
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Substrate co-doping modulates electronic metal–support interactions and significantly enhances single-atom catalysis†
J. L.
Shi
a,
J. H.
Wu
b,
X. J.
Zhao
a,
X. L.
Xue
a,
Y. F.
Gao
cd,
Z. X.
Guo
*ea and
S. F.
Li
*a
aInternational Laboratory for Quantum Functional Materials of Henan, School of Physics and Engineering, Zhengzhou University, Zhengzhou, Henan 450001, China. E-mail: sflizzu@zzu.edu.cn
bDepartment of Physics, Henan Institute of Education, Zhengzhou, 450046, China
cDepartment of Materials Science and Engineering, University of Tennessee, Knoxville, Tennessee 37996, USA
dMaterials Science and Technology Division, Oak Ridge National Laboratory, Oak Ridge, Tennessee 37831, USA
eDepartment of Chemistry, University College London, London WC1H 0AJ, UK
First published on 7th October 2016
Abstract
Transitional metal nanoparticles or atoms deposited on appropriate substrates can lead to highly economical, efficient, and selective catalysis. One of the greatest challenges is to control the electronic metal–support interactions (EMSI) between the supported metal atoms and the substrate so as to optimize their catalytic performance. Here, from first-principles calculations, we show that an otherwise inactive Pd single adatom on TiO2(110) can be tuned into a highly effective catalyst, e.g. for O2 adsorption and CO oxidation, by purposefully selected metal–nonmetal co-dopant pairs in the substrate. Such an effect is proved here to result unambiguously from a significantly enhanced EMSI. A nearly linear correlation is noted between the strength of the EMSI and the activation of the adsorbed O2 molecule, as well as the energy barrier for CO oxidation. Particularly, the enhanced EMSI shifts the frontier orbital of the deposited Pd atom upward and largely enhances the hybridization and charge transfer between the O2 molecule and the Pd atom. Upon co-doping, the activation barrier for CO oxidation on the Pd monomer is also reduced to a level comparable to that on the Pd dimer which was experimentally reported to be highly efficient for CO oxidation. The present findings provide new insights into the understanding of the EMSI in heterogeneous catalysis and can open new avenues to design and fabricate cost-effective single-atom-sized and/or nanometer-sized catalysts.
Introduction
Transitional metal (TM) and noble metal (NM) nanoparticles supported on oxide surfaces have been extensively used as efficient and economical catalysts in many industrial applications, such as CO oxidation in vehicle emission reduction,1–3 water–gas shift reaction,4 and hydrogenation.5 However, the intrinsic catalytic activities and selectivity of these supported metal particles are strongly size- and geometry-dependent, particularly at the nanometer and sub-nanometer scales. In such regimes, the catalysis of TMs/NMs even with a given shape is characterized by a significant size effect, due to the variation of the increasingly large fraction of the low-coordinate metal atoms that often function as active sites for catalysis.6–8 As the ultimate size limit, single-atom catalysts (SACs) deposited uniformly on appropriate substrates are widely expected to maximize the efficiency, activity and selectivity of TM and NM catalysts, particularly for the latter. For example, SACs of Pt, Rh, Pd and Ru on FeOx are reported to exhibit high performance for O2 activation and CO oxidation.9 A single Fe atom embedded in a silica matrix enhances methane activation.10 However, it was also experimentally reported that the catalytic activity for CO oxidation is insignificant for single Pd atoms on TiO2(110), but is substantially higher for Pd ad-dimers and larger adsorbed clusters.3 Similarly, Au monomers show negligible catalytic activity on TiO2(110) for CO oxidation though Au single atoms exhibit excellent catalysis on other substrates.4,11Clearly, the substrate also plays a crucial role in the chemical activity and selectivity of the deposited nanocatalysts,12–14 as first recognized by Tauster et al.13 in 1978, and now well known as the “strong metal–support interactions (SMSI)”. Recently, Bruix et al.14 provided an in-depth analysis of the effect of SMSI on the significantly enhanced activity of Pt particles on ceria in water–gas shift reactions. More specifically, Campbell15 coins the findings of Bruix and co-workers14 as “electronic metal–support interactions” (EMSI) manifested by the chemical bonding and associated charge transfer at the metal–support interface. Namely, the enhanced activity of the deposited particles may be essentially attributed to electronic perturbations that modify the electronic states of the deposited metal and improve their catalytic properties due to the EMSI. To demonstrate more convincingly the importance of the EMSI in the activity of a metal catalyst, it is imperative to focus on SAC systems,16 which can unambiguously rule out other factors, such as the electronic quantum size effect17,18 and structural sensitivity19,20 involved in nanoparticles. In Hu and co-worker's recent experiment the role of the EMSI is established by comparison of the activities of Ag-SACs on two types of supports but with different local geometric structures, which in some sense dilutes the pure electronic effect of the EMSI.
Here, using the state-of-the-art first-principles calculations, we consider a Pd atom supported in the vicinity of the surface oxygen vacancy of the rutile TiO2(110), Pd@TiO2(110), as a representative SAC for CO oxidation, to establish the role of a pure electronic effect in the EMSI in controlling the catalysis of a given SAC. A comparative investigation was carried out for the catalysis of Pd@TiO2(110) and those with metal–nonmetal co-dopant pairs in the sub-layer, while keeping the local geometric structure of the Pd atom almost constant. In contrast to Bruix's model catalyst, the present systems can mostly rule out the geometric effect of the substrates in identifying the “pure” electronic contribution to the EMSI effect. Intriguingly, a nearly linear correlation between the strength of the EMSI and the CO oxidation rate was established in the studied systems, i.e., the stronger the EMSI, the larger the CO oxidation rate.
