
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Solvatofluorochromic, non-centrosymmetric π-expanded diketopyrrolopyrrole†
Marek
Grzybowski
a,
Artur
Jeżewski
a,
Irena
Deperasińska
b,
Daniel H.
Friese
c,
Marzena
Banasiewicz
b,
Vincent
Hugues
d,
Bolesław
Kozankiewicz
*b,
Mireille
Blanchard-Desce
*d and
Daniel T.
Gryko
*a
aInstitute of Organic Chemistry of the Polish Academy of Sciences, 01-224 Warsaw, Poland. E-mail: dtgryko@icho.edu.pl; Fax: +48 22 6326681; Tel: +48 22 3433063
bInstitute of Physics, Polish Academy of Sciences, Al. Lotników 32/46, 02-668 Warsaw, Poland
cUniversitetet i Tromsø - Norges Arktiske Universitet, Centre for Theoretical and Computational Chemistry Tromsø, Norway. E-mail: kenneth.ruud@uit.no
dUniversité de Bordeaux, ISM (UMR5255 CNRS), F 33400, Bordeaux, France. E-mail: m.blanchard-desce@ism.u-bordeaux1.fr; Fax: +33 (0)5 40 00 69 94; Tel: +33 (0)5 40 00 67 32
First published on 5th January 2016
Abstract
A novel non-centrosymmetric π-expanded diketopyrrolopyrrole was designed and synthesized. Strategic placement of tert-butyl groups at the periphery of a diketopyrrolopyrrole allowed us to selectively fuse one moiety via tandem Friedel–Crafts-dehydration reactions, resulting in a non-centrosymmetric dye. The structure of the dye was confirmed by X-ray crystallography, revealing that it contains a nearly flat arrangement of four fused rings. Extensive photophysical studies of this new functional dye revealed that the intensity of its emission strongly depends on solvent polarity, which is typical for dipolar chromophores. In non-polar solvents, the fluorescence quantum yield is high whereas in polar solvents such as MeOH, it is 12%. However, upon two-photon excitation the compound behaves like a centrosymmetric dye, showing a two-photon absorption maximum at significantly shorter wavelengths than twice the wavelength of the one-photon absorption maximum.
Introduction
Current methods for fluorescence imaging rely on classical dyes such as fluorescein, rhodamines and coumarins.1 The advent of new luminescent probes is growing in the literature ranging from organic chromophores,1 quantum dots,2 carbon dots,3 organic nanodots,4 fluorescent organic nanoparticles,5etc. Notwithstanding new options, it is clear that organic chromophores, due to their tunability and possibility to attach to sensing units, continue to prevail in real-world applications.6 Among various targets, non-centrosymmetric π-conjugated systems, whose fluorescence is dependent on solvent polarity, are one of the most important tributes.7 The subtle interplay between centrosymmetric and non-centrosymmetric structures has been recently revealed by a few breakthrough approaches showing that (a) quadrupolar molecules can display solvatofluorochromism due to symmetry breaking in the excited-state8 and (b) the emission properties of dipolar fluorophores in aqueous media can be significantly altered by replacing the donor amino substituent with a very polar one.9Application of diketopyrrolopyrroles (DPPs, 1,4-diketo-2H,5H-pyrrolo[4,3-c]pyrroles) began as red-pigments of unprecedented light-fastness.10 In recent decades however, applications of DPPs have been reinvented as they have found new uses in diverse areas of applications such as optoelectronics and molecular electronics. DPPs’ derivatives have been widely employed in fields such as dye-sensitized solar cells,11 light emitting diodes,12 organic field effect transistors13 and above-all, bulk-heterojunction solar cells.14,15 Certain attention has also been focused on its inversed analogue – pyrrolo[3,2-b]pyrrole-2,5(1H,4H)-dione (iDPP).16 Although replacing phenyl substituents17 with five-membered heterocycles and their π-expanded analogues offers some opportunities in modulating the absorption and emission properties,11c,18 the most significant change in optical properties has been achieved by expanding the DPP-core via fusion with other aromatics/dyes. Four strategies towards making such compounds have been recently revealed respectively by Zumbusch,19 Shimizu,20a,c Würthner20b and the Gryko group.21 Two-photon absorbing properties of DPPs have rarely been studied despite the fact that their electron-poor core seems to be an excellent starting point for the construction of D–A–D or A–D architectures, important for generating large two-photon absorption cross-sections.18e,f,21b,22 Herein we would like to present a strategy towards non-centrosymmetric π-expanded diketopyrrolopyrroles.
Design and synthesis
The C2-symmetry of diketopyrrolopyrroles and their N-alkylated derivatives makes it difficult to design analogous dyes lacking this symmetry. Our recently reported two-step route of the DPP chromophore expansion is not exceptional in this regard.21 Double alkylation of DPP pigments with 2-bromoacetaldehyde diethyl acetal and the subsequent treatment of the intermediate diacetals with acid always led to the centrosymmetric double-cyclized products. This was also the case when ketones – phenacyl bromides – were used as alkylating agents.21b Mixed-alkylation of DPP with 2-bromoacetaldehyde diethyl acetal and n-alkyl bromide is one of the several possible strategies to synthesize non-symmetric analogues,23 however, due to different reactivities of the two alkylating agents, the desired non-symmetrically substituted product may be obtained in low yields. Our strategy towards D–A type π-expanded diketopyrrolopyrroles is based on the use of mild conditions in the final ring-closure step. We expected that after the alkylation of DPP 1 with 1-bromo-3,3-dimethylbutan-2-one, instead of previously used bromoacetaldehyde acetal or phenacyl bromides,21 diketone 2 would be obtained (Scheme 1). We have decided to use 1-bromo-3,3-dimethylbutan-2-one in order to ensure that the final donor–acceptor dye would possess the moderately reactive t-BuCO group rather than the chemically unstable CHO functionality.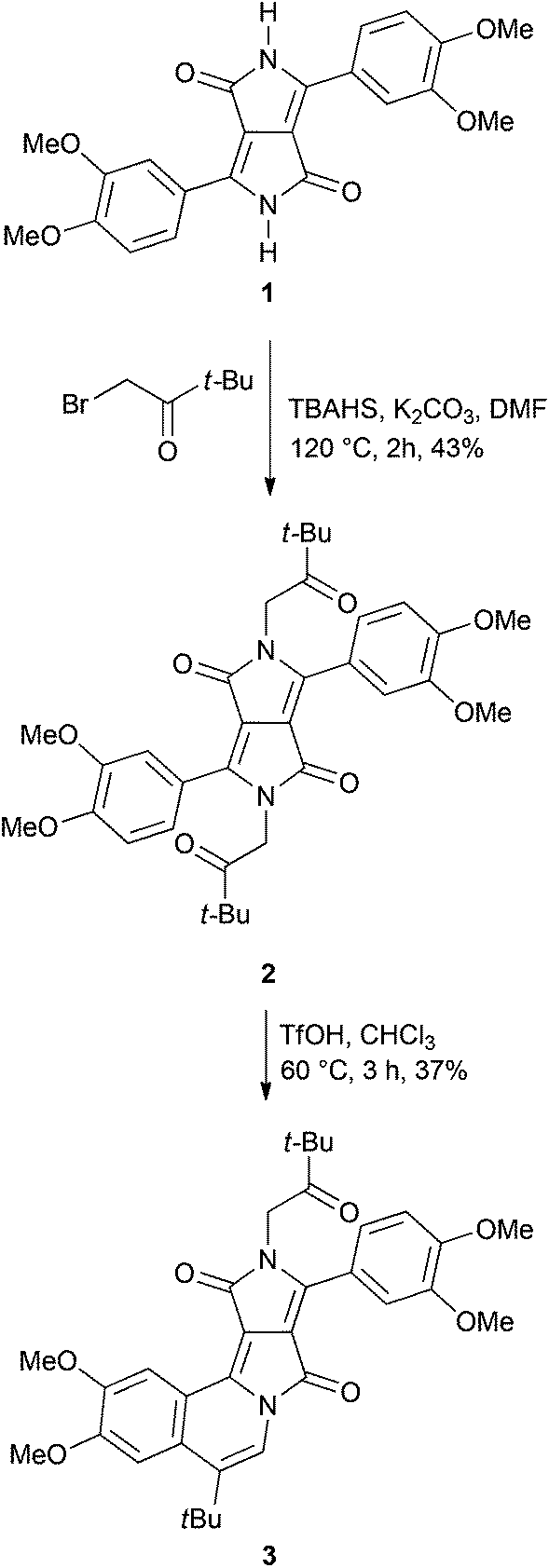 | ||
| Scheme 1 Synthesis of dye 3. | ||
Pigment 1, possessing electron-rich 3,4-dimethoxyphenyl substituents, was prepared according to the literature procedure21avia the condensation of diisopropyl succinate with 3,4-dimethoxybenzonitrile in the presence of sodium tert-amylate (Scheme 1). The alkylation reaction of DPP 1 with commercially available 1-bromo-3,3-dimethylbutan-2-one was performed under similar conditions as developed for the alkylation with 2-bromoacetaldehyde diethyl acetal: at 120 °C in DMF, using potassium carbonate as a base and tetrabutylammonium bisulfate (TBAHS) as a phase-transfer catalyst (Scheme 1).21 As previously noted,21b in contrast to bromoacetaldehyde acetal, bromoketones react much faster and the reaction is finished within 2 hours (acetal required at least 16 h).
