
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Diamondoid-functionalized gold nanogaps as sensors for natural, mutated, and epigenetically modified DNA nucleotides
Ganesh
Sivaraman
a,
Rodrigo G.
Amorim
bc,
Ralph H.
Scheicher
b and
Maria
Fyta
 *a
*a
aInstitute for Computational Physics, Universität Stuttgart, Germany. E-mail: mfyta@icp.uni-stuttgart.de
bDivision of Materials Theory, Department of Physics and Astronomy, Uppsala University, Box 516, SE-751 20 Uppsala, Sweden
cDepartamento de Física, ICEx, Universidade Federal Fluminense – UFF, Volta Redonda/RJ, Brazil
First published on 5th April 2016
Abstract
Modified tiny hydrogen-terminated diamond structures, known as diamondoids, show a high efficiency in sensing DNA molecules. These diamond cages, as recently proposed, could offer functionalization possibilities for gold junction electrodes. In this investigation, we report on diamondoid-functionalized electrodes, showing that such a device would have a high potential in sensing and sequencing DNA. The smallest diamondoid including an amine modification was chosen for the functionalization. Here, we report on the quantum tunneling signals across diamondoid-functionalized Au(111) electrodes. Our work is based on quantum-transport calculations and predicts the expected signals arising from different DNA units within the break junctions. Different gating voltages are proposed in order to tune the sensitivity of the functionalized electrodes with respect to specific nucleotides. The relation of this sensitivity to the coupling or decoupling of the electrodes is discussed. Our results also shed light on the sensing capability of such a device in distinguishing the DNA nucleotides, in their natural and mutated forms.
1. Introduction
Nanometer-sized pores opened in a material have shown high potential as a cheap and ultra-fast third generation sequencing technology.1–9 A nanopore sequencer involves a single molecule technique. This does not require any labels or amplification. Such a device can potentially sequence almost in real time. Nanopores are of low cost and compatible with conventional electronics. They can be rendered into a parallel high-throughput device for reading-out DNA and are able to support long read lengths. Nanopores can electrophoretically translocate biomolecules (DNA, RNA, and proteins) within a salt solution, giving rise to ionic current blockades. The ionic signals can in turn be used to detect the translocation of the biopolymers.10Different sequencing protocols, that is ways to identify the bases along DNA, have been proposed. Among these, the use of transverse current signals across nanopores shows a high promise in view of sequencing applications.11,12 Specifically, in a tunneling recognition scheme a voltage bias which is applied across the electrodes is able to capture the translocating single stranded DNA (ssDNA).13–15 It involves a solid state nanopore including break junction electrodes which can measure the transverse tunneling current, detect the difference in the electronic structure of the translocating nucleobases, and read out the genetic information (i.e. sequence the nucleobases). This method of sensing can be very efficient, but the distinctive coupling of the different nucleotide species to the electrodes needs to be improved and there should be a sufficient sampling in the measurement of each nucleotide located between the electrodes.16,17
A possible improvement is to functionalize the electrodes with small molecules which can form hydrogen bond bridges with the translocating nucleotides.18 In this way, the functionalizing molecule introduces molecular states into the electronic gap of the band structure of the electrodes across which the electrons can hop enhancing the tunneling current.19 Recently, we have shown the potential of amine-derivatives of lower diamondoids to be used as functionalization molecules in a nanogap.20,21 Diamondoids are tiny hydrogenated diamond cages, with tunable electronic properties and a variety of potential applications.22 The smallest diamondoids with a diameter up to a few nanometers are commonly known as ‘lower diamondoids’. The hydrogen bonding between amine-modified diamondoids and DNA nucleobases was proven to be strong enough to be detected, while a comparison of the electronic properties of the diamondoid/nucleobase complexes showed distinguishable characteristics.20 This suggests a possible electronic detection of the nucleobase identity, which needs to be confirmed by an inspection of the tunneling current signals.
To this end, in this work, we perform quantum transport simulations as a proof of concept of the ability of diamondoid-functionalized electrodes to identify different types of DNA nucleotides. Note that nucleotides are the nucleobases attached to a sugar-phosphate group of the DNA backbone. In the following, a lower diamondoid, amantadine (ama), is used as the functionalization molecule of gold electrodes. These electrodes can potentially be embedded in a solid-state nanopore. In order to effectively attach the functionalizing molecule on the gold surface of the electrode, one hydrogen atom of the diamondoid cage has been replaced with a thiol group at the apical position.24 Our investigation will involve four nucleotides, as well as a mutated nucleotide, and an epigenetic marker. We show that diamondoid-functionalized gold electrodes can be used to identify these molecules and differentiate among them.
