
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Non-covalent organocatalysis in asymmetric oxidative C(sp3)–H bond functionalization – broadening C–H bond coupling reactions
Sebastian
Stockerl
a and
Olga García
Mancheño
*ab
aInstitute for Organic Chemistry, University of Regensburg, 93053 Regensburg, Germany. E-mail: olga.garcia-mancheno@chemie.uni-regensburg.de
bStraubing Center of Science for Renewable Resources, 94315 Straubing, Germany
First published on 8th December 2015
Abstract
Among the current huge development activities in C–H functionalization, asymmetric oxidative C(sp3)–H bond coupling strategies have remained underrepresented. Beyond the initial examples using chiral metal complexes, the use of organocatalysis provides a new direction for the design of new asymmetric C–H bond cross-coupling reactions. This highlight focuses on the latest advances in metal-free asymmetric oxidative C(sp3)–H bond functionalization using cooperative non-covalent organocatalysis.
 Olga García Mancheño | Olga García Mancheño received her PhD in 2005 from the Universidad Autónoma de Madrid under the supervision of Prof. Juan C. Carretero. During her PhD she carried out two–three month research-stays with Prof. Manfred T. Reetz (Max-Planck-Institut für Kohlenforschung) and Prof. Karl Anker Jørgensen (University of Aarhus). Next she moved to RWTH-Aachen University for her postdoctoral stay in the group of Prof. Carsten Bolm (2005–2008). At the end of 2008 she started her independent career as Assistant Professor (Habilitand, mentor: Prof. Frank Glorius) at the University of Münster. In 2013 she was appointed as Professor for Organic Chemistry at the University of Regensburg and the Straubing Center of Science for Renewable Resources. Her main research interests include the development of new synthetic methods, with a special focus on catalytic approaches, and their application in the synthesis of bioactive compounds and heterocycles. |
C–H bond functionalization has emerged as a powerful synthetic method for the efficient and straightforward construction of C–C or C–heteroatom bonds.1 In the last decade a large number of practical applications of this C–H coupling methodology have appeared. In this regard, oxidative C(sp3)–H bond functionalization, such as the cross-dehydrogenative coupling (CDC) coined by Li, has attracted great interest.2 This type of chemistry is conceptually very simple and easy to perform. From a mechanistic point of view, it implies an initial substrate oxidation to form a cationic intermediate (or in some cases a radical) that can then undergo a nucleophilic attack to form the new C–Nu bond (Scheme 1).1,2 However, one of the intrinsic selectivity issues in C–H functionalization relies on the ubiquitous presence of C–H bonds in organic molecules. Therefore, in order to attain high levels of regio- and chemoselectivity, substrates such as 1,2,3,4-tetrahydroisoquinolines (THIQ), which possess a distinct prominent easily oxidizable benzylic C–H bond alpha to a N-atom, have extensively been explored.1–3 Although a large variety of nucleophiles have already been enrolled as reaction partners, there is still a limited number of effective and synthetically valuable enantioselective oxidative C(sp3)–H bond functionalization processes.4 In the earlier examples, a Cu–metal catalyst was used in combination with a chiral ligand, such as BOX or PyBOX, providing moderate to good enantioselectivities.5,6 On the other hand, the initial parallel studies by Prof. Nicolaou and by Prof. MacMillan in the area of metal-free asymmetric intramolecular oxidative C(sp3)–H couplings using SOMO activation of aldehydes with MacMillan-type catalysts and CAN or [Fe(phen)3](PF6)3 as oxidants provided moderate to excellent enantioselectivities (70–98% ee).7 However, the intermolecular reactions of C(sp3)–H bonds in the α-position of a nitrogen atom with ketones proved to be more challenging, showing in some cases to be inefficient (<20% ee) and irreproducible due to fast product racemization processes.8 Fortunately, several efforts have proven that the appropriate choice of the substrate, carbonyl nucleophile, oxidant and amino organocatalyst can lead to high levels of enantioinduction (Scheme 1).9
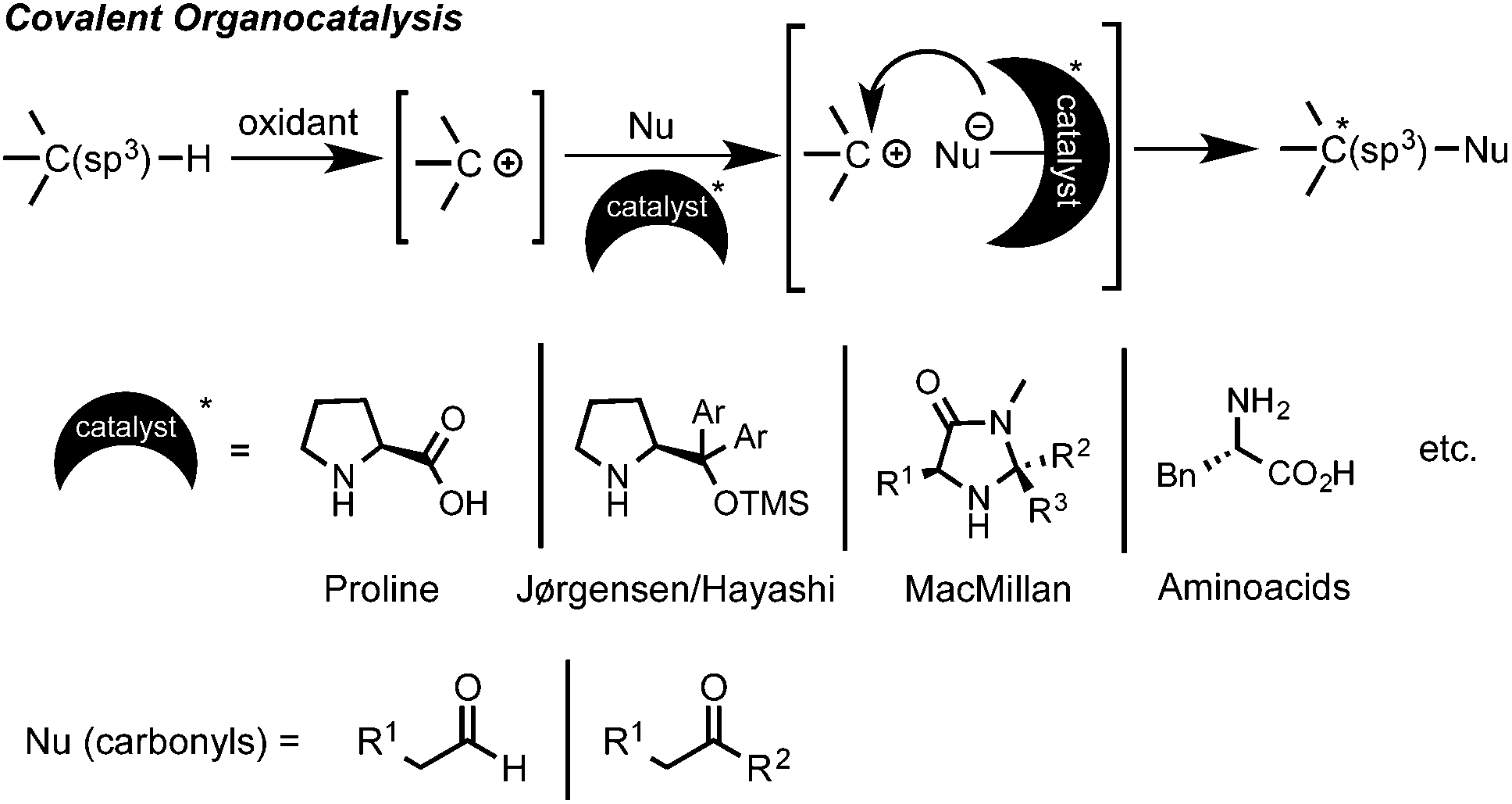 | ||
| Scheme 1 Asymmetric covalent-organocatalysis approach. | ||
Additionally, efficient bicatalytic systems that combine a metal and an organocatalyst have also been recently developed.10 Among those combinations, several amines,10a,b heterocyclic carbenes (NHC)10d and thioureas10e have synergistically been employed with copper salts or metal-based photoredox catalysts. The metal catalyst is generally added to promote the oxidation of the substrate, but in some cases it is also involved in the asymmetric step. The few examples using non-covalent organocatalysis are quite appealing since they opened new opportunities for a broader nucleophile scope and the development of novel enantioselective reactions. Thus, other non-carbonylic nucleophiles can now be also enrolled. However, due to multiple possible positioning and coordination points, the control of non-covalent interactions still represents a great challenge. Consequently, complete metal-free processes are still quite rare within these types of interactions.11,12 Herein, the two latest approaches based on non-covalent bifunctional organocatalysis by hydrogen bonding11 and anion catalysis12 are highlighted (Scheme 2).