Diketone 2 was obtained in moderate yield (43%, Scheme 1). It was well soluble in chlorinated and aromatic solvents and strongly fluorescent in solutions. It is noteworthy that product 2 also exhibited intense fluorescence in the solid-state under UV irradiation.
Dye 2 was added to the reaction with triflic acid (TfOH) at 60 °C in chloroform and the progress was controlled using TLC. When the conversion of the starting material was complete (about 3 h), the main product was purified by silica gel chromatography. NMR spectra revealed that non-centrosymmetric dye 3 was formed exclusively (Scheme 1).
X-ray quality crystals have been obtained for dye 3. Crystallographic analysis fully confirmed the structure (Fig. 1). The four-fused ring system is almost planar (deviations from the calculated plane less than 0.1 Å). Hence the presence of short hydrogen bonding between carbonyl oxygen and the benzene hydrogen atom (distance: 2.39 Å, Fig. 1) does not cause a significant deformation of the polycyclic system, as was also observed in the case of a bis-fused DPP derivative (O⋯H distance 2.45 Å, highest deviations from planarity: 0.34 Å).21b The plane of the benzene ring in the 3,4-dimethoxyphenyl substituent is twisted by 34.5° relative to the polycyclic system. This torsion angle is similar to the corresponding angles in non-fused N,N-dialkyl DPPs.24 In the tetragonal crystal lattice, molecules of 3 form dimers through π–π interactions, see Fig. S1.† The distance between the chromophores (3.73 Å) is noticeably longer than in the case of the bis-cyclized DPP derivative (3.48 Å),21b which is due to the presence of the bulky tert-butyl groups.
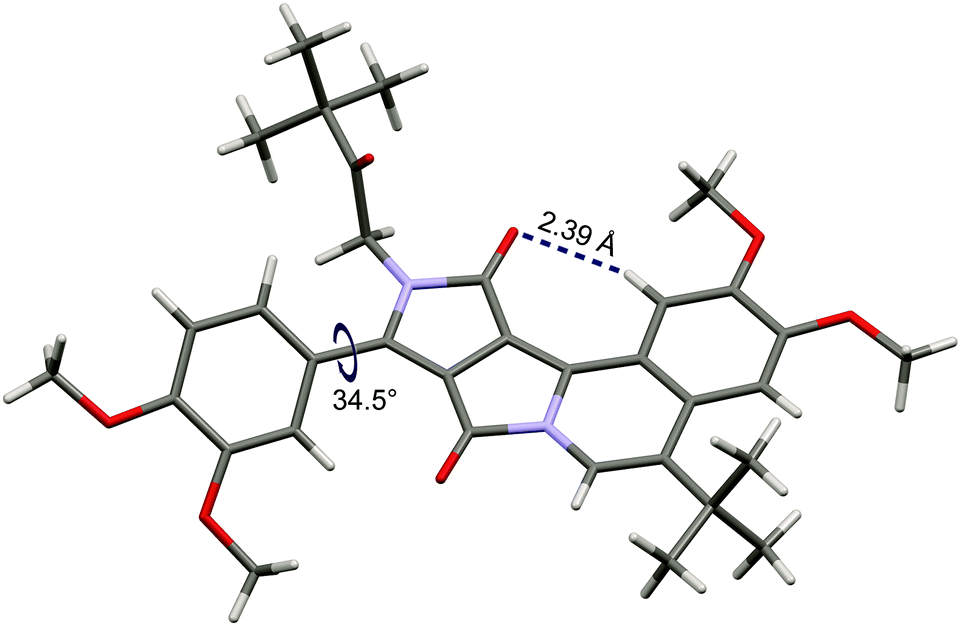 | ||
| Fig. 1 The crystal structure of compound 3 (CCDC 1441247). | ||
Optical properties
The maxima of absorption (λabs) and emission (λem) for 2 are similar to those reported for previously described DPPs having 3,4-dimethoxyphenyl at 3 and 6 positions and 2,2-diethoxyethyl substituents at the nitrogen atoms,21 which indicates negligible perturbation by the nature of the N-substituents (Table 1). In chloroform, mono-fused DPP 3 has an absorption ∼590 nm and an emission at ∼620 nm, which results in a Stokes shift of about 800 cm−1 (Fig. 2, Table 1). The absorption maximum of compound 3 is only 6 nm hypsochromically shifted versus its double-fused analogue 4 (Fig. 3, 587 nm vs. 593 nm),21a whereas emission is even bathochromically shifted (619 nm vs. 597 nm). The Stokes shift, which in the case of centrosymmetric and rigid π-expanded DPPs is typically very small (90–230 cm−1),21a,b increases for compound 3 in CHCl3 to 880 cm−1 (see Table 1). These results are associated with the fact that the rigid and centrosymmetric chromophore in 4 is replaced by the more flexible, non-symmetric, push–pull system of compound 3. The molar absorption coefficient of 3 is 27![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, only slightly higher than that before cyclization (compound 2), but significantly lower than that of the bis-cyclized compound 4 (110000 M−1 cm−1).21a,b
000, only slightly higher than that before cyclization (compound 2), but significantly lower than that of the bis-cyclized compound 4 (110000 M−1 cm−1).21a,b
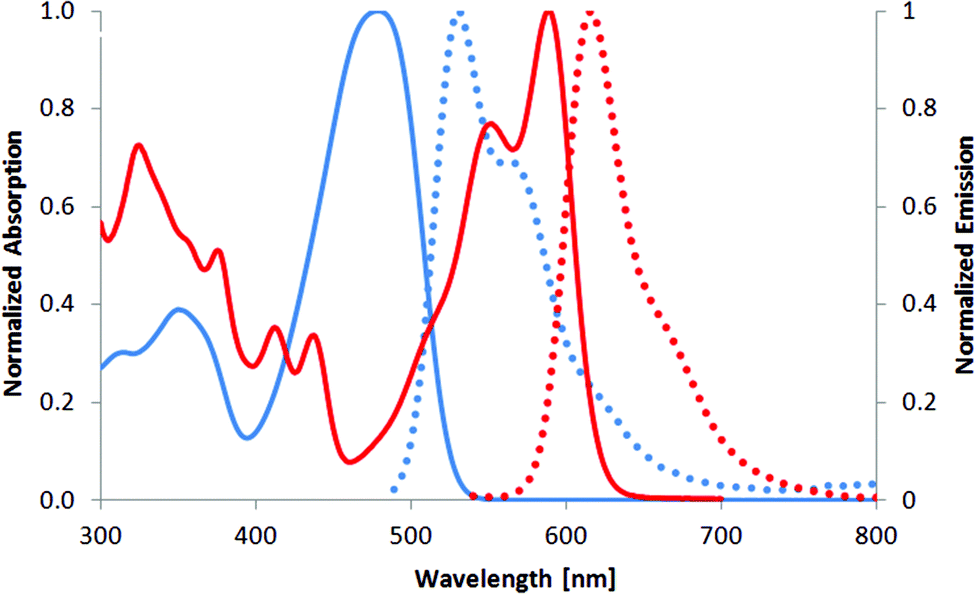 | ||
