
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthetic genetic polymers: advances and applications
Qian
Ma
a,
Danence
Lee
a,
Yong Quan
Tan
b,
Garrett
Wong
b and
Zhiqiang
Gao
*a
aDepartment of Chemistry, National University of Singapore, Singapore 117543. E-mail: chmgaoz@nus.edu.sg; Fax: +6779-1691; Tel: +6516-3887
bDepartment of Biochemistry, National University of Singapore, Singapore 117597
First published on 8th August 2016
Abstract
Synthetic genetic polymers, also known as xeno-nucleic acids (XNAs), are chemically modified or synthesized analogues of natural nucleic acids. Initially developed by synthetic chemists to better understand nucleic acids, XNAs have grown rapidly over the last two decades in both diversity and usefulness. Their tailor-made functionalities allow them to overcome perennial problems in using natural nucleic acids in technical applications. In this article, key milestones in XNA research are reviewed through highlighting representative examples. The advantages of using XNAs over natural nucleic acids are discussed. It is hoped that this article will provide a summary of the advances and current understanding of XNAs in addition to their technical applications, serving as an entry point to those who are interested in the synthesis and application of XNAs. Besides interesting results, challenges encountered may inspire researchers to perfect the synthesis of XNAs and tailor their functionalities.
 Qian Ma | Ms Qian Ma was born in Shannxi, China. She received her B.Sc. in Chemistry from Nanjing University in 2015. She is currently pursuing her Ph.D. at the National University of Singapore (NUS). Her research is primarily on the development of highly sensitive biosensors and bioassays for nucleic acids and proteins. |
 Danence Lee | Mr Danence Lee was born in Singapore. He graduated from NUS in 2010 with a B.Sc. (first class honors) in chemistry. After a 5-year teaching stint with the Ministry of Education, he is back at NUS to pursue his Ph.D. in Chemistry. His current research interest focuses mainly on the development of efficient methodologies for organic synthesis via bimetallic and cooperative catalysis. |
 Yong Quan Tan | Mr Yong Quan Tan was born in Singapore. He received his B.Sc. in Chemistry from NUS in 2015. He is currently pursuing his Ph.D. at NUS. His research is focused on the development of synthetic microcompartments for improving biosynthesis of value-added chemicals in microorganisms. |
 Garrett Wong | Mr Garrett Wong was born in Singapore. He received his B.Sc. (Honors) in Life Science from NUS in 2015. He is currently pursuing his Ph.D. at the NUS and Imperial College London. His research is focused on the metabolic engineering of microorganisms for the production of ergot alkaloids. |
 Zhiqiang Gao | Dr Zhiqiang Gao is an associate professor at the Department of Chemistry, NUS. He received his B.Sc. and Ph.D. in Chemistry from Wuhan University. The following years he worked as a postdoctoral fellow at Åbo Akademi University and the Weizmann Institute of Science. After spending three years in the United States and eight years at the Institute of Bioengineering and Nanotechnology, he joined NUS in April 2011. Research in his laboratory currently includes bioengineering, renewable energy, electrochemistry, analytical chemistry, and materials science. |
1. Introduction
As natural genetic polymers, nucleic acids – deoxyribonucleic acid (DNA) and ribonucleic acid (RNA) – have evolved over geological time to reliably store and transmit hereditary information. Nucleic acids are able to achieve this due to their specific hydrogen bonding patterns between nucleobases. As described by the Watson–Crick rules, adenine (A) always pairs with thymine (T) (or uracil (U) in the case of RNA), while cytosine (C) always pairs with guanine (G).1 Incompatible hydrogen-bonding patterns along with proofreading enzymes ensure high fidelity in the replication and transmission of genetic information.2 The negatively charged phosphate backbones of nucleic acids ensure that they are highly soluble in water irrespective of their sequences. Thus, long and diverse nucleic acid strands can be synthesized without the need to consider their aqueous solubility. Furthermore, the modularity of nucleic acids facilitates ease of synthesis, either enzymatically or chemically.3 These properties have resulted in the ready adoption of natural nucleic acids as convenient and useful materials in various technical applications.Inspired by the elegancy of natural nucleic acids, researchers have been actively pursuing the ultimate goal of emulating natural nucleic acids with totally man-made entities – synthetic genetic polymers or xeno-nucleic acids (XNAs). The term “XNA” was first coined by Herdewijn and Marlière to describe artificial genetic polymers with the potential to emulate natural nucleic acids in information storage and propagation.4 Synthetic genetic polymers, as the name suggests, contain unnatural components with chemical modifications to either the nucleobases, the sugar moieties, the phosphodiester backbones, or a combination of the above.5 The endeavor to create XNAs has resulted in a deepened understanding of natural nucleic acids in terms of their structure, chemical and physical properties, and function. These valuable insights have in turn spun off other developments particularly in the fields of molecular biology, gene therapy, bioassays, diagnostics, and biocatalysis.6 By successfully emulating nucleic acids in information storage and propagation – both prerequisites for evolution and life7,8 – these laboratory synthesized XNAs have also helped scientists to rethink the assumption that nucleic acids and proteins are the only chemicals that power cells, fueling the possibility of the existence of xeno-organisms that utilize a completely different set of biomolecules.9 The explosion of research activities in XNAs also implies that it is impractical to cover every aspect of this exciting field. Therefore, this article focuses on the progress of XNAs research and their technical applications, starting off with an overview detailing various types of XNAs, their fundamental aspects, synthesis and evolution strategies, and ending up with their technical applications, challenges, and outlook.
2. XNAs and their synthesis
2.1. XNAs with modified nucleobases
While essentially living organisms on Earth utilize nucleic acids in storing and transmitting genetic information, researchers have always envisioned the creation of a synthetic self-replicating organism that can incorporate a third base-pairing mode (X–Y) into its genetic polymers (Fig. 1).10 Aided by the advances in organic synthesis and bioanalytical methods, this vision has gradually become a reality.11 One of the main reasons for creating such an organism with an expanded genetic alphabet is for the organism to ribosomally produce novel proteins incorporated with unnatural amino acids. The discovery process of the third base pair capable of replication often revolves around iterating these processes – the chemical synthesis of artificial nucleotides, ascertaining the specificity and efficiency of the base pairing by biochemical methods, and identifying high potential base pairs for further chemical modifications.12 In order to qualify as the third base pair, the bases X and Y must exclusively be paired with each other in the helical structure as well as during nucleic acid synthesis by polymerases.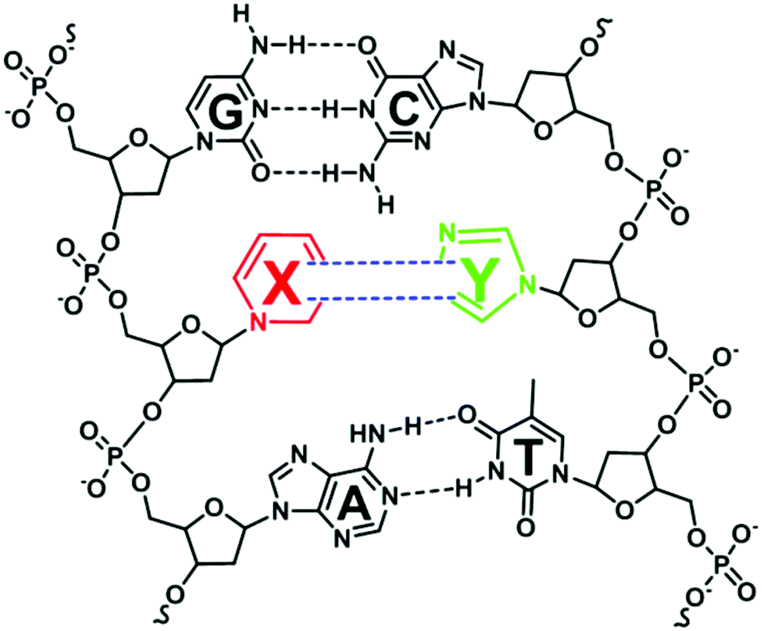 | ||
| Fig. 1 A hypothetically unnatural base pair (X–Y) that can function in DNA replication and transcription, allowing ribosomal incorporation of unnatural amino acids during translation. | ||
An example of such iterations is illustrated by Benner's work in developing unnatural base pairs, revolving around developing base pairs with hydrogen bonding patterns similar to those of natural base pairs. Notable pioneering work by his group includes the development of an isoG–isoC base pair (Fig. 2) and their successful in vitro incorporation into natural nucleic acids for replication and transcription.13,14 However, the potential of the isoG–isoC base pair functioning as the third base pair is seriously limited by several issues concerning its specificity and the most serious of which is the fact that isoG undergoes tautomerization in aqueous media at physiological pH.15,16 Once enolized, isoG (enol) pairs with thymine instead of isoC, thus severely compromising the fidelity of the isoG–isoC base pair. Furthermore, isoC was found to be chemically unstable in alkaline media.15 In order to resolve these issues, one of the strategies developed is to substitute thymine with a thymine analogue with reduced hydrogen bonding ability. Such a thymine analogue still binds to adenine but no longer mispairs with isoG (enol). The methylation of the 5-position of isoC (5-Me-isoC) has also helped to improve the stability of isoC.17 Through such successful iterations of modifications, Benner's group managed to demonstrate that the isoG–5-Me-isoC base pair can be the third base pair for polymerase chain reactions (PCR) with a fidelity up to 98% (Fig. 3).18 More recently, a new base pairing between a triaza-isoG (dP) and nitropyridone (dZ) was developed by his group (Fig. 3).19 This base pair shows enhanced polymerase recognition with improved chemical stability, enabling a fidelity up to 99.8% during PCR. They took their research a step further by designing unnatural nucleobases that pair with natural nucleobases but not with each other. Thus, undesirable primer–primer interactions are minimized with greatly improved specificity in PCR.20
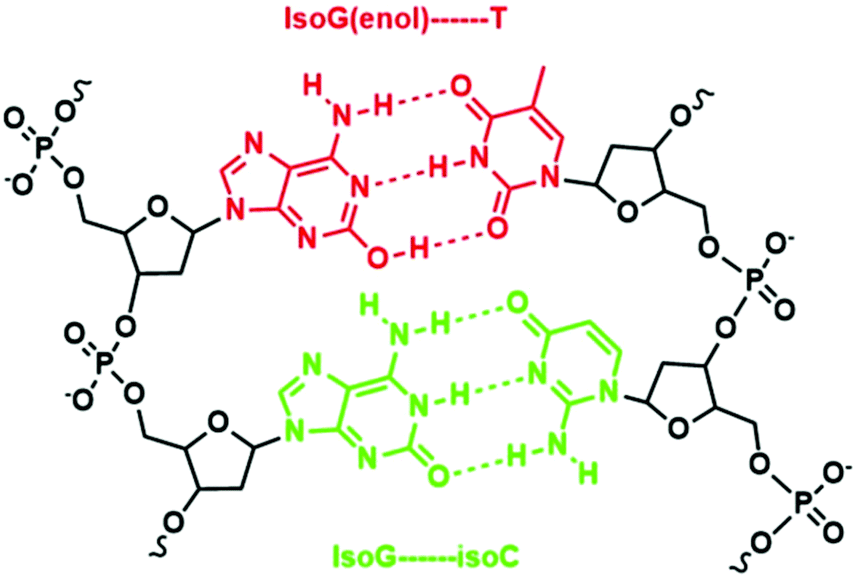 | ||
| Fig. 2 IsoG–isoC base pair and the loss of fidelity after the tautomerization of isoG. | ||
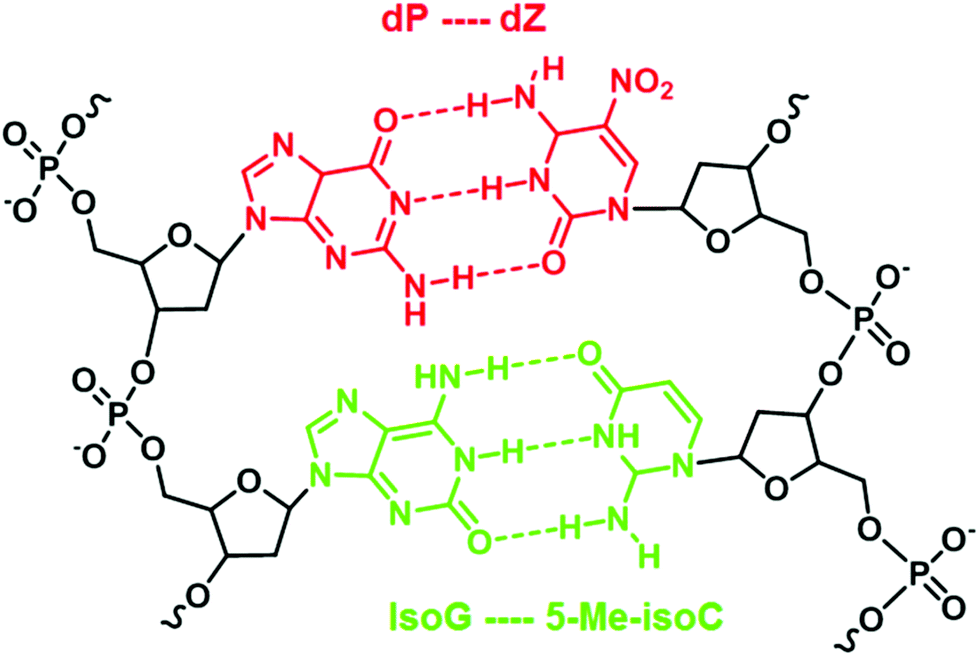 | ||
| Fig. 3 IsoG–5-Me-isoC and dP–dZ base pairs. | ||
In stark contrast to Benner's strategy, Morales et al. developed base pairs that depend on shape complementarity rather than hydrogen bonding patterns, thereby proving that hydrogen bonding is not a definite requirement for replication.21 Although these base pairs are unable to function as the third base pair, this work inspired the development of unnatural base pairs that do not exploit hydrogen bonding to achieve base-pairing fidelity.22–24 Well-known base pairs developed are 5SICS–MMO2,25 5SICS–DMO,26 5SICS–NaM,27 and Ds–Px (Fig. 4).28 For these base pairs, a balance between shape complementarity and size similarity to natural nucleobases is achieved. Through careful design, repeated optimization, and testing, the hydrophobic interactions and stacking between the base pairs have resulted in enhanced fidelity during PCR and transcription. For instance, the Ds–Px pair can undergo a hundred cycles of PCR with 97% fidelity.29
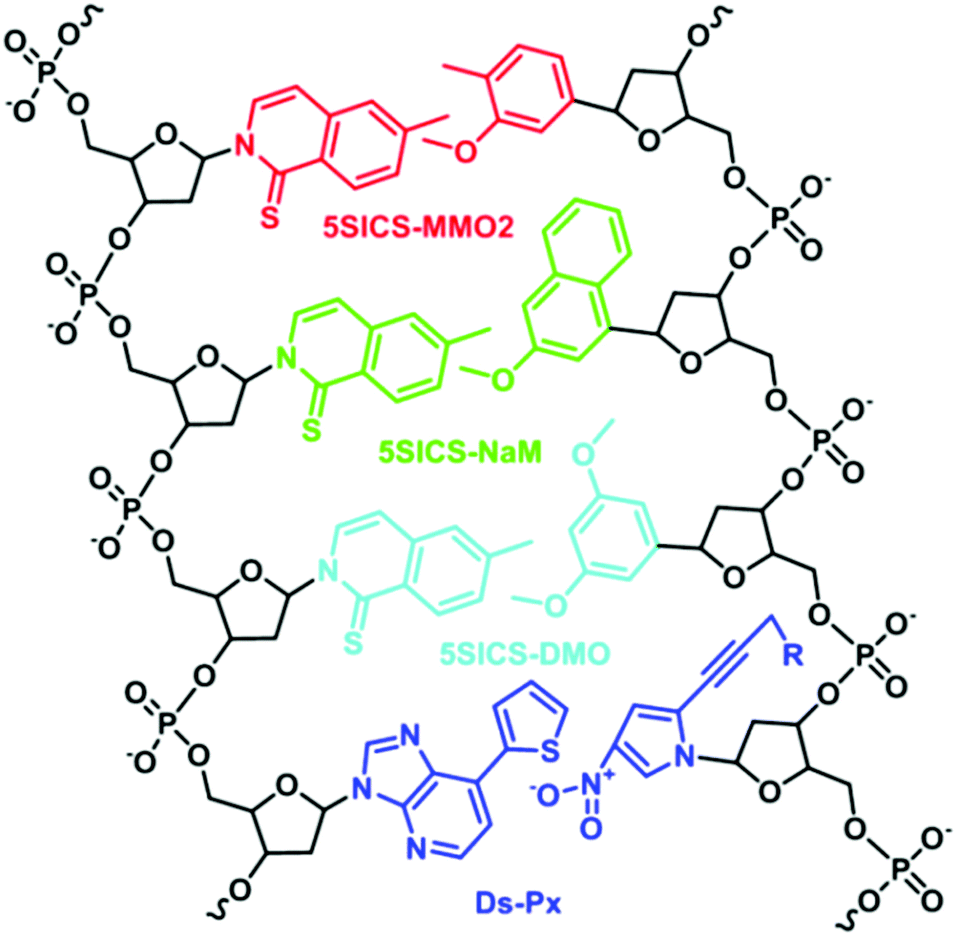 | ||
| Fig. 4 Chemical structures of 5SICS–MMO2, 5SICS–NaM, 5SICS–DMO, and Ds–Px base pairs. | ||
Through extensive research, the genetic alphabet has been expanded to beyond A, C, G, and T. Numerous nucleoside analogues that are highly capable of specific and robust base-pairing have been synthesized and are shown to be successful as the third base pair in PCR (Table 1). The arduous journey in search for the third base pair has furthered the understanding of biology from a chemical perspective.
The evolution of life over geological time has perfected a highly conservative system to reliably store and transmit hereditary information through nucleic acids. Currently, genetic information of all known living organisms is conveyed through the order of the four natural nucleobases – the genetic code. In other words, the arrangement of the four nucleobases along the backbone of DNA/RNA in a specific order underlies the preservation and transmission of genetic information. The creation of extra genetic codes,13,14,20,25–33 together with the development of XNA replicating systems will shake our common belief of the origin of life. It will ultimately lead to the expansion of our concept of life since life does not need to be exclusively based on a certain group of biological entities. The development of xeno-biology could lead to the production of unprecedented lineages of unnatural biological entities for a wide variety of purposes ranging from diagnostics to therapeutics. It could also provide us with an ultimate biosafety toolbox capable of safeguarding any type of genetic interaction between synthetic and natural life forms.34,35 On the other hand, great care must be taken pertaining to the societal aspects of xeno-biology with concerns such as biosafety, biosecurity, intellectual property rights, and governance.36
2.2. XNAs with modified phosphodiester backbones
Besides nucleobases, there have been endeavors in modifying the phosphodiester linkages of nucleic acids.37 Phosphodiester linkages have a huge influence on the physicochemical properties of nucleic acids. Not only do their anionic structures repel each nucleotide to create a sufficient space to facilitate enzymatic access to their sequences, their high polarity ensures high solubility in water. Modifications to the phosphate backbones may therefore significantly perturb the properties of nucleic acids, thus aiding the understanding of nucleic acids. Among nucleotides with modifications to the phosphodiester linkages, two broad classes – phosphorothiolate nucleotides38 and boranophosphate nucleotides39 – stand out (Fig. 5). The phosphorothiolate nucleotides differ from natural nucleotides in that the oxygen atom in the phosphodiester linkage is substituted by a sulfur atom. Such a substitution was found to reduce the probability of phosphorothiolate nucleotides from undergoing nuclease degradation.40 Its markedly reduced polarity, due to the increased polarizability of the sulfur atom, has also been utilized to understand the mechanisms of reactions occurring at phosphodiester linkages.41 The boranophosphate nucleotides are similar to the phosphorothiolate nucleotides in that the oxygen atom in the phosphate backbone is substituted by a boron atom. Both modified nucleotides are capable of undergoing PCR by using an engineered Taq DNA polymerase.42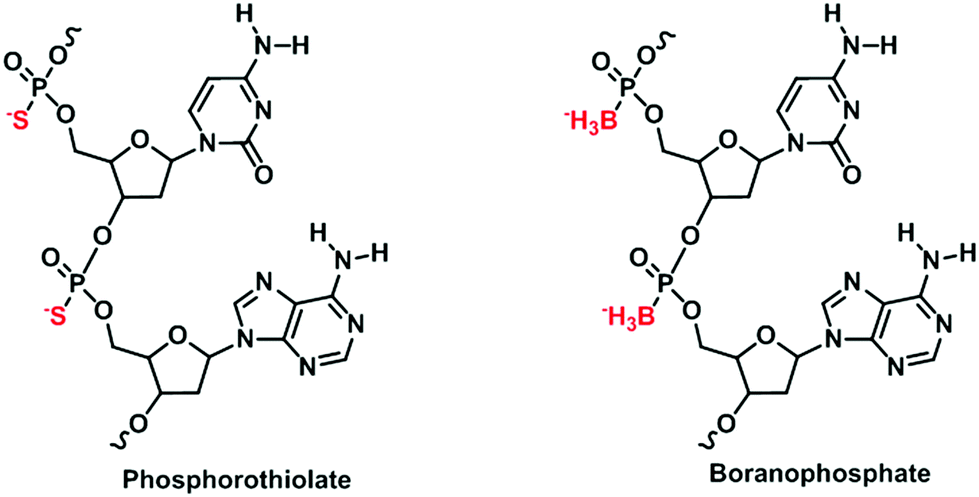 | ||
| Fig. 5 Representative nucleotides with modifications to the phosphodiester backbones. | ||
2.3. XNAs with modified sugar moieties
Representative XNAs with modifications to the sugar moieties of nucleic acids include threose nucleic acid (TNA), hexitol nucleic acid (HNA), and locked nucleic acid (LNA) (Fig. 6). TNA consists of an unnatural four-carbon threose sugar instead of a ribose sugar.43 The consequence of this modification is that the phosphodiester linkage is shortened by a single bond. Despite this difference, TNA is capable of pairing with complementary nucleic acid strands. TNA has also been shown to fold into tertiary structures with the desired chemical functions, driving speculation that TNA could be an RNA progenitor.44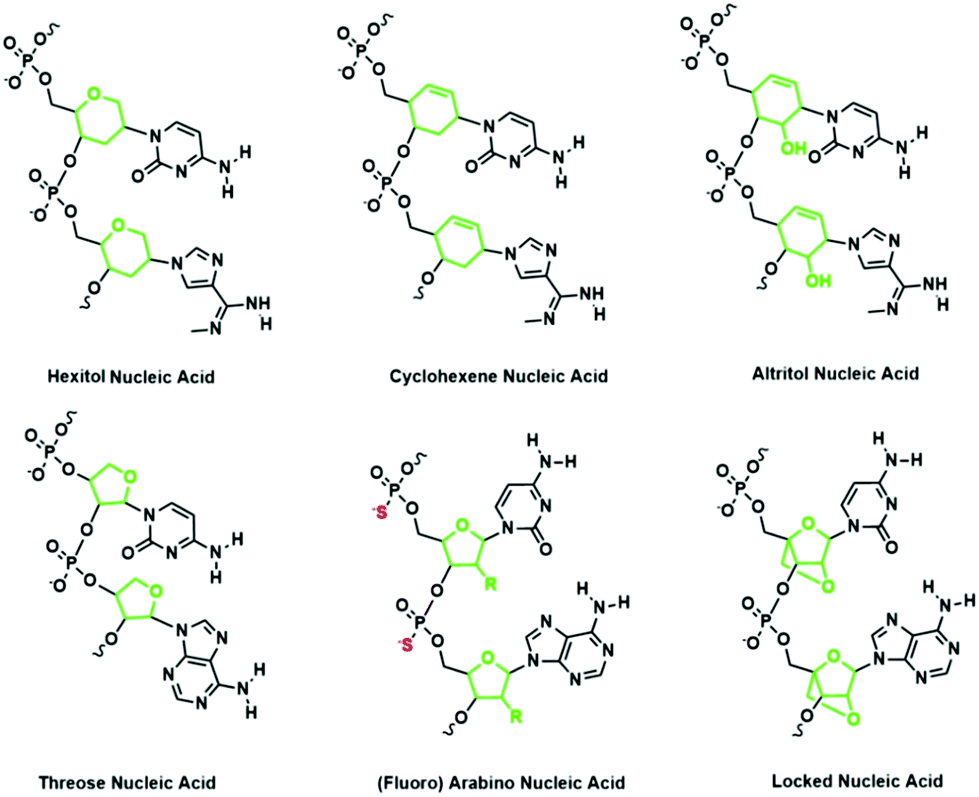 | ||
| Fig. 6 Representative nucleotides with modifications to the sugar moieties. | ||
HNA consists of a six-membered pyranosyl ring with the phosphodiester linkages connected at the 4′ and 5′ positions of the pyranosyl ring.45 Like TNA, HNA can form duplexes with its complementary nucleic acids.45 Also, HNA can form a double helix with itself.46 Recently, Pinheiro et al. sought to identify polymerases that are capable of generating long XNAs to carry significant genetic information by using a compartmentalized self-tagging strategy.47 This strategy involves the construction of a library of mutated polymerases and the use of primers and modified nucleotides such as HNA in selecting the best polymerase. Remarkably, they were successful in identifying a mutant polymerase that can synthesize HNA oligonucleotides from DNA templates and a reverse transcriptase that transcribes HNA oligonucleotides back to DNA. This strategy also enables the identification of polymerases that can transcribe other XNAs with modifications to the sugar moieties such as cyclohexenyl nucleic acid (CeNA), LNA, TNA, altritol nucleic acid, arabinose nucleic acid (ANA), and 2′-fluoro arabinose nucleic acid (FANA) (Fig. 6). This general strategy for identifying polymerases that accept a variety of sugar-modified nucleic acids has great potential in advancing the field of synthetic genetics. Besides TNA and HNA, another representative sugar modified XNA is LNA, which contains a bridging methylene between the 2′ and 4′ position of the ribose. The rigidity of LNA restricts the degree of freedom of LNA strands, thus conferring remarkable hybridization stability. In addition, the fully alkylated 2′-O enhances the resistance of LNA to nuclease degradation in contrast to RNA, which can be easily degraded by nucleases that exploit its free 2′-OH.48
2.4. XNAs with both modified sugar moieties and phosphodiester backbones
A radical modification to the backbones of nucleic acids is to totally replace phosphate and sugar backbones with a peptide – peptide nucleic acid (PNA). PNA contains repeating aminoethylglycine units joined to each other by peptide linkages (Fig. 7).49 The backbone can be easily modified to replace the glycine moiety with lysine,50 arginine,51 or cysteine52 – changes that can alter the physicochemical properties of PNA. The most outstanding feature of PNA is that it is electrically neutral. Therefore, unlike nucleic acids with negatively charged backbones, a PNA strand can form a duplex with either itself or a DNA/RNA duplex without the need for any counter ions for stabilization.53 Furthermore, the abolishment of inter-strand electrostatic repulsion also means that PNA/nucleic acid duplexes are thermodynamically more stable than double-stranded nucleic acids. These traits together with better resistance to nuclease degradation, allow PNA to be used in numerous applications,54,55 even though it remains refractory to PCR.56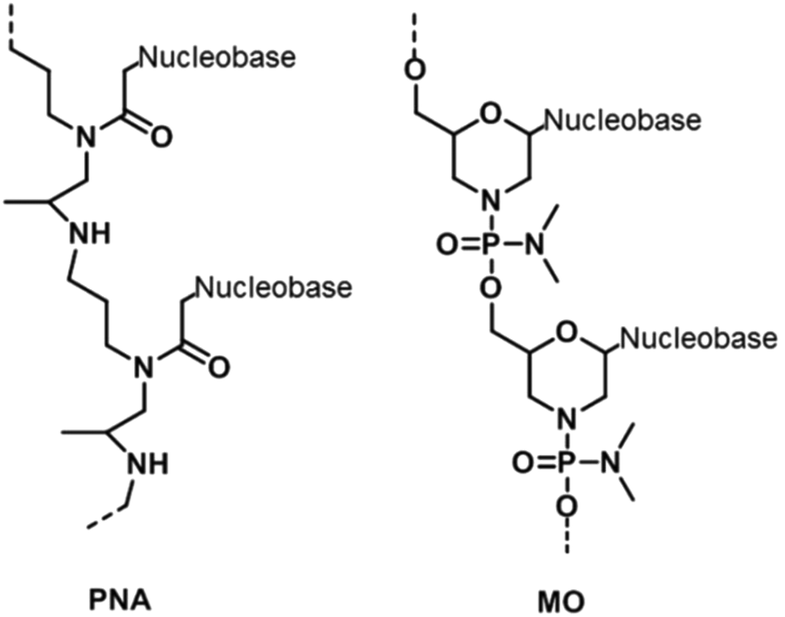 | ||
| Fig. 7 Chemical structures of PNA and MO. | ||
Another type of XNA with modifications to both the sugar moieties and phosphodiester linkages is morpholino (MO) in which riboses are replaced by morpholine moieties while the phosphodiester backbone is replaced by a phosphorodiamidate backbone (Fig. 7).57 Similar to PNA, MO possesses an electrically neutral phosphorodiamidate backbone. Despite its electrical neutrality, MO has good solubility in water due to the hydrophilic morpholine ring.58 MO is also resistant to a wide range of nucleases, which makes MO oligomers very useful as intracellular probes.59 More importantly, MO oligomers sterically prevent enzymes from accessing their target nucleic acids. By doing so, MO is capable of probing and regulating gene expression in vivo. For instance, a MO strand can bind to a region of a target messenger RNA (mRNA), reducing the translation of the protein encoded by the mRNA.60 Therefore, MO allows gene and protein expression to be investigated easily in a cellular context.
3. Applications of XNAs
3.1. XNAs in molecular biology
Because of the scope of this article, only a brief summary of the applications of XNAs in molecular biology is presented in this section since the biological importance of nucleic acids extends beyond the nucleotide sequence. The secondary and tertiary structures of nucleic acids are known to be involved in various functions. For example, the propensity of DNA to form double-stranded helices is generally believed to confer great stability and provide an avenue for proofreading during replication, both of which are important factors in long-term information storage. Other than the familiar double helix, DNA is also capable of forming a few tertiary structures through non-Watson–Crick base-pairing modes, such as a G-quadruplex and an i-motif, which are thought to be involved in gene expression.61 The formation and interactions of G-quadruplexes have been observed by incorporating a fluorescent purine analogue, 2-aminopurine, into a region of human telomere known to form such a structure.62 In addition, several groups have developed PNA probes capable of binding to G-quadruplexes, presumably by binding to the loop region or by strand invasion.63,64 However, such probes are currently limited to in vitro applications.65 In contrast to the limited structural diversities of DNA, RNA can adopt a much greater variety of tertiary structures. This is due to the fact that while DNA is primarily concerned with preserving hereditary information, RNA is involved in many cellular processes, such as the control of transcription, catalysis in protein translation, and alternative splicing.66 RNA molecules involved in these functions are aptly termed “functional RNAs (fRNAs)”.67 A significant proportion of fRNAs are double-stranded or contain double-stranded regions, such as siRNA and riboswitches. XNA triplex-forming probes have therefore been developed to detect double-stranded RNAs. For example, Wang et al. modified RNA with carboxamide-linked pyrene to discriminate between single- and double-stranded RNAs.68 The emission of pyrene excimers was observed for single-stranded RNA but not for double-stranded RNA. More recently, it was observed that PNA modified with thio-pseudoisocytosine nucleobase can form a triplex with double-stranded RNA.69 The neutral backbone of PNA allows a RNA/RNA–PNA triplex to form almost independent of pH and salt concentration, which is an advantage over triplex-forming nucleic acid probes.70 The unnatural nucleobase prevents the formation of PNA–RNA duplex by sterically clashing the Watson–Crick pairing mode and favors the Hoogsteen base-pairing geometry, and therefore triple formation by compatible hydrogen bonds and shape complementarity.Like PNA, MO oligomers with sequences complementary to their target nucleic acids are capable of hybridizing with them according to the Watson–Crick base-pairing mechanism. First studied by Summerton et al.,71 MO oligomers have primarily been used in antisense applications and as a knockdown tool in developmental biology due to their high specificity and ease of cytosolic delivery. Being neutral nucleic acid analogues, PNA and MO are also used for antisense and antigene applications as well as microRNA (miRNA) therapeutics since they are resistant to nuclease and protease digestion whereas nucleic acids are highly susceptible to nuclease degradation.72 They are being used in applications such as correcting splicing errors in pre-mRNAs in cultured cells and in extra-corporal treatments of cells from thalassemic patients. A particularly interesting application in developmental biology is the use of PNA and MO to block the expression of any selected gene throughout the course of embryogenesis.71,72
LNA is best recommended for use in molecular biology where high specificity is required. LNA also has immense therapeutic potential because it can be used for the regulation of gene expression, as it is stable in serum, can be taken up by mammalian cells, and shows low toxicity in vivo.
3.2. XNAs in the study of nucleic acid–protein interaction
Nucleic acids often interact with other biomolecules, most commonly proteins, in order to perform functions related to transcription, translation, and regulation. Classical methods of probing nucleic acid–protein interactions include immunoassays, pull-down assays, and electrophoretic mobility shift assays. To gain a deeper understanding of such interactions, it may be necessary to investigate into the dynamics of such interactions. Nucleic acid dynamics may be elucidated by nuclear magnetic resonance or computational modelling,73 among others. While these approaches have been proven useful, the perennial concern with in vitro and in silico methods is that these methods may not provide an accurate representation of nucleic acid interactions and dynamics in complex cell milieus. It is therefore often useful to obtain in vivo information to complement in vitro and in silico studies. The chemical diversity and bioorthogonality of XNAs are advantageous in this regard. MO antisense probes are well established to form stable and specific duplexes with miRNAs, thereby reducing their interactions with proteins that will exert downstream effects.74 In this manner, the function of a miRNA of interest can be evaluated in a sea of miRNA and other RNA molecules.75 Zielinski et al. developed PNA probes functionalized with a UV-activatable crosslinking amino acid, para-benzoylphenylalanine, to covalently link the probes with RNA-binding proteins (RBP) that interact with the target RNA (Fig. 8).76 The PNA probes can be isolated by antisense oligonucleotides immobilized on magnetic beads, allowing rapid identification of RBPs by mass spectrometry. The key advantage of this approach is that the probes can be used in vivo, allowing weak or transient interactions to be captured. Hence, it is expected that such a technology may reveal novel RBP–RNA interactions previously obscured by sample processing using in vitro assays.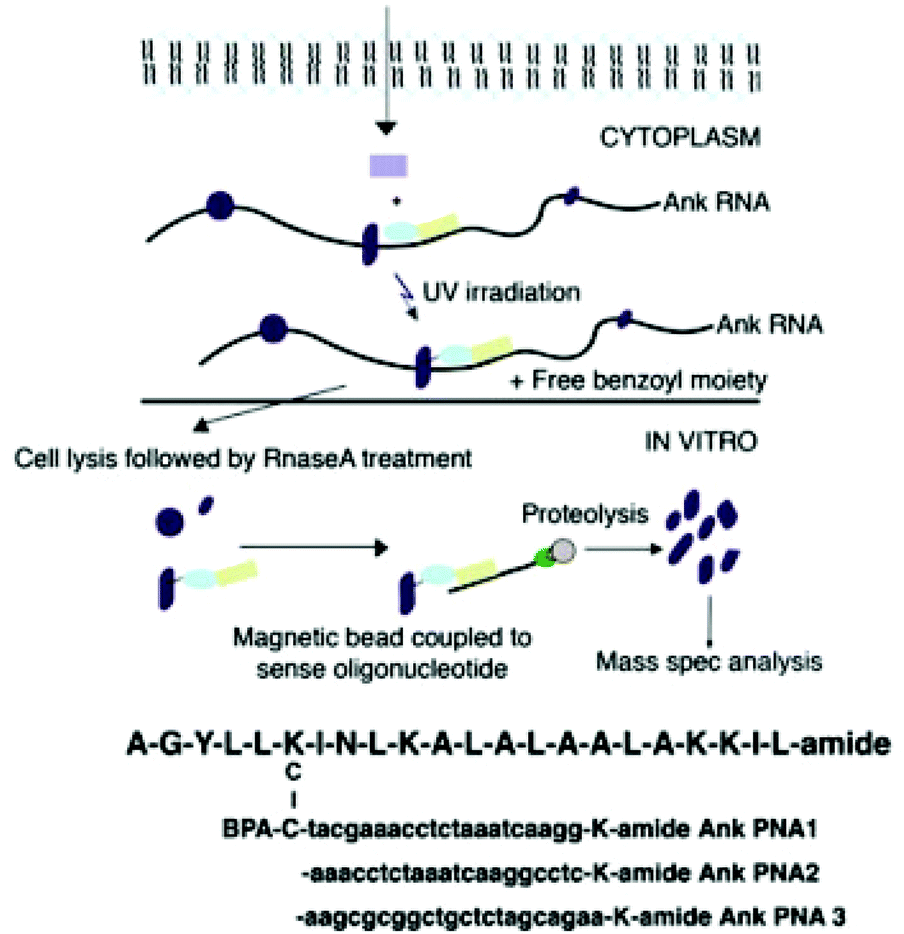 | ||
| Fig. 8 Schematic representation of the study of protein–RNA interaction using the PNA probes (reproduced with permission from ref. 76, Copyright @ 2006, National Academy of Sciences). | ||
A well-known RBP–RNA interaction is that between a short-interfering RNA (siRNA) and an RNA induced silencing complex (RISC) in RNA interference (RNAi), a mechanism of gene knockdown in eukaryotes.77 In order to better understand the mechanism by which siRNA and RISC interact, Hernández et al. developed size-expanded nucleobase RNAs.78 Using these modified guide strands, they found that incorporating the size-expanded nucleobases from positions 2 to 11 results in poor RNAi activity, indicating that RISC should bind very tightly to the RNA strand at these positions. This observation is in concurrence with a known crystal structure of an RISC–siRNA complex.79 Interestingly, modifications at positions 16 to 19 do not have a significant impact on gene silencing, thereby suggesting that there is some degree of flexibility at the 3′ ends of the guide strands in RISC. Consequently, the size-expanded nucleobases can be included at these positions to improve the nuclease resistance of externally administered siRNA.
3.3. XNAs in the study of nucleic acids in vivo
Nucleic acids are highly susceptible to degradation, chemically and enzymatically, and have limited chemical diversity. XNAs circumvent these problems through chemical modifications, thereby providing greater stability, better sensitivity, and more desirable physicochemical properties, while retaining or even improving the specificity of nucleic acids. Consequently, a natural application of XNAs is in their use as hybridization probes. Hybridization probes are short to medium length XNAs, typically between 25- and 100-base-pair long, that detect nucleic acid sequences complementary to the probes.80 Many hybridization probes use fluorophores as reporters due to relative ease of experimental handling and visualization. Well-established examples of hybridization probes are molecular beacons (MBs),81 binary probes (BPs),82 forced-intercalation (FIT) probes83 and fluorescence in situ hybridization (FISH) probes.84 The mechanisms of action of these probes are depicted in Fig. 9. While hybridization probes made from DNA have undoubtedly proven useful for in vitro studies such as real-time PCR, the presence of nucleases and nucleic acid-binding proteins inside cells can cause the fluorophores and quenchers of these hybridization probes to separate even when they are not bound to their target sequences, thus possibly leading to false positives.85 To overcome the shortcomings of MBs made from DNA, Wang et al. synthesized MBs using LNA.86 The presence of a covalent bond between the 2′-O and 4′-C of the ribose moiety in LNA restricts conformational changes in the sugar molecule, resulting in exceptional LNA/DNA duplex stability due to increased base stacking interactions.87,88 In addition, LNA/DNA duplexes obey Watson–Crick base-pairing rules and display greater melting temperature lowering toward mismatches than DNA/DNA duplexes.89,90 Furthermore, Wang et al. found that LNA is highly resistant to nucleases and single-stranded DNA binding proteins in vivo, resulting in an exceptionally low background compared to DNA MBs inside cells.86 In addition, the LNA MBs exhibited superior selectivity for single-nucleotide polymorphisms (SNPs) than their DNA counterparts. It is therefore hardly surprising that LNA MBs quickly found applications in the fields of medical genetics,91–94 biosensors,95–98 and microbiology,99,100 among others.101,102 A perennial problem with DNA MBs is that the stem portion can participate in base pairing with off-target sequences when the MBs open, resulting in false positives. This is especially so in a cellular environment where the genome is present. To remedy this, Crey-Desbiolles and colleagues replaced the nucleotides in the stem portion of MBs with β-D-2′,3′-dideoxyglucopyranosyl (6′ → 4′)-linked nucleotides, also known as homo-DNA.103 In homo-DNA, the pyranose moiety replaces the furanose in natural DNA (Fig. 10). Homo-DNA is similar to HNA, except that the base is attached at the 1′ position of the pyranose instead of 2′ for HNA. Homo-DNA forms a unique double helical structure, thus explaining why homo-DNA is unable to form a duplex with DNA.104 The homo-DNA modified MBs simplify stem design, and have been shown to be significantly more selective than DNA MBs. Not long after, Kim et al. developed MBs with stems made from L-DNA, the enantiomeric form of natural D-DNA (Fig. 10), and loops synthesized with 2′-O-Me modified RNA.105 The modified nucleotides form duplexes only with themselves, eliminating unwanted stem–loop interactions and also improving stability in vivo as the addition of a methyl group at the 2′ position of the RNA ribose abolishes RNase activity,106 ultimately leading to more sensitive and specific MBs. The ability of 2′-O-Me modified RNA to resist nuclease activity is also exploited by Nilsson and co-workers in the development of MBs for monitoring rolling-circle amplification (RCA).107 RCA is an isothermal nucleic acid amplification technique that uses a circular DNA template to generate a very long nucleic strand consisting of many tandem copies of the template and is used in diagnostics and nanotechnology.108,109 Φ29 DNA polymerase is commonly employed in RCA due to its remarkable strand displacing ability and processivity.110 However, the 3′ exonuclease activity of Φ29 DNA polymerase prevents the use of DNA MBs.111 In order to reliably and easily monitor the progress of RCA, Nilsson et al. synthesized MBs from 2′-O-Me RNA, thereby allowing rapid and highly quantitative measurement of RCA kinetics.107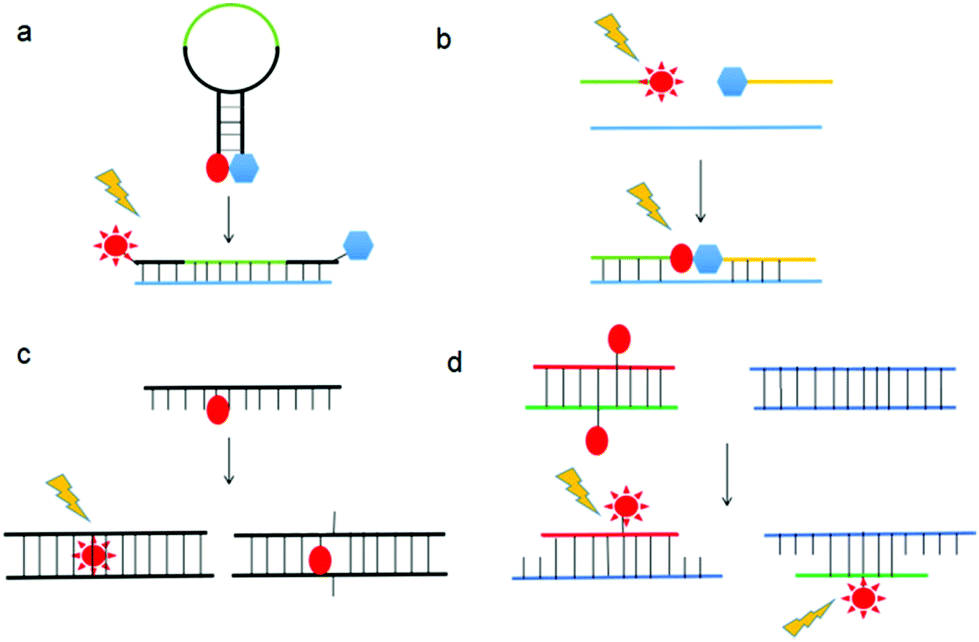 | ||
| Fig. 9 Schematic of mechanisms of action of (a) MB, (b) BP, (c) FIT, and (d) FISH probes. | ||
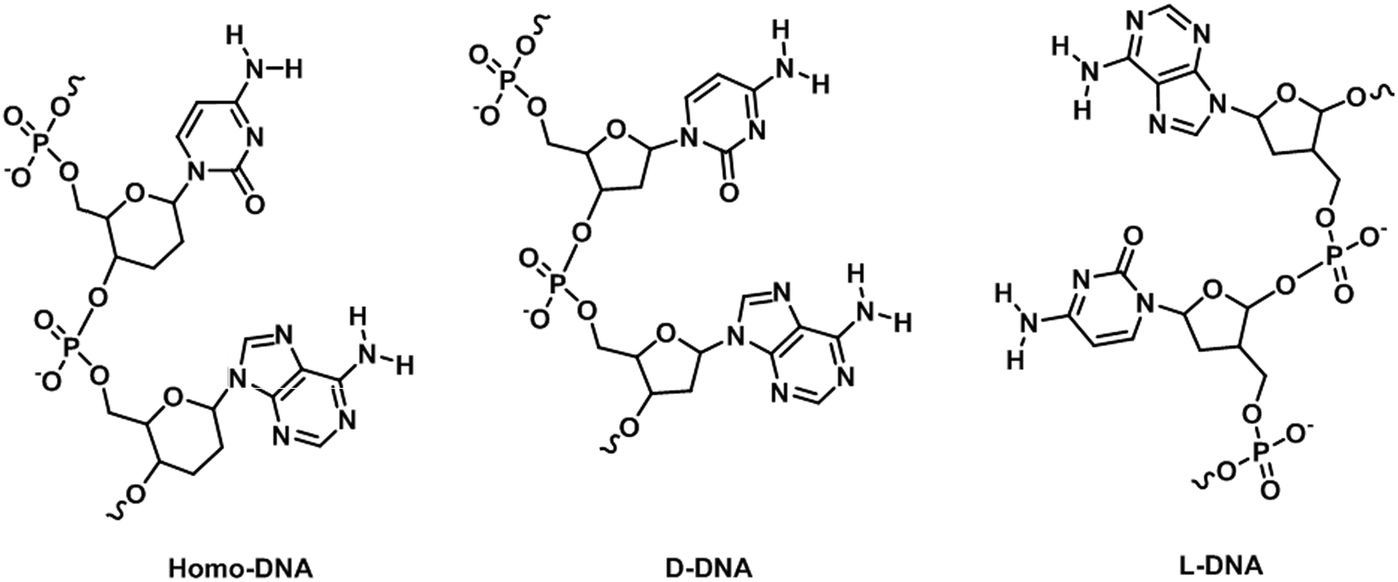 | ||
| Fig. 10 Structures of homo-DNA, D-DNA, and L-DNA. | ||
BPs are an alternative to MBs in the nucleotide sequence detection, especially in SNP discrimination. BPs work by exciting the donor fluorophore on one of the two probes, which results in Förster resonance energy transfer from the donor to an acceptor fluorophore, resulting in fluorescence. BPs are sometimes preferred over MBs as they are reportedly more selective in differentiating between single base changes as the stem region of MBs can result in off-target hybridization.112,113 BPs are also more amenable for mixing and matching, making them more economical than MBs for the detection of a large number of samples of slightly different nucleotide sequences. However, perhaps due to the commercial dominance that MBs possess over BPs, only a handful of attempts have been reported on XNA-substituted BPs. Nevertheless, PNA BPs developed have been found to be superior to DNA BPs because perfectly complementary PNA/DNA duplexes have higher melting temperatures than their DNA/DNA counterparts, but a single mismatch causes a higher melting temperature penalty.114–117
FIT probes are relatively new PNA-based hybridization probes first developed by Kohler and Seitz.83 Instead of the fluorophores being attached on the ends of the probes as with most other hybridization probes, thiazole orange (TO) molecules are incorporated into the probes. TO is a well-established fluorescent DNA intercalator118 and is able to base pair well with all four natural nucleobases.119 The fluorescence of TO is sensitive to its local environment: only when the bases in the FIT probes flanking TO are complementary to the target can TO intercalate into the duplex, reducing rotation around its methylene group, thereby resulting in strong fluorescence.120 The fluorescence of TO is significantly weaker if the central methylene group is allowed to rotate. This happens when there is a base mismatch adjacent to TO. The flexible PNA backbone allows the relatively large TO molecule to be accommodated in a stable PNA/DNA duplex. The inherent discriminatory ability of PNA along with the microenvironment-dependent fluorescence of TO produces highly sensitive probes well suited for SNP detection.121–123
FISH is used to locate nucleotide sequences in formaldehyde-fixed cells. It was first developed to locate genes on a chromosome,84 but can also detect mRNA and miRNA to determine gene expression and localization.124 PNA is the most popular XNA substitute for constructing FISH probes. There are three reasons for its success: (i) the neutral backbone of PNA facilitates diffusion across cell membranes and also leads to increased rate and stability of hybridization with DNA and RNA targets in fixed cells;125 (ii) hybridization can occur at very low ionic strengths, which is advantageous as genomic DNA that has been chemically denatured can re-anneal to each other at high ionic strengths,126 forbidding the binding of probes; and (iii) PNA probes are highly amenable for attachment of a wide selection of fluorophores,127 thus greatly simplifying multiplex imaging. The use of PNA probes in FISH is established, extensive and clinically relevant, from the visualization of genes and genetic elements in human chromosomes128–130 to the identification of microbial pathogens in infected cells.131–133 The broad applicability of PNA FISH probes is perhaps exemplified by the fact that they can even diffuse across the highly hydrophobic and dense cell membrane of Mycobacterium sp.,134 which is a feat that cannot be accomplished by the intrinsically charged natural nucleic acid probes.135 In a similar way, MO-based FISH probes have also been found to be convenient for direct delivery into zebrafish tissues.136 However, the uncharged backbone of PNAs and MOs lowers their hydrophilicity, predisposing these XNAs to aggregation and unspecific hydrophobic interactions with proteins.137 Therefore, it may be prudent to introduce hydrophilic groups on uncharged XNA chains, such as on the nucleobases, to improve aqueous solubility.138
While FISH probes have indubitably been proven valuable for scientists and clinicians, current protocols require fixing the cells in formaldehyde prior to probe delivery and involve extensive washing steps as probes may sometimes fluoresce even when not bound to their targets. It is desirable to perform direct imaging of live cells in order to decrease analysis time, and avoid artefacts that may emerge due to cell fixation and observe cellular dynamics. To this end, hybridization probes that can be delivered into cells with the aid of low detergent concentrations and/or chemical modifications have been developed.139 FISH probes that exhibit low background but high fluorescence only when bound to their targets, i.e. a “turn-on” signal, have been achieved by attaching TO-derived fluorophores to the DNA backbone. Another approach that has been developed is to first deliver a strongly fluorescent probe covalently linked to an azidoether-linked quencher and allow it to hybridize with a part of a target sequence.140 Afterwards, another probe that will bind to the other part of the target sequence is delivered. This probe contains a triphenylphosphine group that will reduce the azidoether to an N,O-acetal through the Staudinger reaction (Fig. 11).141 The acetal is rapidly hydrolyzed to release the quencher, resulting in rapid turn-on fluorescence.142,143
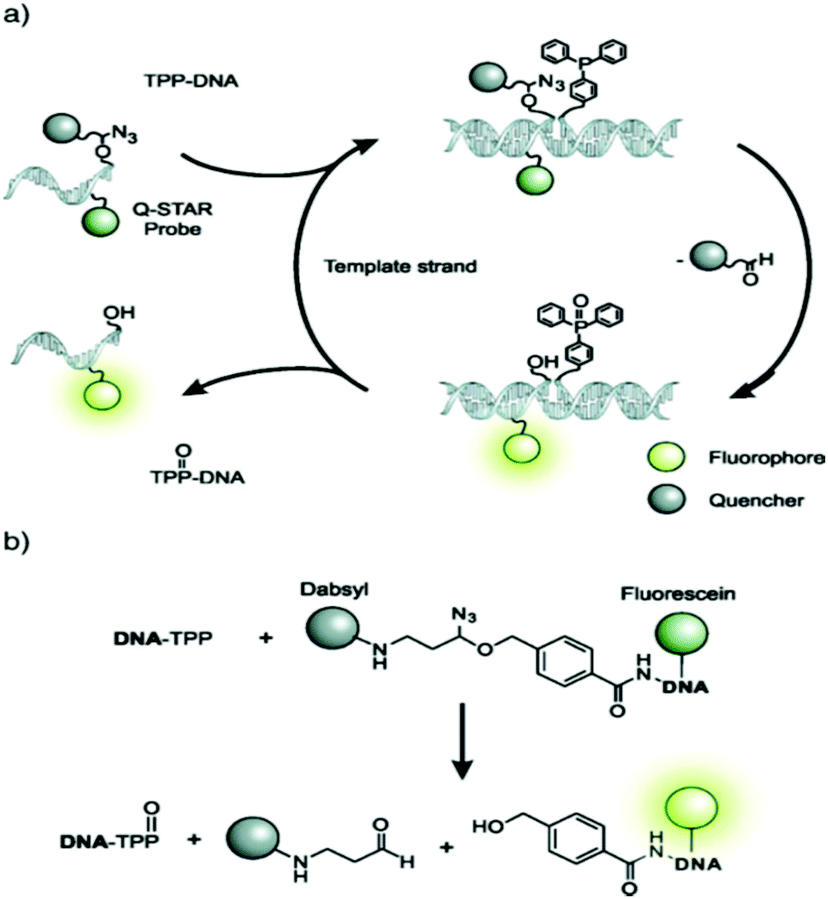 | ||
| Fig. 11 Mechanism of binary FISH probes that exhibit a rapid “turn-on” fluorescence signal (reproduced with permission from ref. 141, Copyright © 2009, American Chemical Society). | ||
3.4. XNAs in the study of nucleic acid–small molecule interactions
While XNAs have been extensively developed for nucleic acids and proteins, there is no reason to limit the applications of XNAs to only biomacromolecules. The strategy for developing highly affinitive XNA-based entities such as aptamers for targeting small molecules is to increase the chemical space of aptamers by using XNAs. Demonstrating this principle, Imaizumi and co-workers incorporated chemically modified uracils (Fig. 12) into DNA and through systematic evolution of ligands by exponential enrichment (SELEX) discovered aptamers that bind to thalidomide and camptothecin with KD values of 1.0 μM and 40 nM, respectively,144 which represents vast improvements over DNA aptamers.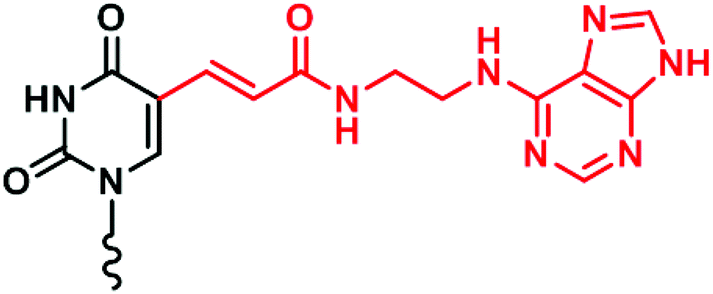 | ||
| Fig. 12 Structure of a chemically modified uracil. | ||
On the other hand, a burgeoning trend is to exploit the modularity and strong signals of oligodeoxyfluorosides (ODFs) to improve current methods for detection of small molecules and ions. ODFs are in essence modified DNA oligonucleotides where the base is substituted for fluorophores (Fig. 13).145 The overall strategy is to generate a combinatorial library of ODF tetramers that incorporate various fluorescent and non-fluorescent monomers. Screening is accomplished with polystyrene beads on which the tetramers are synthesized, thus facilitating deconvolution. ODFs have found applications in detecting and semi-quantifying diverse classes of small molecules and ionic analytes with detection limits commonly found to be in the low micromolar to nanomolar range. Examples of these analytes include: volatile organic molecules,146 anionic water pollutants,147 petroleum products148 and metal ions.149 Recent improvements include the development of sensor arrays to allow a small library of tetrameric ODFs to differentiate a large number of analytes150 and ODFs that can be inexpensively printed on paper.151 These developments place ODFs in a good position to be developed as convenient and economical sensors for widespread applications such as food spoilage and environmental monitoring, especially in areas where laboratory testing is prohibitively expensive and impractical.
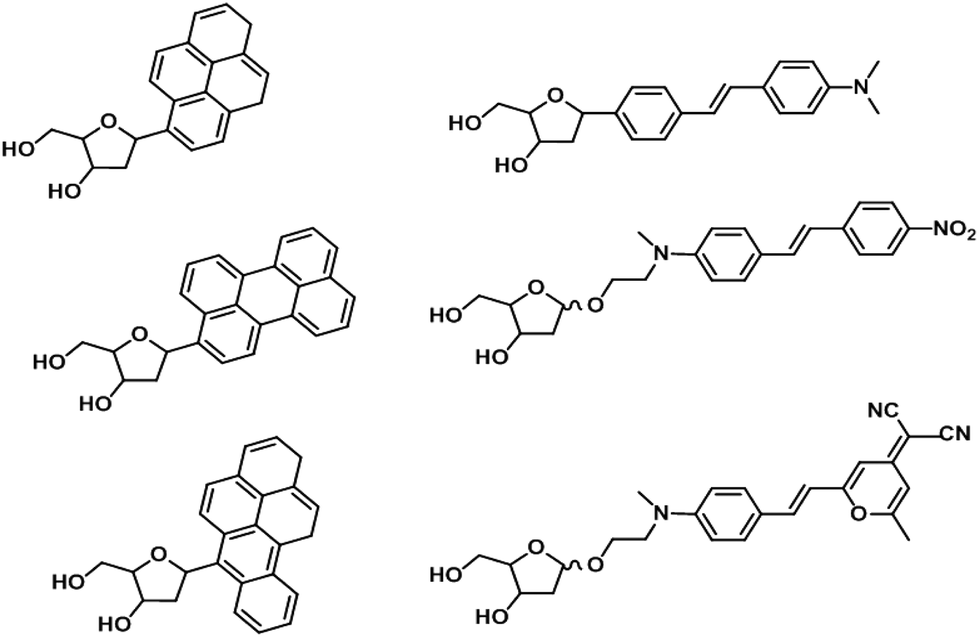 | ||
| Fig. 13 Chemical structures of representative ODF monomers. | ||
3.5. XNAs in bioassays
The most popular XNAs employed in the construction of nucleic acid bioassays are LNA, PNA, and MO. Experimental observations have confirmed that LNA has an extraordinarily enhanced thermal stability, high affinity, low toxicity, improved triplex formation, and nuclease resistance.152–154 LNA probes also show good sensitivity and they are commonly considered for the improvement of the sensitivity of detection.155 LNA probes have been used for simple and specific DNA detection of chronic myeloid leukemia and acute promyelocytic leukemia.156 The signal generated upon hybridization can also be amplified using different approaches.157,158 Recently, an approach on the locking of a furanose ring via a methylene linkage between 4′-C and 2′-O has been developed.159,160PNA has also been widely employed in the construction of nucleic acid bioassays. PNA was firstly investigated by Nielsen and colleagues in 1991 to, specifically, interact with double-stranded DNA in a triple-helix fashion.161,162 As a charge-neutral XNA, PNA has a greater binding affinity to its complementary nucleic acid due to the absence of electrostatic repulsion.163 The high binding affinity also allows the distinction of closely related sequences, even at single-base levels. A representative example is described by Fan et al. in which hybridized anionic target miRNA strands are utilized to attract protonated aniline molecules and consequently their polymerization along those strands (Fig. 14).164 Another example was reported by Su and co-worker,165 in which DNA/PNA hybridization is studied using a PNA-immobilized microwell plate. After hybridizing with sampled DNA strands, the negative charges brought to the microplate are exploited for the introduction of cationic horseradish peroxidase through electrostatic interaction to produce a detectable signal.165 The current trend in the development of PNA-based probes is based on the modification or extension of their backbones or linkers.162 The change in the conformation of PNA strongly impacts on the hybridization, detection, and regeneration. For instance, the design of a PNA backbone can lead to label-free bioassays.166
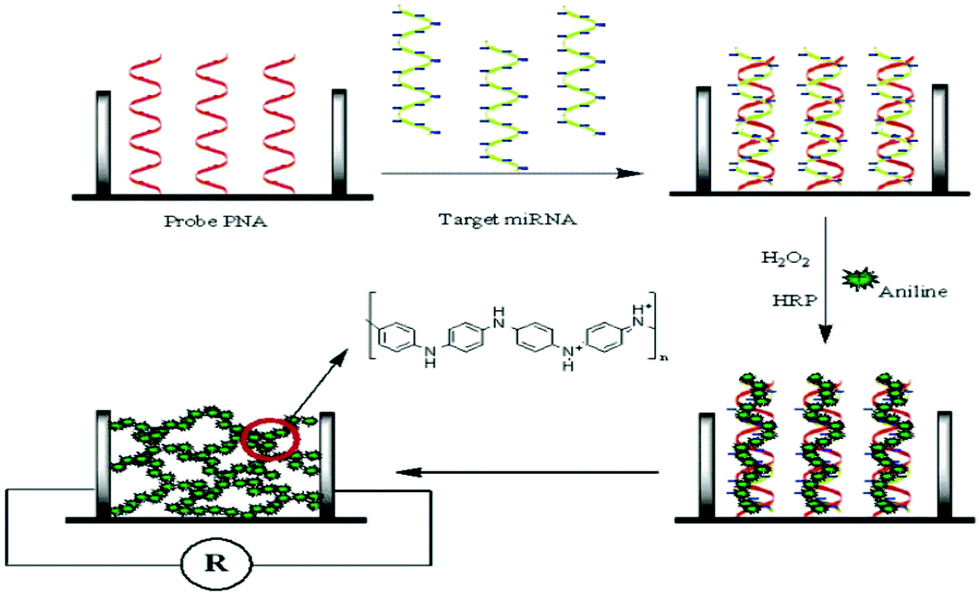 | ||
| Fig. 14 Schematic illustration of the sensing mechanism involving PNA probes (reproduced with permission from ref. 164, Copyright © 2007, American Chemical Society). | ||
Another charge-neutral XNA engaged in nucleic acid bioassays is MO. Several attempts have been made in applying MO in the construction of nucleic acid bioassays. It was shown that the performance of the bioassays is comparable to that of PNA-based ones.167–169 Likewise, accompanying with hybridization, a large number of negative charges are brought into the bioassays, therefore offering a convenient means to introduce signaling units through electrostatic interaction. For example, a cationic redox polymer, acting as a signal generator, is introduced through electrostatic interaction after hybridization for the detection of nucleic acids.167
To effectively alleviate the problem of extensive secondary structures, which severely hinder hybridization, Zu et al. proposed a colorimetric assay for the detection of nucleic acids under extremely low salt conditions (Fig. 15).168 Since the base-pairing of nucleic acids is exclusively dependent on temperature and salt concentration, the secondary structures are less stable and more accessible under low salt conditions at moderate temperatures. To accomplish their goal, practically salt-independent probes such as MO are engaged. It was found that as low as 2.5 mM total salts is sufficient to successfully carry out hybridization. Their assay worked effectively in detecting sequences that are likely to form secondary structures.
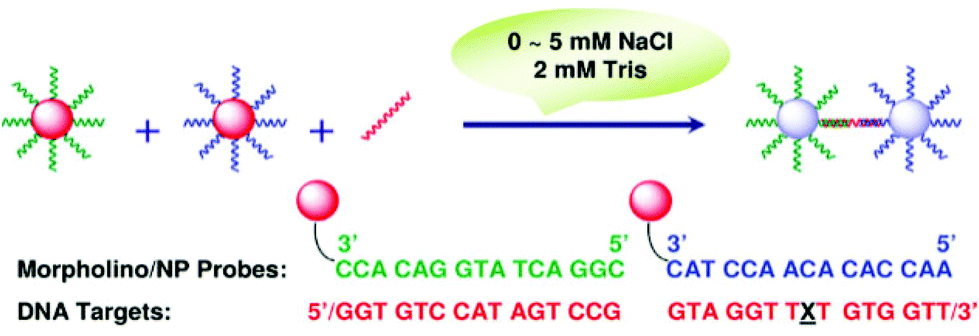 | ||
| Fig. 15 Schematic presentation of the colorimetric detection of nucleic acids using MO probes under extremely low salt conditions (reproduced with permission from ref. 168, Copyright © 2007, American Chemical Society). | ||
Based on experimental evidence gathered so far, the charge-neutral XNAs are better probes for nucleic acid bioassays. There are several distinct advantages to substitute nucleic acids by the charge-neutral XNAs like PNA and MO such as higher hybridization efficiencies, better sequence specificities, greater resistance to enzymatic degradation, and lower salt-dependence of hybridization.170,171 In addition, the negative charges brought into the bioassays by hybridized nucleic acid strands provide an additional strategy of signal generation. However, they have not yet replaced nucleic acid probes primarily because of their high cost.
Unlike other classes of biomolecules, proteins and peptides are inherently more challenging to detect. Widely used techniques in protein detection are antibody-based assays such as enzyme linked immunosorbent assays and Western blotting. The production of antibodies is, however, laborious and expensive.172 It is simply not economically viable or feasible to raise antibodies for every potential target protein in the proteomic universe. While antibodies may be irreversibly denatured when exposed to non-physiological temperature, pH, salt concentration, or solvent, aptamers can, in principle, always be renatured and regain their target binding affinity. The ability of aptamers to be denatured and renatured multiple times is another key advantage they have over antibodies.
Nucleic acid aptamers have therefore been an exciting avenue for the development of protein probes since their discovery as high affinity binding reagents, first described in the 1990s.173,174 Since then a number of nucleic acid aptamers have been generated to bind protein biomarkers of diseases such as VEGF,175,176 thrombin,177 HIV related proteins,178,179 PDGF,180 and NF-κB.181 Also, numerous aptamer-based assays have since been reported and reviewed.182,183 These assays have been reported to have detection limits as low as fg ml−1. However, the key constraints of aptamer-based bioassays are: (i) the development of aptamers with high affinity and specificity due to the limited chemical diversity of natural nucleotides and (ii) the susceptibility of nucleic acids to be enzymatically digested in cells. XNA aptamers are therefore well positioned to circumvent these limitations and several groups have indeed divulged the enhanced binding capabilities of XNA aptamers to various protein targets as summarized in Table 2. Interestingly, it has been demonstrated that the Ds–Px base pair can be used to generate XNA aptamers that could outcompete previously developed nucleic acid aptamers in binding to IFN-γ and VEGF.201 Experimental evidence suggested that the availability of hydrophobic residues on the aptamer facilitates binding by interacting with the hydrophobic domains present on target proteins.
| Nucleic acid modification | Binding target | Ref. |
|---|---|---|
| HNA | sTAR | 47 |
| Hen egg lysozyme | 48 | |
| 2′-NH2-pyrimidine | bFGF | 184 |
| hTSH | 185 | |
| IgE | 186 | |
| 2′-Me/2′-Fluoro | VEGF | 187 |
| EGFR | 188 | |
| PSA | 189 | |
| PSMA | 190 | |
| HIV-1Ba–L glycoprotein 120 | 191 | |
| TFPI | 192 | |
| CD4 | 193 | |
| Phosphorothiolate | E-selectin | 194 |
| NF-κB | 195 | |
| HIV-1 RT | 196 | |
| Dengue-2 envelope protein | 197 | |
| LNA | CD73 | 198 |
| Thrombin | 199 | |
| 2′-OMe A/G | IL-23 | 200 |
| Ds–Px | VEGF, IFN-γ | 201 |
The poster child for protein-binding XNA aptamers would perhaps be the slow off-rate modified aptamers (SOMAmers). Selected by SELEX, SOMAmers are DNA aptamers containing 2′-deoxyuridine nucleotides that have modifications on the C5 position of the base.202 These modifications decorate uracil with a variety of residues that attempt to mimic and even transcend the chemical diversity displayed by amino acids as illustrated in Fig. 16. The first generation of SOMAmers born in 2010 have been shown to be capable of binding to many human protein targets.202 So far, SOMAmers specific to more than one thousand different human proteins have been discovered. Moreover, the SOMAscan™ proteomic assay developed based on these SOMAmers is now commercially available. The principle behind the assay is simple and elegant – use the library of characterized SOMAmers to bind proteins, pull down the protein–SOMAmer complexes, wash off non-specific interactions, and deconvolute the products with a DNA microarray that hybridizes to the coded non-binding ends of SOMAmers.203
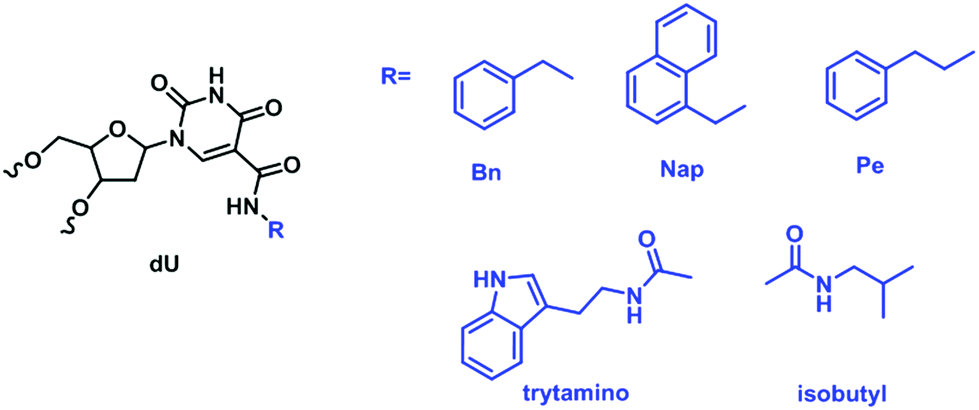 | ||
| Fig. 16 A partial representation of modifications at the C5-position of deoxyuridine available for the preparation of SOMAmers. | ||
To better understand the mechanism of SOMAmers, Davies et al.204 and Gelinas and co-workers205 crystalized complexes of SOMAmers bound to their specific targets – IL-6 and PDGF-BB.206 These structures clearly suggest that the high affinities between SOMAmers and IL-6 and PDGF are largely due to the shape complementarity to the hydrophobic pockets of the proteins. These structures further corroborate with the observation that aptamers with modifications containing aromatic hydrophobic moieties tend to produce the best performing aptamers.207
Since 2012, SOMAscan™ has been successfully employed by many groups to identify biomarkers of various diseases ranging from neurodegeneration to cancer. Of the 1129 human protein targets detectable by the SOMAscan™ assay, a wide range of different protein types involved in various key cellular processes and diseases were covered.208 To further demonstrate the applicability of SOMAscan™ as a proteomic technology relevant in a clinical setting, Gold et al. applied SOMAmers to identify novel biomarkers for chronic kidney disease202 and non-small cell lung cancer.209 They reported a measurable dynamic range of eight orders of magnitude and a median detection limit of 40 fM for the SOMAmers used in the SOMAscan™ assay.210 The number of biomarkers identified by using SOMAscan™ is expected to grow over the coming years as more researchers adopt SOMAscan™ for future proteomic applications due its highly competitive properties and automatable workflow.211,212
DNA microarrays are an indispensable tool for probing the complexity of biological systems since their inception in the 1990s.213 The capability to decipher and individually probe specific targets in highly complex biological matrices in a simple yet massively high-throughput manner is crucial for the advancement of genomics and transcriptomics.214,215 Unfortunately, protein microarrays, on the other hand, have lagged behind, but not for a lack of trying. The most effective technique for proteomics thus far has been mass spectrometry. Even so, complex approaches of tandem mass spectrometry, such as selected reaction monitoring,216 sequential window acquisition of all theoretical fragment-ion spectra,217 and other labelling strategies218,219 have to be incorporated into mass spectrometry in order to be able to make sense of the complex data generated. Being able to apply the microarray technology to proteomics would tremendously aid the deconvolution process. XNA aptamers currently present a highly feasible transduction interface between the two. As previously mentioned, SOMAscan™ technology does this by having the non-binding ends of the SOMAmers code for specific sequences, which can be printed onto microarrays and used for deconvolution.203 In the case of XNA microarrays, PNA tags have been used to essentially “barcode” members of split and mix peptide libraries.220,221 For example, Diaz- Mochón et al. created PNA tagged protease substrate libraries to develop a rapid and high-throughput method for studying protease consensus sequences.221 While XNAs are well-positioned to act as a much-needed bioaffinitive interface to a microarray for proteomic applications, they are also very capable of being refashioned to be used as transducers between complex combinatorial libraries and microarrays. Going along the same train of thought, the same strategy could be expanded for studying other important enzymatic processes. One such example reported by Diaz-Mochón et al. was for identifying Abl protein kinase substrate specificity.221 It is conceivable then to further use a similar strategy for better understanding the consensus sequences that signal for other post-translational modifications.
3.6. XNAs in biocatalysis
XNAs have also shown a great promise in biocatalysis. A good example is the creation of catalytic XNAs – XNAzymes that ligate and cleave RNA.222 Several interesting RNase-like XNAzymes selected through a bimolecular approach efficiently cleave RNA strands (Fig. 17). Being the most promising XNAzyme, F2R17 was further examined. It was observed that the RNase-like activity with reasonable regioselectivity is still observable even F2R17 is reduced to only 39 nucleotides. As compared to the uncatalysed reaction, the truncated F2R17 showed a 104-fold increment in the reaction rate. After structure fine-turning, it was further demonstrated that the XNAzymes also exhibit ligase-like activity. Experimental evidence indicates that only in the presence of XNAzymes will the ligation of RNA take place. Thus, this represents an exciting field that has a great potential to be further developed into new enzyme mimetics that can catalyze unconventional reactions.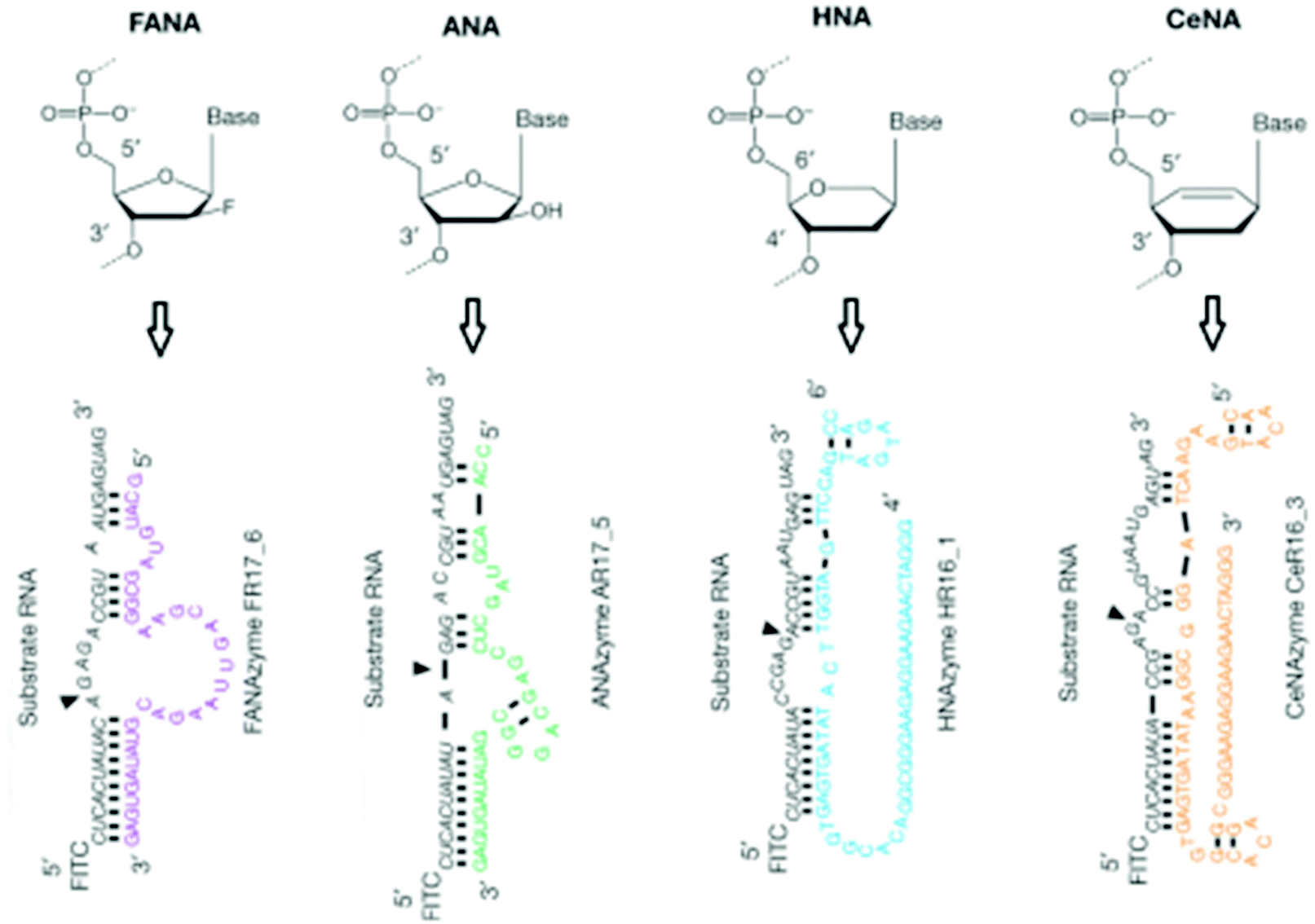 | ||
| Fig. 17 Examples of XNAzymes that have RNA cleavage and ligation properties (reproduced with permission from ref. 222, Copyright © 2015, Nature Publishing Group). | ||
Limitations of the biocatalytic XNAs that are urgently needed to be addressed are their low fidelity and the limited availability of XNA polymerases and modified nucleotides, thus inferring that they make selection processes less stringent, under-sampling of the library, and tedious comparisons between XNAs and nucleic acid sequence spaces. As the fidelity of the XNA system is normally a few orders of magnitude lower than that of nucleic acid systems, it is therefore much more challenging to identify XNA sequences encoding the best catalytic moieties.
4. Conclusions and outlooks
Essentially all XNAs are currently synthesized by solid-phase chemistry that is largely similar or, in cases where the XNAs have phosphate-sugar backbones, the same as the phosphoramidite chemistry used to chemically synthesize nucleic acids. Solid-phase synthesis of nucleic acids is highly efficient with a stepwise yield of >99% routinely achieved.223 However, the yield of oligonucleotide products decreases exponentially with increasing chain length. To illustrate, with a moderate coupling efficiency of 99.5%, the yield after 100 cycles is only 60%. As in the case of nucleic acid synthesis, the yield of XNAs also decreases exponentially with increasing chain length. In some applications where long XNA strands are involved, they can be fettered by the current limitation of synthetic chemistry. In an attempt to overcome this limitation, DNA-templated chemical generation of XNAs has recently been reported,224 but the length and fidelity leave much to be desired for the above applications. It is therefore necessary to turn to Nature's chemists – enzymes – for accurately making long XNAs. However, the mechanisms by which most polymerases implement high fidelity and specificity also prevent the incorporation of chemically modified nucleotides. Hence, every new modification must be laboriously tested with a plethora of natural and engineered polymerases. In other words, conventional biochemistry cannot keep up with the diversity easily generated by chemical synthesis. Additionally, polymerases are cornerstones of most nucleotide sequencing techniques. Current XNA sequencing techniques may give abnormal results225 or involve converting the XNA sequence to DNA, thereby encompassing additional steps.226 More reliable and expeditious sequencing techniques would be immensely beneficial for the deconvolution of XNAs, especially in evolution experiments where large XNA libraries are continuously generated. Indeed, the main advantage of using DNA or RNA over XNAs in SELEX is the wide selection of polymerases available, and the cost of such polymerases is typically the limiting factor. An obvious solution to this limitation would be to evolve new polymerases that would be capable of incorporating these unnatural nucleotides. While several commercially available polymerases are able to incorporate unnatural nucleotides, the processivity and fidelity are less than ideal.227 Nevertheless, these polymerases could serve as starting templates from which newer and more efficient XNA polymerases could be evolved or engineered. In the search for improved XNA polymerases, encouraging results can be found in the work of the Holliger47 and Romesberg groups.228Another way around the synthesis issue would perhaps be the use of XNA oligomers that exhibit ligase activities. It is conceivable to then develop DNAzymes or even self-catalytic XNAzymes to perform the polymerization of XNAs. These XNAzymes would also possibly be readily capable of being incorporated into already existing bioanalytical techniques, such as the ligase chain reaction, which has already been successfully demonstrated with DNAzymes.229–231 Having ligase activity, a bolder though not impossible development would be then to develop polymerases made from XNAs. The main advantages in using catalytic nucleic acids in methods requiring thermal cycling are the inherent thermal stability and capability of nucleic acids to refold and regenerate their desired properties. In contrast, renaturation and restoration of activity after heating are not possible for most enzymes. Promising results have recently been reported by Taylor et al.222 Additionally, XNAzymes with catalytic capabilities could provide an efficient means to amplify analytical signals in bioassays. Extending this idea to XNA nanotechnology and nanostructures, it would be interesting to be able to engineer XNA self-assemblies to produce XNA nanostructures capable of coordinating analytes for the construction of highly sensitive bioassays. Moreover, XNA aptamers have a great potential to be developed into high affinity ligands, it is not too much of a stretch to envision developing toolkits for molecular biology and molecular therapy.
For the most part in this review article, we have highlighted numerous methods that have been established for creative and apt use of XNAs. It is, however, unfortunate that there is a clear lack of new applications for XNAs in purification techniques. While XNA aptamers have a great potential for developing high affinity reagents, their use has not translated into purification technologies. There have been few attempts to develop methodologies for the use of aptamers in purification techniques with limited success. One such example can be found in a report by Javaherian et al., in which a method for developing DNA aptamers against protein targets in cell lysate and using the raised aptamers for purification was described.232 Their technique would be akin to attempting to raise polyclonal antibodies for protein purification, albeit at a faster and cheaper pace with the SELEX principle at its heart. As promising as this technique is, the purification process is still relatively extensive and tedious, but perhaps feasible if coupled to an automation system using XNA aptamers in a manner similar to the SOMALogic methodology.212 To speculate further, should there eventually be a technique for the convenient synthesis of long XNA sequences, it would be plausible to develop XNA purification “gels”. In 2012, Zhao et al. applied RCA to produce long DNA strands of repeating aptamer units specific to protein tyrosine kinases to create a “gel” that is capable of capturing cancer cells at a higher efficiency than with single fixed aptamers or antibodies.233 While the capture target for their purpose were cells, it is not too much of a stretch to envision developing a gel-like material that can be packed onto a column specifically targeting small molecules or other biological macromolecules. Similarly, with enzymatic synthesis, long strands of XNAs or metal ion-incorporated XNAs could be structured using DNA origami methods to fashion a new generation of metal–organic frameworks potentially applicable as chromatographic stationary phases.234
While Nature already provides toolkits for detecting, purifying, and manipulating biological systems, sometimes these tools need to be improved using synthetic chemistry. Nucleic acids are highly amenable to chemical modifications due to their modularity and high coupling efficiency, thereby lowering the technical barrier in adopting XNA technology. Indeed, while XNAs were first developed by researchers seeking to emulate nature; some types of XNAs, such as LNA, PNA, and MO, have already been commercialized and used by biologists with little or no synthetic chemistry experience. On the other hand, XNAs containing man-made nucleobases were initially developed to better understand the physicochemical properties and biology of nucleic acids, but it was quickly realized that they can be tailor-made to overcome common shortcomings of nucleic acids in technical applications, such as in vivo stability and specificity. Although short to medium length XNAs are sufficient for many applications, problems in accurately and affordably synthesizing long XNAs may be the bottleneck in the applications of XNAs, such as XNAzymes and in nanotechnology. Nevertheless, there is intense ongoing research in developing more efficient polymerases for XNAs. With increasing relevance in molecular biology, proteomics, and diagnostics, among other fields, XNAs epitomize one of the key goals of synthetic biology, which is to engineer biology to overcome problems and rise up to greater challenges.
Acknowledgements
Financial support of this work by the Ministry of Education under the grant MOE-2014-T2-081 is gratefully acknowledged.References
- J. D. Watson and F. H. Crick, Nature, 1953, 171, 737–738 CrossRef CAS PubMed.
- S. A. Benner, Acc. Chem. Res., 2004, 37, 784–797 CrossRef CAS PubMed.
- M. D. Matteucci and M. H. Caruthers, J. Am. Chem. Soc., 1981, 103, 3185–3191 CrossRef CAS.
- P. Herdewijn and P. Marlière, Chem. Biodiversity, 2009, 6, 791–808 CAS.
- R. Johnson, Nat. Chem., 2015, 7, 94 CrossRef CAS.
- J. C. Chaput, H. Yu and S. Zhang, Chem. Biol., 2012, 19, 1360–1371 CrossRef CAS PubMed.
- F. H. Crick, Nature, 1970, 277, 561–563 CrossRef.
- G. F. Joyce, PLoS Biol., 2012, 10, e1001323 CAS.
- F. R. Steele and L. Gold, Nat. Biotechnol., 2012, 30, 624–625 CrossRef CAS PubMed.
- I. Hirao, M. Kimoto and R. Yamashige, Acc. Chem. Res., 2012, 45, 2055–2065 CrossRef CAS PubMed.
- D. A. Malyshev, K. Dhami, T. Lavergne, T. Chen, N. Dai, J. M. Foster, I. R. Corrêa and F. E. Romesberg, Nature, 2014, 509, 385–388 CrossRef CAS PubMed.
- A. T. Krueger and E. T. Kool, Chem. Biol., 2009, 16, 242–248 CrossRef CAS PubMed.
- J. A. Piccirilli, S. A. Benner, T. Krauch, S. E. Moroney and S. A. Benner, Nature, 1990, 343, 33–37 CrossRef CAS PubMed.
- C. Switzer, S. E. Moroney and S. A. Benner, J. Am. Chem. Soc., 1989, 111, 8322–8323 CrossRef CAS.
- C. Y. Switzer, S. E. Moroney and S. A. Benner, Biochemistry, 1993, 32, 10489–10496 CrossRef CAS PubMed.
- T. A. Martinot and S. A. Benner, J. Org. Chem., 2004, 69, 3972–3975 CrossRef CAS PubMed.
- Y. Tor and P. B. Dervan, J. Am. Chem. Soc., 1993, 115, 4461–4467 CrossRef CAS.
- A. M. Sismour and S. A. Benner, Nucleic Acids Res., 2005, 33, 5640–5646 CrossRef CAS PubMed.
- Z. Yang, A. M. Sismour, P. Sheng, N. L. Puskar and S. A. Benner, Nucleic Acids Res., 2007, 35, 4238–4249 CrossRef CAS PubMed.
- S. Hoshika, F. Chen, N. A. Leal and S. A. Benner, Angew. Chem., Int. Ed., 2010, 49, 5554–5557 CrossRef CAS PubMed.
- J. C. Morales and E. T. Kool, Nat. Struct. Mol. Biol., 1998, 5, 950–954 CAS.
- D. L. McMinn, A. K. Ogawa, Y. Wu, J. Liu, P. G. Schultz and F. E. Romesberg, J. Am. Chem. Soc., 1999, 121, 11585–11586 CrossRef CAS.
- G. T. Hwang, Y. Hari and F. E. Romesberg, Nucleic Acids Res., 2009, 37, 4757–4763 CrossRef CAS PubMed.
- T. Mitsui, A. Kitamura, M. Kimoto, T. To, A. Sato, I. Hirao and S. Yokoyama, J. Am. Chem. Soc., 2003, 125, 5298–5307 CrossRef CAS PubMed.
- A. M. Leconte, G. T. Hwang, S. Matsuda, P. Capek, Y. Hari and F. E. Romesberg, J. Am. Chem. Soc., 2008, 130, 2336–2343 CrossRef CAS PubMed.
- D. A. Malyshev, D. A. Pfaff, S. I. Ippoliti, G. T. Hwang, T. J. Dwyer and F. E. Romesberg, Chem. – Eur. J., 2010, 16, 12650–12659 CrossRef CAS PubMed.
- K. Betz, D. A. Malyshev, T. Lavergne, W. Welte, K. Diederichs, T. J. Dwyer, P. Ordoukhanian, F. E. Romesberg and A. Marx, Nat. Chem. Biol., 2012, 8, 612–614 CrossRef CAS PubMed.
- M. Kimoto, R. Kawai, T. Mitsui, S. Yokoyama and I. Hirao, Nucleic Acids Res., 2009, 37, e14 CrossRef PubMed.
- R. Yamashige, M. Kimoto, Y. Takezawa, A. Sato, T. Mitsui, S. Yokoyama and I. Hirao, Nucleic Acids Res., 2012, 40, 2793–2806 CrossRef CAS PubMed.
- K. Vastmans, M. Froeyen, L. Kerremans, S. Pochet and P. Herdewijn, Nucleic Acids Res., 2001, 29, 3154–3163 CrossRef CAS PubMed.
- J. K. Ichida, A. Horhota, K. Zou, L. W. McLaughlin and J. W. Szostak, Nucleic Acids Res., 2005, 33, 5219–5125 CrossRef CAS PubMed.
- P. Gu, G. Schepers, J. Rozenski, A. van Aerschot and P. Herdewijn, Oligonucleotides, 2003, 13, 479–489 CrossRef CAS PubMed.
- K. Sefaha, Z. Yang, K. M. Bradley, S. Hoshika, E. Jiménez, L. Zhang, G. Zhu, S. Shanker, F. Yu, D. Turek, W. Tan and S. A. Benner, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 1449–1454 CrossRef PubMed.
- A. M. Sismour, S. Lutz, J. H. Park, M. J. Lutz, P. L. Boyer, S. H. Hughes and S. A. Benner, Nucleic Acids Res., 2004, 32, 728–735 CrossRef CAS PubMed.
- P. Marliere, Syst. Synth. Biol., 2009, 3, 77–84 CrossRef PubMed.
- M. Schmidt, BioEssays, 2010, 32, 322–331 CrossRef CAS PubMed.
- A. Eschenmoser, Science, 1999, 284, 2118–2124 CrossRef CAS PubMed.
- F. Eckstein, Annu. Rev. Biochem., 1985, 54, 367–402 CrossRef CAS PubMed.
- K. W. Porter, J. D. Briley and B. R. Shaw, Nucleic Acids Res., 1997, 25, 1611–1617 CrossRef CAS PubMed.
- Y. Xu and E. T. Kool, Nucleic Acids Res., 1998, 26, 3159–3164 CrossRef CAS PubMed.
- N.-S. Li, J. K. Frederiksen and J. A. Piccirilli, Acc. Chem. Res., 2011, 44, 1257–1269 CrossRef CAS PubMed.
- F. J. Ghadessy, N. Ramsay, F. Boudsocq, D. Loakes, A. Brown, S. Iwai, A. Vaisman, R. Woodgate and P. Holliger, Nat. Biotechnol., 2004, 22, 755–759 CrossRef CAS PubMed.
- K. Schöning, P. Scholz, S. Guntha, X. Wu, R. Krishnamurthy and A. Eschenmoser, Science, 2000, 290, 1347–1351 CrossRef.
- H. Yu, S. Zhang and J. C. Chaput, Nat. Chem., 2012, 4, 183–187 CrossRef CAS PubMed.
- J. D. Vaught, C. Bock, J. Carter, T. Fitzwater, M. Otis, D. Schneider, J. Rolando, S. Waugh, S. K. Wilcox and B. E. Eato, J. Am. Chem. Soc., 2010, 132, 4141–4151 CrossRef CAS PubMed.
- C. Hendrix, H. Rosemeyer, B. D. Bouvere, A. V. Aerschot, F. Seela and P. Herdewijn, Chem. – Eur. J., 1997, 3, 1513–1520 CrossRef CAS.
- V. B. Pinheiro, A. I. Taylor, C. Cozens, M. Abramov, M. Renders, S. Zhang, J. C. Chaput, J. Wengel, S. Y. P. Chew, S. H. McLaughlin, P. Herdewijn and P. Holliger, Science, 2012, 336, 341–344 CrossRef CAS PubMed.
- G. C. K. Roberts, E. A. Dennis, D. H. Meadows, J. S. Cohen and O. Jardetzky, Proc. Natl. Acad. Sci. U. S. A., 1969, 62, 1151–1158 CrossRef CAS.
- P. E. Nielsen, M. Egholm, R. H. Berg and O. Buchardt, Science, 1991, 254, 1497–1500 CAS.
- P. E. Nielsen, G. Haaima, A. Lohse and O. Buchardt, Angew. Chem., Int. Ed. Engl., 1996, 35, 1939–1942 CrossRef.
- P. Zhou, A. D. Andrasi, B. Bhattacharya, H. O'Keefe, P. Vatta, J. J. H. Nielsen and D. H. Ly, Bioorg. Med. Chem. Lett., 2006, 16, 4931–4935 CrossRef CAS PubMed.
- Y. Ura, J. M. Beierle, L. J. Leman, L. E. Orgel and M. R. Ghadiri, Science, 2009, 325, 73–77 CrossRef CAS PubMed.
- A. Sen and P. E. Nielsen, Nucleic Acids Res., 2007, 35, 3367–3374 CrossRef CAS PubMed.
- A. K. L. Teo, C. L. Lim and Z. Gao, Electrochim. Acta, 2014, 126, 19–30 CrossRef CAS.
- B. Yevgeny and D. R. Liu, Chem. Biodiversity, 2009, 16, 265–276 Search PubMed.
- P. E. Nielsen, Chem. Biodiversity, 2010, 7, 786–804 CAS.
- J. Summerton and D. Weller, Antisense Nucleic Acid Drug Dev., 2009, 7, 187–195 CrossRef PubMed.
- H. Kang, P. J. Chou, W. C. Johnson, D. Weller, S. B. Huang and J. E. Summerton, Biopolymers, 1992, 32, 1351–1363 CrossRef CAS PubMed.
- R. M. Hudziak, E. Barofsky, D. F. Barofsky, D. L. Weller, S. Huang and D. D. Weller, Antisense Nucleic Acid Drug Dev., 2009, 6, 267–272 CrossRef PubMed.
- M. T. Howard, R. F. Gesteland and J. F. Atkins, RNA, 2004, 10, 1653–1661 CrossRef CAS PubMed.
- Y. Xu and H. Sugiyama, Nucleic Acids Res., 2004, 34, 949–954 CrossRef PubMed.
- T. Kimura, K. Kawai, M. Fujitsuka and T. Majima, Tetrahedron, 2007, 63, 3585–3590 CrossRef CAS.
- B. Datta and B. A. Armitage, J. Am. Chem. Soc., 2001, 123, 9612–9619 CrossRef CAS PubMed.
- A. Paul, P. Sengupta, Y. Krishnan and S. Ladame, Chemistry, 2008, 14, 8682–8689 CrossRef CAS PubMed.
- I. G. Panyutin, M. I. Onyshchenko, E. A. Englund, D. H. Appella and R. D. Neumann, Curr. Pharm. Des., 2012, 18, 1984–1991 CrossRef CAS PubMed.
- S. Kishore and S. Stamm, Science, 2006, 311, 230–232 CrossRef CAS PubMed.
- S. R. Eddy, Nat. Rev. Genet., 2001, 2, 919–929 CrossRef CAS PubMed.
- G. Wang, G. V. Bobkov, S. N. Mikhailov, G. Schepers, A. V. Aerschot, J. Rozenski, M. V. Auweraer, P. Herdewijn and S. D. Feyter, Nat. Rev. Genet., 2001, 10, 1175–1185 Search PubMed.
- G. Devi, Z. Yuan, Y. Lu, Y. Zhao and G. Chen, Nucleic Acids Res., 2014, 42, 4008–4018 CrossRef CAS PubMed.
- A. S. A. Almakarem, A. I. Petrov, J. Stombaugh, C. L. Zirbel and N. B. Leontis, Nucleic Acids Res., 2011, 40, 1407–1423 CrossRef PubMed.
- J. Summerton, in Discoveries in antisense nucleic acids, ed. C. Brakel, Portfolio Publishing, Woodlands, TX, 1989, vol. 2, pp. 71–80 Search PubMed.
- S. C. Ekker and J. D. Larson, Genesis, 2001, 30, 89–93 CrossRef CAS PubMed.
- A. Y. L. Sim, P. Minary and M. Levitt, Curr. Opin. Struct. Biol., 2012, 22, 273–278 CrossRef CAS PubMed.
- A. Nasevicius and S. C. Ekker, Nat. Genet., 2000, 26, 216–220 CrossRef CAS PubMed.
- W. P. Kloosterman, A. K. Lagendijk, R. F. Ketting, J. D. Moulton and R. H. A. Plasterk, PLoS Biol., 2007, 5, e203 Search PubMed.
- J. Zielinski, K. Kilk, T. Peritz, T. Kannanayakal, K. Y. Miyashiro, E. Eiríksdóttir, J. Jochems, Û. Langel and J. Eberwine, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 1557–1562 CrossRef CAS PubMed.
- G. Meister and T. Tusch, Nature, 2004, 431, 343–349 CrossRef CAS PubMed.
- A. R. Hernández, L. W. Peterson and E. T. Kool, ACS Chem. Biol., 2012, 7, 1454–1461 CrossRef PubMed.
- Y. Wang, S. Juranek, H. Li, G. Sheng, G. S. Wardle, T. Tuschl and D. J. Patel, Nature, 2009, 461, 754–761 CrossRef CAS PubMed.
- C. C. Chou, C. H. Chen, T. T. Lee and K. Peck, Nucleic Acids Res., 2004, 32, e99 CrossRef PubMed.
- S. Tyagi, S. A. E. Marras, J. A. M. Vet and F. R. Kramer, Nat. Biotechnol., 1996, 14, 303–308 CrossRef CAS PubMed.
- R. A. Cardullo, S. Agrawal, C. Flores, P. C. Zamecnik and D. E. Wolf, Proc. Natl. Acad. Sci. U. S. A., 1988, 85, 8790–8794 CrossRef CAS.
- O. Köhlera and O. Seitz, Chem. Commun., 2003, 2938–2939 RSC.
- P. R. L. Safer, M. Levine and D. C. Ward, Proc. Natl. Acad. Sci. U. S. A., 1982, 79, 4381–4385 CrossRef.
- N. Ota1, K. Hirano, M. Warashina, A. Andrus, B. Mullah, K. Hatanaka and K. Taira, Nucleic Acids Res., 1998, 26, 735–743 CrossRef.
- L. Wang, C. J. Yang, C. D. Medley, S. A. Benner and W. Tan, J. Am. Chem. Soc., 2005, 127, 15664–15665 CrossRef CAS PubMed.
- S. K. Singh and J. Wengel, Chem. Commun., 1998, 1247–1248 RSC.
- A. A. Koshkin, P. Nielsen, M. Meldgaard, V. K. Rajwanshi, S. K. Singh and J. Wengel, J. Am. Chem. Soc., 1998, 120, 13252–13253 CrossRef CAS.
- A. A. Koshkin, S. K. Singh, P. Nielsen, V. K. Rajwanshi, R. Kumar, M. Meldgaard, C. E. Olsen and J. Wengel, Tetrahedron, 1998, 54, 3607–3630 CrossRef CAS.
- P. J. Hrdlicka, B. R. Babu, M. D. Sørensen, N. Harrit and J. Wengel, J. Am. Chem. Soc., 2005, 127, 13293–13299 CrossRef CAS PubMed.
- L. Morandi, D. Biase, M. Visani, V. Cesari, G. D. Maglio, S. Pizzolitto, A. Pession and G. Tallini, PLoS One, 2012, 7, e36084 CAS.
- L. Morandi, D. Ferrari, C. Lombardo, A. Pession and G. Tallini, J. Virol. Methods, 2007, 140, 148–154 CrossRef CAS PubMed.
- M. P. Johnson, L. M. Haupt and L. R. Griffiths, Nucleic Acids Res., 2004, 32, e55 CrossRef PubMed.
- P. Sidon, P. Heimann, F. Lambert, B. Dessars, V. Robin and H. E. Housni, Clin. Chem., 2006, 52, 1436–1438 CAS.
- S. Mishra, S. Ghosh and R. Mukhopadhyay, Langmuir, 2012, 28, 4325–4333 CrossRef CAS PubMed.
- W. Han, J. Liao, K. Chen, S. Wu, Y. Chiang, S. G. Lo, C. Chen and C. Chiang, Anal. Chem., 2010, 82, 2395–2400 CrossRef CAS PubMed.
- J. Chen, J. Zhang, K. Wang, X. Lin, L. Huang and G. Chen, Anal. Chem., 2008, 80, 8028–8034 CrossRef CAS PubMed.
- K. Martinez, M. Estevez, Y. Wu, J. A. Phillips, C. D. Medley and W. Tan, Anal. Chem., 2009, 81, 3448–3454 CrossRef CAS PubMed.
- Y. Zhao, G. Li, C. Sun, C. Li, X. Wang, H. Liu, P. Zhang, X. Zhao, X. Wang, Y. Jiang, R. Yang, K. Wan and L. Zhou, PLoS One, 2015, 10, e0143444 Search PubMed.
- M. H. Josefsen, C. Löfström, H. M. Sommer and J. Hoorfar, Mole. Cell. Probes, 2009, 23, 201–203 CrossRef CAS PubMed.
- S. Salvi, F. D. Orso and G. Morelli, J. Agric. Food Chem., 2008, 56, 4320–4327 CrossRef CAS PubMed.
- M. B. Gašparič, K. Cankar, J. Žel and K. Gruden, BMC Biotechnol., 2008, 8, 1–12 CrossRef PubMed.
- C. C. Desbiolles, D. R. Ahn and C. J. Leumann, Nucleic Acids Res., 2005, 33, e77 CrossRef PubMed.
- M. Egli, P. S. Pallan, R. Pattanayek, C. J. Wilds, P. Lubini, G. Minasov, M. Dobler, C. J. Leumann and A. Eschenmoser, J. Am. Chem. Soc., 2006, 128, 10841–10856 CrossRef PubMed.
- Y. Kim, C. J. Yang and W. Tan, Nucleic Acids Res., 2007, 35, 7279–7287 CrossRef CAS PubMed.
- D. R. Yazbeck, K. L. Min and M. J. Damha, Nucleic Acids Res., 2002, 30, 3015–3025 CrossRef CAS PubMed.
- M. Nilsson, M. Gullberg, F. Dahl, K. Szuhai and A. K. Raap, Nucleic Acids Res., 2002, 30, e66 CrossRef PubMed.
- P. M. Lizardi, X. Huang, Z. Zhu, P. B. Ward, D. C. Thomas and D. C. Ward, Nat. Genet., 1998, 19, 225–232 CrossRef CAS PubMed.
- M. M. Ali, F. Li, Z. Zhang, K. Zhang, D. Kang, J. A. Ankrum, X. C. Leb and W. Zhao, Chem. Soc. Rev., 2014, 43, 3324–3341 RSC.
- L. Blanco, A. Bernad, J. M. Lázaro, G. Martín, C. Garmendia and M. Salas, J. Biol. Chem., 1989, 264, 8935–8940 CAS.
- M. Vegaa, J. M. Lázaro, M. Salas and L. Blanco, J. Mol. Biol., 1998, 279, 807–822 CrossRef.
- D. M. Kolpashchikov, J. Am. Chem. Soc., 2006, 128, 10625–10628 CrossRef CAS PubMed.
- D. M. Kolpashchikov, Chem. Rev., 2010, 110, 4709–4723 CrossRef CAS PubMed.
- A. Karadag, M. Riminucci, P. Bianco, N. Cherman, S. A. Kuznetsov, N. Nguyen, M. T. Collins, P. G. Robey and L. W. Fisher, Nucleic Acids Res., 2004, 32, e63 CrossRef PubMed.
- J. D. Luo, E. C. Chan, C. L. Shih, T. L. Chen, Y. Liang, T. L. Hwang and C. C. Chiou, Nucleic Acids Res., 2006, 34, e12 CrossRef PubMed.
- Z. Wang, K. Zhang, Y. Shen, J. Smith, S. Bloch, S. Achilefu, K. L. Wooley and J. S. Taylor, Org. Biomol. Chem., 2013, 11, 3159–3167 CAS.
- E. M. Kyger, M. D. Krevolin and M. J. Powell, Anal. Biochem., 1998, 260, 142–148 CrossRef CAS PubMed.
- A. N. Glazer and H. S. Rye, Nature, 1992, 359, 859–861 CrossRef CAS PubMed.
- D. Loakes, Nucleic Acids Res., 2001, 29, 2437–2447 CrossRef CAS PubMed.
- O. Köhler, D. V. Jarikote and O. Seitz, ChemBioChem, 2005, 6, 69–77 CrossRef PubMed.
- E. Socher and O. Seitz, Methods Mol. Biol., 2008, 429, 187–197 CAS.
- E. Socher, D. V. Jarikote, A. Knoll, L. Röglin, J. Burmeister and O. Seitz, Anal. Biochem., 2008, 375, 318–330 CrossRef CAS PubMed.
- L. Bethge, D. V. Jarikote and O. Seitz, Bioorg. Med. Chem., 2008, 16, 114–125 CrossRef CAS PubMed.
- J. M. Levsky and R. H. Singer, J. Cell Sci., 2003, 116, 2833–2838 CrossRef CAS PubMed.
- S. Rigby, G. W. Procop, G. Haase, D. Wilson, G. Hall, C. Kurtzman, K. Oliveira, S. V. Oy, J. J. H. Nielsen, J. Coull and H. Stender, J. Clin. Microbiol., 2002, 40, 2182–2186 CrossRef CAS PubMed.
- F. Pellestor, P. Paulasova, M. Macek and S. Hamamah, J. Histochem. Cytochem., 2005, 53, 395–400 CrossRef CAS PubMed.
- A. M. Blanco and R. A. Artero, Methods, 2010, 52, 343–351 CrossRef CAS PubMed.
- V. Uhlmann, M. Prasad, I. Silva, K. Luettich, L. Grande, L. Alonso, M. Thisted, K. J. Pluzek, J. Gorst, M. Ring, M. Sweeney, C. Kenny, C. Martin, J. Russell, N. Bermingham, M. O'Donovan, O. Sheils and J. J. O'Leary, Mol. Pathol., 2000, 53, 48–50 CrossRef CAS PubMed.
- P. Paulasova, B. Andréo, J. Diblik, M. Macek and F. Pellestor, Mol. Hum. Reprod., 2004, 10, 467–472 CrossRef CAS PubMed.
- I. E. Agerholm, S. Ziebe, B. Williams, C. Berg, D. G. Crüger, G. Bruun Petersen and S. Kølvraa, Hum. Reprod., 2005, 20, 1072–1077 CrossRef CAS PubMed.
- M. Shinozaki, Y. Okubo, D. Sasai, H. Nakayama, S. Y. Murayama, T. Ide, M. Wakayama, T. Ishiwatari, N. Tochigi, T. Nemoto and K. Shibuya, J. Clin. Microbiol., 2013, 51, 1295–1298 CrossRef PubMed.
- F. Bonvicini, C. Filippone, E. Manaresi, G. A. Gentilomi, M. Zerbini, M. Musiani and G. Gallinella, Clin. Chem., 2006, 52, 6973–6978 Search PubMed.
- M. Shinozaki, Y. Okubo, D. Sasai, H. Nakayama, S. Y. Murayama, T. Ide, M. Wakayama, N. Hiruta and K. Shibuya, J. Clin. Microbiol., 2011, 49, 3808–3813 CrossRef PubMed.
- R. B. Mutalik and H. Nikaido, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 4958–4963 CrossRef PubMed.
- H. Stender, K. Lund, K. H. Petersen, O. F. Rasmussen, P. Hongmanee, H. Miörner and S. E. Godtfredsen, J. Clin. Microbiol., 1999, 37, 2760–2765 CAS.
- A. K. Lagendijk, J. D. Moulton and J. Bakkers, Biol Open., 2012, 15, 566–569 CrossRef PubMed.
- M. Egholm, O. Buchardt, P. E. Nielsen and R. H. Berg, J. Am. Chem. Soc., 1992, 114, 1895–1897 CrossRef CAS.
- R. H. E. Hudson, Y. Liu and F. Wojciechowski, Can. J. Chem., 2007, 85, 302–312 CrossRef.
- G. Bao, W. J. Rhee and A. Tsourkas, Annu. Rev. Biomed. Eng., 2009, 11, 25–47 CrossRef CAS PubMed.
- R. M. Franzini and E. T. Kool, Bioconjugate Chem., 2011, 22, 1869–1877 CrossRef CAS PubMed.
- R. M. Franzini and E. T. Kool, J. Am. Chem. Soc., 2009, 131, 16021–16023 CrossRef CAS PubMed.
- Z. Gao, H. Deng, W. Shen and Y. Ren, Anal. Chem., 2013, 85, 1624–1630 CrossRef CAS PubMed.
- H. Deng, W. Shen, Y. Ren and Z. Gao, Biosens. Bioelectron., 2014, 60, 195–200 CrossRef CAS PubMed.
- Y. Imaizumi, Y. Kasahara, H. Fujita, S. Kitadume, H. Ozaki, T. Endoh, M. Kuwahara and N. Sugimoto, J. Am. Chem. Soc., 2013, 135, 9412–9419 CrossRef CAS PubMed.
- J. N. Wilson, Y. N. Teo and E. T. Kool, J. Am. Chem. Soc., 2007, 129, 15426–15427 CrossRef CAS PubMed.
- F. Samain, N. Dai and E. T. Kool, Chem. – Eur. J., 2011, 17, 174–183 CrossRef CAS PubMed.
- H. Kwon, W. Jiang and E. T. Kool, Chem. Sci., 2015, 6, 2575–2583 RSC.
- F. Samain, N. Dai and E. T. Kool, Chem. – Eur. J., 2011, 17, 174–183 CrossRef CAS PubMed.
- S. S. Tan, S. J. Kim and E. T. Kool, J. Am. Chem. Soc., 2011, 133, 2664–2671 CrossRef CAS PubMed.
- L. H. Yuen, R. M. Franzini, S. S. Tan and E. T. Kool, J. Am. Chem. Soc., 2014, 136, 14576–14582 CrossRef CAS PubMed.
- H. Kwon, F. Samaina and E. T. Kool, Chem. Sci., 2012, 3, 2542–2549 RSC.
- X. Zhang, C. Yu, X. Huang, J. Zheng, X. Guan, D. Luo and L. Li, Electrochim. Acta, 2012, 81, 233–238 CrossRef CAS.
- A. I. Taylor, S. A. Franklin and P. Holliger, Curr. Opin. Chem. Biol., 2014, 22, 79–84 CrossRef CAS PubMed.
- Q. Wang, L. Chen, Y. Long, H. Tian and J. Wu, Theranostics, 2013, 3, 395–408 CrossRef CAS PubMed.
- M. Castoldi, S. Schmidt, V. Benes, M. Noerholm, A. E. Kulozik, M. W. Hentze and M. U. Muckenthaler, RNA, 2006, 12, 913–920 CrossRef CAS PubMed.
- J. Chen, J. Zhang, K. Wang, X. Lin, L. Huang and G. Chen, Anal. Chem., 2008, 80, 8028–8034 CrossRef CAS PubMed.
- A. N. Elayadi, D. A. Braasch and D. R. Corey, Biochemistry, 2002, 41, 9973–9981 CrossRef CAS PubMed.
- P. S. Ng and D. E. Bergstrom, Nano Lett., 2005, 5, 107–111 CrossRef CAS PubMed.
- D. A. Braasch and D. R. Corey, Chem. Biol., 2001, 8, 1–7 CrossRef CAS PubMed.
- B. Vester and J. Wengel, Biochemistry, 2004, 43, 13233–13241 CrossRef CAS PubMed.
- P. E. Nielsen, M. Egholm, R. Berg and O. Buchardt, Science, 1991, 254, 1497–1500 CAS.
- B. Hyrup and P. E. Nielsen, Bioorg. Med. Chem., 1996, 4, 5–23 CrossRef CAS PubMed.
- X. Su, H. F. Teh, K. M. Aung, Y. Zong and Z. Gao, Biosens. Bioelectron., 2008, 15, 1715–1720 CrossRef PubMed.
- Y. Fan, X. Chen, A. D. Trigg, C. Tung, J. Kong and Z. Gao, J. Am. Chem. Soc., 2007, 129, 5437–5443 CrossRef CAS PubMed.
- X. Su, H. F. Teh, X. Lieu and Z. Gao, Anal. Chem., 2007, 79, 7192–7197 CrossRef CAS PubMed.
- P. Wittung, S. K. Kim, O. Buchardt, P. Nielsen and B. Norden, Nucleic Acids Res., 1994, 22, 5371–5377 CrossRef CAS PubMed.
- Z. Gao and B. P. Ting, Analyst, 2009, 134, 952–957 RSC.
- Y. Zu, A. L. Ting, G. Yi and Z. Gao, Anal. Chem., 2011, 83, 4090–4094 CrossRef CAS PubMed.
- Z. Gao, H. Deng, W. Shen and Y. Ren, Anal. Chem., 2013, 85, 1624–1630 CrossRef CAS PubMed.
- O. Brandt and J. D. Hoheisel, Trends Biotechnol., 2004, 22, 617–622 CrossRef CAS PubMed.
- K. Shantanu and B. Deepak, Appl. Microbiol. Biotechnol., 2006, 71, 575–586 CrossRef PubMed.
- V. J. Ruigrok, M. Levisson, M. H. Eppink, H. Smidt and V. D. Oost, Biochem. J., 2011, 436, 1–13 CrossRef CAS PubMed.
- C. Tuerk and L. Gold, Science, 1990, 249, 505–510 CAS.
- A. D. Ellington and J. W. Szostak, Nature, 1990, 346, 818–822 CrossRef CAS PubMed.
- Y. Nonaka, W. Yoshida, K. Abe, S. Ferri, H. Schulze, T. T. Bachmann and K. Ikebukuro, Anal. Chem., 2013, 85, 1132–1137 CrossRef CAS PubMed.
- C. Bell, E. Lynam, D. J. Landfair, N. Janjic and M. E. Wiles, In Vitro Cell. Dev. Biol.: Anim., 1999, 35, 533–542 CrossRef CAS PubMed.
- M. F. Kubik, A. W. Stephens, D. Schneider, R. A. Marlar and D. Tasset, Nucleic Acids Res., 1994, 22, 2619–2626 CrossRef CAS PubMed.
- I. Lebars, P. Legrand, A. Aime, N. Pinaud, S. Fribourg and C. D. Primo, Nucleic Acids Res., 2008, 36, 7146–7156 CrossRef CAS PubMed.
- B. A. Sullenger, H. F. Gallardo, G. E. Ungers and E. Gilboa, Cell, 1990, 63, 601–608 CrossRef CAS PubMed.
- L. S. Green, D. Jellinek, R. Jenison, A. Östman, C. H. Heldin and N. Janjic, Biochemistry, 1996, 35, 14413–14424 CrossRef CAS PubMed.
- D. B. Huang, D. Vu, L. A. Cassiday, J. M. Zimmerman, L. J. Maher and G. Ghosh, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 9268–9273 CrossRef CAS PubMed.
- A. B. Iliuk, L. Hu and W. A. Tao, Anal. Chem., 2011, 83, 4440–4452 CrossRef CAS PubMed.
- J. Zhou, M. R. Battig and Y. Wang, Anal. Bioanal. Chem., 2010, 398, 2471–2480 CrossRef CAS PubMed.
- D. Jellinek, L. S. Green, C. Bell, C. K. Lynott, N. Gill, C. Vargeese, G. Kirschenheuter, D. P. C. McGee and P. Abesinghe, Biochemistry, 1995, 34, 11363–11372 CrossRef CAS PubMed.
- Y. Lin, D. Nieuwlandt, A. Magallanez, B. Feistner and S. D. Jayasena, Nucleic Acids Res., 1996, 24, 3407–3414 CrossRef CAS PubMed.
- T. W. Wiegand, P. B. Williams, S. C. Dreskin, M. H. Jouvin, J. P. Kinet and D. Tasset, J. Immunol., 1996, 157, 221–230 CAS.
- E. W. M. Ng, D. T. Shima, P. Calias, E. T. Cunningham and R. Guyer, Nat. Rev. Drug Discovery, 2006, 5, 123–132 CrossRef CAS PubMed.
- N. Li, H. H. Nguyen, M. Byrom and A. D. Ellington, PLoS One, 2011, 6, e20299 CAS.
- M. Svobodova, D. H. J. Bunka, P. Nadal, P. G. Stockley and C. K. O'Sullivan, Anal. Bioanal. Chem., 2013, 405, 9149–9157 CrossRef CAS PubMed.
- S. E. Lupold, B. J. Hicke, Y. Lin and D. S. Coffey, Cancer Res., 2002, 62, 4029–4033 CAS.
- J. Zhou, P. Swiderski, H. Li, J. Zhang, C. P. Neff, R. Akkina and J. J. Rossi, Nucleic Acids Res., 2009, 37, 3094–3109 CrossRef CAS PubMed.
- E. K. Waters, R. M. Genga, M. C. Schwartz, J. A. Nelson, R. G. Schaub, K. A. Olson, J. C. Kurz and K. E. McGinness, Blood, 2011, 117, 5514–5522 CrossRef CAS PubMed.
- K. A. Davis, Y. Lin, B. Abrams and S. D. Jayasena, Nucleic Acids Res., 1998, 26, 3915–3924 CrossRef CAS PubMed.
- A. P. Mann, A. Somasunderam, R. N. Alicea, X. Li, A. Hu, A. K. Sood, M. Ferrari, D. G. Gorenstein and T. Tanaka, PLoS One, 2010, 5, e13050 Search PubMed.
- D. J. King, S. E. Bassett, X. Li, S. A. Fennewald, N. K. Herzog, B. A. Luxon, R. Shope and D. G. Gorenstein, Biochemistry, 2002, 41, 9696–9706 CrossRef CAS PubMed.
- A. Somasunderam, M. R. Ferguson, D. R. Rojo, V. Thiviyanathan, X. Li, W. A. O'Brien and D. G. Gorenstein, Biochemistry, 2005, 44, 10388–10395 CrossRef CAS PubMed.
- S. H. A. Gandham, D. E. Volk, G. L. R. Lokesh, M. Neerathilingam and D. G. Gorenstein, Biochem. Biophys. Res. Commun., 2014, 453, 309–315 CrossRef CAS PubMed.
- I. C. Elle, Mol. BioSyst., 2015, 11, 1260–1270 RSC.
- Y. Kasaharaa, Y. Irisawaa, H. Ozakia, S. Obikab and M. Kuwaharaa, Bioorg. Med. Chem. Lett., 2013, 23, 1288–1292 CrossRef PubMed.
- P. E. Burmeister, C. Wang, J. R. Killough, S. D. Lewis, L. R. Horwitz, A. Ferguson, K. M. Thompson, P. S. Pendergrast, T. G. McCauley, M. Kurz, J. Diener, S. T. Cload, C. Wilson and A. D. Keefe, Oligonucleotides, 2006, 16, 337–351 CrossRef CAS PubMed.
- M. Kimoto, R. Yamashige, K. Matsunaga, S. Yokoyama and I. Hirao, Nat. Biotechnol., 2013, 31, 453–457 CrossRef CAS PubMed.
- L. Gold, D. Ayers, J. Bertino, C. Bock, A. Bock, E. N. Brody, J. Carter, A. B. Dalby, B. E. Eaton, T. Fitzwater, D. Flather, A. Forbes, T. Foreman, C. Fowler, B. Gawande, M. Goss, M. Gunn, S. Gupta, D. Halladay, J. Heil, J. Heilig, B. Hicke, G. Husar, N. Janjic, T. Jarvis, S. Jennings, E. Katilius and D. Zichi, PLoS One, 2010, 5, e15004 CAS.
- L. Gold, J. J. Walker, S. K. Wilcox and S. Williams, Nat. Biotechnol., 2012, 29, 543–549 CAS.
- D. R. Davies, A. D. Gelinas, C. Zhang, J. C. Rohloff, J. D. Carter, D. O'Connell, S. M. Waugh, S. K. Wolk, W. S. Mayfield, A. B. Burgin, T. E. Edwards, L. J. Stewart, L. Gold, N. Janjic and T. C. Jarvis, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 19971–19976 CrossRef CAS PubMed.
- A. D. Gelinas, D. R. Davies, T. E. Edwards, J. C. Rohloff, J. D. Carter, C. Zhang, S. Gupta, Y. Ishikawa, M. Hirota, Y. Nakaishi, T. C. Jarvis and N. Janjic, J. Biol. Chem., 2014, 289, 8720–8734 CrossRef CAS PubMed.
- E. F. Pettersen, T. D. Goddard, C. C. Huang, G. S. Couch, D. M. Greenblatt, E. C. Meng and T. E. Ferrin, J. Comput. Chem., 2004, 25, 1605–1612 CrossRef CAS PubMed.
- J. C. Rohloff, A. D. Gelinas, T. C. Jarvis, U. A. Ochsner, D. J. Schneider, L. Gold and N. Janjic, Mol. Ther.—Nucleic Acids, 2014, 3, e201 CrossRef CAS PubMed.
- M. R. Mehan, R. Ostroff, S. K. Wilcox, F. Steele, D. Schneider, T. C. Jarvis, G. S. Baird, L. Gold and N. Janjic, in Complement Therapeutics, ed. J. D. Lambris, V. M. Holers and D. Ricklin, Springer, New York, 2013, vol. 735, pp. 283–300 Search PubMed.
- R. M. Ostroff, W. L. Bigbee, W. Franklin, L. Gold, M. Mehan, Y. E. Miller, H. I. Pass, W. N. Rom, J. M. Siegfried, A. Stewart, J. J. Walker, J. L. Weissfeld, S. Williams, D. Zichi and E. N. Brody, PLoS One, 2010, 5, e15003 CAS.
- B. Lollo, F. Steele and L. Gold, Proteomics, 2014, 14, 638–644 CrossRef CAS PubMed.
- J. Webber, T. C. Stone, E. Katilius, B. C. Smith, B. Gordon, M. D. Mason, Z. Tabi, I. A. Brewis and A. Clayton, Mol. Cell. Proteomics, 2014, 13, 1050–1064 CAS.
- T. R. Keeney, C. Bock, L. Gold, S. Kraemer, B. Lollo, M. Nikrad, M. Stanton, A. Stewart, J. D. Vaught and J. J. Walker, J. Lab. Autom., 2009, 14, 360–366 CrossRef CAS.
- E. M. Southern, DNA Arrays, 2001, 170, 1–15 CAS.
- K. M. Wassarman, F. Repoila, C. Rosenow, G. Storz and S. Gottesman, Genes Dev., 2001, 15, 1637–1651 CrossRef CAS PubMed.
- J. J. Diaz-Mochón, L. Bialy, L. Keinickea and M. Bradley, Chem. Commun., 2005, 1384–1386 RSC.
- P. Picotti and R. Aebersold, Nat. Methods, 2012, 9, 555–566 CrossRef CAS PubMed.
- X. Zhu, Y. Chen and R. Subramanian, Anal. Chem., 2014, 86, 1202–1209 CrossRef CAS PubMed.
- M. Bantscheff, M. Schirle, G. Sweetman, J. Rick and B. Kuster, Anal. Bioanal. Chem., 2007, 389, 1017–1031 CrossRef CAS PubMed.
- X. Han, A. Aslanian and J. R. Yates, Curr. Opin. Chem. Biol., 2008, 12, 483–490 CrossRef CAS PubMed.
- N. Winssinger, R. Damoiseaux, D. C. Tully, B. H. Geierstanger, K. Burdick and J. L. Harris, Chem. Biol., 2004, 11, 1351–1360 CrossRef CAS PubMed.
- J. J. Diaz-Mochón, L. Bialya and M. Bradley, Chem. Commun., 2006, 3984–3986 RSC.
- A. I. Taylor, V. B. Pinheiro, M. J. Smola, A. S. Morgunov, S. Peak-Chew, C. Cozens, K. M. Weeks, P. Herdewijn and P. Holliger, Nature, 2015, 518, 427–430 CrossRef CAS PubMed.
- S. L. Beaucage and R. P. Iyer, Tetrahedron, 1992, 48, 2223–2311 CrossRef CAS.
- J. Niu, R. Hili and D. R. Liu, Nat. Chem., 2013, 5, 282–292 CrossRef PubMed.
- I. Hirao, M. Kimoto, T. Mitsui, T. Fujiwara, R. Kawai, A. Sato, Y. Harada and S. Yokoyama, Nat. Methods, 2006, 3, 729–735 CrossRef CAS PubMed.
- Z. Yang, F. Chen, J. B. Alvarado and S. A. Benner, J. Am. Chem. Soc., 2011, 133, 15105–15112 CrossRef CAS PubMed.
- V. B. Pinheiro and P. Holliger, Curr. Opin. Chem. Biol., 2012, 16, 245–252 CrossRef CAS PubMed.
- R. C. Holmberg, A. A. Henry and F. E. Romesberg, Biomol. Eng., 2005, 22, 39–49 CrossRef CAS PubMed.
- L. Lu, X. Zhang, R. Kong, B. Yang and W. Tan, J. Am. Chem. Soc., 2011, 133, 11686–11691 CrossRef CAS PubMed.
- F. Wang, J. Elbaz and I. Willner, J. Am. Chem. Soc., 2012, 134, 5504–5507 CrossRef CAS PubMed.
- H. Dong, C. Wang, Y. Xiong, H. Lu, H. Ju and X. Zhang, Biosens. Bioelectron., 2013, 41, 348–353 CrossRef CAS PubMed.
- S. Javaherian, M. U. Musheev, M. Kanoatov, M. V. Berezovski and S. N. Krylov, Nucleic Acids Res., 2009, 37, e62 CrossRef PubMed.
- W. Zhao, C. H. Cui, S. Bosee, D. Guoa, C. Shen, W. P. Wong, K. Halvorsen, O. C. Farokhzad, G. S. L. Teo, J. A. Phillips, D. M. Dorfman, R. Karnik and J. M. Karp, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 19626–19631 CrossRef CAS PubMed.
- Y. Yu, Y. Ren, W. Shen, H. Deng and Z. Gao, Trends Anal. Chem., 2013, 50, 33–41 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2016 |