2. Methodology
We use the amantadine-functionalized gold electrodes to investigate the quantum transport properties of four natural nucleotides: deoxyadenosine monophosphate (dAMP, in the following abbreviated simply as A), deoxythymidine monophosphate (dTMP, in the following abbreviated simply as T), deoxycytidine monophosphate (dCMP, in the following abbreviated simply as C) and deoxyguanosine monophosphate (dGMP, in the following abbreviated simply as G). These are separately placed between the functionalized electrodes. Here, we investigate only the single-functionalization case, that is only the left electrode has been functionalized by a diamondoid. Only one diamondoid (ama) will be used in this work, as a proof of concept. The choice of this diamondoid is based on a broader analysis (to be published elsewhere) using additional diamondoids as functionalization molecules. In addition to the analysis of the tunneling current signals arising from each nucleotide between the functionalized electrodes, we extend the investigation to study the efficiency of such a functionalized-nanopore device in distinguishing natural nucleotides from mutations (8-oxodeoxyguanosine monophosphate – 8-oxo-Gua, in the following abbreviated simply as d8oG25) and epigenetic markers (5-methyl deoxycytidine monophosphate – 5-mdCMP, in the following abbreviated simply as d5mC26). The diamondoid is attached on a (111) gold surface. For the calculations we have used semi-infinite electrodes.In this work, density functional theory (DFT) based27,28 simulations as implemented in the code SIESTA29 were carried out. The use of quantum-mechanical calculations has been proven very efficient in the field of nanopore sequencing.30 We have used the generalized gradient approximation of Perdew–Burke–Ernzerhof (PBE-GGA),31 and the norm-conserving Troullier–Martins pseudopotentials.32 For expanding the Kohn–Sham states, we have considered a double-ζ with polarization (DZP) basis-set for the molecules, and a single-ζ with polarization (SZP) for the gold atoms (5d10, 6s1). These basis sets have been proven efficient in simulating similar systems.18,20 An energy shift of 0.01 Ry with a real space sampling grid (mesh cutoff) of 200 Ry, and 5 × 5 × 1 k points using the Monkhorst–Pack scheme were also used. The ionic relaxations were performed until the net forces of each atomic component become smaller than 0.01 eV Å−1. The Au(111) unit cell was fully relaxed and we obtained a lattice constant of 4.186 Å which compares well with the data reported in the literature.33
The electronic transport calculations are performed using DFT combined with the non-equilibrium Green's function (NEGF) formalism, as implemented in the TranSIESTA34 code. Fig. 1a shows the electronic transport setup, composed of three parts: the two electrodes (left and right) and the scattering region (device). For solving this transport problem, it is possible to write Green's functions according to:
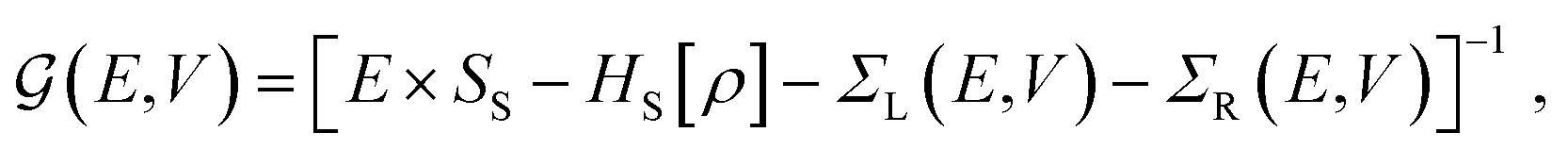 | (1) |
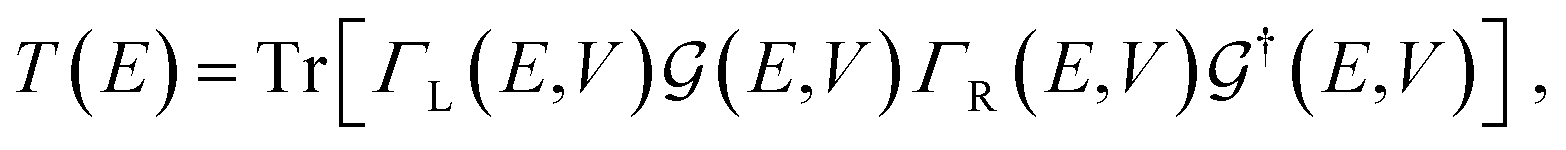 | (2) |
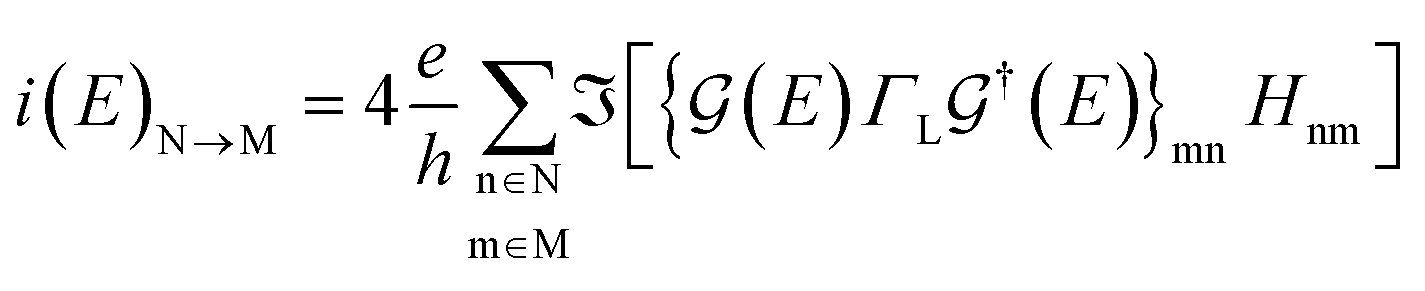 | (3) |
 | ||
| Fig. 1 The computational setup for the transport calculations along the Z direction is shown in panels (a) and (b). Three outer layers of the Au(111) material on either side of the nanogap form semi-infinite leads. The inner five layers of Au(111) on either side along with the embedded molecules form the scattering region. (c) Variation of the zero bias conductance with the capacitor gap size. (d) Local current23 across the nanogap for two different capacitor gaps. In these panels, gold atoms are shown in yellow, carbon in gray, nitrogen in blue, hydrogen in cyan, and sulfur in orange. | ||
Fig. 1c shows the zero bias conductance for different nanogap widths (d). We have noticed a high conductance for distances smaller than 4.5 Å. For distances larger than 5 Å the conductance goes to zero. The local current is shown in Fig. 1d, for two nanogap widths. For the smallest nanogap width of 3 Å, we have seen the highest conductance. Since, no bias voltage was applied across the electrodes, this conductance can lead to the projection of the transmittance and the tunneling current passing across the gap. For a nanogap of 6 Å, the conductance is zero and the local current is observed to diminish.
