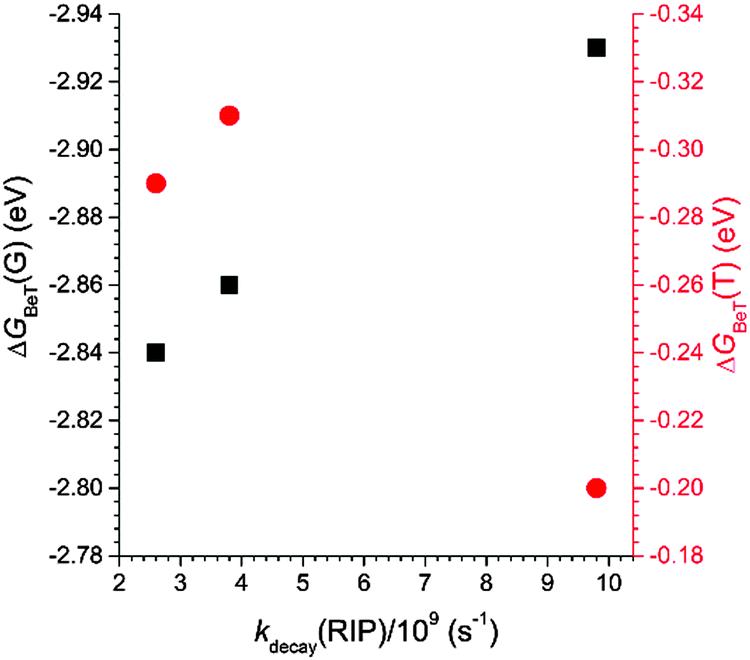 | ||
| Fig. 10 Plots of the free energy change for charge recombination within the radical ion pair [PC˙−PVK˙+] to the ground (ΔGBeT(G)) and triplet (ΔGBeT(T)) states of the coumarin compounds as a function of the rate constant for the decay of the radical ion pair (kdecay(RIP)). | ||
Summary and conclusions
n–π* chromophores are considered to be weak fluorophores, because they undergo fast non-radiative relaxation. The utility of n–π* fluorophores has not been appreciated, and successful applications of these compounds are scarce, with the exception of coumarin compounds bearing strong electron donors. We sought to improve the fluorescence properties of n–π* fluorophores by implementing two photophysical design strategies. These strategies promoted intra- and intermolecular charge transfer to electron-deficient n–π* fluorophores, and generated an ICT transition state and an exciplex, both of which were fluorescent. The archetype 1-benzopyran-2-one (coumarin) was chosen as a model platform. Aryl rings, including phenyl (PC), 1-naphthyl (NC), and 9-anthryl (AC), were introduced at the 7 position of coumarin. AC was found to display strong ICT fluorescence emission, whereas the fluorescence intensities of coumarin and the other derivatives were very low. The increase in fluorescence was ascribed to the reordering of the electronic states through the generation of an ICT transition state and destabilization of the n–π* transition state. The weakly emissive C, PC, and NC became highly fluorescent when doped into PVK films. The fluorescence originated from the exciplex, as demonstrated by the mechanistic studies. Femtosecond laser flash photolysis experiments enabled us to directly identify the electron-transfer species generated during the exciplex de-excitation processes. The relationship between the electron-transfer rates and the driving force for electron transfer revealed that fast charge recombination provided the dominant relaxation pathway. This establishment delivered valuable insights that further improved the fluorescence properties. Recent studies have described the utility of electron-transfer species as emitting centers,71–103 whereas the molecular principles underlying high-efficiency exciplex fluorescence have yet to be fully established. We hope that the results presented here will provide useful insights into the future utilization of charge-transfer fluorescence.Experimental details
Synthetic methods
Commercially available chemicals were used as received. All glassware, syringes, and magnetic stir bars were thoroughly dried in a convection oven. Reactions were monitored using thin layer chromatography (TLC) using commercial TLC plates (silica gel 60 F254, Merck Co.). Silica gel column chromatography was performed with silica gel 60 (particle size 0.063–0.200 mm, Merck Co.). 1H and 13C NMR spectra were collected using Bruker, Ultrashield 300, 400, and 500 NMR spectrometers and were referenced to TMS. High-resolution mass spectra were acquired using JEOL, JMS-600W/JMS-700GC and Applied Biosystems, Tempo Nano HPLC/QSTAR Elite spectrometers. Elemental analysis was performed using a CE Instrument, EA1110 instrument.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :n-hexane = 1:1 (v/v) yielded a white powder in a 34% yield. 1H NMR (400 MHz, CDCl3) δ: 6.44 (d, J = 9.6 Hz, 1H), 7.337.43 (m, 2H), 7.52 (d, J = 2.0 Hz, 1H), 7.66 (d, J = 9.6 Hz, 1H). 13C NMR (75 MHz, CDCl3) δ: 117.01, 117.90, 120.29, 125.93, 128.02, 128.97, 142.92, 154.40, 160.01.