Methods
Our DFT calculations21 were performed using the Vienna ab initio simulation package (VASP)22 with the projector augmented wave (PAW)23 method. For the exchange–correlation energy, we employed the generalized gradient approximation functional of Perdew–Burke–Ernzerhof.24 Simulation details are presented in S1 of the ESI.†Results and discussion
First of all, we confirm that the Pd single atom prefers to be located in the vicinity of the surface oxygen vacancy (VO) site of the TiO2(110) surface, as deduced from both the energetics and low diffusion rates of the Pd atoms observed in the experiment,25 supported also by recent state-of-the-art first-principles calculations.26 Based on this, the O2 adsorption and CO oxidation process are re-examined by extensive additional calculations. The present results are in close agreement with previous calculations,26i.e., the O2 molecule can only weakly adsorb on the Pd@TiO2(110) complex with an adsorption energy, i.e., Eads(O2) = 0.241 eV, and the CO oxidation experiences a large activation barrier of 1.132 eV. Therefore, both the small binding energy and the large CO oxidation barrier together determine the low catalytic activity of Pd@TiO2(110). For more details on the geometric structure of Pd@TiO2(110) and the minimum energy path for CO oxidation on it, see Fig. S1.†To optimize the chemical activity of the intrinsically inert Pd@TiO2(110), co-doping is invoked here to modulate the electronic structures of the TiO2(110) substrate and consequently tune the EMSI between the Pd metal atom and the co-doped substrates. It is widely accepted that the substitutional doping of a binary oxide semiconductor is extremely difficult due to the limited solubility of the dopants, especially for p-type doping.27 Recently, Zhu et al.28 reported that the introduction of donor–acceptor co-dopant pairs is an effective approach to enhance the solubility of dopants in TiO2. In view of this, we choose two typical 3d transition metals, V and Cr, as the n-type dopants to substitute for a Ti atom, and two nonmetal elements, N and C, as the p-type dopants to substitute for a neighboring O site, respectively. Therefore, four metal–nonmetal co-dopant pairs can be obtained, i.e., p-type V–C, compensated V–N and Cr–C, and n-type Cr–N, respectively.
Fig. 1 depicts the most stable configurations of the four co-doped Pd@TiO2(110) complexes. Energetically, it is confirmed that the co-dopant pairs do prefer to form a metal–nonmetal dimer in Pd@TiO2(110), due to the strong Columbic electrostatic attraction between the metal donor and nonmetal acceptor ions. Consequently, the introduction of co-dopant pairs into the TiO2 substrate leads to three important features for the binding of the Pd on the defective TiO2(110) surface. Firstly, without co-doping, the Pd atom favors exactly the VO site, whereas it slightly departs from the VO in the co-doped cases; however, upon O2 and CO co-adsorption, the Pd atom on pristine TiO2(110) also relaxes to the same local site as that in the co-doped cases, which will be discussed later. Secondly, as shown in Fig. 1, the Pd atom possesses similar local supporting structures in all the four co-doped cases. For details on the bond lengths of the Pd atom with the substrates, see S3 of the ESI.† Thirdly, the co-dopant pairs enhance the electronic binding of the Pd atom with the TiO2(110) substrate, i.e., from 1.641 to 1.788, 1.769, 1.768 and 1.768 eV, for the un-doped, V–C, V–N, Cr–C, and Cr–N co-doped Pd@TiO2(110), respectively.
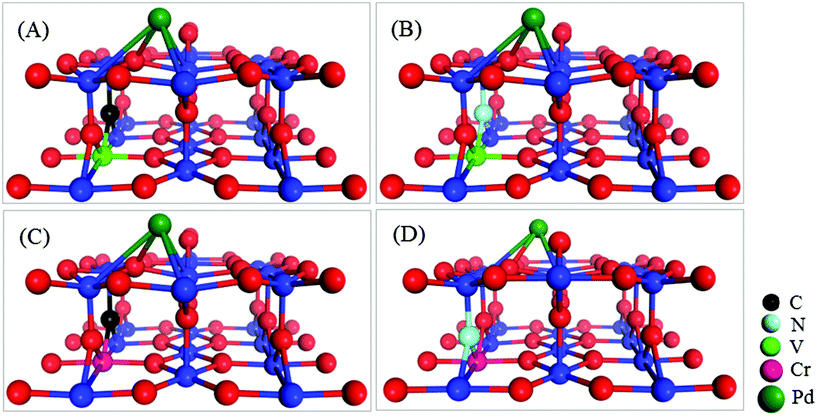 | ||
| Fig. 1 Local geometric structures of co-doped Pd@TiO2(110) complexes with (A) V–C; (B) V–N; (C) Cr–C; and (D) Cr–N co-dopant pairs. | ||
Now, we discuss the doping effect in modulating the electronic structures of the Pd atom through the EMSI. In Fig. 2, the total density of states (DOS) of the co-doped Pd@TiO2(110) species including the local projected DOS (LPDOS) of the dopants and the Pd atom are presented. For comparison, the LPDOS of the un-doped Pd@TiO2(110) are also shown in Fig. 2(A). As shown in Fig. 2(B)–(E), the energy gap between the valence band maximum (VBM) and the conduction band minimum (CBM) is significantly reduced by co-doping. Specifically, in the un-doped Pd@TiO2(110) system, the VBM is dominated by the p orbital of the O atoms and the CBM is located in the vicinity of the lowest-unoccupied-molecular-orbital (LUMO) of the Pd atom, respectively. In addition, the HOMO–LUMO gap (Egap(HOMO–LUMO)) of the Pd atom is about 1.464 eV. Upon doping, both the VBM and CBM are changed due to the introduction of the co-dopant states and the resulting EMSI. Distinctly, in all these four co-doped cases, the VBM is shifted upward and now positioned in the vicinity of the HOMO of the co-dopant pairs (see Fig. 2(B)–(E)), and the CBM is further lowered except for the cases of V–N co-doping (see Fig. 2(C)). Importantly, the frontier orbital (HOMOs) of the Pd atom hybridizes with the co-dopant states by the Fermi level, which effectively shifts the HOMOs of the Pd single atom upwards. For example, in the p-type V–C co-doped complex, the (d-electron dominated) HOMOs of the P atom hybridize with the HOMOs of the V atom and those of the C atom in the energy window of −0.6 to −0.3 eV. Furthermore, the HOMO of the Pd atom is now upward shifted by about 0.5 eV, see Fig. 2(B). Moreover, such an orbital hybridization between the Pd atom and the substrate, particularly with the doped metal cations, is accompanied by charge transfer from the former to the latter via a charge compensation mechanism. Therefore, such a charge transfer leads the Pd atom to depart from its closed-shell (d10) characteristics, and is now positively charged, as confirmed by the Bader charge analysis presented in Table 1. Here, we emphasize that it is just the hybridization and charge transfer that increase the HOMO of the Pd atom and enhance its chemical activity towards oxygen adsorption and activation, which will be illustrated shortly.