For the device functionalization we have used a supercell of 14.8 × 14.8 × 40.8 Å. Five unit cell repetitions in x and y directions, and ten perpendicular to the atomic planes in the z direction with a gap of 19 Å between the gold capacitor were used and are shown in Fig. 1b. A thiolated amantadine is placed close to the left gold layers and was relaxed until the thiol group of the amantadine was bonded to the gold surface. Next, a nucleotide was added close to amantadine with an orientation favoring the formation of hydrogen bonds with the diamondoid. The nucleobase and the sugar-phosphate group point towards the right inner gold layer. The choice of the distance between the gold layers is based on the condition that even the largest two nucleotides (A and G) should fit inside the gap. These specific conformations are chosen for consistency with our earlier studies.20
In real experiments, DNA is a charged molecule, which would react to a small bias voltage across the device and would re-orient with respect to the applied bias direction. Accordingly, a conformational scan would be necessary to capture the dynamics of the nucleotides within the electrodes and include the conformational variability in the analysis. In this work, though, we aim to present a proof of principles of the bio-sensitivity of a diamondoid-functionalized sequencing device and will not focus on the effect of the nucleotide conformations on the transport properties. This should be the subject of a separate study.
3. Results
The geometries obtained after relaxation for all the natural nucleotides (A, C, T, and G), as well as for the two modified nucleotides (d5mC and d8oG) within the diamondoid-functionalized gold electrodes are depicted in Fig. 2. For all cases, the nucleobases point to the diamondoid side, while the phosphate groups are closer to the right electrode. No covalent bonding occurs between the nucleotides and the electrodes. The residual distances on each side are not larger than 2.5 Å. The full relaxation of each geometry gives the best coupling between the molecules and the device. These configurations will be used further in evaluating the transport properties for all nucleotides.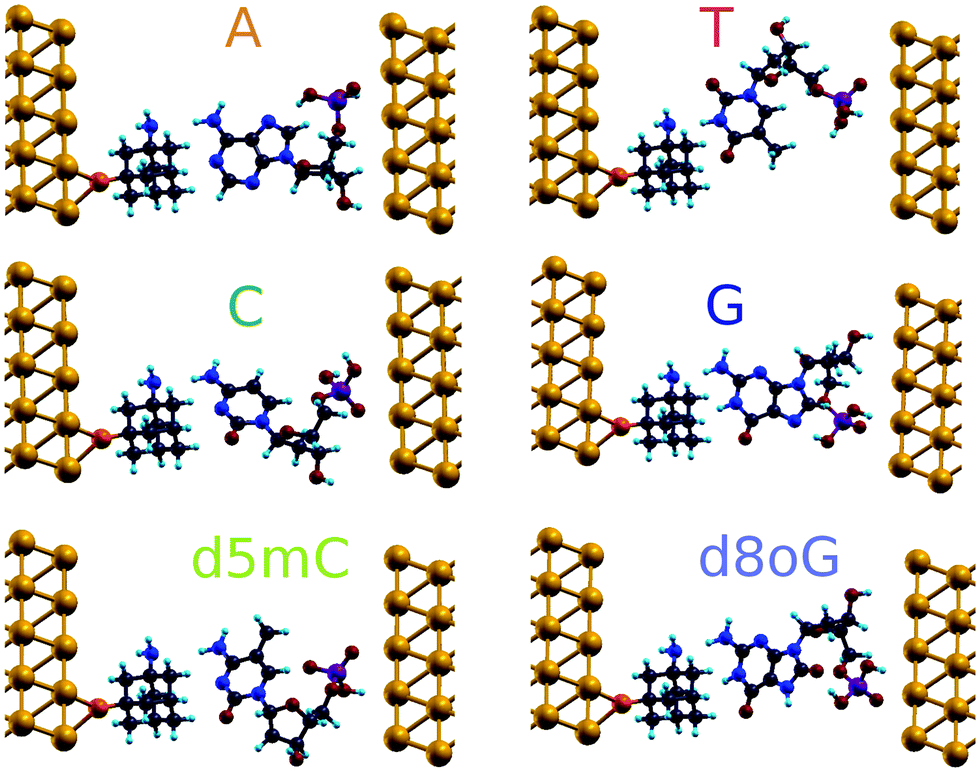 | ||
| Fig. 2 Fully relaxed geometries of diamondoid-functionalized electrodes including the nucleotides. The transport calculations are performed for the systems shown in this figure. The setups are generated for six nucleotide cases: the four natural ones, as well as one methylated and one mutated nucleotide, as denoted by the labels. In these panels, gold atoms are shown in yellow, carbon in gray, nitrogen in blue, hydrogen in cyan, oxygen in red, phosphorus in purple, and sulfur in orange. The same color coding is used throughout this work. | ||
The main challenge for the Au(111)-diamondoid functionalized device is to efficiently distinguish the nucleotides using the tunneling current signals. To this end, Fig. 3 shows the calculated transmission curves for all nucleotides. We observe sharp peaks with T(E) ≈ 10−1, at different energies for each nucleotide in the transmission T(E) spectra. The values for T(E) in the range 10−4–10−6 correspond to energies far from the resonances. In order to evaluate the peak origin, we calculated the projected density of states via Green's functions, which were projected in three atomic groups: (i) gold atoms, (ii) nucleotides, and (iii) diamondoids. We have found that the higher contribution for each peak comes from the nucleotides (not shown here). These results suggest that for specific energies, the transmittance spectral resonances lead to the nucleotide fingerprints. The observed T(E) hierarchy at the Fermi level (G > C > A > T) shows a difference up to one order of magnitude for the nucleotides compared to G. A and T show a very similar transmission close to the Fermi level, which might result in overlapping electronic signals, and a difficulty in distinguishing them from each other. The sensing device proposed here shows better read-out possibilities than other types of functionalizing devices. A qualitative comparison of the transmission peaks shown in Fig. 3 of the present paper to those obtained from cytosine-functionalized gold electrodes (see Fig. 4 in ref. 18) reveals that in the former setup the peaks are energetically further separated and hence more distinguishable than in the latter case.