:2 N K2CO3 (aq.) (2:1, v/v) and refluxed for 2 days. The cooled reaction mixture was poured onto water, and extracted with CH2Cl2 (100 mL × four times). The organic layer was dried over anhydrous MgSO4 and concentrated. Silica gel column chromatography was performed while increasing the polarity of the eluents from CH2Cl2:n-hexane = 1:2 (v/v) to CH2Cl2:n-hexane = 1:1 (v/v) to furnish a yellow powder in a 52% yield. 1H NMR (300 MHz, CDCl3) δ: 6.44 (d, J = 9.5 Hz, 1H), 7.42–7.56 (m, 6H), 7.62–7.65 (m, 2H), 7.75 (d, J = 9.5 Hz, 1H). 13C NMR (126 MHz, CDCl3) δ: 115.26, 116.54, 118.02, 123.53, 127.43, 128.33, 128.75, 129.29, 139.38, 143.25, 145.29, 154.73, 161.01. HR MS (FAB+, m-NBA): calcd for C15H10O2 ([M + H]+), 223.0759; found, 223.0765. Anal. calcd for C15H10O2: C, 81.07; H, 4.54. Found: C, 81.02; H, 4.51.
:n-hexane = 1:1 (v/v) yielded a white powder in a 34% yield. 1H NMR (400 MHz, CDCl3) δ: 6.44 (d, J = 9.6 Hz, 1H), 7.337.43 (m, 2H), 7.52 (d, J = 2.0 Hz, 1H), 7.66 (d, J = 9.6 Hz, 1H). 13C NMR (75 MHz, CDCl3) δ: 117.01, 117.90, 120.29, 125.93, 128.02, 128.97, 142.92, 154.40, 160.01.
:2 N K2CO3 (aq.) (2:1, v/v) and refluxed for 2 days. The cooled reaction mixture was poured onto water, and extracted with CH2Cl2 (100 mL × four times). The organic layer was dried over anhydrous MgSO4 and concentrated. Silica gel column chromatography was performed while increasing the polarity of the eluents from CH2Cl2:n-hexane = 1:2 (v/v) to CH2Cl2:n-hexane = 1:1 (v/v) to furnish a yellow powder in a 52% yield. 1H NMR (300 MHz, CDCl3) δ: 6.44 (d, J = 9.5 Hz, 1H), 7.42–7.56 (m, 6H), 7.62–7.65 (m, 2H), 7.75 (d, J = 9.5 Hz, 1H). 13C NMR (126 MHz, CDCl3) δ: 115.26, 116.54, 118.02, 123.53, 127.43, 128.33, 128.75, 129.29, 139.38, 143.25, 145.29, 154.73, 161.01. HR MS (FAB+, m-NBA): calcd for C15H10O2 ([M + H]+), 223.0759; found, 223.0765. Anal. calcd for C15H10O2: C, 81.07; H, 4.54. Found: C, 81.02; H, 4.51.
Characterization
Acknowledgements
This research was supported by a grant from the Samsung Research Funding Center for Future Technology (SRFCMA1301-01 to Y. Y.).Notes and references
- R. O'Kennedy and R. D. Thornes, Coumarins: Biology, Applications and Mode of Actions, Wiley, Chichester, 1997 Search PubMed.