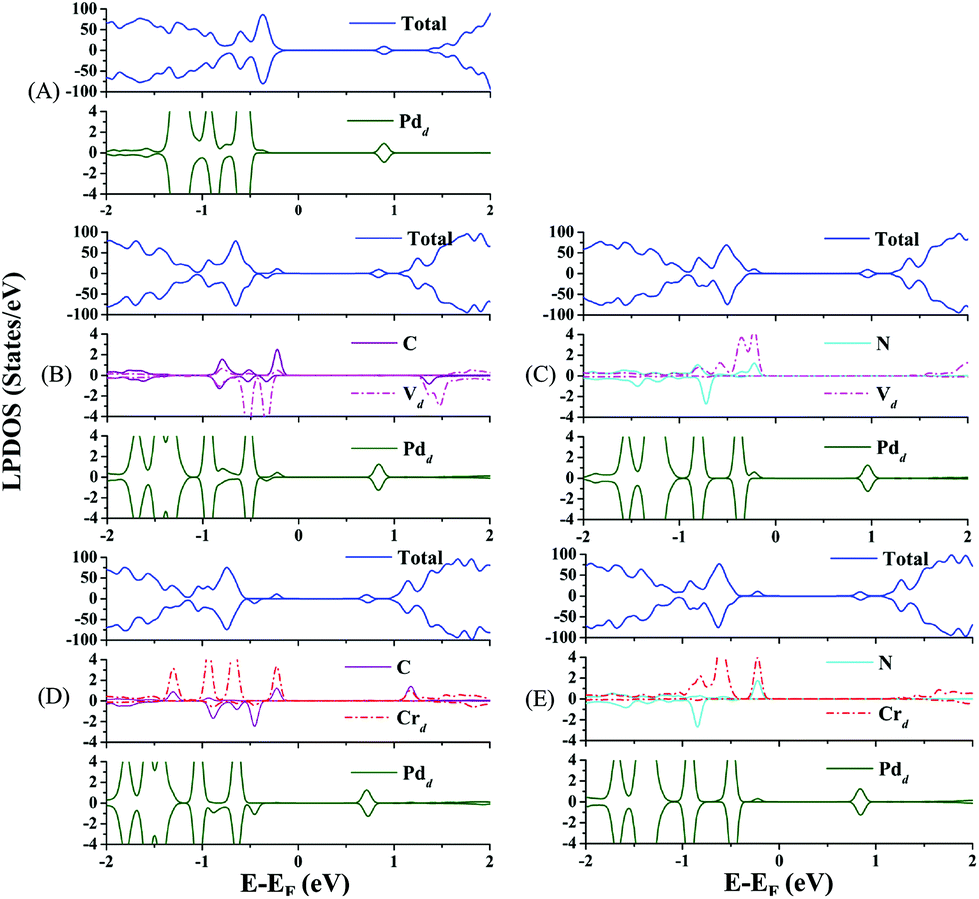 | ||
| Fig. 2 Total and local projected density of states (DOS) of (A) pristine rutile Pd@TiO2(110); and that of (B) V–C, (C) V–N, (D) Cr–C and (E) Cr–N co-doped systems. | ||
| Systems | E b(Pd)(eV) | Q(Pd)(e) | E ads(O2)(eV) | R(O–O)(Å) | Q(O2)(e) | ω(cm−1) |
|---|---|---|---|---|---|---|
| Pd@TiO2(110):V–C | 1.788 | +0.161 | 0.808 | 1.275 | 0.22 | 1296.32 |
| Pd@TiO2(110):V–N | 1.769 | +0.160 | 0.732 | 1.274 | 0.21 | 1302.04 |
| Pd@TiO2(110):Cr–C | 1.768 | +0.164 | 0.794 | 1.278 | 0.22 | 1284.58 |
| Pd@TiO2(110):Cr–N | 1.768 | +0.162 | 0.801 | 1.277 | 0.23 | 1348.51 |
| Pd@TiO2(110) | 1.641 | +0.158 | 0.241 | 1.254 | 0.10 | 1560.83 |
Adsorption of O2 molecules on co-doped Pd@TiO2(110) SACs
To further identify the co-doping effect in modulating the chemical activity of the Pd atom via the EMSI, we now investigate the key process of CO oxidation, i.e., the adsorption and activation of an O2 molecule on the co-doped structures presented in Fig. 1. Upon extensive calculations, we identified the most stable configurations for O2 adsorption on the four co-doped Pd@TiO2(110) complexes, as shown in Fig. 3. Note that in the optimized O2-Pd@TiO2(110) co-doped structures, the Pd atoms only binds directly with one oxygen atom of the O2 molecule, which is very similar to the O2 adsorption on other low dimensional noble metal structures.29,30 In contrast to the case of the un-doped Pd@TiO2(110), the O2 molecule is found to bind strongly to the Pd atom deposited on the doped TiO2(110) substrates. Specifically, as summarized in Table 1, the adsorption energy (Eads(O2)) of the O2 molecule is increased from 0.241 to 0.808, 0.732, 0.794, and 0.801 eV, for pristine Pd@TiO2(110), the V–C, V–N, Cr–C, and Cr–N co-doped complexes, respectively. Meanwhile, in the V–C, V–N, Cr–C, and Cr–N co-doped cases, the stretching vibrational frequency of the adsorbed O2 molecule is further red-shifted from 1560.83 to 1296.32, 1302.04, 1284.58, and 1348.51 cm−1, due to the enlarged O–O bond lengths, i.e., 1.275, 1.274, 1.278, and 1.277 Å, respectively. These data convincingly indicate that the O2 molecule is considerably activated on these co-doped Pd@TiO2(110) SACs due to the enhanced EMSI.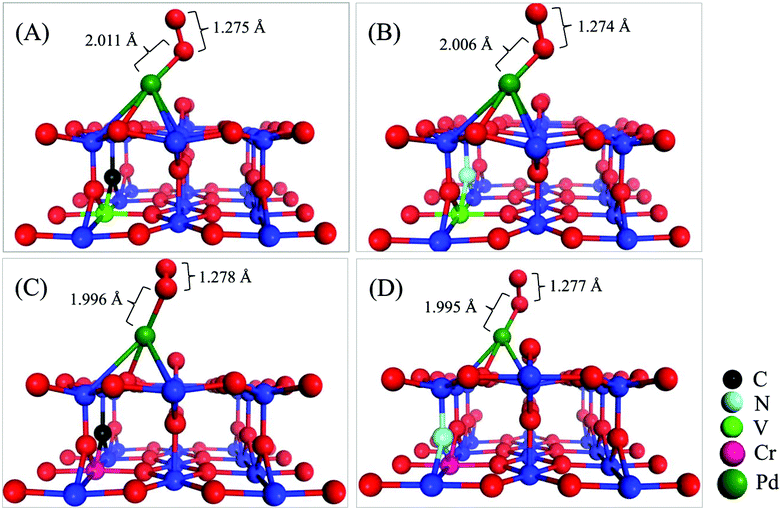 | ||
| Fig. 3 Geometric O2 adsorption structures of co-doped Pd@TiO2(110) complexes with (A) V–C; (B) V–N; (C) Cr–C; and (D) Cr–N co-dopant pairs. | ||