 | ||
| Fig. 3 Transmission curves plotted on a semi-log scale for the six geometries shown in Fig. 2. The shaded region is of interest for sensing (see text). The energy axis has been shifted so as to align zero with the Fermi energy, EF. | ||
In the following, we analyze the energy range (shaded region in Fig. 3), which includes the resonance peaks for all nucleotides studied here, in order to propose a path to clearly identify each nucleobase. Accordingly, we will tune the Fermi energy in a nucleotide-specific way. In practice, in real experiments, this can be done by tuning the gating voltage of an external gate. For example, if we are interested in identifying d8oG to which the first peak on the right side of the shaded region corresponds, we would need to tune the Fermi energy to Vg = −0.76 eV. In order to generalize this analysis, we will assume a similar definition of the gating conductance g (for a small bias) as a function of the gate voltage, Vg:35
| g(Vg) = G0T(μ) | (4) |
 .
.
In order to sense DNA based on the formalism above, a reference system with a specific conductance is needed. Based on this reference, the properties of each nucleotide can be identified. For tunneling systems, the reference device conductance is zero for a large capacitor distance, as is shown in Fig. 1c. Accordingly, another reference system is needed. As a solution to this, we introduce the device sensitivity, considering one nucleotide as a reference with respect to the sampling gating voltage Vg as:
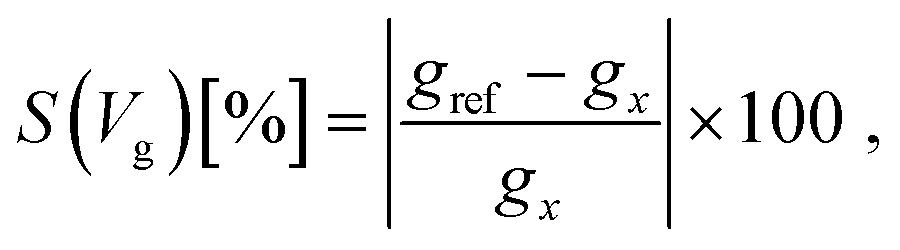
From the sensitivity at a specific sampling gating voltage, it is possible to observe how well resolved the conductance of the reference nucleotide will be with respect to other nucleotides. The device sensitivity according to the definition above and for all nucleotides is summarized in Fig. 4. (Results for the modified nucleotides, d5mC and d8oG, are shown only in panels (c)–(f), since for these the question is whether they can be distinguished from their natural counterparts, C and G.) We begin the analysis with the sensitivity for A (Fig. 4a). For this, A is taken as the reference at a gating voltage of Vg = −0.98 V. It is evident from this figure that the sensitivity of the device in recognizing A is at least five orders of magnitude higher than its natural counterparts (T, C, and G). Accordingly, we can infer that at this gating voltage, the device could clearly identify A.
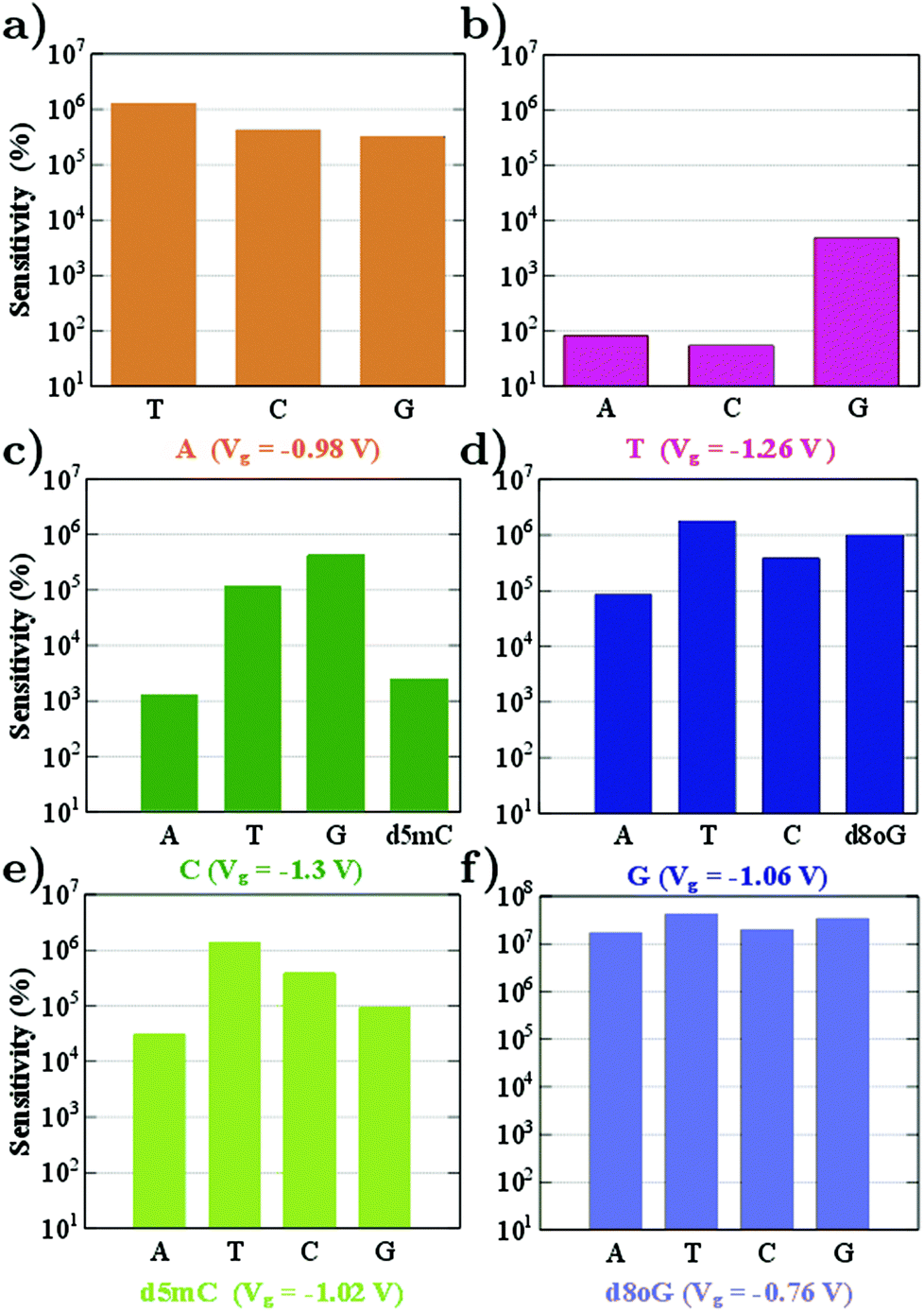 | ||
| Fig. 4 The device sensitivity is plotted on a semi-log scale for different gating voltages corresponding to the transmission peaks of the reference nucleotides. The labels denote the reference nucleotide and the respective gating voltage. | ||