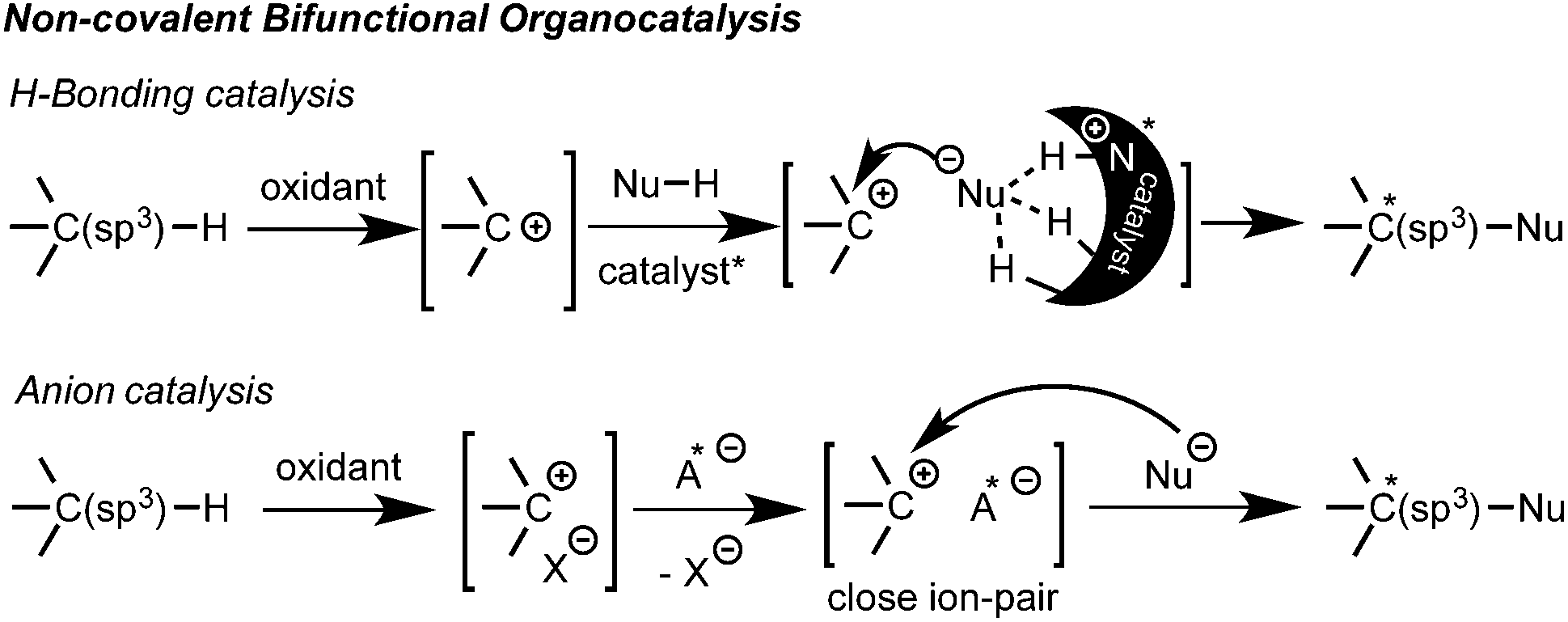 | ||
| Scheme 2 Non-covalent bifunctional organocatalysis approaches. | ||
Intermolecular C–C bond forming reaction
Prof. Wang and coworkers11 reported the use of a bifunctional quinine–thiourea 113 for the coupling of N-aryl-THIQs 2 with α,β-unsaturated N-Boc γ-butyrolactam 3 to produce Morita–Baylis–Hilman (MBH) products 4 (Scheme 3). In this strategy the use of the bifunctional catalyst 1 allowed the simultaneous activation by deprotonation and orientation by H-bonding with the nucleophile. From the tested oxidants DDQ proved to be the most efficient for this transformation, providing good to excellent enantioselectivities (up to 93% ee) with a variety of electron-rich N-aryl THIQs.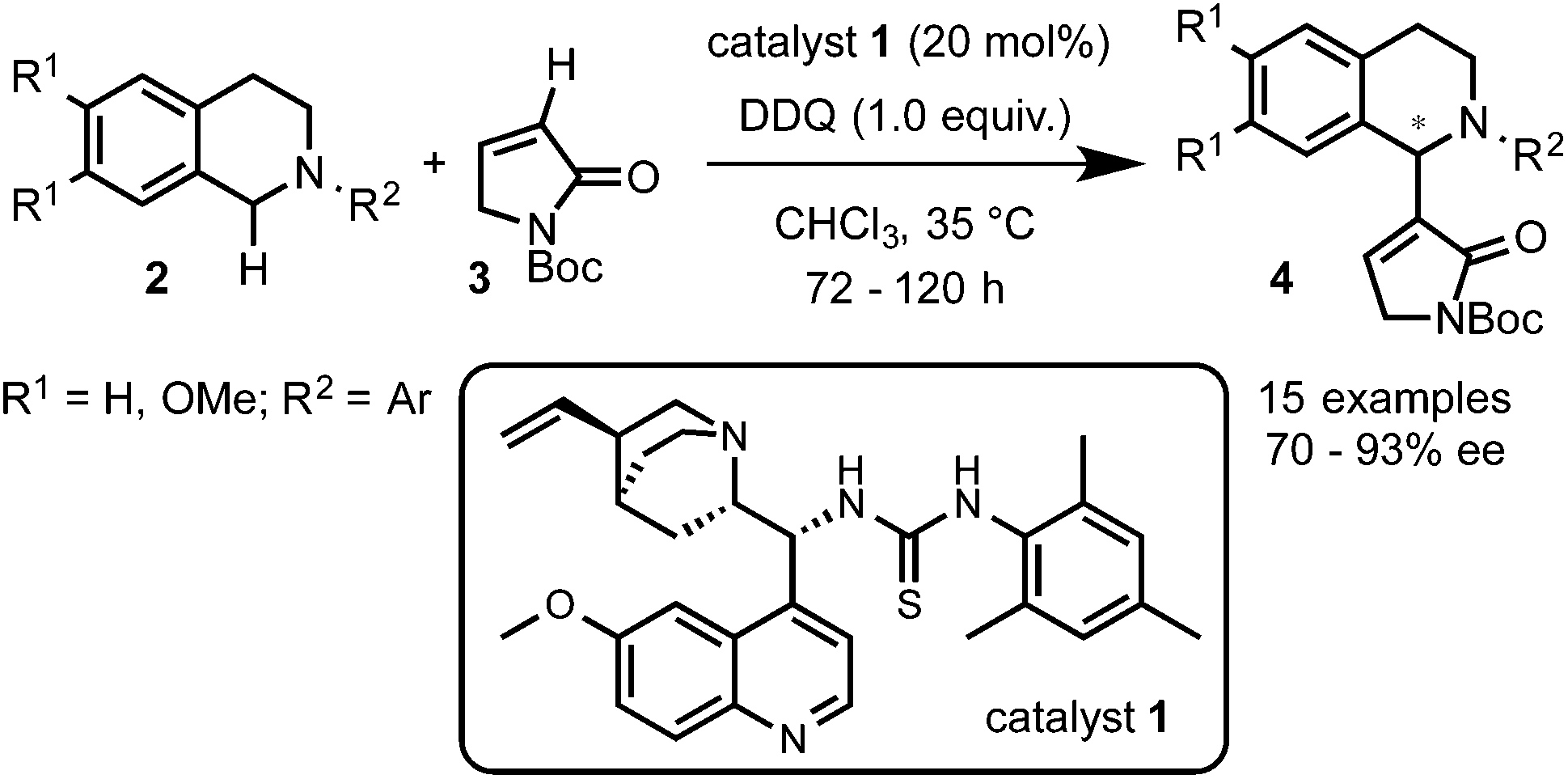 | ||
| Scheme 3 Bifunctional organocatalysis in asymmetric C(sp3)–H functionalization. | ||
Although a highly selective α-alkylation was observed, based on previous work with the nucleophile 3, a MBH mechanism was ruled out.14 In order to explain the regio- and enantioselectivity, the authors proposed a mechanism in which, after deprotonation of the γ-butyrolactam, a H-bonding interaction with the catalyst takes place. However, an unconceivable H-bonding interaction of the thiourea group with the positively charged nitrogen of the THIQ-iminium intermediate I was postulated. A more plausible mechanism implying a multiple hydrogen bonding between the catalyst and the nucleophile is depicted in Scheme 4. In one hand, the oxidation of THIQs with DDQ to form the reactive iminium salt I is well known.1–3 On the other hand, whereas the deprotonation of 3 with tertiary amines such as DABCO or quinuclidine has been described,14 its exact activation by the cinchona-thiourea catalyst is still unclear. A reasonable H-bonding interaction between the protonated quinuclidine unit and the deprotonated reagent can be envisioned. Furthermore, the additional capture of the nucleophile might be possible by H-bonding between the thiourea moiety and its Boc protecting group to form the species II. This might be translated to a more effective orientation and shielding of the reagent for the selective approach to the iminium electrophile, explaining the high enantioselectivities observed.
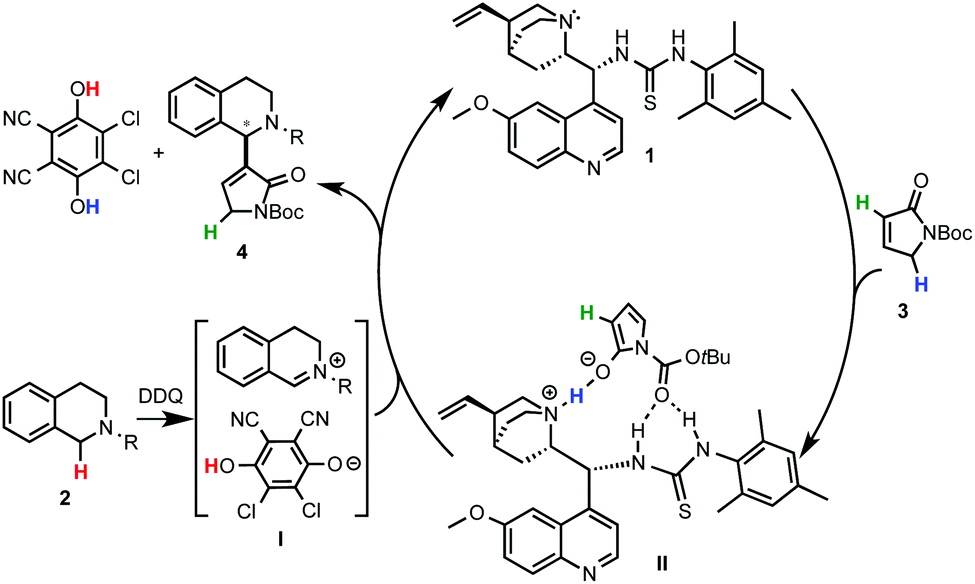 | ||
| Scheme 4 A revisited plausible mechanism for the cinchona thiourea-catalyzed asymmetric C–H coupling of THIQ with 3. | ||
Intramolecular C–N bond forming reaction
Recently, Toste's group made a great breakthrough in asymmetric organocatalyzed C–H functionalization (Scheme 5).12 In this case, the in situ generation of a chiral close ion-pair15 with an anionic catalyst by getting advantage of the cationic character of the oxidation-intermediate 5 was exploited. Thus, a chiral counter ion could be introduced by anion exchange between a chiral anion and an ionic oxidant (e.g. [Ox]+X−) or the iminium salt generated in the first oxidation step. For that reason chiral triazole-containing phosphoric acids (PA) 6 derived from BINOL were employed as precatalysts.16 The active catalyst, the corresponding phosphonate, was generated in the reaction media by addition of a base such as Na3PO4. To prove the concept, the intramolecular amidation of N-aryl substituted THIQs 7 was chosen as the target reaction using as oxidant a TEMPO-derived oxoammonium salt (4-acetamido-2,2,6,6-tetramethyl-1-oxopiperidin-1-ium tetrafluoroborate, 4-AcNHT+BF4−).17,18 The multifunctional nature of the catalysts was carefully designed in order to enhance the positive interactions between the substrate and the catalyst. Thus, the 1,2,3-triazole units present in the catalyst at the 3 and 3′ positions19 could additionally participate in an attractive interaction with the iminium intermediate and, therefore, lead to an efficient chirality transfer. Consequently, moderate to excellent enantioselectivities were achieved (up to 95% ee).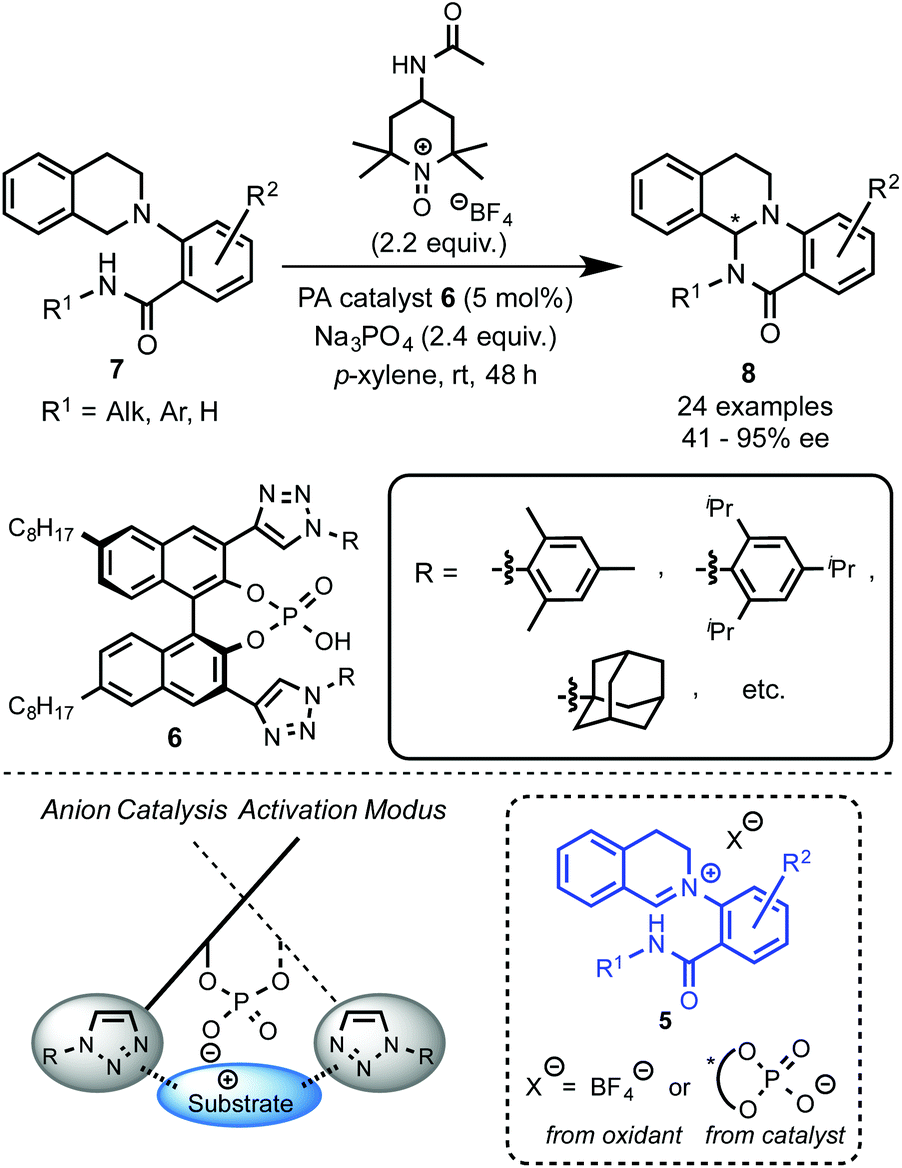 | ||
| Scheme 5 Chiral phosphate catalysis in C(sp3)–H functionalization. | ||
Interestingly, well-established simple PA catalysts designed only to create a sterically hindered environment around the active acid-site failed to provide effective chirality transfer. Although the main task of the substituent at the triazole in the used PA catalyst is also to be steric, positive π–π interactions between the substrate and the triazole ring were also proposed.12b Kinetic isotope effect (KIE) studies and library-trend analysis led to the mechanistic hypothesis shown in Scheme 6. Different KIEs were observed using the (R)- and the (S)-PA, suggesting that the catalyst should be already involved in the oxidation step. Thus, the anion exchange might have occurred between the oxoammonium salt and the catalyst prior the oxidation of the substrate 7 to the iminium intermediate 5 (Scheme 6, left).
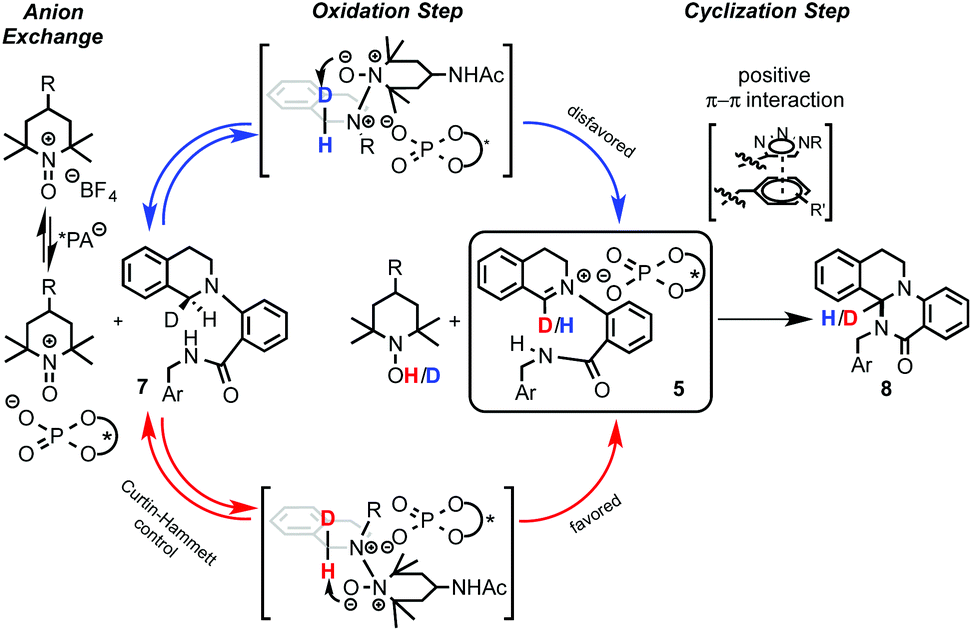 | ||
| Scheme 6 Proposed mechanism for the phosphate-catalyzed reaction. | ||
Conclusions
Considering the current importance of direct C–H bond functionalization towards more efficient and sustainable synthesis, the use of organocatalysis for the development of efficient asymmetric strategies provides a valuable alternative to transition-metal catalyzed approaches. In this highlight, two effective methods based on multiple H-bonding and a synergetic combination of Coulomb and π–π-interactions have been presented. Although the control of supramolecular non-covalent interactions constitutes a fundamental challenge, non-covalent organocatalysis offers high flexibility and unique cooperative features that will allow the design and discovery of novel asymmetric oxidative C–H functionalization reactions. Thus, significant future advances in this field using multifunctional non-covalent organocatalysis are certainly expected in the next few years.Acknowledgements
The Deutsche Forschungsgemeinschaft (DFG) is gratefully acknowledged for generous support.Notes and references
- (a) Handbook of C–H Transformations, ed. G. Dyker, Wiley-VCH, Weinheim, 2005 Search PubMed; (b) J.-Q. Yu and Z.-J. Shi, in C–H. Activation: Topics in Current Chemistry, Springer, Berlin, 2010 Search PubMed.
