
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The molecular steps of citrinin biosynthesis in fungi†
Yi
He‡
ab and
Russell J.
Cox
*bc
aCollege of Food Science and Technology, Huazhong Agricultural University, Wuhan 430070, Hubei Province, P. R. China
bInstitut für Organische Chemie, Leibniz Universität Hannover, Schneiderberg 1B, 30167 Hannover, Germany. E-mail: russell.cox@oci.uni-hannover.de
cSchool of Chemistry, University of Bristol, Cantock's Close, Bristol, UK BS8 1TS, UK
First published on 17th December 2015
Abstract
The individual steps of citrinin 1 biosynthesis in Monascus ruber M7 were determined by a combination of targeted gene knockout and heterologous gene expression in Aspergillus oryzae. The pathway involves the synthesis of an unreduced trimethylated pentaketide 10 by a non-reducing polyketide synthase (nrPKS) known as CitS. Reductive release yields the keto-aldehyde 2 as the first enzyme-free intermediate. The nrPKS appears to be assisted by an as-yet cryptic hydrolysis step catalysed by CitA which was previously wrongly annotated as an oxidase. CitB is a non-heme iron oxidase which oxidises the 12-methyl of 2 to an alcohol. Subsequent steps are catalysed by CitC which oxidises the 12-alcohol to an aldehyde and CitD which converts the 12-aldehyde to a carboxylic acid. Final reduction of C-3 by CitE yields citrinin. The pathway rules out alternatives involving intramolecular rearrangements, and fully defines the molecular steps for the first time and corrects previous errors in the literature. The activity of CitB links the pathway to fungal tropolone biosynthesis and the observation of aminated shunt products links the pathway to azaphilone biosynthesis. Production of citrinin by coordinated production of CitS + CitA–CitE in the heterologous host A. oryzae, in which each gene was driven by a constitutive promoter, was achieved in high yield.
Introduction
Like many mycotoxins, citrinin 1 was discovered by Harold Raistrick and coworkers in the 1930s.1 Its structure was solved by Whalley and coworkers in 1948.21 was isolated first from Penicillium citrinum, but is also known to be produced by other Penicillium species3 as well as species of Monascus including M. ruber and M. purpureus.4 It is a potent mycotoxin with nephrotoxic activities due to inhibition of respiration complex III.5 Citrinin was among the first compounds to be identified as a polyketide and its biosynthesis was investigated by Birch, Whalley and others during the 1950s using radioisotopes,6 and more latterly by Staunton,7 Sankawa8 and others9 using stable isotopes and NMR during the 1980s and 1990s. The gene cluster for citrinin, centered on the 7.9 kb gene pksCT which encodes an iterative type I non-reducing polyketide synthase (nr-PKS), was discovered in M. purpureus during the mid 2000s by Nihira and coworkers.10,11 Near-identical clusters have been discovered in M. ruber, M. aurantiacus and Penicillium expansum.Remarkably little is known, however, of the individual chemical assembly steps of citrinin biosynthesis, and many assumptions and misleading interpretations have appeared in the literature (see ESI†).
Citrinin is widely regarded as a model compound, especially in the arena of fungal polyketide biosynthesis, where it is related to many bioactive metabolites such as the azaphilones,12 sorbicillinoids13 and tropolones.14 We thus sought to definitively clarify its biosynthesis. For this reason we decided to perform a thorough and systematic investigation of the citrinin cluster from Monascus ruber M7 using gene knockout and heterologous expression strategies.
Key questions focus on the identity of the product of the PKS, the order of the post-PKS tailoring steps, the precise limits of the citrinin gene cluster, and the relationship between the citrinin biosynthetic pathway and those of other fungal metabolites. Furthermore, a significant number of errors have appeared in the literature regarding citrinin biosynthesis and we wished to correct these. For example the frequently repeated assignment of ctnB as an oxidoreductase appeared unlikely;7,15 bizarre interpretations of labelling experiments in M. ruber appeared to justify the proposal of unprecedented intramolecular rearrangements during the formation of 1 (see ESI†);16 and errors in diagrams propagate incorrect conclusions about the biosynthetic steps (see ESI†).3
Results
We began by analysing the known gene clusters for citrinin biosynthesis from Penicillium and Monascus species (Fig. 1). Comparisons show that a minimal set of conserved genes likely to be involved in biosynthesis include: citS, encoding an nr-PKS17 (citrinin synthase) with SAT, KS, AT, PT, ACP, C-MeT and R domains; mrl1,18 encoding a serine hydrolase of no known function; mrl2, encoding a non-heme Fe(II) dependent oxygenase similar to TropC from stipitatic acid biosynthesis known to be involved in oxidative ring expansion;14,19mrl4, encoding an NAD(P)+ dependent aldehyde dehydrogenase; mrl6, encoding a short-chain dehydrogenase; and another NAD(P)+-dependent oxidoreductase encoded by mrl7. Other genes nearby such as mrr1 (encoding a likely transporter), mrl3 (encoding a transcription factor) and mrl5 (glyoxylase-like domain) and genes further downstream of mrr1 or upstream of mrl7 appeared to have no likely role in catalysis and were not considered further.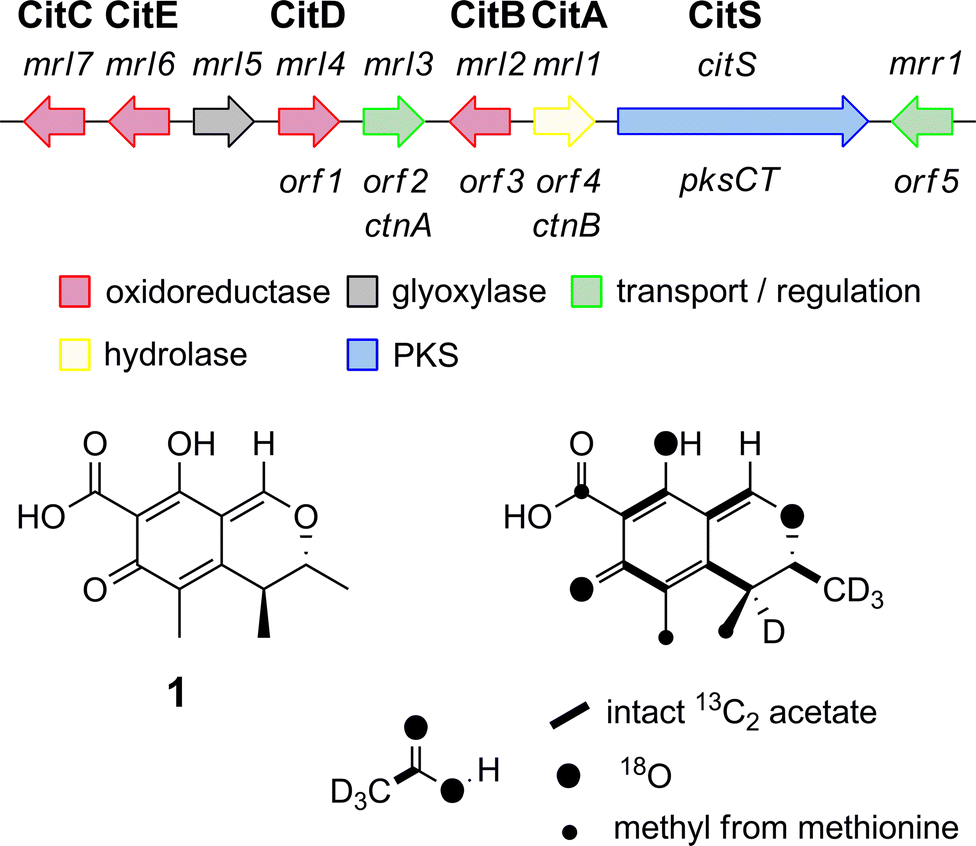 | ||
| Fig. 1 The citrinin gene cluster from Monascus species and the labelling pattern of citrinin deduced from isotopic feeding experiments. | ||
Knockout (KO) of citS encoding the citrinin PKS in Monascus purpureus has already been reported to abolish citrinin biosynthesis.20 We repeated this in M. ruber M7 using a KO strategy based on insertion of a neomycin antibiotic resistance cassette in the target gene via agrobacterium mediated transformation and selection on PDA plates containing the antibiotic G418 (15 μg mL−1). This was facilitated by use of a Δku80 strain as previously reported.21 As expected citrinin biosynthesis was totally abolished (Fig. 2).
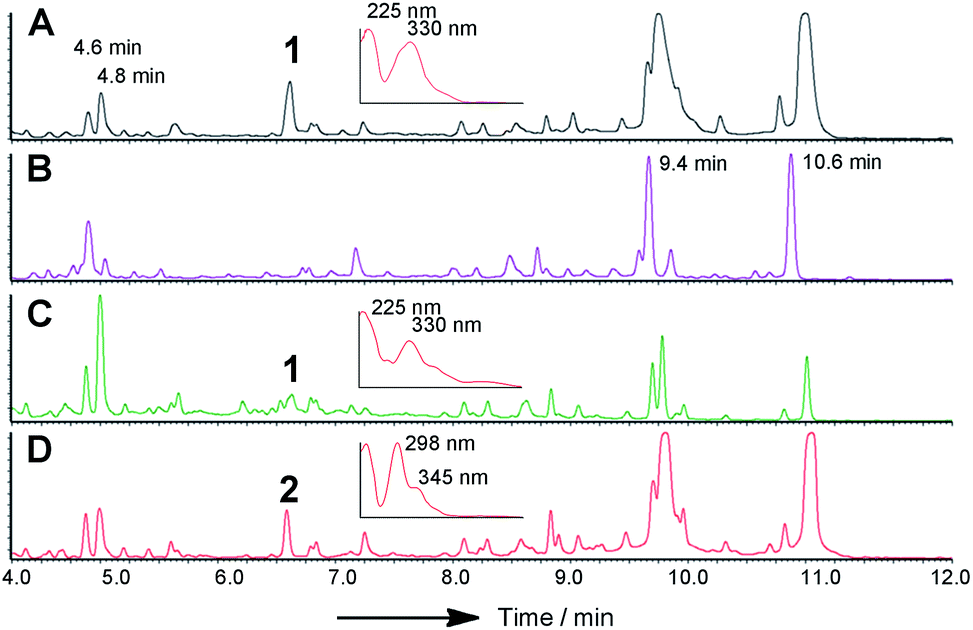 | ||
| Fig. 2 LCMS chromatograms (DAD trace) of organic extracts from targeted KO experiments in M. ruber M7 Δku80: (A) WT, inset shows DAD spectrum of citrinin 6.4 min; (B) ΔcitS; (C) Δmrl1, inset shows DAD spectrum of citrinin 6.4 min; (D) Δmrl2, inset shows DAD spectrum of compound 2, 6.4 min. Peaks at 4.6, 4.8, 9.4 and 10.6 min are due to other M. ruber polyketides unrelated to citrinin. | ||
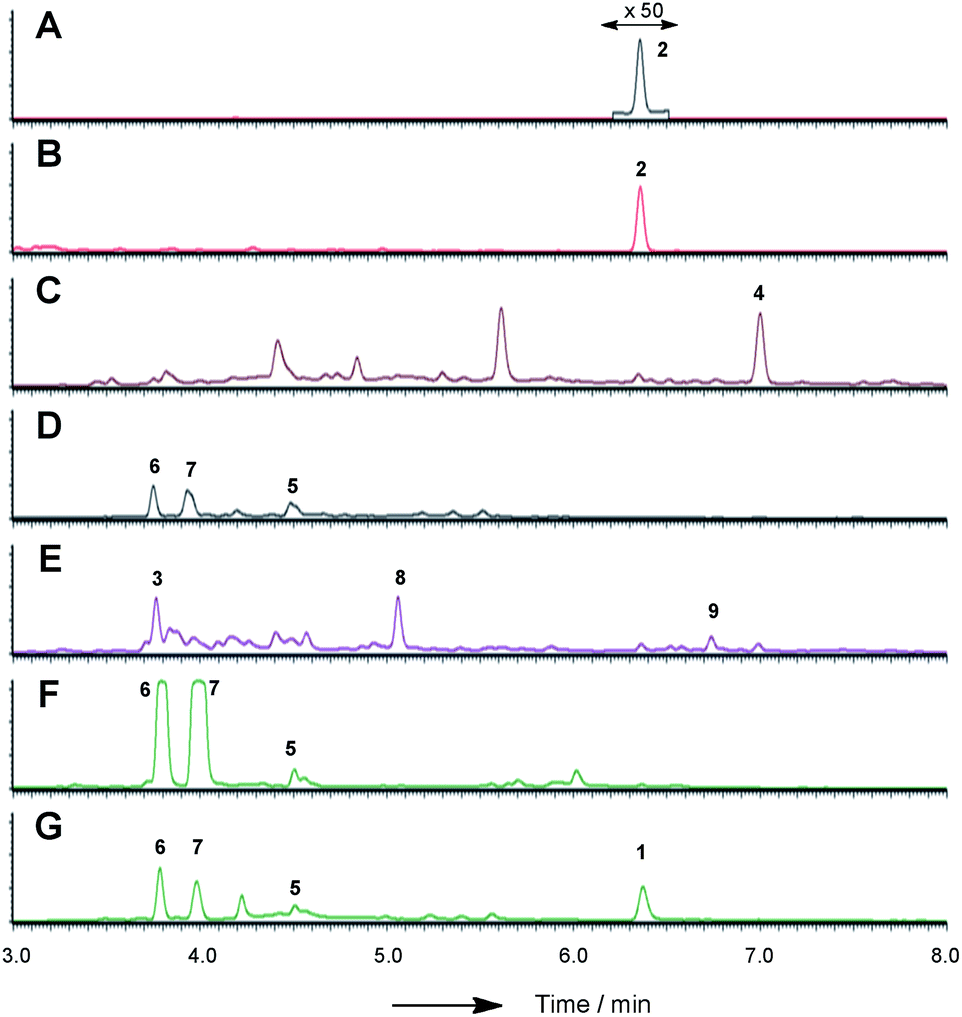 | ||
| Fig. 3 LCMS chromatograms (UV, 350 nm) of organic extracts from heterologous expression experiments in A. oryzae NSAR1: (A) A. oryzae + citS; (B) A. oryzae + citS + mrl1; (C) A. oryzae + citS + mrl1 + mrl2; (D) A. oryzae + citS + mrl1 + mrl2 + mrl4; (E) A. oryzae + citS + mrl1 + mrl2 + mrl7; (F) A. oryzae + citS + mrl1 + mrl2 + mrl7 + mrl4; (G) A. oryzae + citS + mrl1 + mrl2 + mrl7 + mrl4 + mrl6. All chromatograms are presented at the same scale. See ESI† for further detail. | ||
Repeating the procedure with mrl1 led to a significant reduction (>95%), but not abolition, of citrinin biosynthesis, and no new compounds appeared in the LCMS chromatograms of these mutants. KO of mrl2, however, led to complete absence of citrinin and the production of a new compound at the same retention time which was distinguishable by its UV and mass spectra. This new compound was purified and identified by HRMS, 1D and 2D NMR as the ketoaldehyde 2 (see ESI†). Further KO experiments were performed using the same methodology targeted at mrl4, mrl6 and mrl7. In each case abolition of citrinin biosynthesis was observed, but no significant new peaks were detected in the LCMS chromatograms despite genetic analysis showing that the deletions had been created as expected.
In order to gain more detail on the function of the PKS and the tailoring enzymes, the cluster was reconstituted in the heterologous host Aspergillus oryzae using the modular expression system described by Lazarus and coworkers.22 Initial work focussed on the expression of the PKS itself in order to determine the first enzyme-free intermediate. Previous analysis of the PKS gene suggested that it contained a 56 bp intron located at 640 bp of citS. This was removed during yeast-recombination-mediated construction of an expression system (see ESI†). Thus the vector pTYGS arg citS was created in which the inducible promoter PamyB was used to drive expression of citS. Transformation of this system into A. oryzae NSAR1 (ref. 23) led to the production of low levels of the ketoaldehyde 2 (0.8 mg L−1).
In previous heterologous experiments using A. oryzae as a host we have usually observed high yields of secondary metabolites.24 However we are aware that A. oryzae can sometimes have problems in splicing some introns from heterologous genes.25 Although we had removed one intron during cloning we could not be sure that all introns in citS had been located. We therefore sequenced cDNA prepared from the native host M. ruber and found clear evidence for a second intron located between the ACP- and R-domain-encoding regions of the gene. Careful and repeated sequencing indicated this to be 62 base-pairs in length meaning that when spliced the remaining R-domain located downstream would be out-of frame with respect to the rest of the protein and thus unable to form a functional reductive domain (see ESI†). The sequencing results, however, showed a mixed population of transcripts – spliced transcript formed the majority, but unspliced (and thus potentially catalytically active) transcript was clearly present as a minority population. Exhaustive control experiments indicated that this was not caused by contamination with gDNA. We repeated this analysis in the A. oryzae citS expression system and found that A. oryzae treats the intron in the same way, again giving a mixed population of transcripts. Construction of an expression clone in which the 62 bp intron sequence had been removed resulted in no production of 2 or any other new compounds. However, removal of 60 bp (i.e. in-frame) resulted in an active synthase capable of producing 2 at the same titre as the WT system (see ESI†).
We next co-expressed citS with mrl1 (exp. 2, Table 1) using the same promoter system for citS and Padh from A. oryzae26 for mrl1. These clones proved to be very good producers of the ketoaldehyde 2 producing approximately 15 mg L−1.
| Exp. | citS | mrl1/CitA | mrl2/CitB | mrl4/CitD | mrl6/CitE | mrl7/CitC | Products |
|---|---|---|---|---|---|---|---|
| 1 | X | — | — | — | — | — | 2 |
| 2 | X | X | — | — | — | — | 2, 3* |
| 3 | X | X | X | — | — | — | 2*, 3*, 4 |
| 4 | X | X | X | X | — | — | 5*, 6*, 7* |
| 5 | X | X | X | — | — | X | 2*, 3, 4*, 8, 9 |
| 6 | X | X | X | — | X | — | 2 |
| 7 | X | X | X | X | — | X | 5*, 6, 7 |
| 8 | X | X | X | — | X | X | 2*, 3, 4*, 8 |
| 9 | X | X | X | X | X | — | 1*, 5*, 6, 7 |
| 10 | X | X | X | X | X | X | 1, 2*, 4*, 5*, 6, 7 |
In addition shunt compounds were observed such as 3 which was isolated and purified (see ESI†) and identified as the product of aldehyde reduction by full NMR and HRMS analysis.
Bioinformatic analysis of mrl1 indicated two possible start codons differing by 52 codons. We thus also cloned the shorter possibility and expressed this with citS. Since the shorter clone proved to be just as active as the longer clone, and bioinformatic analysis suggested that the initial 52 residues of the longer peptide were not highly homologous to other known proteins we concluded that the shorter ORF is likely to be the true coding region and used this for further experiments.
Introduction of mlr2, encoding the non-heme iron oxygenase, driven by the Padh promoter, then led to the production of the alcohol 4, again in high titre (16 mg L−1, exp. 3), which was purified and fully characterised by HRMS, 1D and 2D NMR (see ESI†). Another compound eluting at 5.6 min showed a similar LCMS profile to 4 but could not be identified due to instability during purification.
We next expressed the citS + mrl1 + mrl2 core genes plus all 7 possible combinations of the remaining genes mrl4, mrl6 and mrl7 (Table 1). Addition of mrl4 alone (exp. 4) to the core genes led to the weak production of compounds 5 (2.5 mg L−1), 6 (3.6 mg L−1) and 7 (4.6 mg L−1). Compound 6 was isolated as a mixture of interconverting diastereomers shown to be the enamine aminal 6 by HRMS and full NMR structural determination (see ESI†). At this stage compound 7 proved highly unstable and could not be identified, but was isolated later (exp. 7, vide infra). Addition of mrl6 to the core genes (exp. 6) produced no additional new compounds that were detected.
Addition of mrl7 alone (exp. 5) gave chromatograms which were substantially similar to WT A. oryzae NSAR, with either very low or zero concentrations of compounds 2 and 4. This showed that 4 was probably converted to a reactive intermediate which was either degraded or converted to numerous shunt products. Close examination of the mass chromatogram revealed a new peak at 5.1 minutes with the same mass as compound 4 (m/z 252, 7.0 min). Purification revealed this to be the hemiacetal 8 (18 mg L−1) which appeared to equilibrate to a mixture of diastereomers. In the presence of methanol (for example during workup) the methyl acetal 9 (as two diastereomers) was also detected by HRMS analysis, but rapid degradation prevented its full characterisation.
Coexpression of mrl7 and mrl4 (exp. 7) with the core genes provided 6 (23 mg L−1) and 7 (30 mg L−1) as the major products in high titre. Although 7, the N-(2-hydroxyethyl) congener, was produced as a major metabolite it proved highly unstable upon purification and NMR solutions always contained diastereomers of 6. Two diastereomers of 7 were evident in the NMR spectra, but only the major diastereomer could be fully characterised. Facile fragmentation of 7 presumably occurs by acid catalysed loss of ethylene oxide, leading to 6 as observed.
Expression of mrl7 and mrl6 together (exp. 8) gave minor amounts of 2 and 4 as well as 3 and 8 (15.6 mg L−1). Coexpression of mrl4 and mrl6 (exp. 9) also generated no new compounds. Finally, coexpression of the full complement of mrl4, mrl6 and mrl7 (exp. 10) led to the production of citrinin 1 in high titre (19.1 mg L−1), accompanied by many of the previously identified intermediates and shunt compounds (Scheme 1).
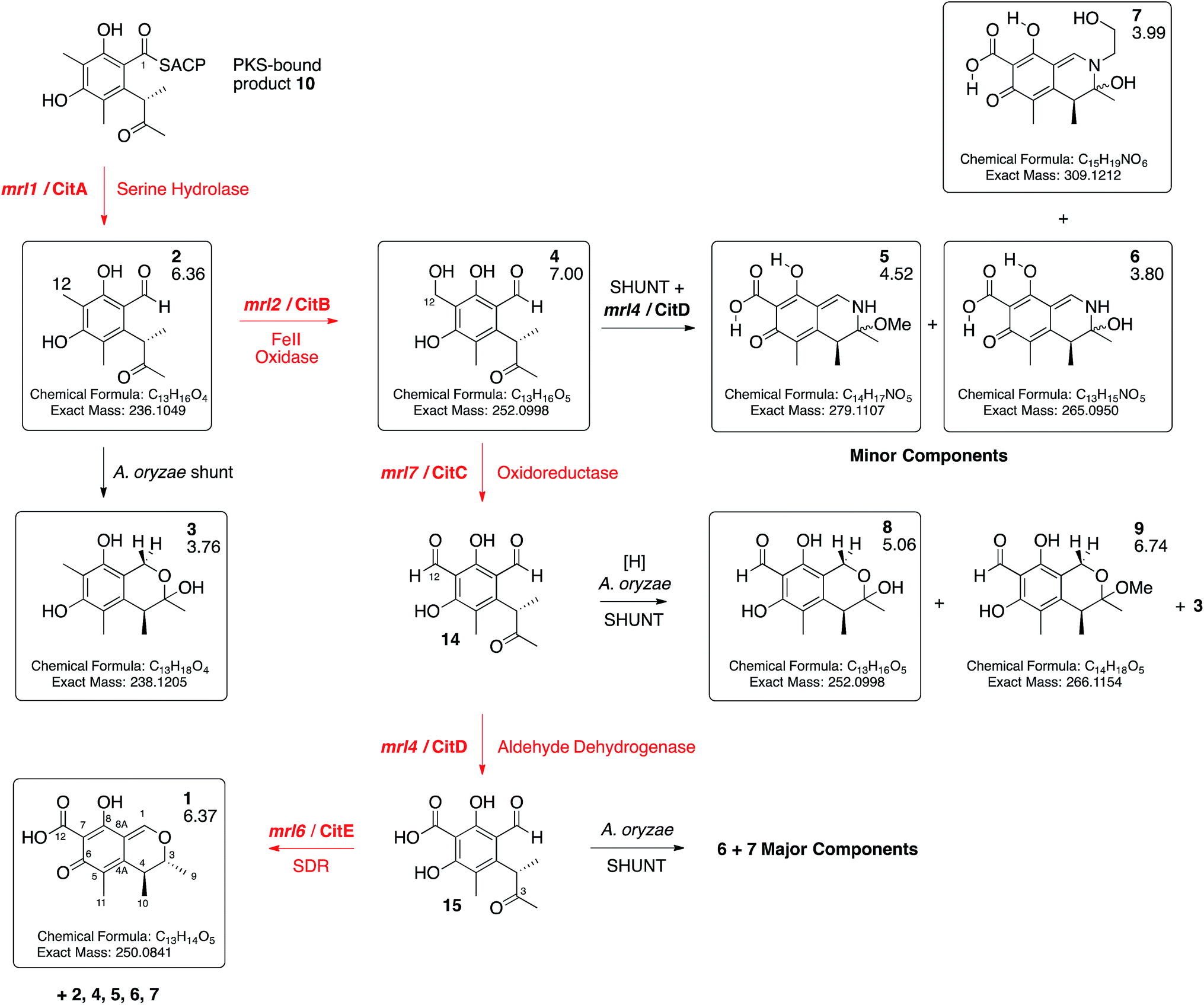 | ||
| Scheme 1 Deduced biosynthetic pathway to citrinin 1 (red steps). Boxed compounds purified and characterised. Black steps represent shunts branching from the main pathway. Figures below compound numbers are LCMS RT values (see ESI† and Fig. 3). Unboxed compounds structures inferred. | ||
The mrl1 gene encodes a protein most likely to be a hydrolase or an acyl transferase. Similar hydrolases are involved in product release during brefeldin biosynthesis, for example. However 2 is an aldehyde, most likely released by reduction of the ACP-bound thiolester 10 (see Scheme 3). We previously observed such a reductive release during biosynthesis of 3-methylorcinaldehyde, the precursor of fungal tropolones, in Acremonium strictum encoded by the aspks1 gene, but in this case no hydrolase is required in A. oryzae.14,27 The role of a hydrolytic protein during citrinin biosynthesis is thus obscure. In order to probe this question further we produced mutants of mrl1. The encoded protein contains a typical catalytic triad of S122, D207 and H235 and each of these residues was individually mutated to Ala. In each case coexpression of citS with the mutated mrl1 genes led to a chemotype identical to that of expression of citS alone showing that the hydrolytic properties of the mrl1 protein are required for the effective formation of 2 (see ESI†). In the cases of the WT protein and each mutant we also searched the LCMS chromatograms for possible product-related compounds released by hydrolysis – however no evidence supporting the existence of such compounds could be found.
Bioinformatic analysis of the CitS R-domain and R-domains of related nrPKS indicate a highly conserved cysteine (C2551, see ESI†). A homology model of the CitS R-domain constructed using the X-ray structure of the recently reported myxalamid R-domain as a template (see ESI†) showed that C2551 is located in the active site close to the catalytically important Y2392 and K2396 residues, in the vicinity of the NADPH binding pocket. In order to probe the role of this cysteine we mutated it to Ala. However we observed no differences in the function of CitS-C2551A vs. the WT protein when expressed either alone, or with either of the mrrl1 long or short proteins (see ESI†).
Discussion
Heterologous expression of citS, the citrinin PKS, yields the trimethylated pentaketide aldehyde 2. Experiments by Staunton and coworkers sought to determine if 2-methyl-3-hydroxy butyrate 11 could be the starter unit for biosynthesis, giving 12 as an intermediate, or whether the PKS itself reduces the C-3 carbonyl giving 12 directly (Scheme 2).28 Other fungal systems, such as the resorcylic acid lactones, are well known in which a reduced polyketide acts as a starter for a non-reducing PKS.29 Our results clearly show that the citrinin PKS produces the ketone 2 as the first enzyme free intermediate meaning that reduction of C-3 must be performed by a tailoring enzyme. This is in accord with the production of 2 by an iterative PKS which is now known to belong to the non-reducing class.17 Release of the aldehyde 2 is consistent with the presence of the C-terminal reductive release domain. The titre of 2 afforded in the heterologous expression experiment was surprisingly low (<1 mg L−1) – in our previous investigation into the related fungal tetraketide PKS methylorcinaldehyde synthase (MOS) we observed high titres from similar expression experiments (ca. 40 mg L−1).28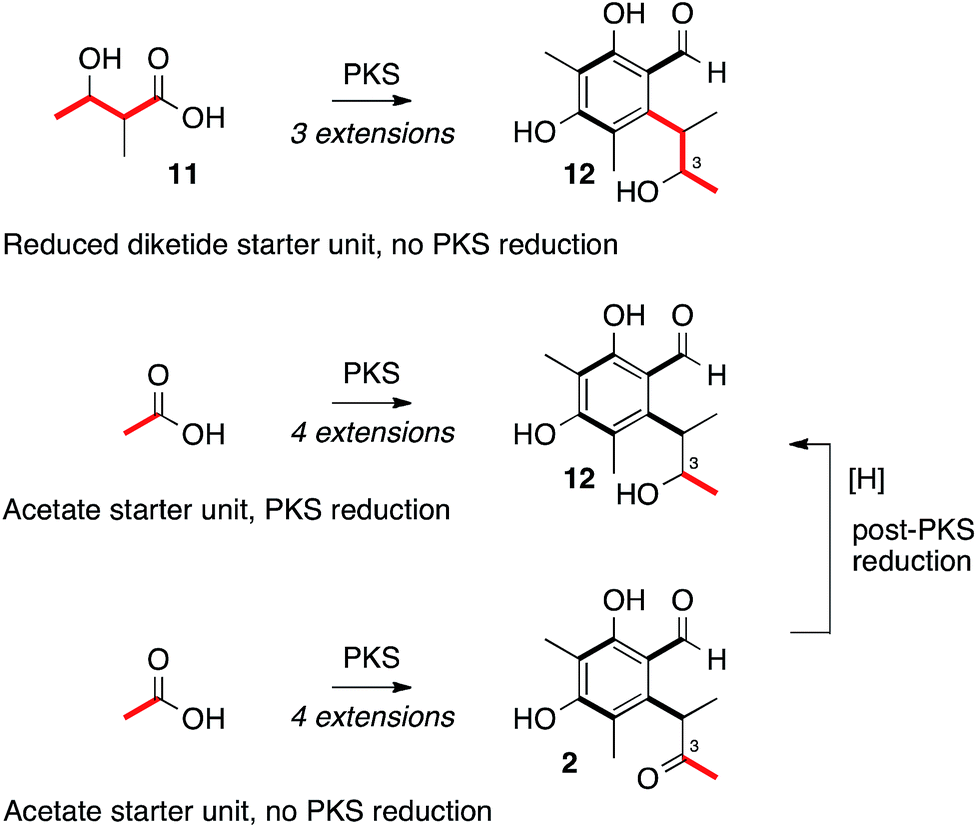 | ||
| Scheme 2 Plausible routes to alcohol oxidation level at C-3. | ||
Immediately adjacent to the citS gene is mrl1 which encodes a hydrolase. This gene, also known as ctnB, is frequently and incorrectly referred to as an oxidoreductase.10 The protein encoded by mrl1 contains no recognisable protein motifs for redox cofactor binding sites and is clearly closely related to the esterase/lipase family (EC 3.1.1) of serine hydrolases.30
We have shown that the putative active site Ser-122, Asp-207 and His-235 residues are involved in a catalytic step during biosynthesis by systematic mutation of each to Ala. Coexpression of citS and mrl1 leads to dramatic increase in titre of 2. Likewise KO of mrl1 in WT M. ruber leads to drastic reduction, but not abolition, of citrinin 1 titres.
In other PKS systems hydrolases are often involved in product release. For example an α/β hydrolase is involved in release of brefeldin intermediates,31 and LovG releases the completed nonaketide dihydromonacolin from the lovastatin nonaketide synthase (LNKS).31 However the release of the final polyketide acyl group 10, bound to the thiol of ACP is reductive, in analogy to the reductions of many other enzyme-bound thiolesters, for example during the biosynthesis of 3-methylorcinaldehyde 13 by the PKS methylorcinaldehyde synthase, and myxalamid 14 by the mixed PKS-NRPS MxaA (Scheme 3).32
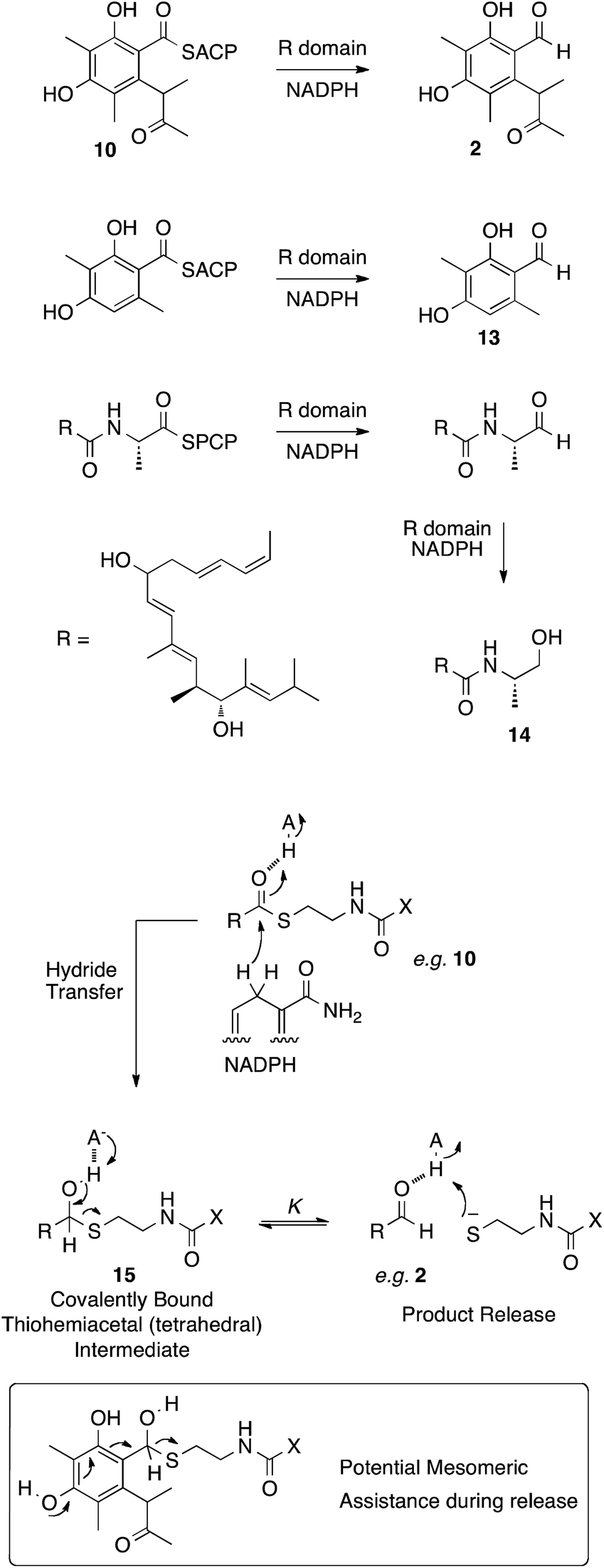 | ||
| Scheme 3 Reductive release mechanisms and a plausible mechanism. X = pantetheine-ACP. | ||
These reactions involve the transfer of hydride from NADPH to form a thiohemiacetal intermediate 15 which is still covalently bound to the enzyme (Scheme 3). Breakdown of the thiohemiacetal 15 then releases the aldehyde (e.g.2) from the active site. In the case of 14 biosynthesis the released aldehyde is reduced a second time.
Previous studies on the thermodynamics of thiohemiacetal formation have shown that these compounds are in equilibrium with the free thiol and aldehyde. The equilibrium constant K for this process does not change significantly with thiol pKa, but does change with the identity of the acyl group. In general, electron-withdrawing groups stabilise the electron-rich thiohemiacetal reducing the value of K (so that K < 1 [Scheme 3], although not usually less than 0.1), favouring thiohemiacetal formation.33 In contrast, electron donating groups favour the separate thiol and aldehyde so that K > 1 (i.e. product release here).34 In the case of CitS the substrate is highly electron rich – the conjugated aromatic would be expected to contribute strongly to collapse of the tetrahedral thiohemiacetal and release of the product (Scheme 3). Thus it seems unlikley that the mrl1 protein could be involved in product release.
Other roles for the mrl1 protein could be as an acyl transferase, possibly responsible for loading starter unit or malonyl extender unit on to the PKS. These roles are normally performed by the PKS SAT and AT domains respectively. Sequence analysis of CitS suggests that these domains are intact, with the expected conserved catalytic residues (see ESI†). Other possibilities include transfer of enzyme-bound thiolester 10 to the mrl1 protein (16) and then on to CoA, forming 17, prior to reduction. Hydrolysis of 16 or 17 could lead to compounds such as 18–20, however LCMS analysis of extracts failed to provide evidence for the presence of these species (Scheme 4).
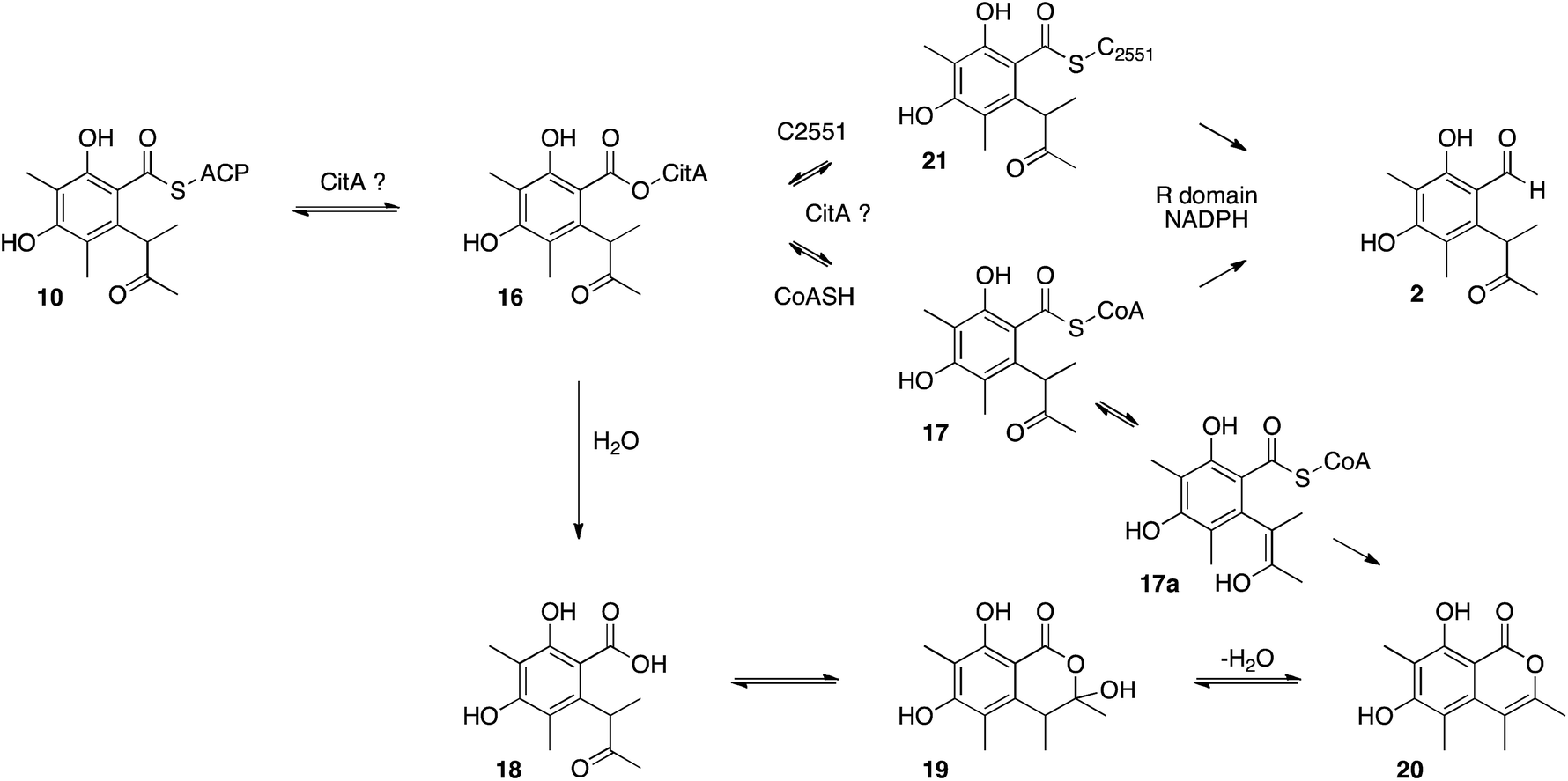 | ||
| Scheme 4 Possible roles of CitA during citrinin biosynthesis. | ||
Another possibility could be transfer of the polyketide to a conserved cysteine thiol in the R-domain active site (e.g.21). Some acyl group reductases, for example aspartate semialdehyde dehydrogenase,35 use an active site thiol rather than a pantetheine thiol to hold the acyl group. One such conserved cysteine (C2551) was identified in the active site of the CitS R-domain, but mutational analysis showed it is not required for reductive release.
Thus a hydrolytic/transferase enzyme activity such as that seemingly catalysed by the mrl1 hydrolase is clearly not expected in this case, or required by closely related polyketide synthases such as those which produce 3-methylorcinaldehyde 13 at high titre without hydrolytic assistance. The exact catalytic role of the mrl1 protein therefore remains mysterious, although it is clear that it increases the productivity of the PKS. We thus rename the mrl1 hydrolase protein as CitA as it is the earliest non-PKS step during citrinin biosynthesis. It should be noted that during the preparation of this manuscript Kwon and coworkers36 reported a similar phenomenon during the biosynthesis of azaphilones also in Monascus species, and the involvement of hydrolytic processes during aromatic polyketide biosynthesis may therefore be a more general phenomenon. We plan further in vitro experiments to probe the interaction of CitA with CitS in future.
Adjacent to the mrl1 gene in M. ruber is mrl2 which encodes a non-heme iron oxidase related to TropC. TropC oxidises a methyl group during tropolone biosynthesis and also catalyses rearrangement of the dearomatised ring (Scheme 5). Here we have shown by KO and by heterologous expression that the mrl2 encoded protein catalyses the oxidation of the C-12 methyl of 2 to an alcohol 4, but in contrast to TropC it does this while the ring is still aromatic, and there is thus no oxidative rearrangement. This is the second step in the pathway and we thus name this protein CitB.
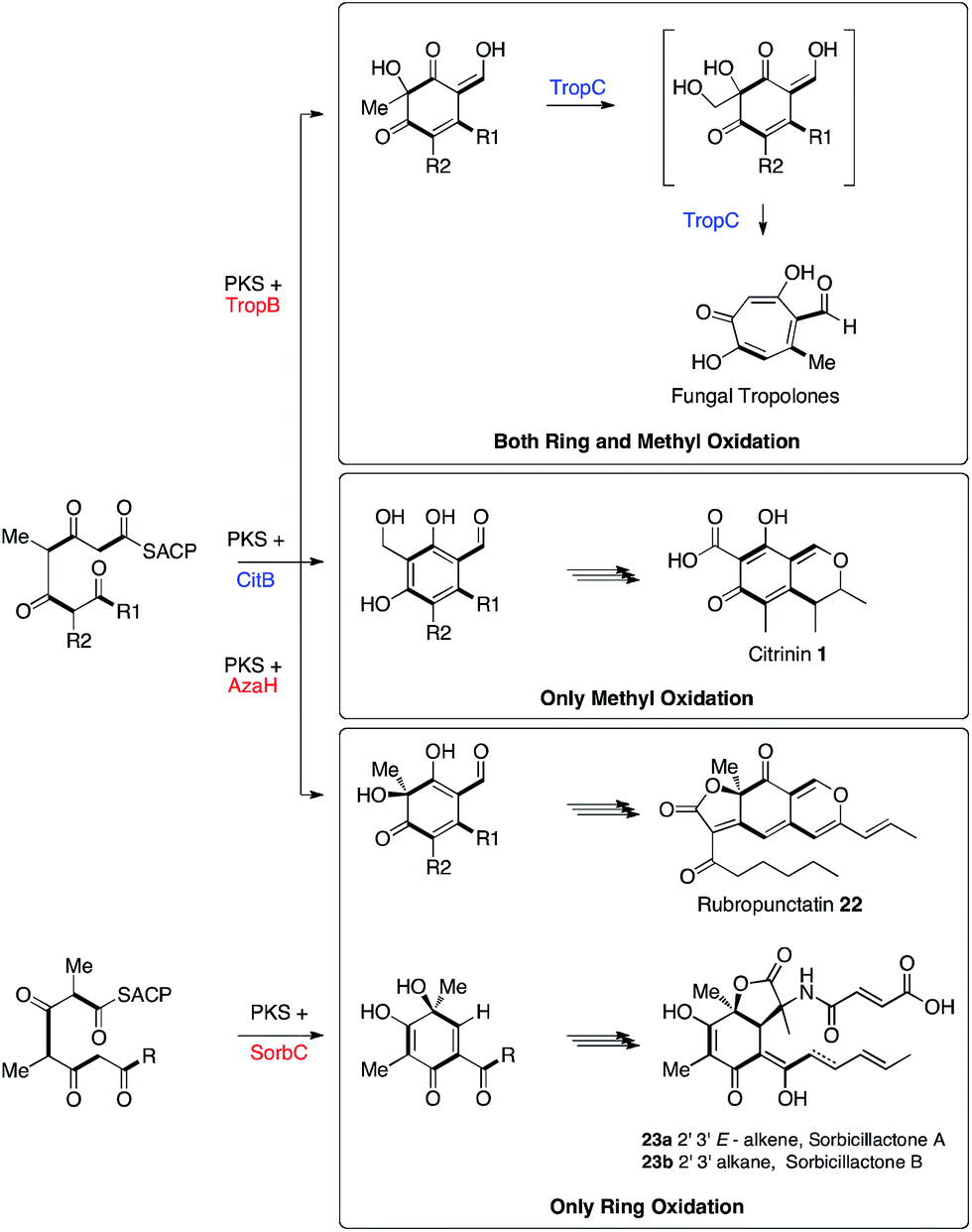 | ||
| Scheme 5 Biosynthetic relationships between the fungal tropolone, citrinin, azaphilone and sorbicillinoid pathways. | ||
Tropolone biosynthesis requires oxidation of both the ring itself and the ring-methyl group of a polyketide aldehyde. The biosynthesis of citrinin, in which only the ring-methyl is oxidised, contrasts with the biosynthesis of azaphilones such as rubropunctatin 2237 and the sorbicillinoids such as sorbicillactones 23ab13 where only ring oxidation is required. Thus the three pathways are clearly differentiated by the presence or absence of these two different oxidative steps. This knowledge will be important for more accurately predicting the products of related fungal biosynthetic gene clusters.
Conversion of 4 to 1 requires two further oxidations of C-12 and a reduction of C-3. The remaining 3 genes in the cluster all encode oxidoreductases and heterologous expression of citS, mrl1 and mrl2 with mrl4, mrl6 and mrl7 leads to good production of 1 in A. oryzae. The data obtained here are consistent with a synthetic scheme in which 4 is first oxidised by the mlr7 oxidoreductase (now renamed CitC) to produce the bisaldehyde 14. In our expression experiments this compound is highly unstable and thus not observed directly, but we do observe the shunt compounds 8 and 9 which result from the expected oxidation accompanied by an adventitious A. oryzae shunt reduction of C-1. In cases where MeOH was used during purification the methyl acetal 9 was formed. The course of oxidation mirrors that observed during stipitatic acid biosynthesis where a ring-methyl is converted to a carboxylic acid.19
The fourth catalytic step is catalysed by the mrl4 enzyme (now renamed CitD) which encodes an aldehyde dehydrogenase. This is expected to give 15, but again we only observe the shunt metabolites 6 and 7 which have incorporated ammonia or ethanolamine respectively – again presumably because 15 is highly reactive. Once again, if MeOH is present during purification then the methylaminal 5 is observed. In this experiment 6 and 7 are produced as the major metabolites. This contrasts with experiments expressing these genes but lacking mrl7 where the same compounds were observed in overall low titre, presumably due to low activity of an adventitious A. oryzae 12-oxidase.
The final transformation is reduction of C-3 by the mrl6 enzyme (now renamed CitE). This protein appears highly selective for its substrate as its presence in any context other than a full complement of CitS + CitA–D does not result in observable new compounds. Reduction by CitE provides the chemically stable citrinin nucleus. Although this experiment afforded 1 in good titre, the conversion is not clean and several of the previously observed intermediates and shunts are also observed. This may occur because of an unnatural stoichiometry of gene expression means that the activities of the pathway enzymes are not properly balanced.
Conclusion
Heterologous expression of the citrinin biosynthetic genes in A. oryzae using strong promoters has elucidated the precise steps of citrinin biosynthesis in fungi and once again demonstrated that full pathways can be reconstituted in fungi to yield metabolites in good titre. The titres obtained here (ca. 20–50 mg L−1) compare favourably with low titres reported (up to 1.5 mg L−1) when a simple transcript containing the citS + citA–D genes was transferred to A. oryzae from M. purpureus, relying on recognition of Monascus promoters by the Aspergillus host.11Our results show that an unprecedented hydrolytic activity appears to be involved during polyketide construction or release by the citrinin PKS. They also show that reduction of C-3 occurs as the final step of the pathway, proving that the PKS itself does not reduce this position and that 2-methyl-3-hydroxybutyrate 11 is not used as a starter unit for subsequent 3-fold extension by the PKS as had been previously hypothesised. The results also show that ctnB/citA does not encode an oxidoreductase as widely reported, but a hydrolase which appears to assist the PKS in generating product efficiently. The next steps are oxidative-hydroxylation of the 12-methyl group of 2 to a carboxylic acid 15via an initial alcohol 4 and an aldehyde 14. The presence or absence of these oxidative genes in other pathways defines tropolone, sorbicillinoid and azaphilone biosynthesis. The intermediates in this oxidative process appear to be highly reactive and, in the absence of the final reduction, are shunted by native A. oryzae enzymes to numerous biproducts, including azaphilones and their aminated products. However, the final ketone reduction catalysed by CitE sets up formation of the stable quinomethide structure of citrinin 1. Thus the heterologous expression strategy also reveals the precise nature and order of the reactions which were not revealed by targeted KO in the native organism, and furthermore rules out the unprecedented rearrangements suggested previously.16 Overall our results have completely defined the citrinin pathway in fungi and as the biosynthesis of this compound serves as a model for that of many other fungal metabolites this knowledge will underpin future advances in fungal biosynthesis and engineering.
Acknowledgements
Prof. Fusheng Chen (Huazhong Agricultural University Wuhan, Hubei Province, P. R. China) is thanked for advice and supply of strains Monascus ruber M7 and MrΔku80 which were constructed in his lab. Funding by the China Scholarship Council (YH), the Fundamental Research Funds for the Central Universities of China (Program No. 2662015PY167, 2014PY034, YH), and the PhD Candidate Research Innovation Project of Huazhong Agricultural University (No. 2014bs37, YH); Leibniz Universität Hannover and DFG (LCMS equipment) are gratefully acknowledged.Notes and references
- H. Raistrick and A. C. Hetherington, Philos. Trans. R. Soc., B, 1931, 220, 269–295 CrossRef.
- J. P. Brown, N. J. Cartwright, A. Robertson and W. B. Whalley, Nature, 1948, 162, 72–73 CAS.
- A.-R. Ballester, M. Marcet-Houben, E. Levin, N. Sela, C. Selma-Lázaro, L. Carmona, M. Wisniewski, S. Droby, L. González-Candelas and T. Gabaldón, Mol. Plant-Microbe Interact., 2015, 28, 232–248 CrossRef CAS PubMed.
- P. J. Blanc, M. O. Loret and G. Goma, Biotechnol. Lett., 1995, 17, 291–294 CrossRef CAS.
- G. M. Chagas, A. P. Campello and M. L. Klüppel, J. Appl. Toxicol., 1992, 12, 123–129 CrossRef CAS PubMed.
- A. J. Birch, P. Fitton, E. Pride, A. Ryan, H. Smith and W. Whalley, J. Chem. Soc., 1958, 4576–4581 RSC.
- J. Barber, R. H. Carter, M. J. Garson and J. Staunton, J. Chem. Soc., Perkin Trans. 1, 1981, 2577–2583 RSC; J. Barber and J. Staunton, J. Chem. Soc., Chem. Commun., 1980, 1163–1164 RSC; J. Barber and J. Staunton, J. Chem. Soc., Perkin Trans. 1, 1980, 2244–2448 RSC; J. Barber and J. Staunton, J. Chem. Soc., Chem. Commun., 1979, 1098–1099 RSC.
- U. Sankawa, Y. Ebizuka, H. Noguchi, Y. Isikawa, S. Kitaghawa, Y. Yamamoto, T. Kobayashi and Y. Iitak, Tetrahedron, 1983, 39, 3583–3591 CrossRef CAS.
- L. Colombo, C. Gennari, D. Potenza, C. Scolastico, F. Aragozzini and C. Merendi, J. Chem. Soc., Perkin Trans. 1, 1981, 2594–2597 RSC; L. Colombo, C. Gennari, C. Scolastico, F. Aragozzini and C. Merendi, J. Chem. Soc., Chem. Commun., 1980, 1132–1134 RSC.
- T. Shimizu, H. Kinoshita and T. Nihira, Appl. Environ. Microbiol., 2007, 73, 5097–5103 CrossRef CAS PubMed.
- K. Sakai, H. Kinoshita, T. Shimizu and T. Nihira, J. Biosci. Bioeng., 2008, 106, 466–472 CrossRef CAS PubMed.
- J.-M. Gao, S.-X. Yang and J.-C. Qin, Chem. Rev., 2013, 113, 4755–4811 CrossRef CAS PubMed.
- A. al Fahad, A. Abood, K. M. Fisch, A. Osipow, J. Davison, M. Avramović, C. P. Butts, J. Piel, T. J. Simpson and R. J. Cox, Chem. Sci., 2014, 5, 523–527 RSC.
- J. Davison, A. al Fahad, M. Cai, Z. Song, S. Y. Yehia, C. M. Lazarus, A. M. Bailey, T. J. Simpson and R. J. Cox, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 7642–7647 CrossRef CAS PubMed.
- Y.-P. Li, Y.-F. Pan, L.-H. Zou, Y. Xu, Z.-B. Huang and Q.-H. He, J. Agric. Food Chem., 2013, 61, 7397–7402 CrossRef CAS PubMed.
- H. Hajjaj, A. Klaebe, M. Loret, G. Goma, P. Blanc and J. Francois, Appl. Environ. Microbiol., 1999, 65, 311–314 CAS.
- R. J. Cox, Org. Biomol. Chem., 2007, 5, 2010–2026 CAS.
- Gene nomenclature – since numerous gene nomenclatures exist for the various known citrinin clusters, and we do not wish to prematurely influence the names of the proteins (which should be named in order of their activity) we prefer to name the genes by their relative position to the PKS – i.e. mrl1 is the first gene to the left of the PKS in the Monascus ruber cluster etc. A full list of gene and protein names is included in the ESI.† The annotated gene cluster has been submitted to genbank with accession number KT781075.
- A. al Fahad, A. Abood, T. J. Simpson and R. J. Cox, Angew. Chem., Int. Ed., 2014, 53, 7519–7523 CrossRef CAS PubMed.
- T. Shimizu, H. Kinoshita, S. Ishihara, K. Sakai, S. Nagai and T. Nihira, Appl. Environ. Microbiol., 2005, 71, 3453–3457 CrossRef CAS PubMed.
- Y. He, Q. Liu, Y. Shao and F. Chen, Appl. Microbiol. Biotechnol., 2013, 97, 4965–4976 CrossRef CAS PubMed.
- K. A. K. Pahirulzaman, K. Williams and C. M. Lazarus, Methods Enzymol., 2012, 517, 241–260 CAS.
- F. J. Jin, J.-I. Maruyama, P. R. Juvvadi, M. Arioka and K. Kitamoto, FEMS Microbiol. Lett., 2004, 239, 79–85 CrossRef CAS PubMed.
- A. M. Bailey, R. J. Cox, K. Harley, C. M. Lazarus, T. J. Simpson and E. Skellam, Chem. Commun., 2007, 4053–4055 RSC.
- Z. Song, W. Bakeer, J. W. Marshall, A. A. Yakasai, R. M. Khalid, J. Collemare, E. Skellam, D. Tharreau, M.-H. Lebrun, C. M. Lazarus, A. M. Bailey, T. J. Simpson and R. J. Cox, Chem. Sci., 2015, 6, 4837–4845 RSC.
- T. Akao, M. Sano, O. Yamada, T. Akeno, K. Fujii, K. Goto, S. Ohashi-Kunihiro, K. Takase, M. Yasukawa-Watanabe, K. Yamaguchi, Y. Kurihara, J.-I. Maruyama, P. R. Juvvadi, A. Tanaka, Y. Hata, Y. Koyama, S. Yamaguchi, N. Kitamoto, K. Gomi, K. Abe, M. Takeuchi, T. Kobayashi, H. Horiuchi, K. Kitamoto, Y. Kashiwagi, M. Machida and O. Akita, DNA Res., 2007, 14, 47–57 CrossRef CAS PubMed.
- A. M. Bailey, K. M. Fisch, E. Skellam, D. Ivison, R. J. Cox, C. M. Lazarus and T. J. Simpson, Chem. Commun., 2010, 46, 5331–5333 RSC.
- R. Carter, M. Garson and J. Staunton, J. Chem. Soc., Chem. Commun., 1979, 1097–1098 RSC.
- Y. Xu, P. Espinosa-Artiles, V. Schubert, Y.-M. Xu, W. Zhang, M. Lin, A. A. L. Gunatilaka, R. Süssmuth and I. Molnár, Appl. Environ. Microbiol., 2013, 79, 2038–2047 CrossRef CAS PubMed.
- A. O. Zabala, Y.-H. Chooi, M. S. Choi, H.-C. Lin and Y. Tang, ACS Chem. Biol., 2014, 9, 1576–1586 CrossRef CAS PubMed.
- W. Xu, Y.-H. Chooi, J. W. Choi, S. Li, J. C. Vederas, N. A. Da Silva and Y. Tang, Angew. Chem., Int. Ed., 2013, 52, 6472–6475 CrossRef CAS PubMed.
- J. F. Barajas, R. M. Phelan, A. J. Schaub, J. T. Kliewer, P. J. Kelly, D. R. Jackson, R. Luo, J. D. Keasling and S.-C. Tsai, Chem. Biol., 2015, 22, 1018–1029 CrossRef CAS PubMed.
- R. J. Cox, A. Hadfield and M. Mayo-Martin, Chem. Commun., 2001, 1710–1711 RSC.
- M. S. Kanchuger and L. D. Byers, J. Am. Chem. Soc., 1979, 101, 3005–3010 CrossRef CAS.
- A. Hadfield, C. Shammas, G. Kryger, D. Ringe, G. Petsko, J. Ouyang and R. Viola, Biochemistry, 2001, 40, 14475–14483 CrossRef CAS PubMed.
- B. Balakrishnan, R. Chandran, S.-H. Park and H.-J. Kwon, J. Microbiol. Biotechnol., 2015, 25, 1648–1652 CrossRef PubMed.
- B. Bijinu, J.-W. Suh, S.-H. Park and H.-J. Kwon, RSC Adv., 2014, 4, 59405–59408 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: All experimental, chromatographic, bioinformatic and spectroscopic details. See DOI: 10.1039/c5sc04027b |
| ‡ College of Food Science and Engineering, Wuhan Polytechnic University, Wuhan 430023, Hubei Province, P. R. China. |
| This journal is © The Royal Society of Chemistry 2016 |