In order to understand the underlying physics in Fig. 4a, the zero bias scattering state eigenchannel wavefunctions (EWF),36 at Vg = −0.98 V, are shown in the lower panel of Fig. 5. Both the real and imaginary contributions to the EWFs are shown. For reference A, the real part of EWF is localized over the whole scattering region. For the non-reference nucleotides (T, C, and G) the EWF is mainly located at the diamondoid site, showing a small contribution to the nucleotide sites. For the non-reference nucleotides, the EWFs clearly decay as we move from the left electrode to the right, resulting in a smaller T(E). An inspection of the imaginary part of the EWFs for A points to states spread almost over the whole device, from the left, over the molecules, to the right electrode. This behavior clearly denotes an enhancement of the electronic coupling between the gold electrodes, the diamondoid, and A. Such a spread contribution is not observed for the non-reference nucleotides. This effect of electronic coupling is reflected in the local currents,23 which are also sketched in the top panel of Fig. 5. According to this figure and based on the above analysis, local currents are observed mainly for the reference A and not for the non-reference nucleotides, for which the local current is negligible. These results indicate that, based on the device sensitivity and the local current measurements for a gating voltage of Vg = −0.98 V, it is possible to identify only A.
 | ||
| Fig. 5 A comparison of four natural nucleotides at a gating voltage of Vg = −0.98 V which corresponds to the first transmission peak for A below the Fermi level. The local currents are plotted in the top panel and the eigenchannel wavefunctions at the bottom panel. For clarity all wavefunctions are plotted for the same isovalues, while positive values of the wavefunctions are shown in purple and negative in blue. The imaginary part is not clearly visible in most of the cases since its too small or below the cutoff. | ||
In Fig. 4 we observe that the device sensitivity spans a range of 102–107 and note that the gating voltage should be carefully chosen in order to identify the nucleotides. At a gating voltage Vg = −1.3 V, the conductance for C is at least three orders of magnitude more resolved compared to its natural non-reference counterparts (A, T, and G). At Vg = −1.06 V, the gating conductance for G is at least four orders of magnitude more resolved than that for the non-reference A, T, and C. For Vg = −0.98 V or Vg = −1.06 V, the sensitivity distributions for the non-reference nucleotides are not considerably broad, but differ only by one order of magnitude. On the other hand, taking C as a reference for Vg = −1.30 V, leads to a sensitivity in the range 103–105 (see Fig. 4c).
Fig. 4b shows the sensitivity histogram for T at Vg = −1.26 V, calculated from the transmission curves in Fig. 3. In the latter figure, the transmittance peak for T is very sharp and smaller than the peaks for the other nucleotides. It is clear that T shows at least one order of magnitude difference in the transmission with respect to the relevant data for A, G, d8oG, and d5mC, and about two orders of magnitude difference from the C case. At this gating voltage, other nucleotides (A and C) have a much higher transmission than T. In order to evaluate the contribution of the nucleotides to the transmission, we have calculated the projected density of states (PDOS). We have observed (not shown) that the transmission peak for T has at least two major contributions: one from the diamondoid and one from the nucleotide T.
