
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRedox tunable viologen-based porous organic polymers†
Carol
Hua
a,
Bun
Chan
a,
Aditya
Rawal
b,
Floriana
Tuna
c,
David
Collison
c,
James M.
Hook
b and
Deanna M.
D'Alessandro
*a
aSchool of Chemistry, The University of Sydney, New South Wales 2006, Australia. E-mail: deanna.dalessandro@sydney.edu.au
bNMR Facility, Mark Wainwright Analytical Centre, The University of New South Wales, New South Wales 2052, Australia
cSchool of Chemistry and Photon Science Institute, The University of Manchester, Manchester M13 9PL, UK
First published on 1st March 2016
Abstract
The use of an organic donor–acceptor polymer containing a viologen electron acceptor and triarylamine electron donor as a platform in the development of multifunctional materials is presented. The highly robust porous organic polymer (POP) system allows for exploration of the interplay between electronic and host–guest interactions in the synthesized polymers, POP-V1, which contains a redox-active triarylamine core and POP-V2, which contains a redox-inactive benzene core, where each of the redox states present can be reversibly accessed. The degree of charge transfer in addition to the H2 and CO2 gas adsorption properties of the polymer are able to be tuned as a function of the electronic state which has important implications for the potential applications of these polymers in optical, electrochromic and solar cell devices.
Introduction
Multifunctional materials, wherein multiple properties can be accessed within the same material in response to external stimuli such as light or a change in redox state, offer significant potential for the development of electroactive, energy storage and magnetoelectric materials. These systems exhibit switchable properties that are able to be turned “on” and “off” in response to external perturbations. Exploiting the properties of the different accessible states offers the potential for lower energy and cost alternatives to existing technologies.Polymeric donor–acceptor systems are particularly versatile platforms for the development of multifunctional materials and have found applications in numerous areas ranging from optical and electrochromic devices,1–9 to solar cells and organic semiconductors.10–12 In these systems, oxidation of the donor substituent and reduction of the acceptor unit offers at a minimum, three distinct accessible redox states within the same material. Despite the potential to access the novel host–guest and physical properties distinctly offered by each state,13 this aspect has, to date, received very limited attention. Importantly, many of the interesting chemical and physical properties of donor–acceptor systems can be attributed to charge-transfer mechanisms which underpin the function of these materials;14,15 the ability to modify these properties as a function of the redox state opens potentially new pathways for the application of these materials.
In this work, the combination of electron donor triarylamine and electron acceptor viologen moieties within porous organic polymer networks was explored as a strategy to produce highly versatile donor–acceptor switchable materials which are responsive to electrochemical and chemical stimuli. The incorporation of two highly stable and reversible processes – the one electron oxidation to the triarylamine radical cation16,17 and the one electron reduction to the viologen radical cation18,19 – was exploited to yield materials with the capacity to reversibly switch between multiple redox states. As shown in Fig. 1, the POP-V1 polymer, which contains the redox-active triarylamine core and the POP-V2 polymer, which contains the redox-inactive benzene core, were compared to understand the nature of the charge-transfer properties in this class of donor–acceptor materials. The electroactive properties of the polymers were interrogated using in situ solid state vis/NIR and EPR spectroelectrochemical experiments which have yielded insights into their function.13,20–25 The ability to tune the host–guest properties of POP-V1 and POP-V2 in their different redox states provides a basis for regulating the uptake and selectivity of CO2, N2 and H2 which has important implications for the use of these materials in applications such as electrical swing adsorption.
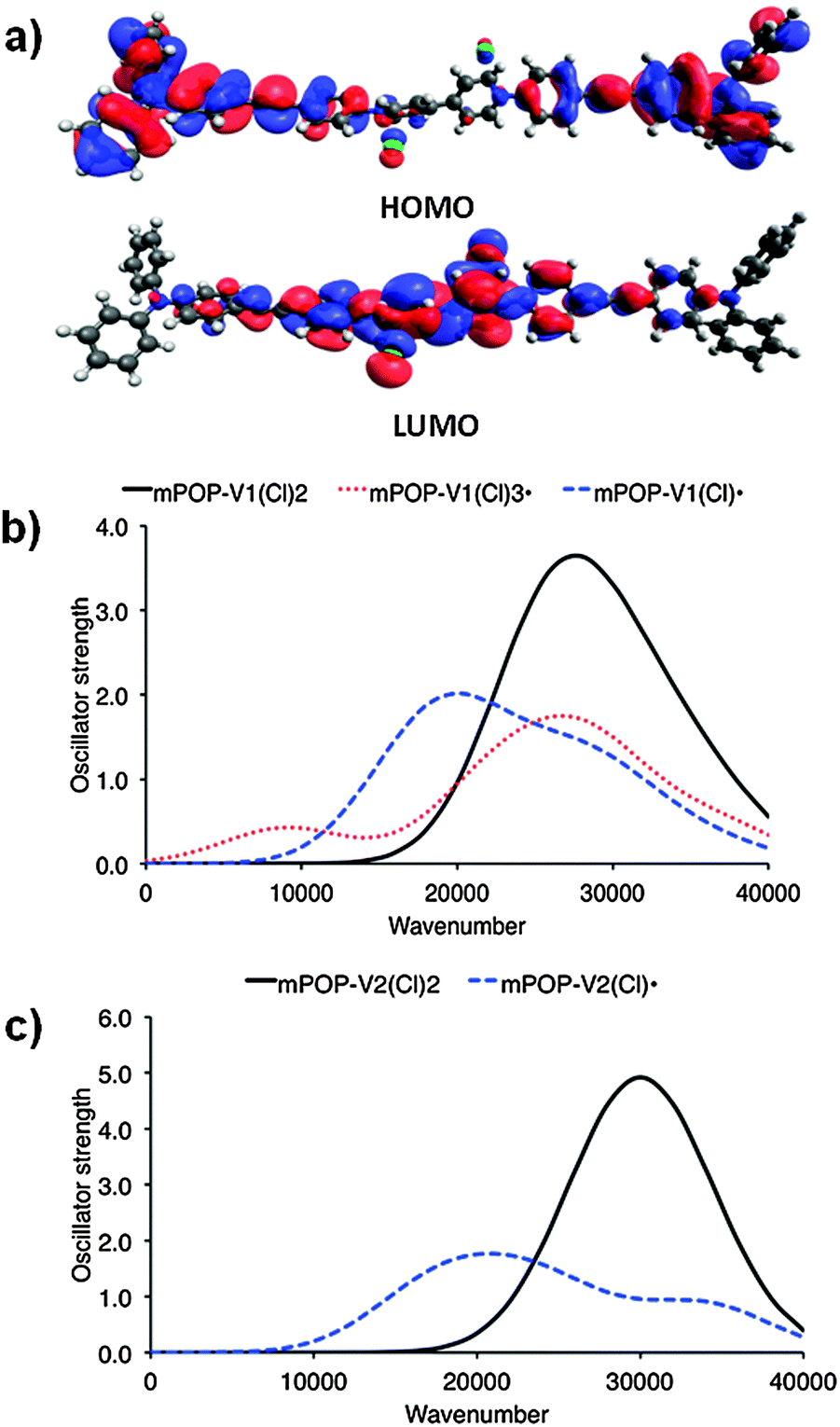 | ||
| Fig. 1 (a) DFT HOMO and LUMO of mPOP-V1(Cl)2 and computed [TDA-BMK/6-31G(d)] UV/vis spectra for the model systems (b) mPOP-V1 and (c) mPOP-V2. | ||
Results and discussion
Synthesis and characterisation
POP-V1 and POP-V2 were synthesized via a Sonogashira–Hagihara palladium catalyzed cross coupling reaction between tris(p-ethynylphenyl)amine or 1,3,5-tris(4-ethynylphenyl)benzene and 1,1′-bis(4-bromophenyl)-4,4′-bipyridinium chloride to yield POP-V1 and POP-V2 as dark yellow/brown and yellow solids, respectively (Scheme 1). The polymers were characterized using solid state NMR 13C CPMAS and 13C CP non-quaternary suppression (NQS) spectra (Fig. S1, ESI†) where several resonances in the alkynyl region (∼90 ppm) were indicative of successful cross coupling of the monomer units. The broadness of the peaks in the solid state NMR spectra was attributed to the amorphous nature of these materials. The thermal gravimetric analysis (TGA) indicates that both of the polymers displayed high thermal stability up to 300 °C (Fig. S2, ESI†).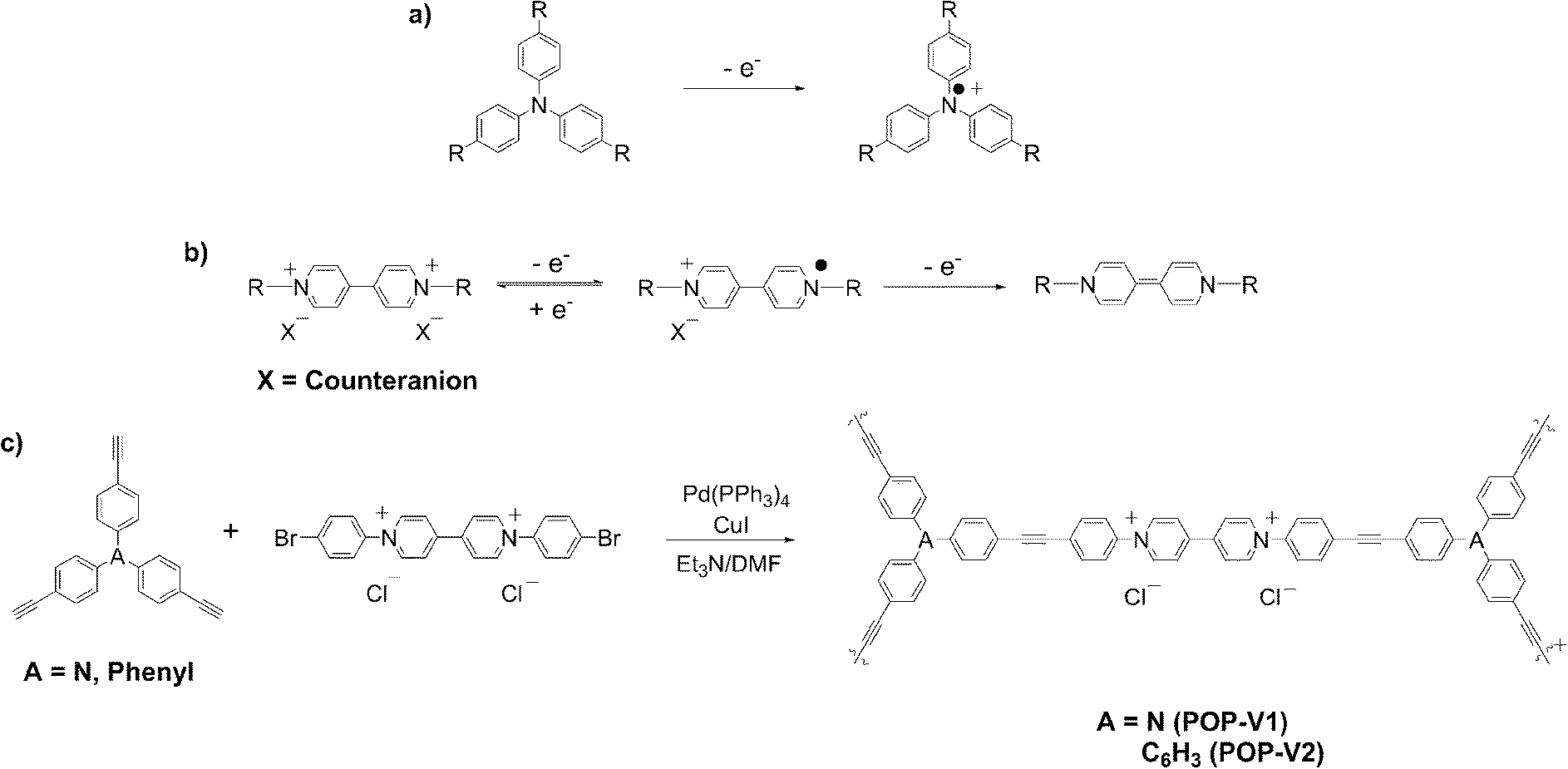 | ||
| Scheme 1 (a) One electron oxidation of triarylamine to the radical cation, (b) reduction processes of viologen and (c) synthesis scheme for POP-V1 and POP-V2. | ||
Redox-state dependent spectral properties
Solid state cyclic voltammetry experiments in [(n-C4H9)4N]PF6/CH3CN electrolyte for POP-V1 revealed one broad anodic process at ∼1.0 V vs. Fc/Fc+ which was assigned to oxidation of the triarylamine core and a quasi-reversible cathodic process at −1.0 V vs. Fc/Fc+ which was assigned to the reduction of the viologen moiety to its radical cation (Fig. S3a, ESI†). These assignments are supported by our analysis of the DFT orbitals of the model system [mPOP-V1(Cl)2], which shows that the HOMO is largely centered around the amines, while the LUMO is primarily viologen-based (Fig. 2b). We attribute the second cathodic process at −2.0 V vs. Fc/Fc+ to the irreversible reduction of the viologen radical cation to its doubly reduced neutral quinoid form. The voltammogram of POP-V2 was characterized by a reversible cathodic process at ∼1.25 V vs. Fc/Fc+ due to reduction of the viologen (Fig. S3b, ESI†), which is consistent with the computed LUMO of the model system, mPOP-V2(Cl)2, centered around this moiety. The second cathodic process was not clearly defined and may indicate a comproportionation reaction between the neutral viologen and the doubly charged species to yield two monoradical cation species.18 No anodic processes were observed for POP-V2, which was expected given the absence of the triarylamine core in this polymer.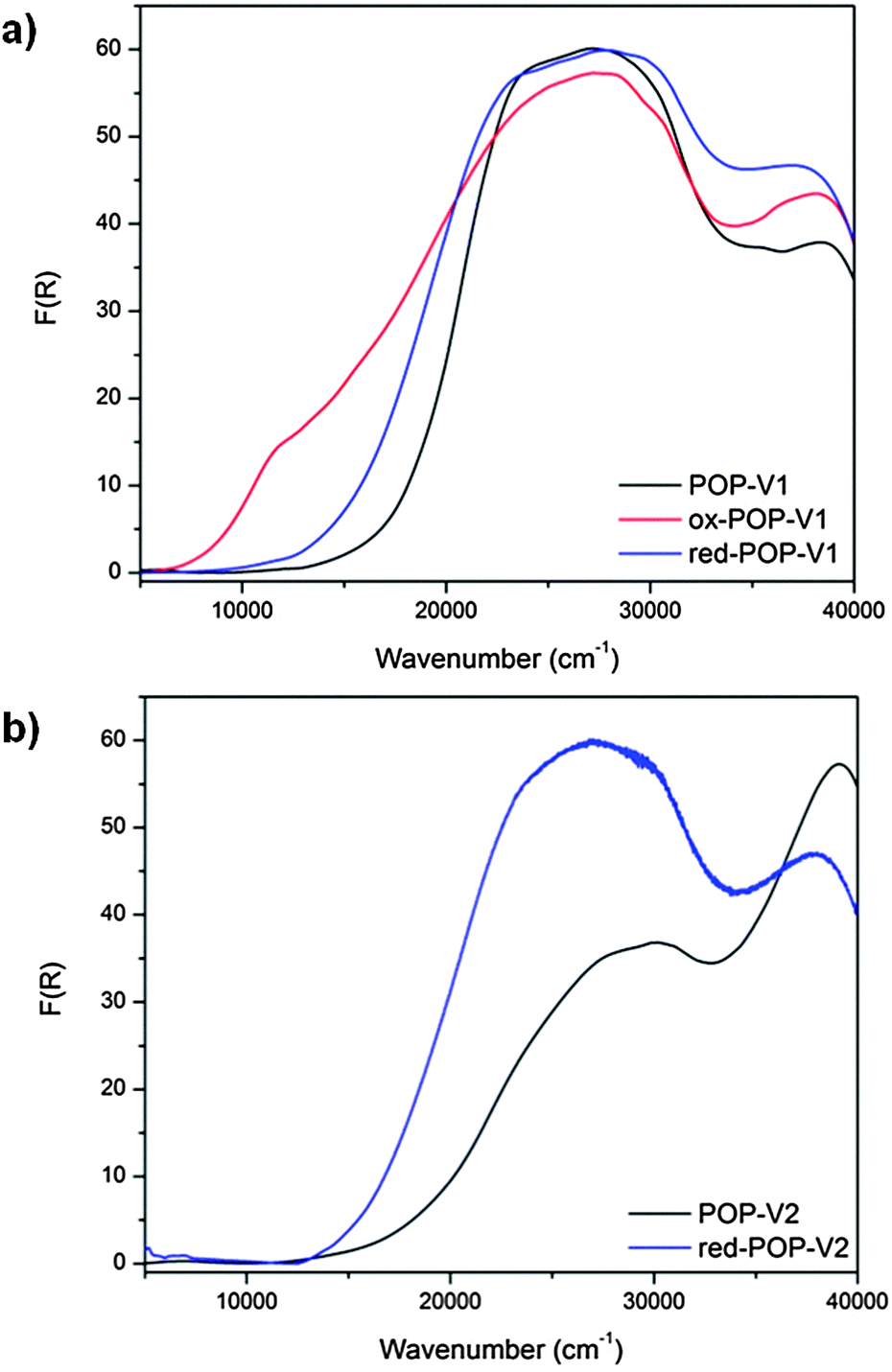 | ||
Fig. 2 Solid state UV/vis/NIR spectra of (a) POP-V1, ox-POP-V1 and red-POP-V1, (b) POP-V2 and red-POP-V2 over the range 5000–40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cm−1. 000 cm−1. | ||
The UV/vis/NIR spectra of POP-V1 and POP-V2 were characterized by intense absorption bands above 20000 cm−1 which were assigned to overlapping π to π* transitions of the aromatic moieties present (Fig. 1). Our computed spectra (Fig. 1a) for the neutral model systems, as well as those for the corresponding oxidized and reduced states, are qualitatively consistent with the experimental spectra. The dark brown POP-V1, when compared to the light brown POP-V2, displayed an intense absorption band >16000 cm−1 (Fig. 2a) which was attributed to the donor–acceptor interactions within the polymer. This assignment is supported by TDA-DFT calculations, which associate the corresponding absorption in mPOP-V1(Cl)2 with predominantly the HOMO–LUMO transition. As mentioned earlier, the HOMO in our DFT model is mainly associated with the amines and the LUMO with the viologen, the transition is therefore related to a charge transfer from the triarylamine to the viologen. This resulted in a smaller optical band gap for POP-V1 (2.17 eV) than POP-V2 (2.24 eV). In comparison, the calculated HOMO–LUMO gaps for our model systems, mPOP-V1(Cl)2 and mPOP-V2(Cl)2, are 3.9 and 4.5 eV, respectively. The weak EPR signal observed for POP-V1 in the neutral state, which was absent in POP-V2, can be attributed to the donor–acceptor properties of the polymer, as the electron was spontaneously transferred from the triarylamine electron donor to the viologen acceptor. Indeed, our DFT calculations show that within POP-V1(Cl)2, which is overall neutral, there is a net charge of +0.23 associated with the two terminal triarylamine units.
A darkening in color of POP-V1 from dark brown to black was observed upon chemical oxidation with nitrosonium tetrafluoroborate and was consistent with the appearance of a band at 12000 cm−1 (Fig. 2a). This is assigned to the localized D0 to D1 transition of the triarylamine radical cation,17 with our DFT calculations for mPOP-V1(Cl)3˙ (where an additional Cl has been added for charge balance) suggesting a transition from a low-lying triarylamine-based occupied orbital to the SOMO. In addition, a red shift of the band at 20000 cm−1 was also observed.
The chemical reduction of POP-V1 and POP-V2 with lithium naphthalenide resulted in a darkening of the solids to dark brown. The UV/vis/NIR spectra for the reduced forms of POP-V1 and POP-V2 were characterized by absorption bands due to the reduced viologen between 22000–25000 cm−1 (Fig. 2).26–28 The optical band gap for the reduced species was lower than that of the neutral polymer system (POP-V1: 2.17 eV, red-POP-V1: 1.81 eV, POP-V2: 2.24 eV, red-POP-V2: 1.94 eV) which was in accordance with computational results from the literature where the HOMO–LUMO separation decreases upon reduction of the dication to the monocation, then to the neutral species,29 as well as the results of our DFT calculations for the model systems mPOP-V1(Cl)2 (HOMO–LUMO gap = 3.9 eV) and mPOP-V1(Cl)˙ (3.4 eV), and mPOP-V2(Cl)2 (4.5 eV) and mPOP-V2(Cl)˙ (3.3 eV).
In the EPR spectra of the polymers, ox-POP-V1 exhibited a smaller g-factor (2.001) than POP-V1 (2.003) (Fig. S6a, ESI†) whilst red-POP-V1 had a larger g-factor (2.004). The shift in the value of g from ge (=2.0023) indicates that the SOMO of the oxidized radical undergoes coupling with an empty orbital, and conversely, that the SOMO of the reduced radical couples with a filled orbital of the polymer. Red-POP-V2 exhibited a smaller g-factor (1.999) than POP-V2 (2.002) (Fig. S6b, ESI†). Interestingly, the trend for POP-V2 and its reduced analogue was opposite to that observed with POP-V1, indicating that reduction of POP-V2 results in coupling of the SOMO of the radical with an empty orbital. The difference in the trends for the g-factors between POP-V1 and POP-V2 indicates that the energies of each of the orbitals differ due to the presence of either an N-centered (triarylamine) in POP-V1 or phenyl-centered core in POP-V2.
Probing the interplay between electronic and optical properties
Application of a potential of 1.75 V to POP-V1 during the solid state vis/NIR spectroelectrochemical experiment resulted in a color change from dark brown to black and the formation of two bands in the NIR/vis spectrum (Fig. 3b). As with our assignment for the chemically oxidized species, the band at ∼12000 cm−1 was assigned to the formation of the triarylamine radical cation and corresponds to the localized π to π* transition while the band at ∼14000 cm−1 corresponded to a charge transfer process from the triarylamine to the viologen acceptor. As the bands were spectrally reversible upon application of a negative potential, the charge transfer interactions present in the polymer could be tuned in response to an electrical stimulus. The reduced state of the polymer was obtained upon application of a potential of −2.0 V (Fig. 3a), where the band at ∼21000 cm−1 increased in intensity whilst the band at ∼23000 cm−1 decreased in intensity due to reduction of the viologen linker in the POP-V1 polymer.
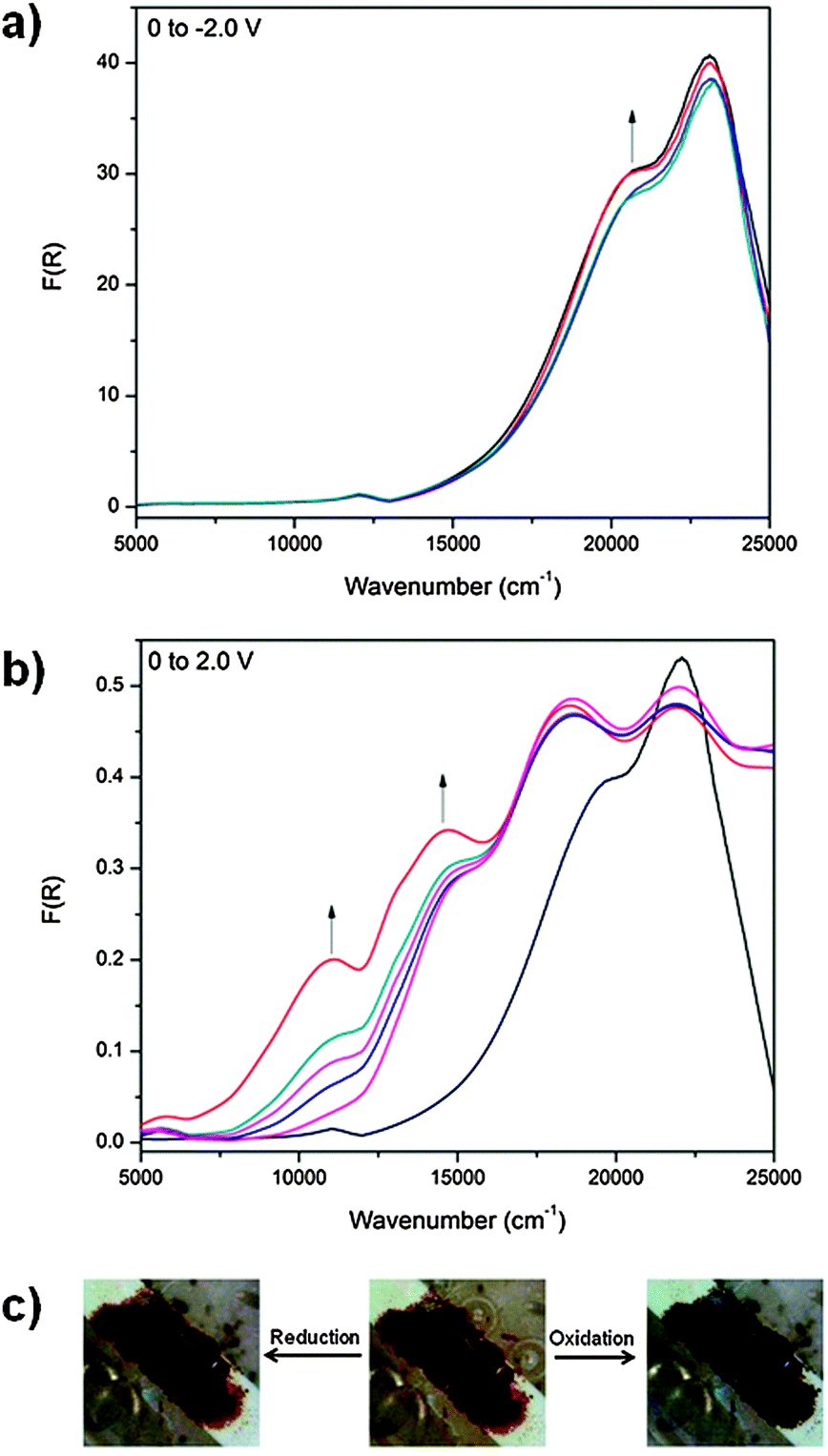 | ||
| Fig. 3 In situ solid state vis/NIR spectroelectrochemical experiment in [(n-C4H9)4N]PF6/CH3CN for POP-V1 where (a) oxidation (0 to 2.0 V), (b) reduction (0 to −2.0 V) and (c) photos of the oxidation/reduction. | ||
In the neutral state, a small EPR signal was observed for POP-V1 which may be a consequence of the donor–acceptor properties of the polymer, where the electron was spontaneously transferred from the triarylamine electron donor to the viologen electron acceptor. Upon application of a potential of −0.9 V in the solid state EPR spectroelectrochemical experiment for POP-V1, the EPR signal was significantly enhanced from the neutral state indicating formation of the viologen radical cation (Fig. 4). The intensity of this signal decreased upon application of a potential of 0 V to reform the neutral state; a potential of 1.7 V then resulted in an enhancement of the signal intensity due to formation of the triarylamine radical cation, which was able to be reduced upon application of a potential of 0 V.
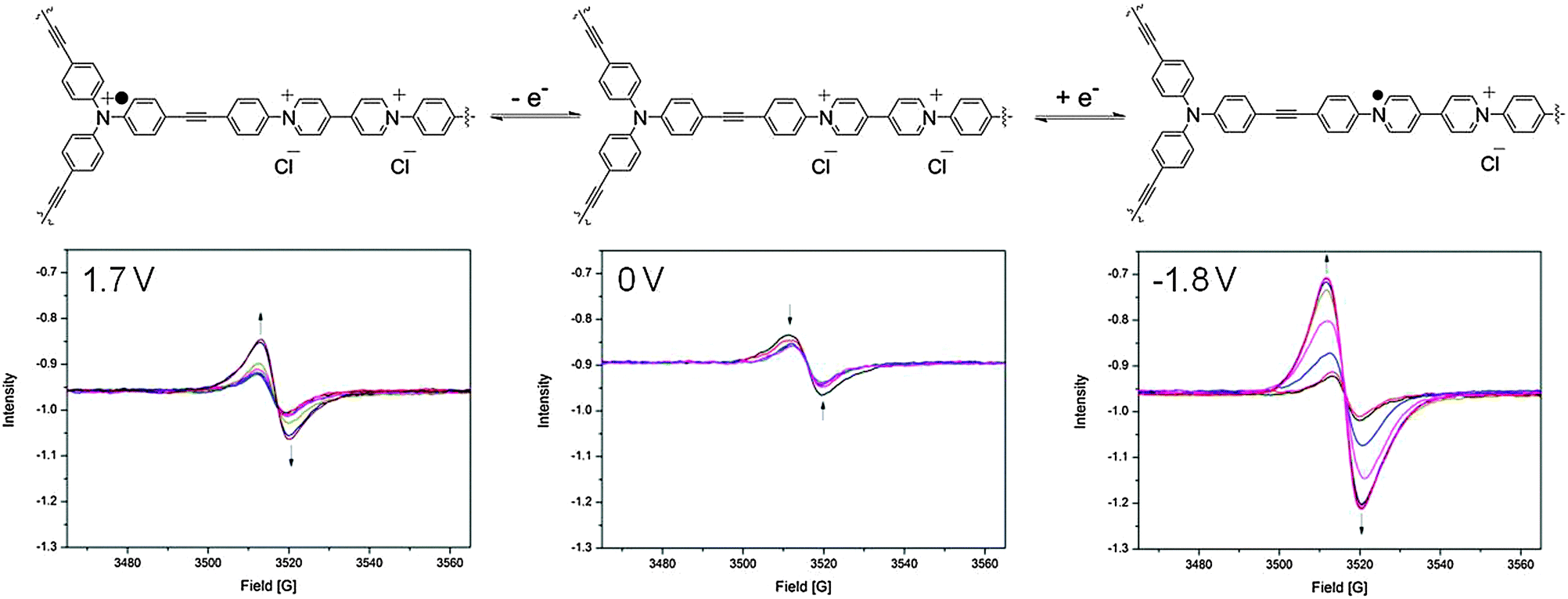 | ||
| Fig. 4 Solid state EPR spectroelectrochemical data for POP-V1 in [(n-C4H9)4N]PF6/CH3CN electrolyte. The arrows indicate the direction of spectral progression throughout the course of the experiment. | ||
The viologen radical cation exhibited a different g-factor (2.004) when compared to the triarylamine radical cation (2.002), which corresponds to the trend observed for the chemically oxidized and reduced species for POP-V1 (Fig. S11, ESI†). The EPR signal due to the viologen radical cation displayed more anisotropy than that for the triarylamine radical cation, presumably due to the reduced symmetry of the viologen moiety. Upon application of a potential of −1.8 V (Fig. 4), the signal intensity was significantly increased when compared to the first reduction (Fig. S9, ESI†). The first reduction and oxidation processes within the framework likely resulted in structural changes within the polymeric system, which allowed the counter-ions to diffuse with greater ease into the pores of the network. Consistent with this argument, our DFT-optimized structures for mPOP-V1(Cl)˙ and mPOP-V1(Cl)3˙ have Cl− ions that are more loosely bound than those in mPOP-V1(Cl)2. Thus, the Cl− ions in mPOP-V1(Cl)2 have a distance of 2.26 Å to the closest carbon atom, whereas in the oxidized and reduced analogs the corresponding distances are 2.50 Å and 2.84 Å, respectively. This increased the degree of reduction obtainable, which made the system more amenable to changes in redox state.
For POP-V2, application of a negative potential of −2.0 V resulted in a color change from yellow to brown with an increase in the intensity of the band at ∼18000 cm−1 and decrease in the band at 22500 cm−1 (Fig. S7, ESI†). These processes can be attributed to formation of the viologen radical cation30–32 and were spectrally reversible upon application of a positive potential.
The signal intensity increase in the solid state EPR spectroelectrochemical experiment for POP-V2 upon application of a potential of −1.7 V was due to formation of the viologen radical cation (Fig. S10, ESI†). As the potential was increased to 1.5 V, the signal intensity decreased to a point where it remained constant, as expected given the absence of the redox-active triarylamine core. During the second reduction process, the signal shape changed from one with axial anisotropy to one higher in symmetry, suggesting that the radical environment had been altered during the first reduction process, presumably due to structural change within the polymer system in an analogous fashion to POP-V1.
Exploiting tunable redox states to modulate H2 and CO2 uptake
The nitrogen sorption isotherms at 77 K for POP-V1, ox-POP-V1, red-POP-V1, POP-V2 and red-POP-V2 displayed a significant hysteresis upon desorption which points towards flexibility within the different interlinked polymer networks (Fig. 5a). The step in the N2 desorption isotherm at ca. 542 mbar for POP-V1, red-POP-V1, POP-V2 and red-POP-V2 indicated the presence of mesopores within the polymer structures, whilst the absence of this step for ox-POP-V1 was likely to arise from the tetrafluoroborate counterion from the chemical oxidant (NOBF4). The pore size analysis indicated several different sized pores at 6, 10, 12.5 and 22 Å for the polymer systems (Fig. S13, ESI†). Ox-POP-V1 lacked the 22 Å size pore which correlated well with the absence of a step in the N2 desorption isotherm.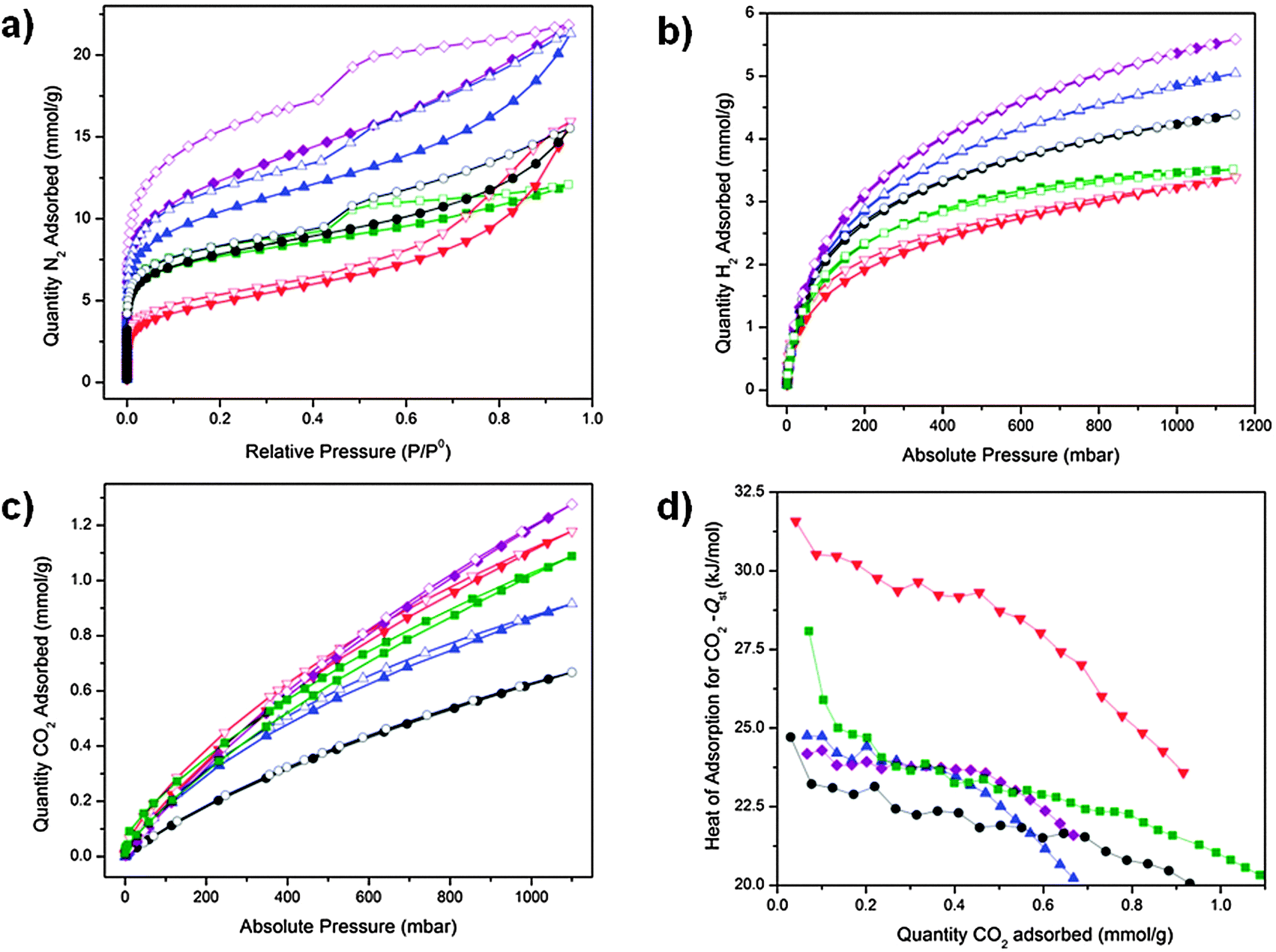 | ||
Fig. 5 Gas sorption isotherms of POP-V1 ( ), ox-POP-V1 ( ), ox-POP-V1 ( ), red-POP-V1 ( ), red-POP-V1 ( ) and POP-V2 ( ) and POP-V2 ( ), red-POP-V2 ( ), red-POP-V2 ( ) for (a) N2 at 77 K, (b) H2 at 77 K, (c) CO2 at 298 K and (d) the isosteric heat of adsorption for CO2 where adsorption = filled symbols and desorption = open symbols. The lines joining each point are provided as a guide to the reader. ) for (a) N2 at 77 K, (b) H2 at 77 K, (c) CO2 at 298 K and (d) the isosteric heat of adsorption for CO2 where adsorption = filled symbols and desorption = open symbols. The lines joining each point are provided as a guide to the reader. | ||
All polymer systems displayed moderate BET surface areas of 390–960 m2 g−1 (Table 1). The significant drop in surface area for ox-POP-V1 was presumably due to additional occupancy of the pores by the BF4− counter-ions from the chemical oxidant. A decrease in the BET surface area upon reduction was contrary to the expectation that the reduced polymer systems should exhibit higher surface areas as the doubly reduced quinone state of the viologen moiety is neutral, negating the need for counterions in the pores for charge balance. A plausible rationale for this observation is that removal of the chloride counterions results in a partial collapse of the networks due to the loss of stability which is provided by the counterion in the neutral polymers. In the latter case, electrostatic repulsion within the positively charged networks would be expected to support increased network porosity.
| BET surface area [m2 g−1] | |
|---|---|
| POP-V1 | 812 |
| Ox-POP-V1 | 390 |
| Red-POP-V1 | 606 |
| POP-V2 | 960 |
| Red-POP-V2 | 591 |
Hydrogen isotherms at 77 K for the neutral polymers (POP-V1: 5.04 mmol g−1, POP-V2: 5.59 mmol g−1 at 1150 mbar) displayed a significantly higher uptake than the reduced polymers (red-POP-V1: 4.39 mmol g−1, red-POP-V2: 3.51 mmol g−1 at 1150 mbar) (Fig. 5b). Likewise, POP-V1 also displayed a significantly higher uptake of hydrogen (5.04 mmol g−1 at 1150 mbar) when compared to ox-POP-V1 (3.38 mmol g−1 at 1150 mbar) (Fig. 5b). These observations are consistent with the lower surface area of the polymer upon oxidation or reduction.
The CO2 uptake was also dependent on the redox state. A lower uptake of CO2 at 298 K was observed for the reduced networks (red-POP-V1: 0.92 mmol g−1, red-POP-V2: 1.09 mmol g−1 at 1100 mbar) when compared to their neutral analogues (POP-V1: 0.67 mmol g−1, POP-V2: 1.27 mmol g−1 at 1100 mbar). However, ox-POP-V1 displayed a higher CO2 uptake (1.18 mmol g−1 at 1100 mbar) when compared to POP-V1, despite having a significantly lower surface area (Fig. 5c). To understand the origins of these observations, we have computed the mPOP-V1(Cl)n–CO2 complexes (n = 0–3) and the corresponding binding energies. The calculated structures suggest that the interaction of CO2 with a Cl− ion is important for its binding within the materials (Fig. 6). It is presumed that reduction of the polymers with LiNp (−3.10 vs. Fc/Fc+)33 resulted in the formation of the neutral viologen quinone state in red-POP-V1 and red-POP-V2. Indeed, for mPOP-V1 that represents a model of the fully-reduced polymer with no Cl−, the binding energy is significantly smaller (3.1 kJ mol−1vs. 11.4 kJ mol−1 for mPOP-V1(Cl)2). Upon one-electron oxidation to mPOP-V1(Cl)3 (16.6 kJ mol−1) or one-electron reduction to mPOP-V1(Cl) (16.8 kJ mol−1), the binding becomes somewhat stronger. The presence of the BF4− counterion is presumed to have a similar affect towards the binding energy of the ox-POP-V1 polymer with CO2 as that of Cl− due to the interaction of the negatively charged ion with the partially positive carbon of CO2. It thus appears that the increased CO2 adsorption upon polymer oxidation can be attributed to both an increase in the binding, as well as an increase in the number of anionic binding sites. On the other hand, the decreased CO2 binding in the reduced form may be a consequence of the removal of the counter-ions from the polymer.
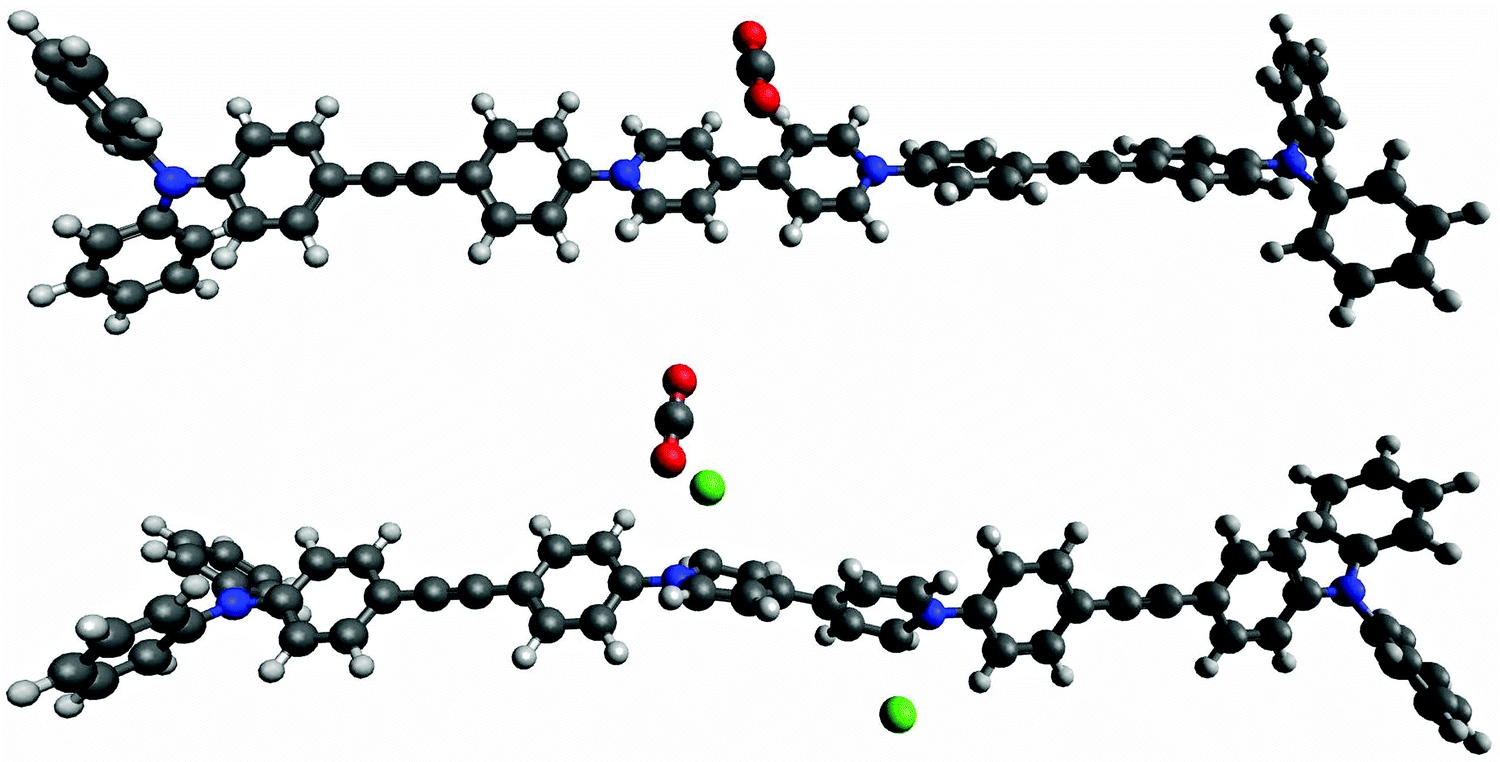 | ||
| Fig. 6 DFT HOMO (−0.20977) and LUMO (−0.06623) of mPOP-V1(Cl) with BMK/6-31G(d) orbital energies (hartree) in parentheses of the monomer POP-V1 polymer fragments, mPOP-V1 and mPOP-V1(Cl)3 with CO2 where C = grey, N = blue, H = white, O = red, Cl = green. The interaction of CO2 with Cl- in mPOP-V1(Cl)3 is observed. | ||
The CO2 isosteric heats of adsorption for all polymers are indicative of a physisorption process (Fig. 5d), which is in agreement with our calculated complex structures. The reduced polymers exhibited lower or comparable heats of adsorption relative to the neutral systems whilst the enhanced affinity for CO2 by ox-POP-V1 (−32 kJ mol−1 at low gas loadings) was evident from the significantly higher heat of adsorption for CO2 when compared to POP-V1 (−25 kJ mol−1 at low gas loadings).
The single point selectivity improved as the temperature was increased due to the higher interaction of the quadrupolar CO2 molecules with the surface of the polymer (Fig. S14, ESI†). In all cases, the selectivity for CO2 over N2 increased upon reduction of the framework at 295 K whilst the selectivity was significantly decreased for ox-POP-V1. For the neutral and oxidized species, the selectivity for CO2vs. N2 was enhanced upon increasing the temperature to 308 and 318 K. The increased kinetic energy allowed the stronger electrostatic interaction of the polar CO2 molecule to dominate over the weaker interaction of the non-polar N2 molecule with the material. These results demonstrate the ability to tune the selectivity parameters for CO2/N2 as both a function of the redox state and temperature in these materials.
Conclusions
The combination of the electron acceptor viologen with the electron donor triarylamine has yielded donor–acceptor polymers, POP-V1 and POP-V2, with differential electronic and physical properties in their distinct redox states. The reversible nature of the redox switching provides access to different charge transfer processes, optical and host–guest properties which can be exploited to control the adsorption of gases such as H2 and CO2. The ability to access multiple switchable properties within the same material demonstrates significant potential for the use of these systems in applications ranging from molecular electronics to electrical swing adsorption processes.34–36Experimental section
All chemicals and solvents were used as obtained and used without further purification. Tris(p-iodophenyl)amine,37 1,3,5-tris(p-ethynylphenyl)benzene,38 1,1′-bis(4-bromophenyl)-4,4′-bipyridinium chloride,39 Pd(PPh3)440 and lithium naphthalenide33 were synthesized according to literature procedures. Tris(p-ethynylphenyl)amine41 and 1,1′-bis(2,4-dinitrophenyl)-4,4′-bipyridine-1,1′-diium chloride42 were synthesized according to modified literature procedures. Acetonitrile and triethylamine were dried over CaH2 and dimethylformamide dried over activated CaSO4. Solution state 1H and 13C{1H} NMR spectra were recorded on Bruker AVANCE200, AVANCE300 or AVANCE500 spectrometers operating at 200, 300, 500 MHz for 1H and 50, 75, 125 MHz for 13C respectively. 1H and 13C{1H} NMR chemical shifts were referenced internally to residual solvent resonances. Spectra were recorded at 298 K and chemical shifts (δ), with uncertainties of ±0.01 Hz for 1H and ±0.05 Hz for 13C are quoted in ppm. Coupling constants (J) are quoted in Hz and have uncertainties of ±0.05 Hz for 1H–1H. Deuterated solvents were obtained from Cambridge Stable Isotopes and used as received.Synthesis of POP-V1
Tris(p-ethynylphenyl)amine (0.500 g, 1.57 mmol), 1,1′-bis(4-bromophenyl)-4,4′-bipyridinium chloride (0.679 g, 1.26 mmol) and Pd(PPh3)4 (14.6 mg, 0.0126 mmol) were suspended in a mixture of DMF (5 mL) and triethylamine (5 mL) and stirred under nitrogen for 5 minutes prior to the addition of CuI (4.80 mg, 0.0252 mmol). The reaction mixture was observed to darken from a bright yellow suspension to a dark yellow suspension. The reaction was then heated at 90 °C for 40 hours to yield a dark red/brown solid. The reaction mixture was cooled and filtered. The solid was washed consecutively with DMF, chloroform, methanol, water, methanol and acetone before being air dried. The solid was then washed by soxhlet extraction with methanol until the washings turned colorless before being dried under vacuum to yield the product as a dark yellow-red solid (820 mg). Elemental analysis, found: C, 77.33; H, 4.33 and N, 5.35%.Synthesis of POP-V2
1,3,5-Tris(p-ethynylphenyl)benzene (0.176 g, 0.464 mmol), 1,1′-bis(4-bromophenyl)-4,4′-bipyridinium chloride (0.200 g, 0.371 mmol) and Pd(PPh3)4 (4.2 mg, 3.71 × 10−6 mol) were suspended in a mixture of DMF (5 mL) and triethylamine (5 mL) and stirred under nitrogen for 5 minutes. CuI (1.4 mg, 7.42 × 10−6 mol) was then added and the reaction mixture heated at 90 °C for 40 hours resulting in a color change from pale green to dark brown/green. The reaction mixture was cooled, filtered and worked up as for POP-V1 to yield the product as a yellow solid (250 mg). Elemental analysis, found: C, 83.45; H, 4.48 and N, 1.18%.Chemical oxidation
The polymer (100 mg) was suspended in dry and distilled acetonitrile (2 mL) and the mixture thoroughly degassed. A saturated solution of nitrosonium tetrafluoroborate (10 mL) in acetonitrile (10 mL) was added slowly dropwise with vigorous bubbling of nitrogen to yield an immediate color change to dark brown. The mixture was left to stir at room temperature with vigorous bubbling of nitrogen for 30 minutes, the solid filtered off and dried.Chemical reduction
The polymer (20 mg) was suspended in dry THF (2 mL) and a solution of lithium naphthalenide (0.1 M in THF) added slowly dropwise under argon with stirring. The solid was observed to change color from yellow to brown. The reaction mixture was stirred for 2 hours under argon before the solid was filtered, washed with THF and dried.Computational calculations
Standard DFT calculations were carried out with Gaussian 09.43 The molecules containing the viologen moiety capped with two triarylamines [mPOP-V1(Cl)n] or triarylbenzene [mPOP-V2(Cl)n] (Fig. 3a), and with an appropriate number of chloride ions for charge balance, were employed to model the polymers in their neutral (2 Cl−), oxidized (3 Cl−), singly-reduced (1 Cl−) and doubly-reduced (0 Cl−) states. Geometries were obtained at the B-PW91/6-31G(d) level.44,45 The vibrational frequencies of stationary points were inspected to ensure that they corresponded to minima on the potential energy surface. The UV/vis spectra of the model systems were computed with the TDA-DFT procedure, i.e., time-dependent DFT with the Tamm–Dancoff approximation46 using the BMK functional47 in conjunction with the 6-31G(d) basis set. Molecular orbitals reported in the text correspond to those obtained with these TDA-BMK calculations. To examine the binding of CO2 with mPOP-V1(Cl)n, we carried out constrained optimization (at the B-PW91/6-31G(d) level) of mPOP-V1(Cl)n–CO2 complexes, in which the fully-optimized mPOP-V1(Cl)n geometry is employed but frozen in the calculation. Refined single-point energies were obtained with the MN12L procedure48 together with the 6-31+G(d) basis set. Complexation energies were reported as vibrationless values in kJ mol−1.Solid state NMR
The 13C CPMAS solid state NMR experiments were carried out on a wide-bore Bruker Biospin Avance III solids-300 MHz spectrometer operating at a frequency of 75 MHz for the 13C nucleus. Approximately 80 mg of sample was packed into 4 mm zirconia rotors fitted with Kel-f® caps and spun in a double resonance H–X probehead at 8 kHz MAS (Magic Angle Spinning). The 13C and 1H 90° radio frequency pulse lengths were optimized to 3.5 μs each. The 13C CPMAS spectra were acquired with 1 ms cross polarization contact time with a total suppression of spinning sidebands (TOSS) scheme, followed by 1H decoupling at 75 kHz field strength using Spinal-64 decoupling. The 13C NQS (non-quaternary carbon suppression) spectra were recorded by turning off the 1H decoupling for 40 μs during the TOSS period. For sufficient signal to noise, ca. 10k transient were acquired for each sample with recycle delays of 3.0 s in between to ensure sufficient relaxation of the 1H magnetization. The spectra were obtained at room temperature. The 13C chemical shifts were referenced to the glycine CO peak at 176 ppm.Electrochemistry
Solid state electrochemical measurements were performed using a Bioanalytical Systems Epsilon Electrochemical Analyser. Argon was bubbled through solutions of 0.1 M [(n-C4H9)4N]PF6 dissolved in distilled CH3CN. The cyclic voltammograms (CVs) for POP-V1 were recorded using a glassy carbon working electrode (1.5 mm diameter), a platinum wire auxiliary electrode and an Ag/Ag+ wire quasi reference electrode. Solid state samples were mounted on the glassy carbon working electrode by dipping the electrode into a paste made of the powder sample in acetonitrile. The CVs for POP-V2 were recorded using a 1 mm wide strip of graphite as the working electrode. A piece of platinum wire was adhered to the top of the graphite strip with conductive silver paint. The sample was immobilized on the working electrode by smearing a paste of the sample suspended in acetonitrile to the surface of the graphite strip and allowing the acetonitrile to evaporate. Ferrocene was added as an internal standard upon completion of each experiment. All potentials are quoted in V versus Fc/Fc+.UV/vis/NIR
UV/vis/NIR spectroscopy was performed on a CARY5000 Spectrophotometer equipped with a Harrick Praying Mantis accessory, where dried BaSO4 was used for the baseline. The spectra of the chemically reduced species were recorded under an atmosphere of argon. Spectra are reported as the Kubelka–Munk transform F(R) as a function of wavenumber (cm−1). The optical band gap was estimated by drawing a tangential line along the slope to the curve and determining the wavelength (in nm) where this line intersected the x-axis. The wavelength was then converted to eV.Solid state spectroelectrochemistry (vis/NIR)
In the solid state, the diffuse reflectance spectra of the electrogenerated species were collected in situ in 0.1 M [(n-C4H9)4N]PF6/CH3CN electrolyte over the range 5000–25000 cm−1 using a Harrick Omni Diff Probe attachment and a custom built solid state spectroelectrochemical cell described previously.21 The cell consisted of a Pt wire counter electrode and an Ag/Ag+ quasi-reference electrode. The solid sample was immobilized onto a 0.1 mm thick indium-tin-oxide (ITO) coated quartz slide (which acted as the working electrode) using a thin strip of Teflon tape. The applied potential (from −2.0 to 2.0 V) was controlled using an eDAQ potentiostat. Continuous scans of the sample were taken and the potential increased gradually until a change in the spectrum was observed.
Electron paramagnetic resonance (EPR) spectroscopy
EPR spectroscopy (Bruker EMX-X band) was performed at room temperature on freshly synthesized materials (20 mg). The microwave frequency was between 9.75 and 9.85 GHz for all measurements and the microwave power well below that required for saturation. All other parameters remained constant: modulation frequency, 100 kHz; modulation amplitude, 1.00 G; sweep width, 100 G; and each spectrum was averaged over eight scans. The EPR signals obtained were referenced against Strong Pitch to obtain the g-factor value.Solid state spectroelectrochemistry (EPR)
The procedure and cell set up used were as previously reported for a solution state EPR spectroelectrochemical experiment.49 A three-electrode assembly consisting of simple narrow wires (A–M Systems) as electrodes was used. Teflon coated platinum (0.20 and 0.13 mm coated and uncoated diameters, respectively) and silver wires (0.18 and 0.13 mm coated and uncoated diameters, respectively) were used for the working and quasi-reference electrodes respectively, while a naked platinum wire (0.125 mm) attached to a small piece of rolled platinum mesh (∼5 mm × 4 mm) played the role of the counter electrode. The bottom 1 cm of the Teflon coated wires were stripped (using an Eraser International Ltd, RT2S fine wire stripper). The sample of interest was wrapped in a small piece of platinum mesh (∼5 mm × 3 mm) lengthwise and twisted to ensure the sample remained immobilized. The exposed end of the working electrode was carefully wrapped in a spiral manner around the platinum mesh where the working electrode was positioned lowest such that the redox product of interest was generated at the bottom of the tube and was well separated from the counter electrode. The electrodes were soldered to a narrow three-core flexible wire/cable. The cell used was made by flame sealing the tip of a glass pipette. The potential was controlled with a portable μAutolab II potentiostat and the EPR spectra obtained using an EMX Micro X-band EPR spectrometer equipped with a 1.0 T electromagnet. The EPR signals obtained were referenced against Strong Pitch to obtain the g-factor value, and the response of the EPR sample cavity and that of the sample holder and wire-setup was checked prior each experiment. In all cases the samples were EPR silent prior electrochemistry unless otherwise stated. A 0.1 M [(n-C4H9)4N]PF6/CH3CN supporting electrolyte solution was used for all experiments.Thermal gravimetric analysis (TGA)
TGA was performed under a flow of nitrogen (0.1 L min−1) on a TA Instruments Hi-Res Thermogravimetric Analyser from 25–700 °C at 1 °C min−1.Gas adsorption
Gas adsorption measurements over the 0–1 bar range were conducted using a Micromeritics Accelerated Surface Area and Porosity (ASAP) 2020 instrument. Approximately 50–100 mg of the polymer sample was degassed under vacuum at 70 °C for approximately 14 hours. Nitrogen adsorption isotherms were measured at 77 K via the incremental dosing of nitrogen from 0–1 bar, and surface areas determined via the BET method using the ASAP2020 V4.01 software. CO2 isotherms at 298, 308 and 318 K were measured on the same instrument, with temperature controlled by a Julabo F25 Circulating Heating and Cooling bath. The enthalpy of adsorption for CO2 was calculated using data from the three CO2 isotherms and the Clausius–Clapeyron equation. An estimation of the porosity distribution in the POPs was obtained using the Tarazona Non-Local Density Functional Theory (with cylindrical pores) implemented within the Micromeritics ASAP 2020 software (Version 4.0). The single component selectivity factors for CO2vs. N2 for the POP systems were calculated using eqn (1) at 0.75 bar N2vs. 0.15 bar CO2 (0.1 bar other gases), which are industrially relevant pressures for the capture of CO2 from post combustion flue gas streams.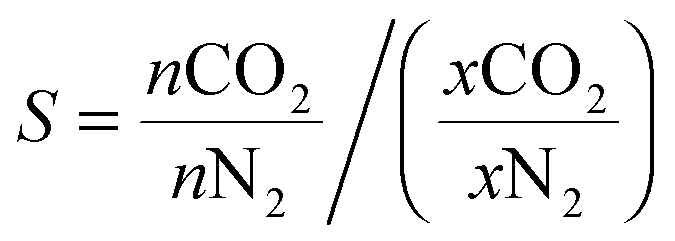 | (1) |
Acknowledgements
We gratefully acknowledge support from the Australian Research Council and the Science and Industry Endowment Fund. We thank the EPSRC UK National Electron Paramagnetic Resonance Service at the University of Manchester.Notes and references
- F. C. Krebs, Nat. Mater., 2008, 7, 766–767 CrossRef CAS PubMed.
- Y. Li, T. Liu, H. Liu, M.-Z. Tian and Y. Li, Acc. Chem. Res., 2014, 47, 1186–1198 CrossRef CAS PubMed.
- A. K. Bakhshi and S. Kaul, Appl. Biochem. Biotechnol., 2001, 96, 125–133 CrossRef CAS PubMed.
- H. A. M. van Mullekom, J. A. J. M. Vekemans, E. E. Havinga and E. W. Meijer, Mater. Sci. Eng., R, 2001, 32, 1–40 CrossRef.
- K. Yoshino, K. Tada, A. Fujii, E. M. Conwell and A. A. Zakhidov, IEEE Trans. Electron Devices, 1997, 44, 1315–1324 CrossRef CAS.
- H. A. M. van Mullekom, J. A. J. M. Vekemans and E. W. Meijer, Chem. – Eur. J., 1998, 4, 1235–1243 CrossRef CAS.
- A. Ajayaghosh, Chem. Soc. Rev., 2003, 32, 181–191 RSC.
- E. Espíldora, J. L. Delgado and N. Martín, Isr. J. Chem., 2014, 54, 429–439 CrossRef.
- V. K. Praveen, C. Ranjith, E. Bandini, A. Ajayaghosh and N. Armaroli, Chem. Soc. Rev., 2014, 43, 4222–4242 RSC.
- P. M. Beaujuge, C. M. Amb and J. R. Reynolds, Acc. Chem. Res., 2010, 43, 1396–1407 CrossRef CAS PubMed.
- L. Beverina, G. A. Pagani and M. Sassi, Chem. Commun., 2014, 50, 5413–5430 RSC.
- C.-L. Liu and W.-C. Chen, Polym. Chem., 2011, 2, 2169–2174 RSC.
- C. Hua, A. Rawal, T. B. Faust, P. D. Southon, R. Babarao, J. M. Hook and D. M. D'Alessandro, J. Mater. Chem. A, 2014, 2, 12466–12474 CAS.
- F. Caruso, V. Atalla, X. Ren, A. Rubio, M. Scheffler and P. Rinke, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 90, 085141 CrossRef.
- E. Kampar and O. Neilands, Russ. Chem. Rev., 1986, 55, 334 CrossRef.
- A. Heckmann and C. Lambert, Angew. Chem., Int. Ed., 2012, 51, 326–392 CrossRef CAS PubMed.
- S. Amthor, B. Noller and C. Lambert, Chem. Phys., 2005, 316, 141–152 CrossRef CAS.
- C. L. Bird and A. T. Kuhn, Chem. Soc. Rev., 1981, 10, 49–82 RSC.
- D. Bongard, M. Möller, S. N. Rao, D. Corr and L. Walder, Helv. Chim. Acta, 2005, 88, 3200–3209 CrossRef CAS.
- F. J. Rizzuto, T. B. Faust, B. Chan, C. Hua, D. M. D'Alessandro and C. J. Kepert, Chem. – Eur. J., 2014, 20, 17597–17605 CrossRef CAS PubMed.
- P. M. Usov, C. Fabian and D. M. D'Alessandro, Chem. Commun., 2012, 48, 3945–3947 RSC.
- C. F. Leong, T. B. Faust, P. Turner, P. M. Usov, C. J. Kepert, R. Babarao, A. W. Thornton and D. M. D'Alessandro, Dalton Trans., 2013, 42, 9831 RSC.
- C. F. Leong, B. Chan, T. B. Faust and D. M. D'Alessandro, Chem. Sci., 2014, 5, 4724–4728 RSC.
- C. Hua, P. Turner and D. M. D'Alessandro, Dalton Trans., 2013, 42, 6310–6313 RSC.
- C. Hua and D. M. D'Alessandro, CrystEngComm, 2014, 16, 6331–6334 RSC.
- J.-K. Sun, X.-H. Jin, L.-X. Cai and J. Zhang, J. Mater. Chem., 2011, 21, 17667–17672 RSC.
- R. Kannappan, C. Bucher, E. Saint-Aman, J.-C. Moutet, A. Milet, M. Oltean, E. Metay, S. Pellet-Rostaing, M. Lemaire and C. Chaix, New J. Chem., 2010, 34, 1373–1386 RSC.
- S. Asaftei, A. M. Lepadatu and M. Ciobanu, Helv. Chim. Acta, 2011, 94, 1091–1101 CrossRef CAS.
- M. E. Alberto, B. C. De Simone, S. Cospito, D. Imbardelli, L. Veltri, G. Chidichimo and N. Russo, Chem. Phys. Lett., 2012, 552, 141–145 CrossRef CAS.
- I. Okura, M. Kobayashi, N. Kim-Thuan, S. Nakamura and K.-I. Nakamura, J. Mol. Catal., 1980, 8, 385–390 CrossRef CAS.
- M. M. Matsushita, M. Morikawa, T. Kawai and T. Iyoda, Mol. Cryst. Liq. Cryst. Sci. Technol., Sect. A, 2000, 343, 87–96 CrossRef CAS.
- E. M. Kosower and J. L. Cotter, J. Am. Chem. Soc., 1964, 86, 5524–5527 CrossRef CAS.
- N. G. Connelly and W. E. Geiger, Chem. Rev., 1996, 96, 877–910 CrossRef CAS PubMed.
- C. A. Grande and A. E. Rodrigues, Int. J. Greenhouse Gas Control, 2008, 2, 194–202 CAS.
- C. A. Grande, R. P. P. L. Ribeiro and A. E. Rodrigues, ChemSusChem, 2010, 3, 892–898 CrossRef CAS PubMed.
- N. Tlili, G. Grévillot, A. Latifi and C. Vallières, Ind. Eng. Chem. Res., 2012, 51, 15729–15737 CrossRef CAS.
- Y. Jiang, Y. Wang, J. Yang, J. Hua, B. Wang, S. Qian and H. Tian, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 1830–1839 CrossRef CAS.
- M. Yang, S. Shi, M. Wang, Y. Wang, Z. Luo, Z. Feng and T. Zhao, J. Appl. Polym. Sci., 2010, 117, 714–719 CrossRef CAS.
- F. Biedermann and O. A. Scherman, J. Phys. Chem. B, 2012, 116, 2842–2849 CrossRef CAS PubMed.
- D. R. Coulson, L. C. Satek and S. O. Grim, Inorganic Syntheses, John Wiley & Sons, Inc., 2007, pp. 121–124 Search PubMed.
- N. Niamnont, N. Kimpitak, K. Wongravee, P. Rashatasakhon, K. K. Baldridge, J. S. Siegel and M. Sukwattanasinitt, Chem. Commun., 2013, 49, 780–782 RSC.
- A. S. Abouelwafa, V. Mereacre, T. S. Balaban, C. E. Anson and A. K. Powell, CrystEngComm, 2010, 12, 94–99 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, Gaussian 09, Revision C.01, Wallingford CT, 2009 Search PubMed.
- A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098 CrossRef CAS.
- J. P. Perdew, Electronic Structure of Solids '91, Akademie Verlag, Berlin, 1991 Search PubMed.
- M. Head-Gordon and S. Hirata, Chem. Phys. Lett., 1999, 314, 291 CrossRef.
- A. D. Boese and J. M. Martin, J. Chem. Phys., 2004, 108, 3085 CrossRef CAS.
- R. Peverati and D. G. Truhlar, Phys. Chem. Chem. Phys., 2012, 14, 13171–13174 RSC.
- Y. Liu, J.-R. Li, W. M. Verdegaal, T.-F. Liu and H.-C. Zhou, Chem. – Eur. J., 2013, 19, 5637–5643 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional synthetic procedures, solid state NMR data, thermal gravimetric analysis, electrochemistry, EPR and gas sorption data. See DOI: 10.1039/c6tc00132g |
| This journal is © The Royal Society of Chemistry 2016 |