Now, we further explore the underlying mechanism of the EMSI in improving the chemical activity of the Pd atom via co-doping. In doing so, we analyze the DOS of the optimized final states of the O2 molecule adsorbed on the Pd atom on TiO2(110) co-doped by V–C, V–N, Cr–C, and Cr–N pairs, respectively. In Fig. 4(B)–(E), the LPDOS of all the Ti atoms, Pd atoms, co-dopant pairs, and the adsorbed O2 species are presented. In addition, the DOS for O2 on the un-doped Pd@TiO2(110) is also shown as a comparison in Fig. 4(A). Here, to illustrate more clearly the role of the EMSI, we only focus on an energy window of −2.0 to 2.0 eV by the Fermi level in analyzing the DOS. As shown in Fig. 4(A), for O2 adsorption on the un-doped Pd@TiO2(110), there is negligible hybridization between the HOMO of the deposited Pd atom and the LUMO of the O2 molecule, due to the relatively deep energy level of the HOMO and the large Egap(HOMO(Pd)–LUMO(O2)). Therefore, O2 can only weakly bind to the Pd@TiO2(110) SAC with a minor charge transfer obtained by Bader charge analysis, i.e., about 0.1e from the Pd to the adsorbed O2 species. However, upon co-doping, the d-electron dominated HOMO of the Pd atom hybridizes with that of the co-dopant pairs in the vicinity of the Fermi level. These hybridized filled states serve as an effective electron reservoir for the incoming O2 molecule to capture the electron charge. More specifically, taking the O2 adsorption on the Cr–N co-doped case as an example, one can see that though the Pd atom only slightly hybridizes with the HOMO (which is about 0.2 eV below the Fermi level) of the Cr atom, in the final state, such an orbital hybridization is significantly enhanced, see Fig. 4(E); meanwhile, for the adsorbed O2 molecule there exists considerable spin-minority LPDOS hybridization with that of the Pd atom around 0.7 eV below the Fermi level. We emphasize that when the O2 molecule is far away from the Pd atom, the spin-minority LPDOS (O2) is about 1.2 eV above the Fermi level and totally unfilled. These results confirm an evident charge transfer (about 0.3e) from the SAC to the O2 molecule. Therefore, one can conclude that the n-type Cr–N pair (essentially the metal Cr) plays an important role in serving as the charge reservoir, compensating for the charge of the Pd atom, and thus enhancing the interaction with the O2 molecule. Subsequently, such a charge transfer weakens the O–O bond and further strengthens the O2 activation as manifested by the enlarged Eads(O2) of 0.801 eV and red-shifted stretching vibration frequency of 1348.51 cm−1, see Table 1.
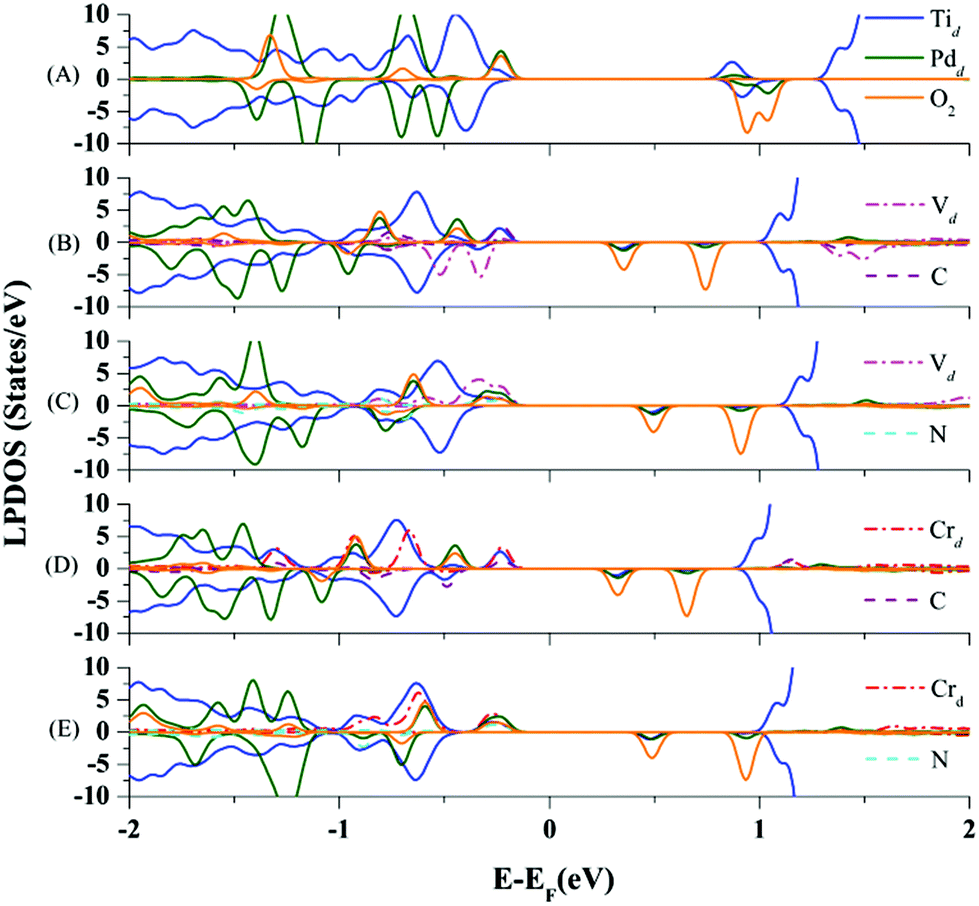 | ||
| Fig. 4 Local projected density of states of optimized structures for O2 molecule adsorption on (A) pristine rutile Pd@TiO2(110); and that of (B) V–C, (C) V–N, (D) Cr–C and (E) Cr–N co-doped complexes. | ||
Note that the p-type V–C, and the two compensated V–N and Cr–C couplings also significantly facilitate the O2 adsorption and activation via a similar mechanism due to the enhanced EMSI. As shown in Fig. 3(B)–(D), the co-dopant-assisted enhanced hybridizations between the Pd atom and the O2 molecule are also observed in the energy range of −1.0 to 1.0 eV by the Fermi levels. Such interactions stimulate the charge transfer between the O2 molecule and SAC, and enhance the O2 activation, as seen from the calculated data summarized in Table 1. Based on the present findings, we can conclude that the chemical activity of the intrinsically inert Pd atom can be effectively optimized by the EMSI via the co-doping approach. To further identify the dominated intrinsic parameter in such an optimization rule, we analyze the DOS of the O2 molecule far (about 5.0 Å) above these co-doped Pd@TiO2(110) SACs, including other n-type co-doping, such as the Cr–S pair; for details, see S4 of the ESI.† We find that, overall, the smaller the Egap(HOMO(Pd)–LUMO(O2)), the larger the Eads(O2), as presented in Fig. 5, in line with the well-known d-band theory.31,32 Note again that, as presented in the ESI (S4)† and the above discussion, the reduced Egap(HOMO(Pd)–LUMO(O2)) originates from an enhanced EMSI through n–p co-doping, i.e., the stronger the EMSI, the smaller the Egap(HOMO(Pd)–LUMO(O2)); and consequently the stronger the O2 activation.
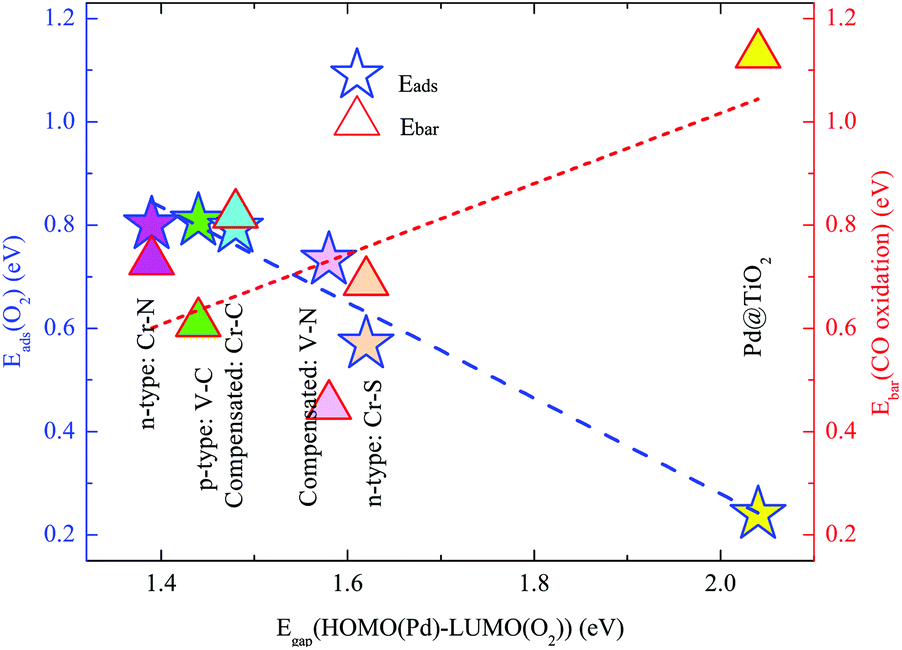 | ||
| Fig. 5 Adsorption energy of the O2 molecule, Eads(O2) (activation barrier for CO oxidation, Ebar(CO oxidation)) on Pd@TiO2(110) and co-doped counterparts with V–C, V–N, Cr–C, Cr–N, and Cr–S co-dopant pairs. | ||
CO oxidation on n–p co-doped Pd@TiO2(110)
Having clearly illustrated the critical step of O2 activation on the n–p co-doped Pd@TiO2(110) systems, we continue to investigate the kinetic processes of CO oxidation on the improved Pd@TiO2(110) SACs. Typically, taking the p-type V–C and the n-type Cr–N co-doped cases as prototypical examples, we investigate the EMSI in optimizing CO oxidation rates by performing substantial simulations using the NEB method.33First, three CO oxidation mechanisms proposed by recent theoretical studies34,35 have been re-examined in the present work. Interestingly, we confirm that the CO oxidation prefers the Langmuir–Hinshelwood (L–H) process in both cases, i.e., CO can also adsorb on the single Pd atom nearby the O2 molecule and the co-adsorbed molecules undergo a bimolecular reaction through the formation of a CO2 precursor, and then releasing the CO2 molecule upon further activation. Note also that for both the pristine and the co-doped Pd@TiO2(110) complexes, upon CO and O2 co-adsorption, the Pd atom is located in almost the same position close to the surface VO site, see also Fig. S1(B)† and Fig. 6. The detailed pathways and energetics for both cases are shown in Fig. 6. More specifically, as shown in Fig. 6(A), in the case of the p-typed V–C co-doped Pd@TiO2(110) system (see structure (i)), we find that the first CO molecule easily adsorbs in the vicinity of the O2 molecule on the Pd catalyst (structure (ii)) via the well-known back-donation charge transfer mechanism,32,36i.e., donation of CO 5σ electrons to the Pd@TiO2(110) substrate and back-donation from the Pd metal atom into the unoccupied 2π* orbital of CO. Here, we note that, as shown in Fig. 2, the upward-shifted HOMO and slightly downward shifted LUMO orbitals of the Pd atom promotes the back-donation process due to the enhanced EMSI. Consequently, the bond length of the adsorbed CO species is slightly enlarged to 1.154 Å from 1.143 Å of the gas phase, and correspondingly the C–O vibrational frequency is red shifted to 2037.56 cm−1 from 2120.64 cm−1. Such a back-donation interaction effectively weakens the C–O bond strength and facilitates the formation of a bent CO2 intermediate species when the CO molecule attacks the adsorbed O2 species. Note that the modest activation of the CO as manifested by the red-shifted vibration frequency renders the Pd atom to be a good SAC candidate to avoid poisoning. An endothermic process with a low activation barrier of Ebar = 0.608 (see Fig. 6(A)-(iii)) for CO2 formation and Ebar = 0.182 eV for CO2 desorption are observed, respectively. Furthermore, when the first CO molecule is oxidized to release a CO2, as presented in Fig. 6(A)-(iv), on the Pd atom there is still one O atom left which can be directly attached by the second incoming CO molecule during the formation of the second CO2, i.e., via the Eley–Rideal (E–R) mechanism (see Fig. 6(A)-(v)), with an activation barrier of only 0.211 eV. After this, a second round of O2 activation and CO oxidation can be continued.
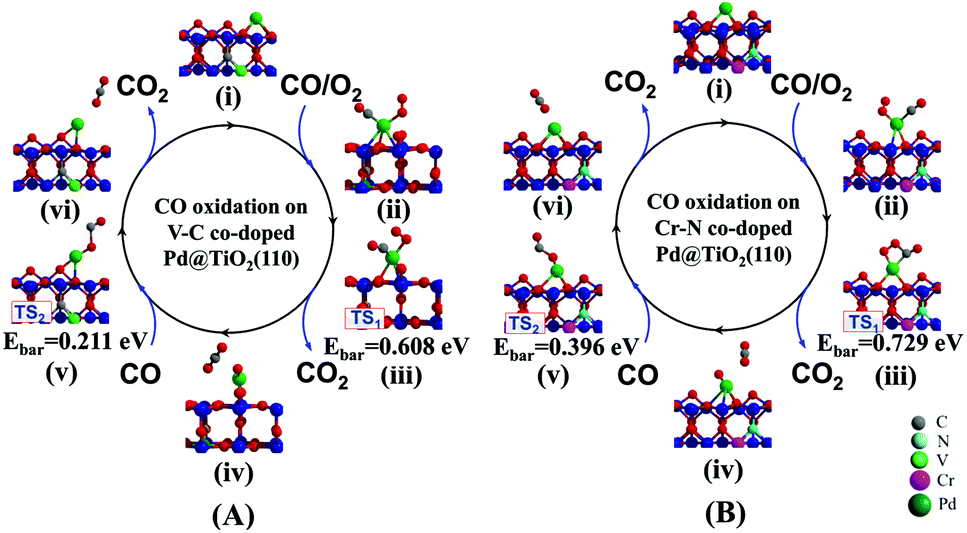 | ||
| Fig. 6 Minimum energy paths for CO oxidation on Pd@TiO2(110) co-doped with prototypical metal–nonmetal pairs. (A) p-Type V–C and (B) n-type Cr–N co-dopants. | ||
For the case of n-type Cr–N co-doping, see Fig. 6(B)–(i), a slightly higher energy barrier of 0.729 eV (see structure (iii)) is detected for the first CO molecule oxidation via the L–H mechanism and 0.396 eV (see structure (v)) for the second CO molecule oxidation via the E–R process, respectively. Such similar rate-determining kinetic processes of the CO oxidation in these two cases can be ascribed to the comparable O2 activations, as indicated by the negligible differences in the Eads(O2) and enlarged O–O bond lengths, as well as the very close values of the red-shifted O–O vibrational frequencies. Here, we also emphasize that in the un-doped Pd@TiO2(110), the large activation barrier for CO oxidation and the significantly small Eads(O2) together dominated the low catalysis of the Pd SAC in the vicinity of the VO. However, the calculated enlarged Eads(O2)/Eads(CO) of 0.808/0.705, 0.801/0.792, 0.794/0.606, and 0.732/0.443 eV on the p-type V–C, n-type Cr–N, compensated Cr–C, and compensated V–N co-doped Pd@TiO2(110), along with the reduced Ebar values of 0.608, 0.729, 0.822 and 0.423 eV for CO oxidation render the Pd atom to be effective for CO oxidation on the same site as that in the un-doped TiO2(110), demonstrating the crucial role of the EMSI in tuning the catalysis of the Pd atom. From Fig. 5, one can see that, the calculated values of the Ebar for the case of pristine Pd@TiO2(110) and those of five co-doped cases establish an overall linear scale behavior as a function of the HOMO(Pd)–LUMO(O2) gap, except for the case of the V–N co-doping, which slightly departs from the fitted linear trend due to the relatively large energy gain upon the local geometric relaxation around the Pd single atom during the CO oxidation, see S5 of the ESI.† Importantly, we emphasize that an activation barrier of about 0.68 eV is calculated for CO oxidation on the Pd2@TiO2(110),26 which was reported to possess good performance for CO oxidation in the experiment.3 Therefore, the present results suggest that upon co-doping with low-cost metal–nonmetal pairs, the intrinsically inert noble Pd SAC is now expected to possess highly efficient catalysis for CO oxidation.
Conclusions
In conclusion, using the state-of-the-art first-principles calculations, we performed a comparative study on the electronic structures of a Pd single ad-atom over defective and metal–nonmetal co-doped TiO2(110) substrates to establish the key role of the EMSI in optimizing the catalysis of SACs. Via the enhanced EMSI by co-doping, the intrinsically inert Pd atom on pristine TiO2(110) can be tuned to serve as an effective electron charge reservoir sustained by the hybridized states of the co-dopant pairs to facilitate O2 activation and CO oxidation. Interestingly, a close-linear correlation between the strength of the EMSI and the activation of the adsorbed O2 molecule, as well as the energy barrier for CO oxidation, is observed in the cases of the Pd atoms deposited on both pristine TiO2(110) and co-doped substrates with low-cost metal–nonmetal pairs. The present findings provide new insights into the understanding of the EMSI in heterogeneous catalysis and can open new avenues to design and fabricate highly efficient and cost-effective single-atom- and/or nanometer-sized catalysts.Acknowledgements
We thank Professor Jun-Hyung Cho, Professor Yu Jia and Professor Zhenyu Zhang for helpful discussion. This work was supported by the NSFC (Grant No. 11074223, 11034006 and 11674289), and partly by the UK EPSRC (EP/L0183301/1) and US NSF (CMMI-1300223).References
- B. Qiao, A. Wang, X. Yang, L. F. Allard, Z. Jiang, Y. Cui, J. Liu, J. Li and T. Zhang, Nat. Chem., 2011, 3, 634–641 CrossRef CAS PubMed.
- M. Moses-DeBusk, M. Yoon, L. F. Allard, D. R. Mullins, Z. Wu, X. Yang, G. Veith, G. M. Stocks and C. K. Narula, J. Am. Chem. Soc., 2013, 135, 12634–12645 CrossRef CAS PubMed.
- W. E. Kaden, T. Wu, W. A. Kunkel and S. L. Anderson, Science, 2009, 326, 826–829 CrossRef CAS PubMed.
- Q. Fu, H. Saltsburg and M. Flytzani-Stephanopoulos, Science, 2003, 301, 935–938 CrossRef CAS PubMed.
- G. Vilé, D. Albani, M. Nachtegaal, Z. Chen, D. Dontsova, M. Antonietti, N. López and J. Pérez-Ramírez, Angew. Chem., Int. Ed., 2015, 54, 11265–11269 CrossRef PubMed.
- N. Lopez, T. V. W. Janssens, B. S. Clausen, Y. Xu, M. Mavrikakis, T. Bligaard and J. K. Nørskov, J. Catal., 2004, 223, 232–235 CrossRef CAS.
- H. Zhang, T. Watanabe, M. Okumura, M. Haruta and N. Toshima, Nat. Mater., 2012, 11, 49–52 CrossRef PubMed.
- L. Li, A. H. Larsen, N. A. Romero, V. A. Morozov, C. Glinsvad, F. Abild-Pedersen, J. Greeley, K. W. Jacobsen and J. K. Nørskov, J. Phys. Chem. Lett., 2013, 4, 222–226 CrossRef CAS PubMed.
- F. Li, Y. Li, X. C. Zeng and Z. Chen, ACS Catal., 2015, 5, 544–552 CrossRef CAS.
- X. Guo, G. Fang, G. Li, H. Ma, H. Fan, L. Yu, C. Ma, X. Wu, D. Deng, M. Wei, D. Tan, R. Si, S. Zhang, J. Li, L. Sun, Z. Tang, X. Pan and X. Bao, Science, 2014, 344, 616–619 CrossRef CAS PubMed.
- S. Lee, C. Fan, T. Wu and S. L. Anderson, J. Am. Chem. Soc., 2004, 126, 5682–5683 CrossRef CAS PubMed.
- J. Zhang and A. N. Alexandrova, J. Phys. Chem. Lett., 2013, 4, 2250–2255 CrossRef CAS.
- S. J. Tauster, S. C. Fung and R. L. Garten, J. Am. Chem. Soc., 1978, 100, 170–175 CrossRef CAS.
- A. Bruix, J. A. Rodriguez, P. J. Ramírez, S. D. Senanayake, J. Evans, J. B. Park, D. Stacchiola, P. Liu, J. Hrbek and F. Illas, J. Am. Chem. Soc., 2012, 134, 8968–8974 CrossRef CAS PubMed.
- C. T. Campbell, Nat. Chem., 2012, 4, 597–598 CrossRef CAS PubMed.
- P. Hu, Z. Huang, Z. Amghouz, M. Makkee, F. Xu, F. Kapteijn, A. Dikhtiarenko, Y. Chen, X. Gu and X. Tang, Angew. Chem., Int. Ed., 2014, 53, 3418–3421 CrossRef CAS PubMed.
- O. Lopez-Acevedo, K. A. Kacprzak, J. Akola and H. Häkkinen, Nat. Chem., 2010, 2, 329–334 CrossRef CAS PubMed.
- M. Valden, X. Lai and D. W. Goodman, Science, 1998, 281, 1647–1650 CrossRef CAS PubMed.
- S. Dahl, A. Logadottir, R. C. Egeberg, J. H. Larsen, I. Chorkendorff, E. Törnqvist and J. K. Nørskov, Phys. Rev. Lett., 1999, 83, 1814–1817 CrossRef.
- J. K. Norskov, T. Bligaard, B. Hvolbaek, F. Abild-Pedersen, I. Chorkendorff and C. H. Christensen, Chem. Soc. Rev., 2008, 37, 2163–2171 RSC.
- P. Hohenberg and W. Kohn, Phys. Rev. B, 1964, 136, B864 CrossRef.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter, 1994, 49, 14251 CrossRef CAS.
- P. E. Blochl, Phys. Rev. B: Condens. Matter, 1994, 50, 17953 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- C. Xu, X. Lai, G. W. Zajac and D. W. Goodman, Phys. Rev. B: Condens. Matter, 1997, 56, 13464–13482 CrossRef CAS.
- S. Li, X. Zhao, J. Shi, Y. Jia, Z. Guo, J.-H. Cho, Y. Gao and Z. Zhang, Phys. Chem. Chem. Phys., 2016, 18, 24872 RSC.
- S. B. Zhang, J. Phys.: Condens. Matter, 2002, 14, R881 CrossRef CAS.
- W. Zhu, X. Qiu, V. Iancu, X.-Q. Chen, H. Pan, W. Wang, N. M. Dimitrijevic, T. Rajh, H. M. Meyer, M. P. Paranthaman, G. M. Stocks, H. H. Weitering, B. Gu, G. Eres and Z. Zhang, Phys. Rev. Lett., 2009, 103, 226401 CrossRef PubMed.
- H. Häkkinen, S. Abbet, A. Sanchez, U. Heiz and U. Landman, Angew. Chem., Int. Ed., 2003, 42, 1297–1300 CrossRef PubMed.
- I. N. Remediakis, N. Lopez and J. K. Nørskov, Angew. Chem., Int. Ed., 2005, 44, 1824–1826 CrossRef CAS PubMed.
- B. Hammer, Y. Morikawa and J. K. Norskov, Phys. Rev. Lett., 1996, 76, 2141–2144 CrossRef CAS PubMed.
- L. P. A. Nilsson and J. K. Nørskov, Chemical Bonding at Surfaces and Interfaces, Elsevier, Amsterdam, 2008 Search PubMed.
- G. Henkelman, B. P. Uberuaga and H. Jónsson, J. Chem. Phys., 2000, 113, 9901–9904 CrossRef CAS.
- H. Y. Kim, H. M. Lee and G. Henkelman, J. Am. Chem. Soc., 2012, 134, 1560–1570 CrossRef CAS PubMed.
- C. Zhang, A. Michaelides and S. J. Jenkins, Phys. Chem. Chem. Phys., 2011, 13, 22–33 RSC.
- R. Hoffmann, Rev. Mod. Phys., 1988, 60, 601–628 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Details on the simulation methods, additional data are presented in Fig. S1–S5. See DOI: 10.1039/c6nr04292a |
| This journal is © The Royal Society of Chemistry 2016 |