Up to this point, we have shown that it is possible to identify the nucleotides by choosing the respective gating voltage. In the following, we consider another possibility to distinguish the nucleobases based on the respective differential conductance 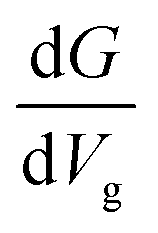 . This can resolve peaks of the nucleotides in cases when these are not easily distinguishable. This can for example be the case for T, according to our previous analysis. The advantage in using the differential conductance is that this quantity can lead to information on the conductivity changes for a range of gating voltages Vg. Fig. 6 shows how well resolved the conductance change for each natural nucleotide is. For A and C, the differential conductance does not overlap with that for the other nucleotides in the range Vg < −1.3 V and −1.06 V < Vg < −0.97 V. In the range Vg = −1.26 ± 0.01 V the differential conductance for T is highly resolved, as it does not overlap with any of the other nucleotides. This observation indicates the possibility to distinguish T for this gating voltage range. The differential conductance for G is very broad and overlaps with A and C. A further inspection of Fig. 6 reveals three ranges for the gating voltage, in which no overlapping between the curve for G and those for the other nucleotides occurs. These results point to the possibility to use the information arising from the differential conductance in order to identify the natural nucleotides within a range of the gating voltage.
. This can resolve peaks of the nucleotides in cases when these are not easily distinguishable. This can for example be the case for T, according to our previous analysis. The advantage in using the differential conductance is that this quantity can lead to information on the conductivity changes for a range of gating voltages Vg. Fig. 6 shows how well resolved the conductance change for each natural nucleotide is. For A and C, the differential conductance does not overlap with that for the other nucleotides in the range Vg < −1.3 V and −1.06 V < Vg < −0.97 V. In the range Vg = −1.26 ± 0.01 V the differential conductance for T is highly resolved, as it does not overlap with any of the other nucleotides. This observation indicates the possibility to distinguish T for this gating voltage range. The differential conductance for G is very broad and overlaps with A and C. A further inspection of Fig. 6 reveals three ranges for the gating voltage, in which no overlapping between the curve for G and those for the other nucleotides occurs. These results point to the possibility to use the information arising from the differential conductance in order to identify the natural nucleotides within a range of the gating voltage.
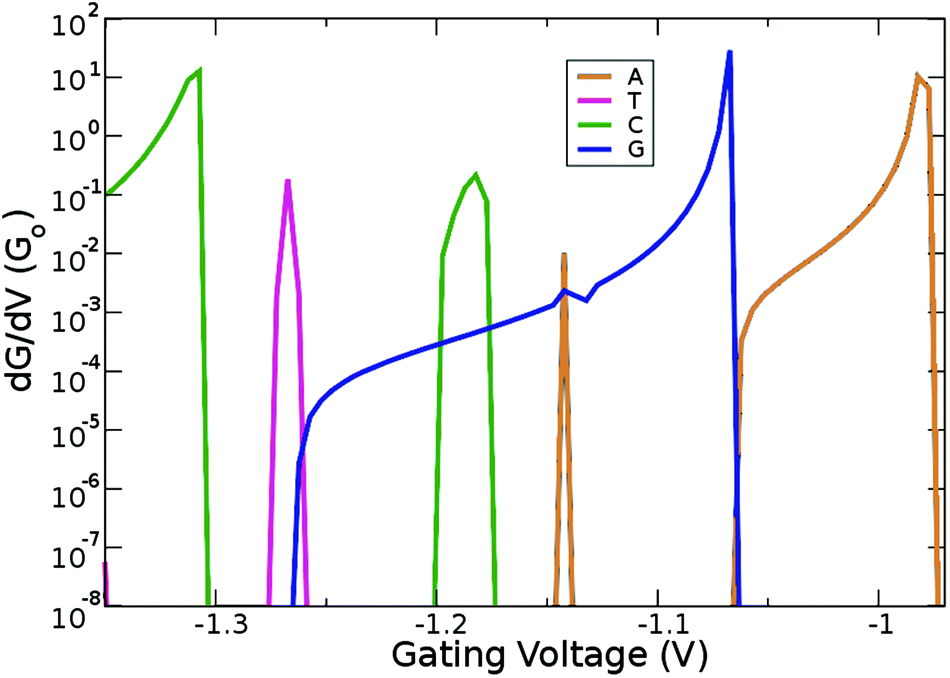 | ||
| Fig. 6 Derivative of the conductance with respect to the gating voltage plotted in the gating window for the four natural nucleotides. | ||
Another important issue in the field of DNA sequencing is the possibility to distinguish pristine nucleotides (like C and G) from their modified forms (like d5mC and d8oG). The transmission curves in Fig. 3 show defined peaks for these two modifications, which do not overlap with the curves for the pristine nucleotides. In Fig. 4c and e, we show the sensitivity for pristine C and its mutant d5mC, and in Fig. 4d and f the data for dGMP and d8oG, respectively. At the chosen gating voltage values the modified nucleotides can be easily distinguished from their pristine counterparts. The EWF and local currents are shown in Fig. 7. For a gating voltage Vg = −1.3 V, corresponding to the peak for C in the transmission spectra, C can be distinguished from d5mC. For d5mC and a gating voltage of Vg = −1.02 V, corresponding to the transmission peak for this nucleotide, the sensitivity difference from the pristine C is about five orders of magnitude. The respective EWFs for d5mC and C are presented in the left panels of Fig. 7 for a gating voltage of Vg = −1.3 V. The real part of the EWFs is localized in both cases. In the pristine C, though, a stronger coupling to the right electrode occurs. This difference is mapped on the local currents, also shown, which are negligible for d5mC, but are finite for C.