- M. Tasior, D. Kim, S. Singha, M. Krzeszewski, K. H. Ahn and D. T. Gryko, J. Mater. Chem. C, 2015, 3, 1421–1446 RSC.
- J. S. Seixas de Melo, R. S. Becker and A. L. Macanita, J. Phys. Chem., 1994, 98, 6054–6058 CrossRef CAS.
- T. Wolff and H. Goerner, Phys. Chem. Chem. Phys., 2004, 6, 368–376 RSC.
- J. Seixas de Melo, R. S. Becker, F. Elisei and A. L. Macanita, J. Chem. Phys., 1997, 107, 6062–6069 CrossRef CAS.
- M. A. El-Sayed, J. Chem. Phys., 1962, 36, 573–574 CrossRef CAS.
- C. M. Krauter, J. Moehring, T. Buckup, M. Pernpointner and M. Motzkus, Phys. Chem. Chem. Phys., 2013, 15, 17846–17861 RSC.
- P. A. Vadola and D. Sames, J. Org. Chem., 2012, 77, 7804–7814 CrossRef CAS PubMed.
- E. Soleimani, M. M. Khodaei, N. Batooie and S. Samadi, J. Heterocycl. Chem., 2012, 49, 409–412 CrossRef CAS.
- X.-G. Liu, S.-S. Zhang, C.-Y. Jiang, J.-Q. Wu, Q. Li and H. Wang, Org. Lett., 2015, 17, 5404–5407 CrossRef CAS PubMed.
- N. Sepay, C. Guha, A. Kool and A. K. Mallik, RSC Adv., 2015, 5, 70718–70725 RSC.
- S. Yaragorla, A. Pareek and R. Dada, Tetrahedron Lett., 2015, 56, 4770–4774 CrossRef CAS.
- L. Calmus, A. Corbu and J. Cossy, Adv. Synth. Catal., 2015, 357, 1381–1386 CrossRef CAS.
- Y. Yang, J. Han, X. Wu, S. Xu and L. Wang, Tetrahedron Lett., 2015, 56, 3809–3812 CrossRef CAS.
- S. K. Gadakh, S. Dey and A. Sudalai, J. Org. Chem., 2015, 80, 11544–11550 CrossRef CAS PubMed.
- C. R. Sinu, D. V. M. Padmaja, U. P. Ranjini, K. C. Seetha Lakshmi, E. Suresh and V. Nair, Org. Lett., 2013, 15, 68–71 CrossRef CAS PubMed.
- K. Li, Y. Zeng, B. Neuenswander and J. A. Tunge, J. Org. Chem., 2005, 70, 6515–6518 CrossRef CAS PubMed.
- D. C. Dittmer, Q. Li and D. V. Avilov, J. Org. Chem., 2005, 70, 4682–4686 CrossRef CAS PubMed.
- J. Donovalova, M. Cigan, H. Stankovicova, J. Gaspar, M. Danko, A. Gaplovsky and P. Hrdlovic, Molecules, 2012, 17, 3259–3276 CrossRef CAS PubMed.
- A. C. Morteani, P. Sreearunothai, L. M. Herz, R. H. Friend and C. Silva, Phys. Rev. Lett., 2004, 92, 247402 CrossRef PubMed.
- M. Lor, J. Thielemans, L. Viaene, M. Cotlet, J. Hofkens, T. Weil, C. Hampel, K. Müllen, J. W. Verhoeven, M. V. d. Auweraer and F. C. D. Schryver, J. Am. Chem. Soc., 2002, 124, 9918–9925 CrossRef CAS PubMed.
- A. Weller, in The Exciplex, ed. M. Gordon and W. R. Ware, 1975, ch. 23 Search PubMed.
- A. C. Morteani, A. S. Dhoot, J.-s. Kim, C. Silva, N. C. Greenham, C. Murphy, E. Moons, S. Cina, J. H. Burroughes and R. H. Friend, Adv. Mater., 2003, 15, 1708–1712 CrossRef CAS.
- S. A. Jenekhe and J. A. Osaheni, Science, 1994, 265, 765–768 CAS.
- X. Zhang, D. M. Kale and S. A. Jenekhe, Macromolecules, 2002, 35, 382–393 CrossRef CAS.
- M. M. Alam and S. A. Jenekhe, J. Phys. Chem. B, 2001, 105, 2479–2482 CrossRef CAS.
- T. Offermans, P. A. van Hal, S. C. J. Meskers, M. M. Koetse and R. A. J. Janssen, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 72, 045213 CrossRef.
- C. Yin, T. Kietzke, D. Neher and H.-H. Horhold, Appl. Phys. Lett., 2007, 90, 092117 CrossRef.
- H. Sakai, A. Itaya and H. Masuhara, J. Phys. Chem., 1989, 93, 5351–5353 CrossRef CAS.
- V. Cherpak, P. Stakhira, B. Minaev, G. Baryshnikov, E. Stromylo, I. Helzhynskyy, M. Chapran, D. Volyniuk, Z. Hotra, A. Dabuliene, A. Tomkeviciene, L. Voznyak and J. V. Grazulevicius, ACS Appl. Mater. Interfaces, 2015, 7, 1219–1225 CAS.
- J. H. Kim, B.-K. An, S.-J. Yoon, S. K. Park, J. E. Kwon, C.-K. Lim and S. Y. Park, Adv. Funct. Mater., 2014, 24, 2746–2753 CrossRef CAS.
- T. Granlund, L. A. A. Pettersson, M. R. Anderson and O. Inganas, J. Appl. Phys., 1997, 81, 8097–8103 CrossRef CAS.
- J. F. Wang, Y. Kawabe, S. E. Shaheen, M. M. Morrell, G. E. Jabbour, P. A. Lee, J. Anderson, N. R. Armstrong, B. Kippelen, E. A. Mash and N. Peyghambarian, Adv. Mater., 1998, 10, 230–233 CrossRef CAS.
- S. L. Lai, M. Y. Chan, Q. X. Tong, M. K. Fung, P. F. Wang, C. S. Lee and S. T. Lee, Appl. Phys. Lett., 2008, 93, 143301 CrossRef.
- R. M. Siegoczynski and W. Ejchart, Macromol. Symp., 2004, 212, 575–580 CrossRef CAS.
- K.-H. Kim, C.-K. Moon, J. W. Sun, B. Sim and J.-J. Kim, Adv. Opt. Mater., 2015, 3, 895–899 CrossRef CAS.
- J. W. Sun, J. Y. Baek, K.-H. Kim, C.-K. Moon, J.-H. Lee, S.-K. Kwon, Y.-H. Kim and J.-J. Kim, Chem. Mater., 2015, 27, 6675–6681 CrossRef CAS.
- G. Li, C. H. Kim, Z. Zhou, J. Shinar, K. Okumoto and Y. Shirota, Appl. Phys. Lett., 2006, 88, 253505 CrossRef.
- T. Zhang, B. Zhao, B. Chu, W. Li, Z. Su, X. Yan, C. Liu, H. Wu, Y. Gao, F. Jin and F. Hou, Sci. Rep., 2015, 5, 10234 CrossRef CAS PubMed.
- T. Zhang, B. Chu, W. Li, Z. Su, Q. M. Peng, B. Zhao, Y. Luo, F. Jin, X. Yan, Y. Gao, H. Wu, F. Zhang, D. Fan and J. Wang, ACS Appl. Mater. Interfaces, 2014, 6, 11907–11914 CAS.
- Y.-S. Park, K.-H. Kim and J.-J. Kim, Appl. Phys. Lett., 2013, 102, 153306 CrossRef.
- X.-K. Liu, Z. Chen, C.-J. Zheng, C.-L. Liu, C.-S. Lee, F. Li, X.-M. Ou and X.-H. Zhang, Adv. Mater., 2015, 27, 2378–2383 CrossRef CAS PubMed.
- X.-K. Liu, Z. Chen, C.-J. Zheng, M. Chen, W. Liu, X.-H. Zhang and C.-S. Lee, Adv. Mater., 2015, 27, 2025–2030 CrossRef CAS PubMed.
- X.-K. Liu, Z. Chen, J. Qing, W.-J. Zhang, B. Wu, H. L. Tam, F. Zhu, X.-H. Zhang and C.-S. Lee, Adv. Mater., 2015, 27, 7079–7085 CrossRef CAS PubMed.
- J. Li, H. Nomura, H. Miyazaki and C. Adachi, Chem. Commun., 2014, 50, 6174–6176 RSC.
- A. P. Kulkarni and S. A. Jenekhe, J. Phys. Chem. C, 2008, 112, 5174–5184 CAS.
- K. Goushi, K. Yoshida, K. Sato and C. Adachi, Nat. Photonics, 2012, 6, 253–258 CrossRef CAS.
- K. Goushi and C. Adachi, Appl. Phys. Lett., 2012, 101, 023306 CrossRef.
- D. Chen, Z. Wang, D. Wang, Y.-C. Wu, C.-C. Lo, A. Lien, Y. Cao and S.-J. Su, Org. Electron., 2015, 25, 79–84 CrossRef CAS.
- Z. Chen, X.-K. Liu, C.-J. Zheng, J. Ye, C.-L. Liu, F. Li, X.-M. Ou, C.-S. Lee and X.-H. Zhang, Chem. Mater., 2015, 27, 5206–5211 CrossRef CAS.
- V. Jankus, P. Data, D. Graves, C. McGuinness, J. Santos, M. R. Bryce, F. B. Dias and A. P. Monkman, Adv. Funct. Mater., 2014, 24, 6178–6186 CrossRef CAS.
- Y. O. Lee, J. Y. Lee, D. T. Quang, M. H. Lee and J. S. Kim, Bull. Korean Chem. Soc., 2006, 27, 1469–1472 CrossRef CAS.
- K. H. Ibnaouf, S. Prasad, A. S. Aldwayyan, M. S. Al Salhi and V. Masilamani, Spectrochim. Acta, Part A, 2012, 97, 1145–1151 CrossRef CAS PubMed.
- V. Masilamani and A. S. Aldwayyan, Spectrochim. Acta, Part A, 2004, 60, 2099–2106 CrossRef CAS PubMed.
- H. N. Ghosh, J. Phys. Chem. B, 1999, 103, 10382–10387 CrossRef CAS.
- A. Furube, R. Katoh, T. Yoshihara, K. Hara, S. Murata, H. Arakawa and M. Tachiya, J. Phys. Chem. B, 2004, 108, 12583–12592 CrossRef CAS.
- G. Heinrich, S. Schoof and H. Gusten, J. Photochem., 1974, 3, 315–320 CrossRef CAS.
- The glass transition temperature of PMMA (105 C) was significantly higher than the room temperature, effectively prohibiting intramolecular motions of the coumarin compounds.
- Y. Tsujii, A. Tsuchida, M. Yamamoto and Y. Nishijima, Macromolecules, 1988, 21, 665–670 CrossRef CAS.
- J. J. Benson-Smith, J. Wilson, C. Dyer-Smith, K. Mouri, S. Yamaguchi, H. Murata and J. Nelson, J. Phys. Chem. B, 2009, 113, 7794–7799 CrossRef CAS PubMed.
- C. Dyer-Smith, J. J. Benson-Smith, D. D. C. Bradley, H. Murata, W. J. Mitchell, S. E. Shaheen, S. A. Haque and J. Nelson, J. Phys. Chem. C, 2009, 113, 14533–14539 CAS.
- Y. Tsujii, A. Tsuchida, Y. Onogi and M. Yamamoto, Macromolecules, 1990, 23, 4019–4023 CrossRef CAS.
- M. Murakami, K. Ohkubo, T. Nanjo, K. Souma, N. Suzuki and S. Fukuzumi, ChemPhysChem, 2010, 11, 2594–2605 CrossRef CAS PubMed.
- D. R. Worrall, S. L. Williams, F. Wilkinson, J. E. Crossley, H. Bouas-Laurent and J.-P. Desvergne, J. Phys. Chem. B, 1999, 103, 9255–9261 CrossRef CAS.
- D. R. Worrall, S. L. Williams and F. Wilkinson, J. Phys. Chem. B, 1997, 101, 4709–4716 CrossRef CAS.
- S. Fukuzumi, T. Okamoto and K. Ohkubo, J. Phys. Chem. A, 2003, 107, 5412–5418 CrossRef CAS.
- S. Fukuzumi, I. Nakanishi and K. Tanaka, J. Phys. Chem. A, 1999, 103, 11212–11220 CrossRef CAS.
- M. Fujitsuka, Y. Yahata, A. Watanabe and O. Ito, Polymer, 2000, 41, 2807–2812 CrossRef CAS.
- G. W. Haggquist and R. D. Burkhart, Macromolecules, 1995, 28, 2465–2471 CrossRef CAS.
- G. W. Haggquist, R. D. Burkhart and K. R. Naqvi, J. Phys. Chem., 1991, 95, 7588–7594 CrossRef CAS.
- X.-L. Chen, R. Yu, Q.-K. Zhang, L.-J. Zhou, X.-Y. Wu, Q. Zhang and C.-Z. Lu, Chem. Mater., 2013, 25, 3910–3920 CrossRef CAS.
- R. Czerwieniec, J.-B. Yu and H. Yersin, Inorg. Chem., 2011, 50, 8293–8301 CrossRef CAS PubMed.
- J. C. Deaton, S. C. Switalski, D. Y. Kondakov, R. H. Young, T. D. Pawlik, D. J. Giesen, S. B. Harkins, A. J. M. Miller, S. F. Mickenberg and J. C. Peters, J. Am. Chem. Soc., 2010, 132, 9499–9508 CrossRef CAS PubMed.
- T. Gneuß, M. J. Leitl, L. H. Finger, N. Rau, H. Yersin and J. Sundermeyer, Dalton Trans., 2015, 44, 8506–8520 RSC.
- T. Hofbeck, U. Monkowius and H. Yersin, J. Am. Chem. Soc., 2015, 137, 399–404 CrossRef CAS PubMed.
- S. Igawa, M. Hashimoto, I. Kawata, M. Yashima, M. Hoshino and M. Osawa, J. Mater. Chem. C, 2013, 1, 542–551 RSC.
- M. J. Leitl, V. A. Krylova, P. I. Djurovich, M. E. Thompson and H. Yersin, J. Am. Chem. Soc., 2014, 136, 16032–16038 CrossRef CAS PubMed.
- M. Osawa, Chem. Commun., 2014, 50, 1801–1803 RSC.
- M. Osawa, M. Hoshino, M. Hashimoto, I. Kawata, S. Igawa and M. Yashima, Dalton Trans., 2015, 44, 8369–8378 RSC.
- A. Tsuboyama, K. Kuge, M. Furugori, S. Okada, M. Hoshino and K. Ueno, Inorg. Chem., 2007, 46, 1992–2001 CrossRef CAS PubMed.
- H. Yersin, A. F. Rausch, R. Czerwieniec, T. Hofbeck and T. Fischer, Coord. Chem. Rev., 2011, 255, 2622–2652 CrossRef CAS.
- D. M. Zink, M. Bächle, T. Baumann, M. Nieger, M. Kühn, C. Wang, W. Klopper, U. Monkowius, T. Hofbeck, H. Yersin and S. Bräse, Inorg. Chem., 2013, 52, 2292–2305 Search PubMed.
- D. M. Zink, T. Grab, T. Baumann, M. Nieger, E. C. Barnes, W. Klopper and S. Bräse, Organometallics, 2011, 30, 3275–3283 CrossRef.
- A. Endo, K. Sato, K. Yoshimura, T. Kai, A. Kawada, H. Miyazaki and C. Adachi, Appl. Phys. Lett., 2011, 98, 083302 CrossRef.
- H. Hirai, K. Nakajima, S. Nakatsuka, K. Shiren, J. Ni, S. Nomura, T. Ikuta and T. Hatakeyama, Angew. Chem., Int. Ed., 2015, 54, 13581–13585 CrossRef CAS PubMed.
- S. Hirata, Y. Sakai, K. Masui, H. Tanaka, S. Y. Lee, H. Nomura, N. Nakamura, M. Yasumatsu, H. Nakanotani, Q. Zhang, K. Shizu, H. Miyazaki and C. Adachi, Nat. Mater., 2015, 14, 330–336 CrossRef CAS PubMed.
- H. Kaji, H. Suzuki, T. Fukushima, K. Shizu, K. Suzuki, S. Kubo, T. Komino, H. Oiwa, F. Suzuki, A. Wakamiya, Y. Murata and C. Adachi, Nat. Commun., 2015, 6, 8476 CrossRef CAS PubMed.
- M. Kim, S. K. Jeon, S.-H. Hwang and J. Y. Lee, Adv. Mater., 2015, 27, 2515–2520 CrossRef CAS PubMed.
- Y. Kitamoto, T. Namikawa, D. Ikemizu, Y. Miyata, T. Suzuki, H. Kita, T. Sato and S. Oi, J. Mater. Chem. C, 2015, 3, 9122–9130 RSC.
- D. R. Lee, S.-H. Hwang, S. K. Jeon, C. W. Lee and J. Y. Lee, Chem. Commun., 2015, 51, 8105–8107 RSC.
- J. Lee, K. Shizu, H. Tanaka, H. Nakanotani, T. Yasuda, H. Kaji and C. Adachi, J. Mater. Chem. C, 2015, 3, 2175–2181 RSC.
- S. Y. Lee, T. Yasuda, I. S. Park and C. Adachi, Dalton Trans., 2015, 44, 8356–8359 RSC.
- G. Mehes, H. Nomura, Q. Zhang, T. Nakagawa and C. Adachi, Angew. Chem., Int. Ed., 2012, 51, 11311–11315 CrossRef CAS PubMed.
- T. Nakagawa, S.-Y. Ku, K.-T. Wong and C. Adachi, Chem. Commun., 2012, 48, 9580–9582 RSC.
- M. Numata, T. Yasuda and C. Adachi, Chem. Commun., 2015, 51, 9443–9446 RSC.
- Y. Sakai, Y. Sagara, H. Nomura, N. Nakamura, Y. Suzuki, H. Miyazaki and C. Adachi, Chem. Commun., 2015, 51, 3181–3184 RSC.
- K. Sato, K. Shizu, K. Yoshimura, A. Kawada, H. Miyazaki and C. Adachi, Phys. Rev. Lett., 2013, 110, 247401 CrossRef PubMed.
- K. Shizu, H. Noda, H. Tanaka, M. Taneda, M. Uejima, T. Sato, K. Tanaka, H. Kaji and C. Adachi, J. Phys. Chem. C, 2015, 119, 26283–26289 CAS.
- K. Suzuki, S. Kubo, K. Shizu, T. Fukushima, A. Wakamiya, Y. Murata, C. Adachi and H. Kaji, Angew. Chem., Int. Ed., 2015, 54, 15231–15235 CrossRef CAS PubMed.
- H. Tanaka, K. Shizu, J. Lee and C. Adachi, J. Phys. Chem. C, 2015, 119, 2948–2955 CAS.
- H. Tanaka, K. Shizu, H. Miyazaki and C. Adachi, Chem. Commun., 2012, 48, 11392–11394 RSC.
- M. Taneda, K. Shizu, H. Tanaka and C. Adachi, Chem. Commun., 2015, 51, 5028–5031 RSC.
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234–238 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian, Inc., Wallingford CT, 2013.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1–S18 display the predicted isodensity plot of the molecular orbitals, the Lippert–Mataga plot, cyclic and differential pulse voltammograms, UV-vis absorption spectra, photoluminescence spectra obtained at 78 K, photoluminescence decay traces, transient absorption spectra, and copies of 1H and 13C NMR spectra; Tables S1 and S2 list the summary of the TD-DFT calculation results and the photophysical data for PMMA films doped with the coumarin compounds. See DOI: 10.1039/c6tc00639f |
| This journal is © The Royal Society of Chemistry 2016 |