- Selected recent reviews on oxidative C–H functionalization: (a) C. J. Scheuermann, Chem. – Asian J., 2010, 5, 436 CrossRef CAS PubMed; (b) M. Klussmann and D. Sureshkumar, Synthesis, 2011, 353 CrossRef CAS; (c) C. S. Yeung and V. M. Dong, Chem. Rev., 2011, 111, 1215 CrossRef CAS PubMed; (d) C. Liu, H. Zhang, W. Shi and A. Lei, Chem. Rev., 2011, 111, 1780 CrossRef CAS PubMed; (e) R. Rohlmann and O. García Mancheño, Synlett, 2013, 6 CAS; (f) S. A. Girard, T. Knauber and C. J. Li, Angew. Chem., Int. Ed., 2014, 53, 74 CrossRef CAS PubMed; (g) R. Narayan, K. Matcha and A. P. Antonchick, Chem. – Eur. J., 2015, 21, 14678 CrossRef CAS PubMed , and references cited therein.
- Recent review on asymmetric couplings of α-C(sp3)–H of amines: (a) Y. Qin, J. Lv and S. Luo, Tetrahedron Lett., 2014, 55, 551 CrossRef CAS. See also: (b) W. Liu, S. Liu, R. Jin, H. Guo and J. Zhao, Org. Chem. Front., 2015, 2, 288 RSC. For a mechanistic study, see: E. Boess, C. Schmitz and M. Klussmann, J. Am. Chem. Soc., 2012, 134, 5317 CrossRef CAS PubMed.
- Recent review on asymmetric C–H bond functionalization: C. Zheng and S.-L. You, RSC Adv., 2014, 4, 6173 RSC.
- Pioneer work: Z. Li and C.-J. Li, Org. Lett., 2004, 6, 4997 CrossRef CAS PubMed.
- See also: (a) N. Sasamoto, C. Dubs, Y. Hamashima and M. Sodeoka, J. Am. Chem. Soc., 2006, 128, 14010 CrossRef CAS PubMed; (b) O. Basléand and C.-J. Li, Org. Lett., 2008, 10, 3661 CrossRef PubMed; (c) G. Zhang, Y. Zhang and R. Wang, Angew. Chem., Int. Ed., 2011, 50, 10429 CrossRef CAS PubMed; (d) W. Cao, X. Liu, R. Peng, P. He, L. Lin and X. Feng, Chem. Commun., 2013, 49, 3470 RSC; (e) J. Yu, Z. Li, K. Jia, Z. Jiang, M. Liu and W. Su, Tetrahedron Lett., 2013, 54, 2006 CrossRef CAS; (f) I. Perepichka, S. Kundu, Z. Hearne and C.-J. Li, Org. Biomol. Chem., 2015, 13, 447 RSC , and references cited herein.
- (a) K. C. Nicolaou, R. Reingruber, D. Sarlah and S. Bräse, J. Am. Chem. Soc., 2009, 131, 2086 CrossRef CAS PubMed; (b) J. C. Conrad, J. Kong, B. N. Laforteza and D. W. C. MacMillan, J. Am. Chem. Soc., 2009, 131, 11640 CrossRef CAS PubMed.
- (a) A. Sud, D. Sureshkumarz and M. Klussmann, Chem. Commun., 2009, 3169 RSC; (b) Y. Pan, C. W. Kee, L. Chen and C.-H. Tan, Green Chem., 2011, 13, 2682 RSC.
- For a recent review, see: (a) Y.-L. Zhao, Y. Wang, Y.-C. Luo, X.-Z. Fu and P.-F. Xu, Tetrahedron Lett., 2015, 56, 3703 CrossRef CAS. See also: (b) F. Benfatti, M. G. Capdevila, L. Zoli, E. Benedetto and G. Cozzi, Chem. Commun., 2009, 5919 RSC; (c) X. H. Ho, S. I. Mho, H. Kang and H. Y. Jang, Eur. J. Org. Chem., 2010, 4436 CAS; (d) B. Zhang, S. K. Xiang, L. H. Zhang, Y. X. Cui and N. Jiao, Org. Lett., 2011, 13, 5212 CrossRef CAS PubMed; (e) J. Zhang, B. Tiwari, C. Xing, X. Chen and Y. G. Chi, Angew. Chem., Int. Ed., 2012, 51, 3649 CrossRef CAS PubMed; (f) Z. L. Meng, S. T. Sun, H. Q. Yuan, H. X. Lou and L. Liu, Angew. Chem., Int. Ed., 2014, 53, 553 CrossRef.
- Amino/Cu: (a) J. Zhang, B. Tiwari, C. Xing, X. Chen and Y. R. Chi, Angew. Chem., Int. Ed., 2012, 51, 3649 CrossRef CAS PubMed; (b) G. Zhang, Y. Ma, S. Wang, W. Kong and R. Wang, Chem. Sci., 2013, 4, 2645 RSC. Alcohol/Cu: (c) G. Zhangh, Y. Ma, S. Wang, Y. Zhang and R. Wang, J. Am. Chem. Soc., 2012, 134, 12334 CrossRef PubMed. NHC/Ru: (d) D. A. DiRocco and T. Rovis, J. Am. Chem. Soc., 2012, 134, 8094 CrossRef CAS PubMed. Thiourea/Ru: (e) G. Bergonzini, C. S. Schindler, C.-J. Wallentin, E. N. Jacobsen and C. R. J. Stephenson, Chem. Sci., 2014, 5, 112 RSC.
- Y. Ma, G. Zhang, D. Yang and R. Wang, Org. Lett., 2014, 16, 5358 CrossRef CAS PubMed.
- (a) A. J. Neel, J. P. Hehn, P. F. Tripet and F. D. Toste, J. Am. Chem. Soc., 2013, 135, 14044 CrossRef CAS PubMed; (b) A. Milo, A. J. Neel, F. D. Toste and M. S. Sigman, Science, 2015, 347, 737 CrossRef CAS PubMed.
- Bifunctional cinchona-thioureas: (a) C. E. Song, in Cinchona Alkaloids in Synthesis and Catalysis, Wiley-VCH, Weinheim, 2009 Search PubMed; (b) Q. L. Zhou, in Privileged Chiral Ligands and Catalysts, Wiley-VCH, Weinheim, 2011 Search PubMed. For the pioneering work, see: (c) B. Vakulya, S. Varga, A. Csmpai and T. Soos, Org. Lett., 2005, 7, 1967 CrossRef CAS PubMed; (d) S. H. McCooey and S. J. Connon, Angew. Chem., Int. Ed., 2005, 44, 6367 CrossRef CAS PubMed; (e) J. Yea, D. J. Dixon and P. S. Hynes, Chem. Commun., 2005, 4481 RSC.
- (a) Z. Duan, Z. Zhang, P. Qian, J. Han and Y. Pan, RSC Adv., 2013, 3, 10127 RSC; (b) J. Zhang, X. Liu, X. Ma and R. Wang, Chem. Commun., 2013, 49, 3300 RSC; (c) N. Ramireddy and J. C.-G. Zhao, Tetrahedron Lett., 2014, 55, 706 CrossRef CAS.
- (a) M. Mahlau and B. List, Angew. Chem., Int. Ed., 2013, 52, 518 CrossRef CAS PubMed; (b) K. Brak and E. N. Jacobsen, Angew. Chem., Int. Ed., 2013, 52, 534 CrossRef CAS PubMed.
- Selected reviews: (a) T. Akiyama, Chem. Rev., 2007, 107, 5744 CrossRef CAS PubMed; (b) M. Terada, Synthesis, 2010, 1929 CrossRef CAS; (c) R. J. Phipps, G. L. Hamilton and D. F. Toste, Nat. Chem., 2012, 4, 603 CrossRef CAS PubMed; (d) D. Parmar, E. Sugiono, S. Raja and M. Rueping, Chem. Rev., 2014, 114, 9047 CrossRef CAS PubMed.
- Reviews on N-oxoammonium chemistry: (a) J. M. Bobbitt and M. C. L. Flores, Heterocycles, 1988, 27, 509 CrossRef CAS; (b) J. M. Bobbitt, C. Brückner and N. Merbouh, Oxoammonium- and nitroxide-catalyzed oxidations of alcohols, in Org. Reactions, ed. S. E. Denmark, Wiley, New York, 2009, vol. 74, p. 103 Search PubMed; (c) T. Stopka and O. García Mancheño, Synthesis, 2013, 1602 Search PubMed. See also: (d) M. A. Mercadante, C. B. Kelly, J. M. Bobbitt, L. J. Tilley and N. E. Leadbeater, Nat. Protoc., 2013, 8, 666 CrossRef CAS PubMed.
- TEMPO oxoammonium salts as oxidants in C–H functionalization: (a) H. Richter and O. Garcia Mancheño, Eur. J. Org. Chem., 2010, 4460 CAS; (b) H. Richter and O. Garcia Mancheño, Org. Lett., 2011, 13, 6066 CrossRef CAS PubMed; (c) S. Wetz, S. Kodama and A. Studer, Angew. Chem., Int. Ed., 2011, 50, 11511 CrossRef PubMed; (d) H. Richter, R. Fröhlich, C. G. Daniliuc and O. García Mancheño, Angew. Chem., Int. Ed., 2012, 51, 8656 CrossRef CAS PubMed; (e) R. Rohlmann, T. Stopka, H. Richter and O. García Mancheño, J. Org. Chem., 2013, 78, 6050 CrossRef CAS PubMed; (f) C. Yan, Y. Liu and Q. Wang, RSC Adv., 2014, 4, 60075 RSC; (g) S. Sun, C. Li, P. E. Floreancig, H. Lou and L. Liu, Org. Lett., 2015, 17, 1684 CrossRef CAS PubMed.
- For the first syntheses of 1,2,3-triazole BINOLs and they application in catalysis, see: (a) S. Beckendorf and O. Garcia Mancheño, Synlett, 2012, 2162 CAS; (b) C. Recsei and S. P. McErlean, Tetrahedron, 2012, 68, 464 CrossRef CAS.
| This journal is © the Partner Organisations 2016 |