 | ||
| Fig. 7 A comparison of C vs. d5mC can be seen in the left panels for Vg = −1.3 V (corresponding to the second transmission peak below the Fermi level for C). A comparison of G vs. d8oG is made in the right panels for Vg = −1.06 V (corresponding to the second transmission peak below the Fermi level for G). For each case, the local currents are sketched in the top panel and the EWFs at the bottom panel. For clarity all wave-functions are plotted for the same isovalue and are color coded according to their positive (purple) and negative (blue) signs. | ||
Finally, by sampling at Vg = −1.06 V, the gating voltage corresponding to the transmission peak for G, it is possible to distinguish G from the mutation d8oG. For Vg = −0.76 V, which corresponds to the transmission peak for d8oG, the sensitivity for d8oG is seven orders of magnitude higher than that for dGMP. Going back to Vg = −1.06 V, at this voltage, the real part of the EWF is localized along the entire device for G and at the functionalized left electrode for the d8oG case, as shown in the right panels of Fig. 7 for G and d8oG. For this gating voltage, the electrodes in the pristine case (G) are coupled and there is a respective local current. In the mutated nucleotide (d8oG) there is no current at this gating voltage. These results again strongly indicate the possibility to distinguish the pristine G over its mutation d8oG. An inspection of the EWFs for d8oG shows a significant difference in the symmetry of the eigenchannel compared to that of G, which suggests a larger asymmetry in the I–V curves for the case of d8oG with respect to G at a low applied bias.37 This asymmetry indicates that d8oG can potentially reveal its rectifying behaviour when the gating voltage is tuned so that the conduction due to these orbitals moves closer to the Fermi level.
4. Conclusions
Using quantum-mechanical calculations, we were able to provide a proof of concept regarding the feasibility of a bio-sensor based on amantadine-functionalized gold electrodes. Our work has shown that such a device can detect different nucleobases and their modifications based on quantum tunneling measurements across the electrodes. Different gating voltages can tune the sensitivity of the functionalized electrodes with respect to specific nucleotides having a high transmission peak at these voltages. We have also revealed the sensing mechanism for one of the pristine nucleotides (T) by analyzing the derivatives of the conductance with respect to the values of the gating voltage. The sensitivity at different gating voltages is also related to the coupling or decoupling of the functionalized electrodes, and the observation or lack of observation of local currents. In order to reveal the physics underlying the observed transport properties, the scatter state wave functions at a higher transmission, as well as the local current plots were presented and discussed.Our results involve the zero bias voltage case, but can give an indication of the I–V characteristics of the diamondoid functionalized device at a small applied bias. The current as a function of applied voltage in the low bias regime (up to 0.1 V) for d5mC and d8oG should be significantly different compared to their respective natural nucleotides. Our data support this assumption, as a low applied bias should not prevent the possibility of distinguishing the different nucleotides based on their I–V response.38 It is not trivially clear how the transmission peaks would shift with respect to an applied bias. However, Fig. 5 and 7 indicate that the EWFs in all cases are more associated with the same (left) electrode implying that the transmission peaks would shift towards the same direction to more negative energies. Accordingly, at a low bias and for a moderate coupling of the electrodes, we can assume that all the transmission peaks do not shift too strongly and that the shift is similar in all cases. The sharp transmission peaks for G and d8oG at −0.28 eV and −0.75 eV, respectively would then show a sharp rise in the current for G between −0.2 V and −0.3 V. The sharp rise in the current for d8oG would be expected at more negative bias voltages. For C and d5mC having their transmission peaks below −1 eV, a bias of about −1 V beyond the low bias limit would be needed. At such a high applied bias the situation is expected to change, as the peaks would shift in a different way, possibly leading to overlapping and indistinguishable transmission peaks. Overall, the amount of increase in the current for each nucleotide is related to the gating voltage needed to shift the transmission peak closer to the Fermi energy and the applied bias voltage. Note, that within our methodology, calculations at a very small bias are still possible as the system is still not out of equilibrium. The application of a large bias, though could also lead to a breakdown of the system.
The focus of this work was to reveal the suitability of diamondoid-functionalized electrodes as biosensors. There are of course many complex issues that have not been taken into account at this point. Some of these involve the effect of conformational variability and dynamics in the transport properties across the electrodes and the corresponding sensitivity of the device. The effect of the surrounding medium, i.e. water and ions is also expected to play a major role. The question is whether these factors will only shift the transport spectra shown in this work or will make the whole picture more complex. Some of these points we aim to address in follow-up studies in the near future. A conformational scan of the DNA orientation in the nanopore and the role of a solvent in the pore can be unveiled in view of the electronic current signatures in diamondoid-functionalized nanopores. The role of the solvent is expected to influence the transport properties. Currently, the focus of a parallel investigation is the comparison of the performance of different diamondoid derivatives as functionalization molecules in a nanopore device, which is also expected to shed light onto pathways to enhance the sensitivity of functionalized nanopores.
Acknowledgements
G. S and M. F. acknowledge financial support from the collaborative network SFB 716 “Dynamic simulations of systems with large particle numbers” funded by the German Funding Agency (Deutsche Forschungsgemeinschaft-DFG). This work was performed on the computational resource ForHLR Phase I funded by the Ministry of Science, Research and the Arts Baden-Württemberg and DFG. The initial benchmark calculations were performed on the computational resource bwUniCluster funded by the Ministry of Science, Research and the Arts Baden-Württemberg and the Universities of the State of Baden-Württemberg, Germany, within the framework program bwHPC. R. G. A. acknowledges financial support from the Carl Tryggers Stiftelse and R. H. S. thanks the Swedish Research Council for a Junior Research Position grant (VR, 621-2009-3628).References
- E. L. van Dijk, H. Auger, Y. Jaszczyszyn and C. Thermes, Trends Genet., 2014, 30, 418 CrossRef CAS PubMed.