| Fig. 2 Absorption (solid) and emission (dotted) spectra of compounds 2 (blue line) and 3 (red line) measured in chloroform. | ||
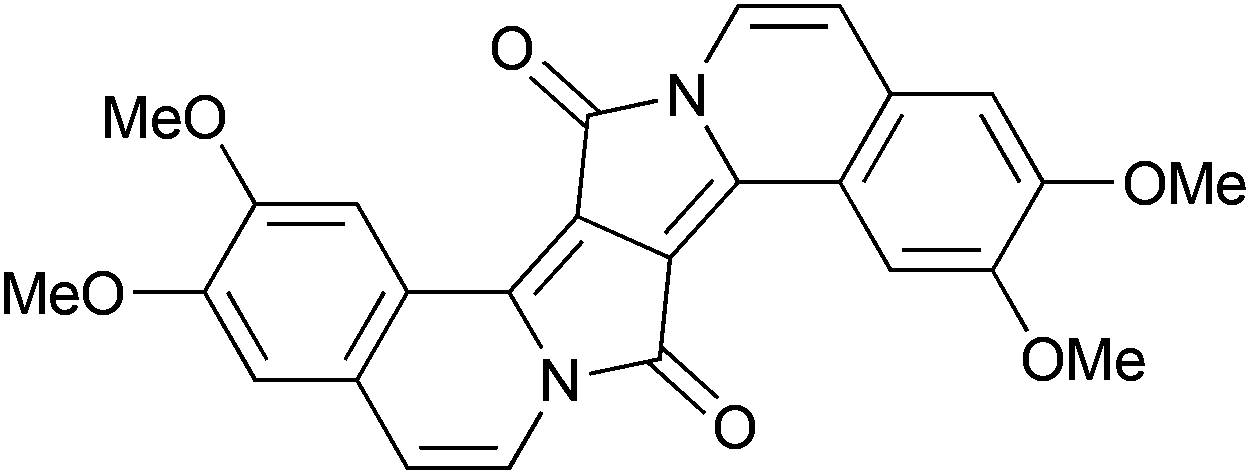 | ||
| Fig. 3 The chemical structure of compound 4. | ||
| Compound | Solvent | λ maxabs [nm] | λ maxem [nm] | Stokes shift [cm−1] | Φ fl |
|---|---|---|---|---|---|
| a Standard: rhodamine 6G in EtOH (Φ = 0.94). b Standard: cresyl violet in methanol (Φ = 0.54). c Ref. 21a. | |||||
| 2 | CHCl3 | 478 | 531 | 2100 | 0.86a |
| 3 | Pentane | 544, 584 | 605, 655 | 600 | 0.66b |
| 3 | Cyclohexane | 544, 585 | 605, 657 | 570 | 0.82b |
| 3 | Toluene | 550, 589 | 615, 655 | 720 | 0.72b |
| 3 | CHCl3 | 587 | 619 | 880 | 0.43b |
| 3 | MeCN | 545, 580 | 619 | 1090 | 0.39b |
| 3 | MeOH | 543, 579 | 619 | 1120 | 0.12b |
| 4 | CHCl3c | 593 | 597 | 110 | 0.85 |
| 4 | DMFc | 593 | 602 | 250 | 0.70 |
Absorption and emission (fluorescence) spectra of compound 3 in n-hexane are presented in Fig. S2.† The Stokes shift in this solvent is relatively small, ∼450 cm−1. Freezing of the matrix to 5 K does not lead to noticeable sharpening of the fluorescence spectrum. The fluorescence quantum yield of 3 in n-hexane is 0.56, whereas the decay time of this emission is 8.37 ns (see the ESI†).
Whereas absorption does not undergo notable changes from non-polar solvents to MeCN and MeOH, the emission quantum yield drastically decreases from 0.66–0.82 in hydrocarbon solvents to 0.12 in MeOH (Table 1). Simultaneously, the emission maximum is slightly bathochromically shifted by 14 nm, when moving from non-polar pentane to polar methanol. The slight red-shift of emission seems to be related more to inhomogeneous broadening with the loss of a fine structure in more polar solvents (starting from CHCl3). The slight red-shift cannot explain the decrease in quantum yield as it does for a push–pull system (only 4 nm from toluene to methanol).
Two-photon absorption (2PA) measurements of both of the new compounds 2 and 3 were performed using the two-photon excited fluorescence (TPEF) method (see the ESI†). Non-fused 2 possesses a moderate 2PA cross-section located around 700 nm (Table 2) resulting most probably from its quadrupolar structure. Interestingly, the new π-expanded DPP 3 possessing a dimethoxyphenyl unit has a larger maximum 2PA cross-section than centrosymmetric compound 4 studied previously (Table 2).20b
| Compound | 2λmax1PA [nm] | λ max2PA [nm] |
σ
max2a [GM] |
σ max2 Φ fl [GM] |
|---|---|---|---|---|
| 2 | 974, 708 | 720 | 260 | 220 |
| 3 | 1176, 1102, 874 | ≤740 | ≥270 | ≥120 |
| 4 | 1186 | 820 | 130 | 110 |
Its 2PA response is also slightly higher than that of the centrosymmetric derivative 2 and does not show a clear maximum. In addition, the 2PA maximum (≤740 nm) of 3 is located at much shorter wavelengths than the doubled wavelength of the one-photon absorption maximum (1176 nm, see Fig. 4). This is an interesting and unusual result, because such an effect is typically observed for centrosymmetric chromophores, for which one-photon allowed excitations are two-photon forbidden and vice versa, whereas for non-symmetric dyes both processes obey the same quantum selection rules and 2PA is usually recorded at approximately twice the wavelength of the 1PA maximum. Therefore, under the two-photon excitation, non-symmetric compound 3 behaves more like a centrosymmetric chromophore.
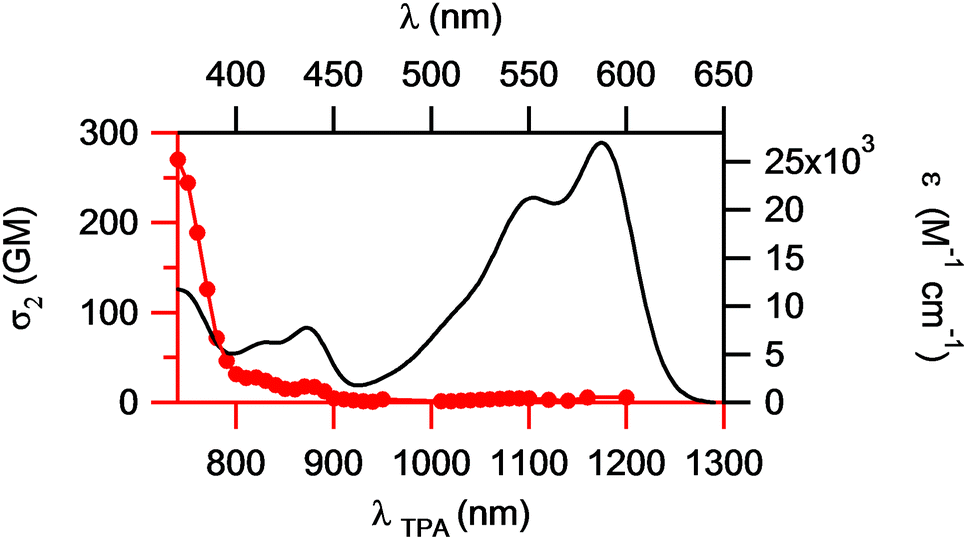 | ||
| Fig. 4 One-photon absorption (solid black line) and two-photon absorption (red line) spectra of compound 3 in CHCl3. | ||
Calculations
Calculated excitation energies, oscillator strengths and 2PA cross-sections are listed in Table 3. We note that the strongest oscillator strength in 1PA is found for the S1 state, and this transition is almost dark in 2PA. In contrast to this, the S5 state exhibits a large 2PA cross-section. At the same time, this state is dark in 1PA. However, for the other states we do not find such a strong alternation between one- and two-photon absorption. Indeed the S2 state shows weak activity in both processes, i.e., 1PA and 2PA whereas the S3 state shows reasonable 1PA and 2PA activities, in good agreement with the experimental data (Fig. 4). Evaluating the orbital transitions involved in the excitation to the S1 state, we note that this state is dominated by the HOMO–LUMO transition. Both the HOMO and the LUMO are located mainly on the DPP backbone (see Fig. 5).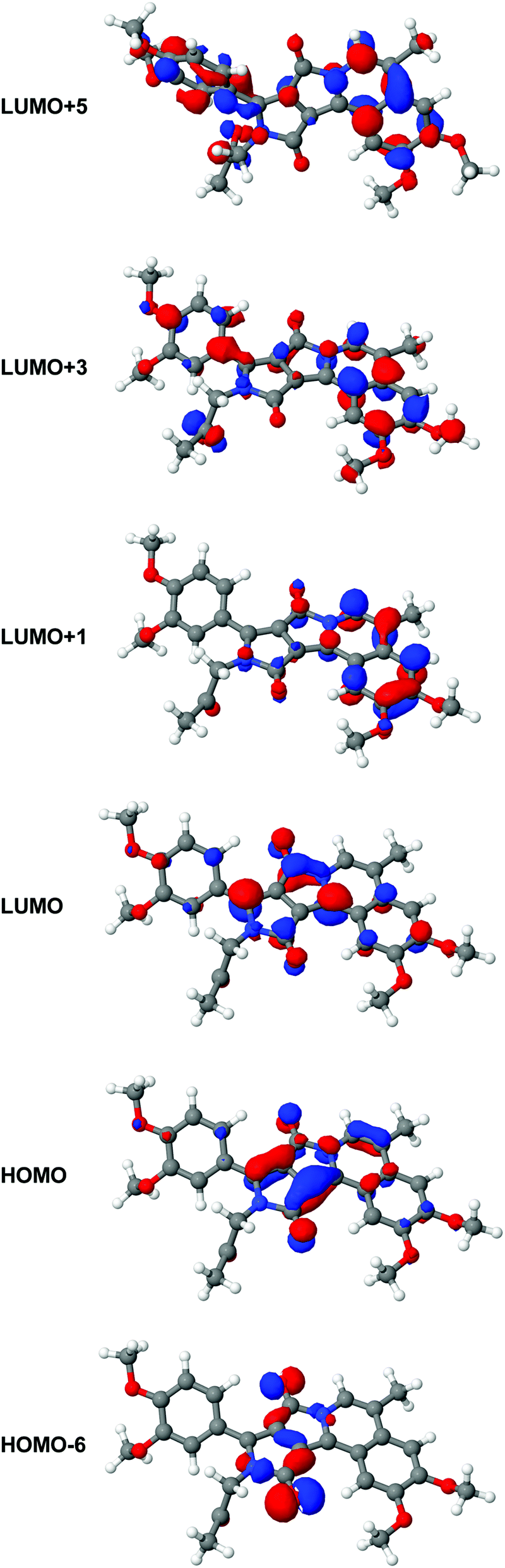 | ||
| Fig. 5 From the top to bottom: LUMO+5, LUMO+3, LUMO+1, LUMO, HOMO and HOMO−6 of 3. The orbital plots have been generated from a CAM-B3LYP calculation using the aug-cc-pVDZ basis set. | ||
| Excited state | 2PA energy (eV) | 2PA energy (nm) | 2PA cross-section (GM) | 1PA oscillator strength |
|---|---|---|---|---|
| 1 | 2.51 | 988.7 | 1.87 | 0.4151 |
| 2 | 3.32 | 747.5 | 42.1 | 0.1242 |
| 3 | 3.87 | 641.2 | 234.0 | 0.1759 |
| 4 | 4.17 | 595.1 | 8.9 | 0.1304 |
| 5 | 4.22 | 588.0 | 1020 | 0.0024 |
| 6 | 4.35 | 570.4 | 479 | 0.0454 |
| 7 | 4.41 | 562.7 | 252 | 0.0727 |
| 8 | 4.49 | 552.7 | 40 | 0.0017 |
| 9 | 4.56 | 544.2 | 228 | 0.0328 |
| 10 | 4.64 | 534.8 | 357 | 0.2408 |
These parts of the HOMO are gerade with respect to the inversion center of the backbone while the corresponding contributions to the LUMO are ungerade. Hence the orbitals show pseudo-centrosymmetry explaining the strong 1PA and weak 2PA character of the states. In contrast to the S1 state, higher electronic states Si (in particular S3 and S5) involve different molecular orbital transitions. Fig. 5 shows the molecular orbitals involved in the S1, S3 and S5 states. While the S3 state is dominated by the HOMO > LUMO+1 transition, the S5 state involves the HOMO−6 > LUMO, HOMO > LUMO+5 and HOMO > LUMO+3-transitions. By evaluating the shape of the orbitals, we note that the HOMO−6 and the HOMO are tendentially gerade with regard to the inversion center of the DPP-backbone (as 3 is not a centrosymmetric molecule, the orbitals cannot be gerade or ungerade in the whole molecule but parts of them can show a local pseudosymmetric behavior). Also the LUMO+3 and the LUMO+5 are pseudo-gerade while the LUMO is pseudo-ungerade. The LUMO+1 cannot be classified as either gerade or ungerade as it is mostly located on the isoquinoline moiety. This explains why the S3 state shows both 1PA and 2PA activities while the S1 state is only active in 1PA and the S5 state only in 2PA. Both states have charge-transfer character. The result of such transitions is to change the dipole moment of a molecule in the excited state, and it promotes a greater cross-section for two-photon transitions.25
Conclusions
In summary, we have proven that cascade processes leading to π-expanded diketopyrrolopyrroles can be controlled to lead to a donor–acceptor type product. The resulting heterocycle possesses four conjugated rings and lacks a center of symmetry. However, 1PA and 2PA measurements revealed that the final dye simultaneously exhibits photophysical characteristics of both symmetric and non-symmetric chromophores. This behavior is in agreement with the results of DFT calculations, and can be explained by analysis of the symmetry of the molecular orbitals in this molecule. This provides a synthetic entry to bright, red-emitting two-photon fluorophores. The other notable finding is that breaking the overall symmetry of a molecule does not necessarily extinguish the symmetry completely. Some molecular properties and excited states still can show “pseudosymmetric behavior”. This study is complementary to the investigation of solvatofluorochromism for centrosymmetric bis(thienyl)DPPs18d and at the same time it offers opportunities in fluorescence imaging.Experimental section
General
All chemicals were used as received unless otherwise noted. All reported 1H NMR and 13C NMR spectra were recorded on a 200, 400, 500 or 600 MHz spectrometer. Chemical shifts (δ ppm) were determined with TMS as the internal reference; J values are given in Hz. UV-Vis and fluorescence spectra were recorded in chloroform. Chromatography was performed on silica (Kieselgel 60, 200–400 mesh). 1,4-Diketo-3,6-bis(3,4-dimethoxyphenyl)pyrrolo[3,4-c]pyrrole (1)21a was prepared according to the literature procedures.:acetone 49:1 → 19:1). Finally, the product was recrystallized by slow addition of pentane to the solution of the product in a small amount of CHCl3. Yield: 258 mg (43%). Red powder. Mp 203–204 °C (CHCl3/pentane). 1H NMR (500 MHz, CDCl3) δ 7.38 (d, J = 2.0 Hz, 2H, 2-H at Ar), 7.19 (dd, J = 8.5, 2.0 Hz, 2H, 6-H at Ar), 6.91 (d, J = 8.5 Hz, 2H, 5-H at Ar), 4.75 (s, 4H, CH2), 3.92 (s, 6H, OCH3), 3.91 (s, 6H, OCH3), 1.19 (s, 18H, tBu). 13C NMR (126 MHz, CDCl3) δ 209.3, 162.5, 151.5, 149.2, 148.1, 121.8, 120.7, 112.0, 111.0, 109.2, 56.2, 56.0, 47.1, 43.4, 26.4. HRMS (ESI) calcd for C34H40N2O8Na (M + Na+): 627.2682, found: 627.2678.
Optical studies
One-photon absorption spectra were measured with a Perkin Elmer Lambda 35 UV/VIS spectrometer. Fluorescence spectra at room and at 5 K were measured with the aid of a home-built set-up equipped with a liquid helium optical cryostat, a McPherson 207 monochromator, an EMI 9659 photomultiplier and an electronic card inserted into a PC. The excitation source was a Coherent Verdi-V5 (532 nm) and a Coherent 700 dye laser (with the simplified optics for a cw operation at 568.5 nm).Fluorescence decay curves were monitored with the aid of a “time correlated” single photon counting technique (in inverted time mode). The system composed of a mode-locked Coherent Mira-HP laser pumped by a Verdi 18 laser, an APE Pulse selector reducing the repetition of Mira laser pulses to 2 MHz, and a frequency doubling crystal. Fluorescence photons, dispersed with a McPherson 207 monochromator, were detected with a HMP-100-50 hybrid detector and a SPC-150 module inserted into a PC, both from Becker&Hickl GmbH.
Two-photon absorption cross-sections of 10−4 M solutions were measured relative to fluorescein in 0.01 M aqueous NaOH for 700–800 nm,26 using the well-established method described by Xu and Webb26b and the appropriate solvent-related refractive index corrections.27 The reference values between 700 and 715 nm for fluorescein were taken from the literature.28 The quadratic dependence of the fluorescence intensity on the excitation power was checked for each sample and all wavelengths. To span the 700–980 nm range, a Nd:YLF-pumped S2 Ti:sapphire oscillator was used generating 150 fs pulses at a 76 MHz rate. To span the 1000–1400 nm range, an OPO (PP-BBO) was added to the setup to collect and modulate the output signal of the Ti:sapphire oscillator.
Computational studies
To simplify the computational treatment, the two t-butyl groups in molecule 3 have been replaced by methyl groups. The t-butyl groups are not expected to influence the absorption properties of the molecule, but to increase the computational complexity because of the increases in the number of atoms and increases in the conformational freedom. The modified structure will be referred to as molecule 3a. The structure of 3a has been optimized using the B3LYP density functional29 and the TZVP basis set30 using the TURBOMOLE suite of programs.31 Two-photon absorption calculations have been carried out using DALTON32 using the CAM-BL3LYP density functional33 and the aug-cc-pVDZ basis set from the Dunning family of basis sets.34D. T. G. thanks the National Science Centre of the Republic of Poland (MAESTRO-2012/06/A/ST5/00216) and the Global Research Laboratory Program (2014K1A1A2064569) through the National Research Foundation (NRF) funded by the Ministry of Science, ICT & Future Planning, Korea. This work was partially supported by the European Commission (TOPBIO ITN) and BASF-Schweiz. MBD gratefully acknowledges financial support from Conseil Régional d'Aquitaine (chaire d'accueil grant and fellowship to VH). D. H. F. acknowledge support from the Research Council of Norway through a Centre of Excellence Grant (Grant No. 179568/V30) and from the Norwegian Supercomputing Program (Grant No. NN4654 K). We also thank Eli M. Espinoza (UC Riverside) and Kenneth Ruud (University of Tromsø).
Notes and references
- (a) R. Y. Chao, M.-F. Ding, J.-Y. Chen, C.-C. Lee, S.-T. Lin and J. Chin, Chem. Soc., 2010, 57, 213–221 Search PubMed; (b) I. Kim, D. Kim, S. Sambasivan and K. H. Ahn, Asian J. Org. Chem., 2012, 1, 60–64 CrossRef; (c) J. Avó, S. Martins, A. J. Parola, J. C. Lima, P. S. Branco, J. P. Prates Ramalho and A. Pereira, ChemPlusChem, 2013, 78, 789–792 CrossRef; (d) T. Mineno, T. Ueno, Y. Urano, H. Kojima and T. Nagano, Org. Lett., 2006, 8, 5963–5966 CrossRef CAS; (e) T. Hirano, K. Kikuchi, Y. Urano, T. Higuchi and T. Nagano, J. Am. Chem. Soc., 2000, 122, 12399–12400 CrossRef CAS; (f) N. Boens, V. Leen and W. Dehaen, Chem. Soc. Rev., 2012, 41, 1130–1172 RSC; (g) G. Ulrich, R. Ziessel and A. Harriman, Angew. Chem., Int. Ed., 2008, 47, 1184–1201 CrossRef PubMed; (h) T. A. Wood and A. Thompson, Chem. Rev., 2007, 107, 1831–1861 CrossRef PubMed.
- (a) O. S. Wolfbeis, Chem. Soc. Rev., 2015, 44, 4743–4768 RSC; (b) Z. Li, Q. Sun, Y. Zhu, B. Tan, Z. P. Xu and S. X. Dou, J. Mater. Chem. B, 2014, 2, 2793–2818 RSC; (c) K. D. Wegner and N. Hildebrandt, Chem. Soc. Rev., 2015, 44, 4792–4834 RSC.
- (a) P. G. Luo, F. Yang, S.-T. Yang, S. K. Sonkar, L. Yang, J. J. Broglie, Y. Liu and Y.-P. Sun, RSC Adv., 2014, 4, 10791–10807 RSC; (b) P. G. Luo, S. Sahu, S.-T. Yang, S. K. Sonkar, J. Wang, H. Wang, G. E. LeCroy, L. Cao and Y.-P. Sun, J. Mater. Chem. B, 2013, 1, 2116–2127 RSC.
- (a) A. Shakhbazau, M. Mishra, T. Ho Chu, C. Brideau, K. Cummins, S. Tsutsui, D. Shcharbin, S. Mignani, J.-P. Majoral, M. Blanchard-Desce, M. Bryszewska, V. W. Yong, P. Stys and J. van Minnen, Macromol. Biosci., 2015, 15, 1523–1534 CrossRef CAS PubMed; (b) R. K. Tathavarty, M. Parent, M. H. V. Werts, S. Gmouh, A.-M. Caminade, L. Moreaux, S. Charpak, J.-P. Majoral and M. Blanchard-Desce, Angew. Chem., Int. Ed., 2006, 45, 4645–4648 CrossRef PubMed.
- (a) S. Fery-Forgues, Nanoscale, 2013, 5, 8428–8442 RSC; (b) E. Genin, Z. Gao, J. A. Varela, J. Daniel, T. Bsaibess, I. Gosse, L. Groc, L. Cognet and M. Blanchard-Desce, Adv. Mater., 2014, 26, 2258–2261 CrossRef CAS PubMed.
- (a) L. D. Lavis and R. T. Raines, ACS Chem. Biol., 2008, 3, 142–155 CrossRef CAS PubMed; (b) R. N. Kumar, V. Bhalla and M. Kumar, Chem. Commun., 2015, 51, 15614–15628 RSC; (c) C. Ji, Y. Zheng, J. Li, J. Shen, W. Yang and M. Yin, J. Mater. Chem. B, 2015, 3, 7494–7498 RSC; (d) B. A. D. Neto, P. H. P. R. Carvalho and J. R. Correa, Acc. Chem. Res., 2015, 48, 1560–1569 CrossRef CAS PubMed; (e) I. López-Duarte, P. Chairatana, Y. Wu, J. Pérez-Moreno, P. M. Bennett, J. E. Reeve, I. Boczarow, W. Kaluza, N. A. Hosny, S. D. Stranks, R. J. Nicholas, K. Clays, M. K. Kuimova and H. L. Anderson, Org. Biomol. Chem., 2015, 13, 3792–3802 RSC; (f) A. Romieu, Org. Biomol. Chem., 2015, 13, 1294–1306 RSC; (g) H. M. Kim and B. R. Cho, Chem. Rev., 2015, 115, 5014–5055 CrossRef CAS PubMed; (h) A. T. Wrobel, T. C. Johnstone, A. D. Liang, S. J. Lippard and P. Rivera-Fuentes, J. Am. Chem. Soc., 2014, 136, 4697–4705 CrossRef CAS PubMed; (i) Z. Guo, S. Park, J. Yoon and I. Shin, Chem. Soc. Rev., 2014, 43, 16–29 RSC.
- (a) G. S. Loving, M. Sainlos and B. Imperiali, Trends Biotechnol., 2010, 28, 73–83 CrossRef CAS PubMed; (b) A. S. Klymchenko and Y. Mely, Prog. Mol. Biol. Transl. Sci., 2013, 113, 35–58 CAS; (c) Z. Yang, J. Cao, Y. He, J. H. Yang, T. Kim, X. Peng and J. S. Kim, Chem. Soc. Rev., 2014, 43, 4563–4601 RSC; (d) G. Weber and F. J. Farris, Biochemistry, 1979, 18, 3075–3078 CrossRef CAS PubMed; (e) D. J. Cowley, Nature, 1986, 319, 14–15 CrossRef; (f) R. B. MacGregor and G. Weber, Nature, 1986, 319, 70–73 CrossRef CAS PubMed; (g) A. Jacobson, A. Petric, D. Hogenkamp, A. Sinur and J. R. Barrio, J. Am. Chem. Soc., 1996, 118, 5572–5579 CrossRef CAS; (h) B. E. Cohen, T. B. McAnaney, E. S. Park, Y. N. Jan, S. G. Boxer and L. Y. Jan, Science, 2002, 296, 1700–1703 CrossRef CAS PubMed; (i) K. Gaus, E. Gratton, E. P. W. Kable, A. S. Jones, I. Gelissen, L. Kritharides and W. Jessup, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 15554–15559 CrossRef CAS PubMed; (j) H. M. Kim, B. H. Jeong, J.-Y. Hyon, M. J. An, M. S. Seo, J. H. Hong, K. J. Lee, C. H. Kim, T. Joo, S.-C. Hong and B. R. Cho, J. Am. Chem. Soc., 2008, 130, 4246–4247 CrossRef CAS PubMed; (k) H. M. Kim, M. J. An, J. H. Hong, B. H. Jeong, O. Kwon, J.-Y. Hyon, S.-C. Hong, K. J. Lee and B. R. Cho, Angew. Chem., Int. Ed., 2008, 47, 2231–2234 CrossRef CAS PubMed; (l) A. S. Klymchenko and R. Kreder, Chem. Biol., 2014, 21, 97–113 CrossRef CAS PubMed.
- (a) F. Terenziani, A. Painelli, C. Katan, M. Charlot and M. Blanchard-Desce, J. Am. Chem. Soc., 2006, 128, 15742–15755 CrossRef CAS PubMed; (b) C.-H. Zhao, A. Wakamiya, Y. Inukai and S. Yamaguchi, J. Am. Chem. Soc., 2006, 128, 15934–15935 CrossRef CAS; (c) J. Do, J. Huh and E. Kim, Langmuir, 2009, 25, 9405–9412 CrossRef CAS PubMed; (d) E. L. Spitler, J. M. Monson and M. M. Haley, J. Org. Chem., 2008, 73, 2211–2223 CrossRef CAS PubMed; (e) W. V. Moreshead, O. V. Przhonska, M. V. Bondar, A. D. Kachkovski, I. H. Nayyar, A. E. Masunov, A. W. Woodward and K. D. Belfield, J. Phys. Chem. C, 2013, 117, 23133–23147 CrossRef CAS; (f) D. H. Friese, A. Mikhaylov, M. Krzeszewski, Y. M. Poronik, A. Rebane, K. Ruud and D. T. Gryko, Chem. – Eur. J., 2015, 21, 18364–18374 CrossRef CAS PubMed; (g) A. Rebane, M. Drobizhev, N. S. Makarov, G. Wicks, G. P. Wnuk, Y. Stepanenko, J. E. Haley, D. M. Krein, J. L. Fore, A. R. Burke, J. E. Slagle, D. G. McLean and T. M. Cooper, J. Phys. Chem. A, 2014, 118, 3749–3759 CrossRef CAS PubMed.
- S. Singha, D. Kim, B. Roy, S. Sambasivan, H. Moon, A. S. Rao, J. Y. Kim, T. Joo, J. W. Park, Y. M. Rhee, T. Wang, K. H. Kim, Y. H. Shin, J. Jung and K. H. Ahn, Chem. Sci., 2015, 6, 4335 RSC.
- (a) A. C. Rochat, L. Cassar and A. Iqbal, Preparation of pyrrolo-(3,4-c) pyrroles, Eur. Pat. Appl, 94911, 1983 Search PubMed; (b) A. Iqbal, M. Jost, R. Kirchmayr, J. Pfenninger, A. Rochat and O. Wallquist, Bull. Soc. Chim. Belg., 1988, 97, 615 CrossRef CAS; (c) Z. M. Hao and A. Iqbal, Chem. Soc. Rev., 1997, 26, 203 RSC; (d) J. S. Zambounis, Z. Hao and A. Iqbal, Nature, 1997, 388, 131 CrossRef CAS; (e) O. Wallquist and R. Lenz, Macromol. Symp., 2002, 187, 617 CrossRef CAS; (f) A. Iqbal, L. Cassar, A. C. Rochat, J. Pfenninger and O. Wallquist, J. Coat. Technol., 1988, 60, 37 CAS; (g) D. G. Farnum, G. Mehta, G. G. I. Moore and F. P. Siegal, Tetrahedron Lett., 1974, 29, 2549 CrossRef.
- (a) S. Qu, C. Qin, A. Islam, Y. Wu, W. Zhu, J. Hua, H. Tian and L. Han, Chem. Commun., 2012, 48, 6972 RSC; (b) S. Qu and H. Tian, Chem. Commun., 2012, 48, 3039 RSC and references cited therein; (c) C. B. Nielsen, M. Turbiez and I. McCulloch, Adv. Mater., 2013, 13, 1859 CrossRef PubMed and references cited therein; (d) S.-Y. Liu, M.-M. Shi, J.-C. Huang, Z.-N. Jin, X.-L. Hu, J.-Y. Pan, H.-Y. Li, A. K.-Y. Jen and H.-Z. Chen, J. Mater. Chem. A, 2013, 1, 2795 RSC; (e) Y.-H. Jeong, C.-H. Lee and W.-D. Jang, Chem. – Asian J., 2012, 7, 1562 CrossRef PubMed; (f) E. Q. Guo, P. H. Ren, Y. L. Zhang, H. C. Zhang and W. J. Yang, Chem. Commun., 2009, 5859 RSC.
- M. Vala, M. Weiter, J. Vynuchal, P. Toman and S. Lunak, J. Fluoresc., 2008, 18, 1181 CrossRef CAS PubMed.
- (a) L. Burgi, M. Turbiez, R. Pfeiffer, F. Bienewald, H. J. Kirner and C. Winnewisser, Adv. Mater., 2008, 20, 2217 CrossRef CAS; (b) M. Tantiwiwat, A. Tamayo, N. Luu, X. D. Dang and T. Q. Nguyen, J. Phys. Chem. C, 2008, 112, 17402 CrossRef CAS; (c) S. L. Suraru, U. Zschieschang, H. Klauk and F. Würthner, Chem. Commun., 2011, 47, 1767 RSC; (d) L. Burgi, M. Turbiez, R. Pfeiffer, F. Bienewald, H.-J. Kirner and C. Winnewisser, Adv. Mater., 2008, 20, 2217 CrossRef CAS; (e) W. S. Yoon, S. K. Park, I. Cho, J.-A. Oh, J. H. Kim and S. Y. Park, Adv. Funct. Mater., 2013, 23, 3519 CrossRef CAS.
- (a) B. Tieke, A. R. Rabindranath, K. Zhang and Y. Zhu, Beilstein J. Org. Chem., 2010, 6, 830 CrossRef PubMed; (b) M. M. Wienk, M. Turbiez, J. Gilot and R. A. J. Janssen, Adv. Mater., 2008, 20, 2556 CrossRef CAS; (c) K. H. Hendriks, W. Li, M. M. Wienk and R. A. J. Janssen, Adv. Energy Mater., 2013, 3, 674 CrossRef CAS; (d) A. L. Kanibolotsky, F. Vilela, J. C. Forgie, S. E. T. Elmasly, P. J. Skabara, K. Zhang, B. Tieke, J. McGurk, C. R. Belton, P. N. Stavrinou and D. D. C. Bradley, Adv. Mater., 2011, 23, 2093 CrossRef CAS PubMed; (e) H. Y. Woo, D. Korystov, A. Mikhailovsky, T.-Q. Nguyen and G. C. Bazan, J. Am. Chem. Soc., 2005, 127, 13794 CrossRef CAS PubMed; (f) H. Y. Woo, B. Liu, B. Kohler, D. Korystov, A. Mikhailovsky and G. C. Bazan, J. Am. Chem. Soc., 2005, 127, 14721 CrossRef CAS PubMed; (g) Y. Li, P. Sonar, L. Murphy and W. Hong, Energy Environ. Sci., 2013, 6, 1684 RSC; (h) K. H. Hendriks, G. H. L. Heintges, V. S. Gevaerts, M. M. Wienk and R. A. J. Janssen, Angew. Chem., Int. Ed., 2013, 52, 8341 CrossRef CAS PubMed; (i) R. C. Coffin, J. Peet, J. Rogers and G. C. Bazan, Nat. Chem., 2009, 1, 657 CrossRef CAS PubMed.
- (a) T.-J. Ha, P. Sonar and A. Dodabalapur, Phys. Chem. Chem. Phys., 2013, 15, 9735 RSC; (b) P. Sonar, T.-J. Ha and A. Dodabalapur, Phys. Chem. Chem. Phys., 2013, 15, 7475 RSC; (c) P. Sonar, J.-M. Zhuo, L.-H. Zhao, K.-M. Lim, J. Chen, A. J. Rondinone, S. P. Singh, L.-L. Chua, P. K. H. Ho and A. Dodabalapur, J. Mater. Chem., 2012, 22, 17284 RSC; (d) Y. Li, P. Sonar, S. P. Singh, Z. E. Ooi, E. S. H. Lek and M. Q. Y. Loh, Phys. Chem. Chem. Phys., 2012, 14, 7162 RSC; (e) P. Sonar, S. P. Singh, E. van L. Williams, Y. Li, M. S. Soh and A. Dodabalapur, J. Mater. Chem., 2012, 22, 4425 RSC; (f) M. Tantiwiwat, A. Tamayo, N. Luu, X. D. Dang and T. Q. Nguyen, J. Phys. Chem. C, 2008, 112, 17402 CrossRef CAS; (g) O. Kwon, J. Jo, B. Walker, G. C. Bazan and J. H. Seo, J. Mater. Chem. A, 2013, 1, 7118 RSC.
- M. Kirkus, S. Knippenberg, D. Beljonne, J. Cornil, R. A. J. Janssen and S. C. J. Meskers, J. Phys. Chem. A, 2013, 117, 2782 CrossRef CAS PubMed.
- (a) T. Potrawa and H. Langhals, Chem. Ber., 1978, 120, 1075 CrossRef; (b) H. Langhals, T. Potrawa, H. Nöth and G. Linti, Angew. Chem., Int. Ed. Engl., 1989, 101, 497 CrossRef CAS; (c) H. Langhals, S. Demmig and T. Potrawa, J. Prakt. Chem., 1991, 333, 733 CrossRef CAS; (d) P. Edman, L. B.-A. Johansson and H. Langhals, J. Phys. Chem., 1995, 99, 8504 CrossRef CAS; (e) M. Vala, M. Weiter, J. Vyňuchal, P. Toman and S. Luňák Jr., J. Fluoresc., 2008, 18, 1181 CrossRef CAS PubMed; (f) S. Luňák Jr., M. Vala, J. Vyňuchal, I. Ouzzane, P. Horáková, P. Možíšková, Z. Eliáš and M. Weiter, Dyes Pigm., 2011, 91, 269 CrossRef; (g) I.-P. Lorenz, M. Limmert, P. Mayer, H. Piotrowski, H. Langhals, M. Poppe and K. Polborn, Chem. – Eur. J., 2002, 8, 4047 CrossRef CAS PubMed; (h) J. Zhang, D.-Y. Kang, S. Barlow and S. R. Marder, J. Mater. Chem., 2012, 22, 21392 RSC; (i) A. Purc, M. Banasiewicz, E. Glodkowska-Mrowka and D. T. Gryko, J. Mater. Chem. C, 2016, 4 10.1039/C5TC03190G.
- (a) S. Stas, J.-Y. Balandier, V. Lemaur, O. Fenwick, G. Tregnago, F. Quist, F. Cacialli, J. Cornil and Y. H. Geerts, Dyes Pigm., 2013, 97, 198 CrossRef CAS; (b) H. Bürckstümmer, A. Weissenstein, D. Bialas and F. Würthner, J. Org. Chem., 2011, 76, 2426 CrossRef PubMed; (c) J. Liu, B. Walker, A. Tamayo, Y. Zhang and T.-Q. Nguyen, Adv. Funct. Mater., 2013, 23, 47 CrossRef CAS; (d) R. S. Szabadai, J. Roth-Barton, K. P. Ghiggino, J. M. White and D. J. D. Wilson, Aust. J. Chem., 2014, 67, 1330–1337 CAS; (e) A. Purc, K. Sobczyk, Y. Sakagami, A. Ando, K. Kamada and D. T. Gryko, J. Mater. Chem. C, 2015, 3, 742 RSC; (f) M. Grzybowski, E. Glodkowska-Mrowka, V. Hugues, W. Brutkowski, M. Blanchard-Desce and D. T. Gryko, Chem. – Eur. J., 2015, 21, 9101–9110 CrossRef CAS PubMed.
- (a) G. M. Fischer, A. R. Ehlers, A. Zumbusch and E. Daltrozzo, Angew. Chem., Int. Ed., 2007, 46, 3750 CrossRef CAS PubMed; (b) G. M. Fischer, M. Isomaki-Krondahl, I. Gottker-Schnetmann, E. Daltrozzo and A. Zumbusch, Chem. – Eur. J., 2009, 15, 4857 CrossRef CAS PubMed; (c) G. M. Fischer, C. Jungst, M. Isomaki-Krondahl, D. Gauss, H. M. Moller, E. Daltrozzo and A. Zumbusch, Chem. Commun., 2010, 46, 5289 RSC; (d) G. M. Fischer, M. K. Klein, E. Daltrozzo and A. Zumbusch, Eur. J. Org. Chem., 2011, 3421 CrossRef CAS; (e) S. Wiktorowski, G. M. Fischer, M. J. Winterhalder, E. Daltrozzo and A. Zumbusch, Phys. Chem. Chem. Phys., 2012, 14, 2921 RSC.
- (a) S. Shimizu, T. Iino, Y. Araki and N. Kobayashi, Chem. Commun., 2013, 49, 1621 RSC; (b) W. Yue, S.-L. Suraru, D. Bialas, M. Muller and F. Würthner, Angew. Chem., Int. Ed., 2014, 53, 6159 CrossRef CAS PubMed; (c) S. Shimizu, T. Iino, A. Saeki, S. Seki and N. Kobayashi, Chem. – Eur. J., 2015, 21, 2893–2904 CrossRef CAS PubMed.
- (a) M. Grzybowski, E. Glodkowska-Mrowka, T. Stoklosa and D. T. Gryko, Org. Lett., 2012, 14, 2670 CrossRef CAS PubMed; (b) M. Grzybowski, V. Hugues, M. Blanchard-Desce and D. T. Gryko, Chem. – Eur. J., 2014, 20, 12493 CrossRef CAS PubMed; (c) D. T. Gryko, M. Grzybowski, P. Hayoz and A. Jeżewski, BASF SE, Patent Appl, PCT/EP2014/054060, 2013 Search PubMed.
- (a) E. Q. Guo, P. H. Ren, Y. L. Zhang, H. C. Zhang and W. J. Yang, Chem. Commun., 2009, 5859 RSC; (b) C. Yang, M. Zheng, Y. Li, B. Zhang, J. Li, L. Bu, W. Liu, M. Sun, H. Zhang, Y. Tao, S. Xue and W. Yang, J. Mater. Chem. A, 2013, 1, 5172 RSC; (c) H. Ftouni, F. Bolze and J.-F. Nicoud, Dyes Pigm., 2013, 97, 77 CrossRef CAS; (d) H. Ftouni, F. Bolze, H. de Rocquigny and J.-F. Nicoud, Bioconjugate Chem., 2013, 24, 942 CrossRef CAS PubMed.
- B. Chen, Y. Yang, P. Cheng, X. Chen, X. Zhan and J. Qin, J. Mater. Chem. A, 2015, 3, 6894 CAS.
- M. Grzybowski and D. T. Gryko, Adv. Opt. Mater., 2015, 3, 280–320 CrossRef CAS.
- D. F. Friese, R. Bast and K. Ruud, ACS Photonics, 2015, 2, 572 CrossRef CAS PubMed.
- (a) M. A. Albota, C. Xu and W. W. Webb, Appl. Opt., 1998, 37, 7352–7356 CrossRef CAS PubMed; (b) C. Xu and W. W. Webb, J. Opt. Soc. Am. B, 1996, 13, 481–491 CrossRef CAS.
- M. H. V. Werts, N. Nerambourg, D. Pélégry, Y. Le Grand and M. Blanchard-Desce, Photochem. Photobiol. Sci., 2005, 4, 531–538 CAS.
- C. Katan, S. Tretiak, M. H. V. Werts, A. J. Bain, R. J. Marsh, N. Leonczek, N. Nicolaou, E. Badaeva, O. Mongin and M. Blanchard-Desce, J. Phys. Chem. B, 2007, 111, 9468–9483 CrossRef CAS PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS.
- A. Schäfer, C. Huber and R. Ahlrichs, J. Chem. Phys., 1989, 100, 5829 CrossRef.
- TURBOMOLE V6.6 2014, a development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989–2007, TURBOMOLE GmbH, since 2007; available from http://www.turbomole.com Search PubMed.
- K. Aidas, C. Angeli, K. L. Bak, V. Bakken, R. Bast, L. Boman, O. Christiansen, R. Cimiraglia, S. Coriani, P. Dahle, E. K. Dalskov, U. Ekstrøm, T. Enevoldsen, J. J. Eriksen, P. Ettenhuber, B. Fernández, L. Ferrighi, H. Fliegl, L. Frediani, K. Hald, A. Halkier, C. Hättig, H. Heiberg, T. Helgaker, A. C. Hennum, H. Hettema, E. Hjertenæs, S. Høst, I.-M. Høyvik, M. F. Iozzi, B. Jansík, H. J. Aa. Jensen, D. Jonsson, P. Jørgensen, J. Kauczor, S. Kirpekar, T. Kjærgaard, W. Klopper, S. Knecht, R. Kobayashi, H. Koch, J. Kongsted, A. Krapp, K. Kristensen, A. Ligabue, O. B. Lutnæs, J. I. Melo, K. V. Mikkelsen, R. H. Myhre, C. Neiss, C. B. Nielsen, P. Norman, J. Olsen, J. M. H. Olsen, A. Osted, M. J. Packer, F. Pawlowski, T. B. Pedersen, P. F. Provasi, S. Reine, Z. Rinkevicius, T. A. Ruden, K. Ruud, V. V. Rybkin, P. Sałek, C. C. M. Samson, A. Sánchez de Merás, T. Saue, S. P. A. Sauer, B. Schimmelpfennig, K. Sneskov, A. H. Steindal, K. O. Sylvester-Hvid, P. R. Taylor, A. M. Teale, E. I. Tellgren, D. P. Tew, A. J. Thorvaldsen, L. Thøgersen, O. Vahtras, M. A. Watson, D. J. D. Wilson, M. Ziolkowski and H. Ågren, The Dalton quantum chemistry program system, WIREs Comput. Mol. Sci., 2013, 4, 269 CrossRef PubMed.
- T. Yanai, D. P. Tew and N. C. Handy, Chem. Phys. Lett., 2004, 393, 51 CrossRef CAS.
- T. H. Dunning, J. Chem. Phys., 1989, 90, 1007 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Full synthetic and analytical data of compounds 2 and 3 as well as copies of 1H NMR and 1C NMR spectra. CCDC 1441247. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ob02583d |
| This journal is © The Royal Society of Chemistry 2016 |