- L. J. Steinbock and A. Radenovic, Nanotechnology, 2015, 26, 074003 CrossRef CAS PubMed.
- S. Harrer, S. C. Kim, C. Schieber, S. Kannam, N. Gunn, S. Moore, D. Scott, R. Bathgate, S. Skafidas and J. M. Wagner, Nanotechnology, 2015, 26, 182502 CrossRef CAS PubMed.
- M. Fyta, J. Phys.: Condens. Matter, 2015, 27, 273101 CrossRef PubMed.
- Editorial, Nature Nanotechnology, 2016, 11, 105.
- U. F. Keyser, Nat. Nanotechnol., 2016, 11, 106 CrossRef CAS PubMed.
- S. Lindsay, Nat. Nanotechnol., 2016, 11, 109 CrossRef CAS PubMed.
- M. di Ventra and M. Taniguchi, Nat. Nanotechnol., 2016, 11, 117 CrossRef CAS PubMed.
- S. J. Heerema and C. Dekker, Nat. Nanotechnol., 2016, 11, 127 CrossRef CAS PubMed.
- A. Meller, L. Nivon and D. Branton, Phys. Rev. Lett., 2001, 86, 3435 CrossRef CAS PubMed.
- M. Zwolak and M. Di Ventra, Nano Lett., 2005, 5, 421 CrossRef CAS PubMed.
- H. S. Kim and Y.-H. Kim, Biosens. Bioelectron., 2015, 69, 186 CrossRef CAS PubMed.
- J. Lagerqvist, M. Zwolak and M. Di Ventra, Nano Lett., 2006, 6, 779 CrossRef CAS PubMed.
- M. Tsutsui, M. Taniguchi, K. Yokota and T. Kawai, Nat. Nanotechnol., 2010, 5, 286 CrossRef CAS PubMed.
- B. N. Miles, A. P. Ivanov, K. A. Wilson, F. Dogan, D. Japrung and J. B. Edel, Chem. Soc. Rev., 2013, 42, 15 RSC.
- S. Carson and M. Wanunu, Nanotechnology, 2015, 26, 074004 CrossRef PubMed.
- P. Krstić, B. Ashcroft and S. Lindsay, Nanotechnology, 2015, 26, 084001 CrossRef PubMed.
- H. He, R. H. Scheicher, R. Pandey, A. R. Rocha, S. Sanvito, A. Grigoriev, R. Ahuja and S. P. Karna, J. Phys. Chem. C, 2008, 112, 3456 CAS.
- D. Branton, Nat. Biotechnol., 2008, 26, 1146 CrossRef CAS PubMed.
- G. Sivaraman and M. Fyta, Nanoscale, 2014, 6, 4225 RSC.
- G. Sivaraman and M. Fyta, EPL, 2014, 108, 17005 CrossRef.
- H. Schwertfeger, A. A. Fokin and P. R. Schreiner, Angew. Chem., Int. Ed., 2008, 47, 1022 CrossRef CAS PubMed.
- N. Okabayashi, M. Paulsson, H. Ueba, Y. Konda and T. Komeda, Phys. Rev. Lett., 2010, 104, 077801 CrossRef PubMed.
- T. M. Willey, J. D. Fabbri, J. R. I. Lee, P. R. Schreiner, A. A. Fokin, B. A. Tkachenko, N. A. Fokina, J. E. P. Dahl, R. M. K. Carlson, A. L. Vance, W. Yang, L. J. Terminello, T. van Buuren and N. A. Melosh, J. Am. Chem. Soc., 2008, 130, 10536 CrossRef CAS PubMed.
- S. Kanvah, J. Joseph, G. B. Schuster, R. N. Barnett, C. L. Cleveland and U. Landman, Acc. Chem. Res., 2010, 43, 280 CrossRef CAS PubMed.
- P. W. Laird, Nat. Rev. Cancer, 2003, 3, 253 CrossRef CAS PubMed.
- P. Hohenberg and W. Kohn, Phys. Rev., 1964, 136, B864 CrossRef.
- W. Kohn and L. J. Sham, Phys. Rev., 1965, 140, A1133 CrossRef.
- J. M. Soler, E. Artacho, J. D. Gale, A. García, J. Junquera, P. Ordejón and D. Sánchez-Portal, J. Phys.: Condens. Matter, 2002, 14, 2745 CrossRef CAS.
- R. Scheicher, A. Grigoriev and R. Ahuja, J. Mater. Sci., 2012, 47, 7439 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- N. Troullier and J. L. Martins, Phys. Rev. B: Condens. Matter, 1991, 43, 1993 CrossRef CAS.
- N. W. Ashcroft and N. D. Mermin, Solid State Physics, Holt, Rinehart and Winston, New York, 1976 Search PubMed.
- M. Brandbyge, J. Mozos, P. Ordejón, J. Taylor and K. Stokbro, Phys. Rev. B: Condens. Matter, 2002, 65, 165401 CrossRef.
- S. K. Min, W. Y. Kim, Y. Cho and K. S. Kim, Nat. Nanotechnol., 2011, 6, 162 CrossRef CAS PubMed.
- M. Paulsson and M. Brandbyge, Phys. Rev. B: Condens. Matter, 2007, 76, 115117 CrossRef.
- W. Ding, C. F. Negre, L. Vogt and V. S. Batista, J. Chem. Theory Comput., 2014, 10, 3393 CrossRef CAS PubMed.
- A. Batra, P. Darancet, Q. Chen, J. S. Meisner, J. R. Widawsky, J. B. Neaton, C. Nuckolls and L. Venkataraman, Nano Lett., 2013, 13, 6233 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |