
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAgeing phenomena in high-voltage aqueous supercapacitors investigated by in situ gas analysis
Minglong
He
a,
Krzysztof
Fic
b,
Elżbieta
Fr![[a with combining cedilla]](https://www.rsc.org/images/entities/char_0061_0327.gif) ckowiak
b,
Petr
Novák
a and
Erik J.
Berg
*a
ckowiak
b,
Petr
Novák
a and
Erik J.
Berg
*a
aElectrochemistry Laboratory, Paul Scherrer Institut, 5232 Villigen PSI, Switzerland. E-mail: erik.jaemstorp-berg@psi.ch
bInstitute of Chemistry and Technical Electrochemistry, Poznan University of Technology, 60-695 Poznan, Poland
First published on 11th December 2015
Abstract
High-voltage aqueous electrolyte based supercapacitors (U > 1.23 V) attract significant attention for next-generation high power, low cost and environmentally friendly energy storage applications. Cell ageing is however markedly pronounced at elevated voltages and results in accelerated overall performance fade and increased safety concerns. Online electrochemical mass spectrometry, combined with cell pressure analysis, is for the first time shown to provide a powerful means for in situ investigation of degradation mechanisms in aqueous electrolyte/carbon based supercapacitors. The activated carbon electrodes possess high specific surface area and oxygen-based surface functionalities (mainly phenol, lactone and anhydride groups), which are oxidized already at a cell voltage of 0.6 V to provoke the evolution of minor amounts of CO and CO2. Noticeable water decomposition starts at a high voltage of 1.6 V with the evolution of H2 on the negative electrode and carbon corrosion on the positive electrode with the generation of predominantly CO. In this paper we also report that short-term cycling leads to partly reversible gas evolution/consumption side-reactions giving negligible capacitance. On the other hand, long-term cycling causes irreversible side-reactions, deteriorates the electrochemical performance, and increases the internal pressure of the cell. Repeated cycling (U < 2 V) is confirmed as a more harmful technique for the electrode integrity compared to the voltage holding in a floating test. In situ gas analysis is shown to provide valuable insights into the electrochemical cell ageing aspects, such as the nature and potential onsets of side-reactions, hence paving the way for fundamental understanding and mitigating the performance and safety loss of high-energy aqueous supercapacitors.
Broader contextGlobal energy consumption and environmental concerns steadily increase with the growth of the world's population and the technical development of our society. Future sustainability is largely dependent upon the widespread implementation of renewable energy generation and storage. Supercapacitors constitute a crucial part in the effective energy storage and delivery, e.g. in electric vehicles (start–stop systems) and wind mills (balancing the energy provided to the grid), because of their exceptional power capability and long cycle life. Supercapacitors with high surface area carbon electrodes operating in aqueous electrolytes offer high power rates, low cost, safe and environmental benign devices, but at moderate energy densities. Significant energy enhancement can be achieved by applying higher operating cell voltages (U > 1.23 V) when using sulphate-based electrolytes. However, for practical implementations a fundamental understanding of their premature ageing as well as related safety issues are required. In the present work, state-of-the-art in situ gas analysis is shown to deliver valuable insights into the governing mechanisms behind the ageing of these devices. Assignment of safe operating voltage limits provides an important knowledge to preserve the chemical integrity of cell components for long-term cycling of aqueous supercapacitors at high voltages without detrimental gas evolution. |
Introduction
Supercapacitors, known also as electrochemical or electrical double layer capacitors (EDLCs), are nowadays widely implemented in applications requiring a rapid electric power supply as the energy in the electrochemical double layer can be accumulated and delivered quickly.1–3 In order to address the growing needs of rapid power sources, e.g. for hybrid electric vehicles, further improvements on several aspects of EDLCs are imperative.4–6 Aqueous electrolyte based EDLCs are attractive due to their relatively high capacitance, excellent power rates, low cost and environmental friendliness. The major disadvantage of aqueous EDLCs, especially based on either acidic or alkaline electrolytes, is the moderate operating voltage window restricted by the thermodynamic stability of water.7 The practical maximum voltage of aqueous EDLCs could however be increased by tuning the physicochemical properties of the electrolyte (pH or conductivity values) and/or surface chemistry of activated carbons (AC), which in turn affect the over-potential for hydrogen and/or oxygen evolution.8Recently, an activated carbon based supercapacitor with 1 mol L−1 Li2SO4 aqueous electrolyte (pH 6–7) demonstrated stable operation at a surprisingly high cell voltage of 2.2 V.6 However, such extraordinary voltage was achieved with noble (gold) current collectors, hence, several parasitic phenomena (e.g. corrosion) influencing final performance were excluded. The significant enhancement of the operating voltage of aqueous EDLCs is achieved by employing neutral solutions of specific alkali salts, e.g. Na2SO4, NaNO3 and Li2SO4,6,9–11 that stabilize the electrode/electrolyte interface. Cell ageing issues are however considerably pronounced at high voltages (and elevated temperatures),12 which cause gradual performance loss (lower capacitance and/or increased internal resistance) and ultimately cell failure and increased safety concerns.13 Ageing processes include mainly side-reactions that lead to electrolyte depletion, electrode pore blocking, and internal cell pressure increase because of volatile side-products. Studies of electrochemically induced gas evolution and associated cell pressure build-up have already provided important insights into degradation mechanisms of activated carbon-based EDLCs, operating in either aqueous12–15 or non-aqueous16–18 electrolytes. Several approaches, such as Tafel plot based estimations,15 gas chromatography14 and cell pressure characterization,12,13 were previously employed to probe the origin of gas evolution in EDLCs.
Brousse et al. investigated the electrochemically induced gas evolution during cycling of a hybrid electrochemical capacitor composed of MnO2 and activated carbon as positive and negative electrodes, respectively, operating in 0.65 mol L−1 K2SO4 aqueous electrolyte.15 O2 evolution was proposed to occur on the MnO2 surface (+) while H2 evolution was attributed to the activated carbon surface (−) upon charging of the cell; the respective volumes of evolved gases were calculated from the corresponding current–potential Tafel plots.
Nakamura et al. studied the influence of the oxygen content of the carbon surface functional groups on the gas evolution onsets in an activated carbon-based EDLC operating in 30 wt% H2SO4 electrolyte.14 A higher oxygen content was suggested to result in a lower gas evolution onset, and higher concentration of acidic functional surface groups on activated carbon brought about more gas evolution. The major components of the evolved gases were determined by gas chromatography to be CO and CO2 during anodic polarization and H2 during cathodic polarization.
Ratajczak et al. recently investigated the ageing and internal pressure build-up of symmetric EDLC cells containing 1 mol L−1 Li2SO4 aqueous electrolyte during accelerated ageing tests.12,13 An internal cell pressure increase was measured by a pressure transducer at a cell voltage of 1.5 V, together with the increase of cell impedance and carbon electrode/current collector corrosion. In non-aqueous EDLC systems, Hahn et al. used a pressure transducer to record both reversible and irreversible cell pressure changes at voltages up to 3 V.16 The authors continued to show that the extent of pressure build-up was related to the stability of the electrolyte solvents, but the occurrence of faradic currents were rather devoted to the formation of soluble electrolyte decomposition products than to gas evolution.17
Online electrochemical mass spectrometry (OEMS) combines characteristics of both gas chromatography and cell pressure analysis by enabling both quantitative and qualitative in situ analysis of gaseous species evolved in the electrochemical cell during cycling.19 OEMS has in recent years become a commonly used tool to investigate side-reactions in several energy storage/conversion systems, e.g. fuel cells and batteries, because of the high sensitivity and specificity of the mass spectrometer.20–27 To the best of our knowledge, OEMS dedicated to EDLCs has only been reported once, but for a non-aqueous electrolyte system.18
The aim of the present study is to identify potential onsets and govern mechanisms of performance limiting side-reactions in a high-voltage aqueous EDLC by performing a comprehensive in situ analysis of gas evolving side-reactions.
Experimental
Electrode and electrolyte preparation
Self-standing circular carbon electrodes were punched (∅ 14 mm for the pressure cell, and ∅ 18 mm for the OEMS cell) from the activated carbon fabric (ACC 507-20, Kynol, Germany). 1 mol L−1 Li2SO4 aqueous electrolyte was prepared by dissolving analytical quality Li2SO4 salt (>99.99%, Sigma-Aldrich, Switzerland) into distilled water. A pH value of ca. 10.2 was measured by a pH-meter (SevenMulti, Mettler Toledo, Switzerland) for the as-prepared Li2SO4 electrolyte at room temperature. The slightly alkaline Li2SO4 electrolyte was believed to be caused by its manufacturing process. In order to secure reproducibility and to avoid additional ionic moieties in the solution, no neutralization of the electrolyte to pH 7 was performed, since the OH− concentration of ∼10−4 mol L−1 was relatively low, and electrode potentials were cautiously defined.Electrochemical cell assembling
Both the in situ pressure and OEMS cells were assembled in a symmetric electrode configuration containing two activated carbon electrodes (mC = 18 mg for the pressure cell, mC = 30 mg for the OEMS cell, mass discrepancy <0.3 mg), a glass fiber separator (∅ 18 mm for pressure cell, and ∅ 28 mm for OEMS cell; GF/A, Whatman, Switzerland) and a fixed amount of 1 mol L−1 Li2SO4 electrolyte (200 μL for the pressure cell, and 250 μL for the OEMS cell). Three hours of equilibrium time was allowed for all measurements before electrochemical cycling, for a stable pressure background.Carbon electrode characterization
The nitrogen adsorption isotherm of the activated carbon electrode was recorded by an ASAP 2460 analyzer (Micromeritics, USA) at 77 K. The specific surface area of the carbon material was calculated using Brunauer–Emmett–Teller (BET) equations. Pore size distribution and pore volumes were calculated using the non-local density functional theory (NLDFT) method.28 The oxygenated surface functionalities of activated carbon were determined by the temperature-programed desorption (TPD) method coupled with a quadrupole mass spectrometer (QMS 403 D Aëolos, Netzsch-Gerätebau, Germany).13 Carbon samples were heated up from 25 °C to 950 °C at a constant heating rate of 20 °C min−1 under a helium flow (50 mL min−1). Quantitative analysis of CO and CO2 groups was calibrated with calcium oxalate monohydrate.In situ pressure and OEMS characterization
The working principles of the in situ pressure and OEMS analysis have been introduced in our previous studies.29 Briefly, the pressure cell is a standard Paul Sherrer Institute coin-type electrochemical test cell,30 to which a pressure transducer (PA-33X, Keller, Switzerland) is directly connected. Internal pressure development of the electrochemical cell is uninterruptedly monitored upon cycling. Specific pressure increase rates inside EDLC pressure cells can be computed by taking the first order derivative of the relevant pressure curve and normalized by mass of two carbon electrodes, which is hereafter named as specific dP/dt.The OEMS setup consists of a quadrupole mass spectrometer (Prisma QME 200, Pfeiffer, Germany), two flowmeters (Red-Y smart Series from Vögtlin Instruments and EL-FLOW Select Series from Bronkhorst AG, Germany), stainless steel pipes, Swagelok fittings and an OEMS cell. Evolved gases were continuously purged out from the head space of the OEMS cell by a controlled Ar flow (0.05 mL min−1), and further diluted in a major Ar flow (0.8 mL min−1) before reaching the inlet of mass spectrometry via a thin capillary. Partial pressure of gas species in Ar flow was recorded as ion-current intensity using a mass spectrometer. The time-lag between gas evolutions on the carbon electrode surface and mass signal detection by OEMS were estimated about 30 seconds31 corresponding to a considerable potential shift of 0.3 V at 10 mV s−1 scan rate, and thus, a slower scan rate of 0.1 mV s−1 was complementarily employed for OEMS measurements to study the potential dependence of the gas evolving reactions.
Electrochemical techniques
Three major electrochemical techniques were conducted in a computer controlled potentiostat/galvanostat (CCCC hardware, Astrol Electronic, Switzerland), including potentiodynamic, potentiostatic and accelerated ageing protocols. The potentiodynamic process was consecutively performed in a cell voltage window of 0–1, 0–1.2, 0–1.4, 0–1.6, 0–1.8 and 0–2 V (3 repeated cycles for each window) at different scan rates of 10, 1 and 0.1 mV s−1. The potentiostatic process step-wise increases the cell voltage by 0.2 V from 0 V to 2 V in every 2 h. The accelerated ageing protocol combined the galvanostatic cycling (0.2 A g−1) with potentiostatic steps (voltage cut-off at 1.8 V for 2 h), in order to simulate the practical ageing process.12 Potentials of the positive and negative electrodes were measured in a three electrode configuration cell containing two symmetric activated carbon electrodes and an Ag/AgCl reference electrode (Sigma-Aldrich, Switzerland) during galvanostatic cycling (0.2 A g−1). All potential values in this work were referred to as the standard hydrogen electrode (SHE) unless otherwise noted.Results and discussion
Activated carbon electrode characterization
Fig. 1a shows the nitrogen adsorption isotherm of the employed activated carbon (ACC 507-20) and its pore size distribution (inset) according to NLDFT calculations. The activated carbon has a specific BET surface area of 2231 m2 g−1 and a narrow pore size distribution with a large fraction of micropores of an average pore diameter of 0.99 nm leading to a high double-layer capacitance.7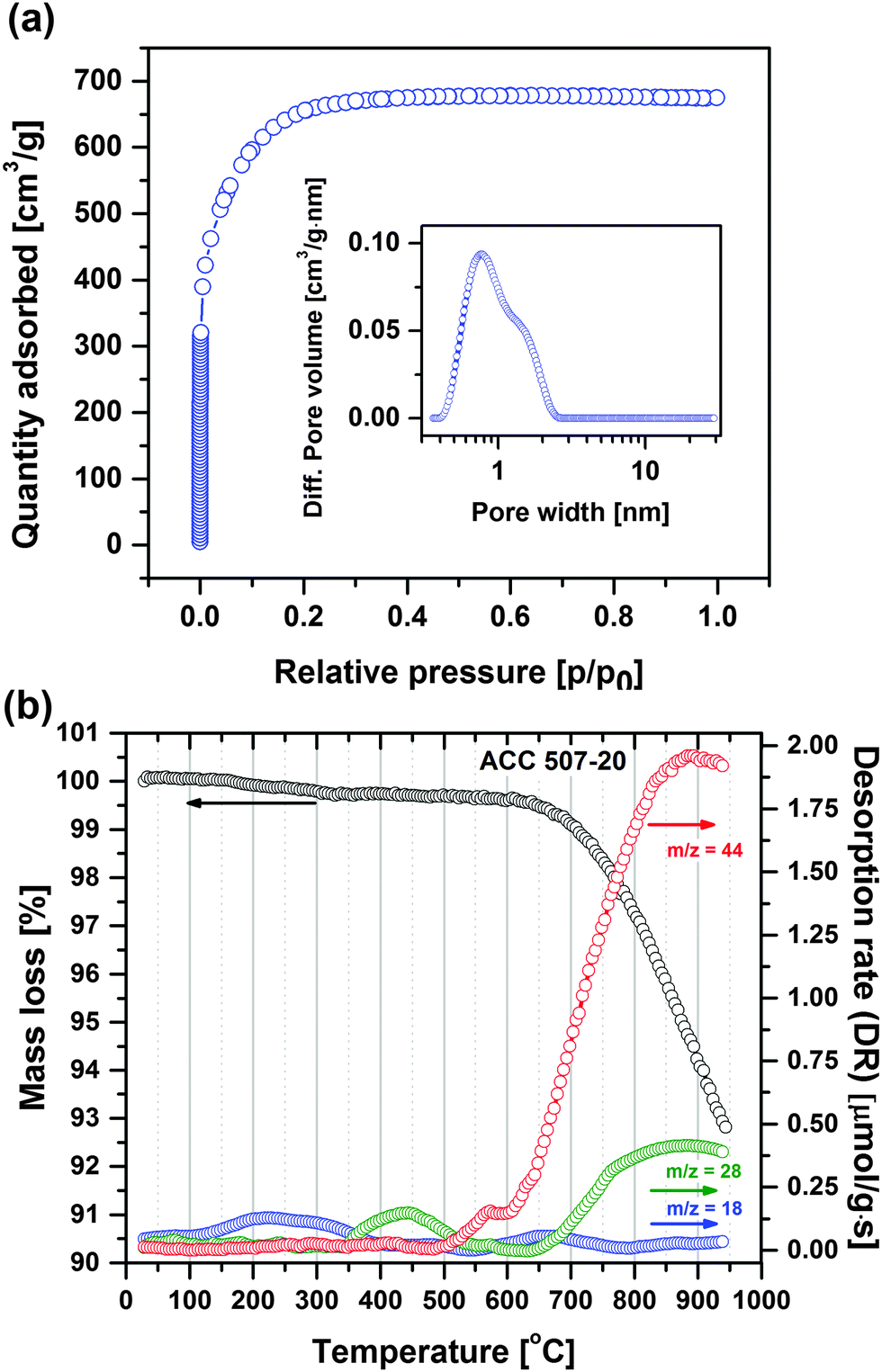 | ||
| Fig. 1 (a) Nitrogen adsorption isotherm profile of ACC 507-20 carbon at 77 K. Inset: Pore size distribution plot. (b) TPD profile of ACC 507-20 carbon (25–950 °C, heating rate: 20 °C min−1). | ||
Fig. 1b displays the mass loss and volatile species desorption profiles of the activated carbon obtained by the TPD technique when the carbon is heated from RT to 950 °C.
The total mass loss is close to 7.2 wt%, including trace of water (m/z = 18, 0.13 mmol g−1) and carbon surface functional group desorption leading to CO (m/z = 28, 0.33 mmol g−1) and CO2 (m/z = 44, 1.34 mmol g−1) gas, which all can be identified according to their particular desorption temperature ranges, respectively.32 The primary mass loss at temperatures lower than 350 °C originates from the moisture adsorbed on the carbon surface. The first desorption peak of m/z = 28 corresponding to CO is detected at temperatures between 350 and 650 °C, and may primarily be related to phenol (R–OH) functionalities.32,33 The first desorption peak of CO2 (m/z = 44) occurs at temperatures between 500 and 600 °C and is probably related to lactone (R1COOR2) functionalities.32,33 The peak positions of CO and CO2 desorption overlap when the temperature increases from 650 to 950 °C, and is believed to be mainly related to anhydride functionalities (R1COOCOR2).32,33 Other surface functionalities which desorb at >650 °C are quinone (R1COCOR2) and carbonyl (RCO) groups. The specific oxygen content (in terms of functional groups) quantified by TPD is significant (ca. 4.9 wt%). Oxygenated species on the carbon surface has a direct and remarkable influence on the electrode characteristics by modifying the electrostatic field on the surface.13 Notably, carbon with higher oxygen content has better wettability in aqueous electrolytes.34
In summary, the employed electrode material is a microporous and high surface area activated carbon with significant amounts of mainly phenol, anhydride and lactone surface functionalities, which generate both CO and CO2 during decomposition.
Electrochemical characterization of carbon electrodes
Fig. 2 shows the cyclic voltammograms (CV, 1 mV s−1) of the symmetric activated carbon EDLC cell containing 1 mol L−1 Li2SO4 electrolyte. The cell voltage limit is increased 200 mV with every 3rd cycle in the voltage range from 1 to 2 V. Up to 1 V, the main electrochemical response is the typical rectangular double-layer profile with a specific capacitance of ∼106 F g−1. A minor amount of faradaic current is however discernible, e.g. possibly deriving from the oxidation and reduction of carbon surface groups.35,36 The stepwise voltage increase up to 2 V significantly intensifies the faradaic currents, being rather a response for water decomposition. However, these faradaic current contributions decrease with cycles in the same voltage range, indicating a gradual change of the activated carbon functionalities.13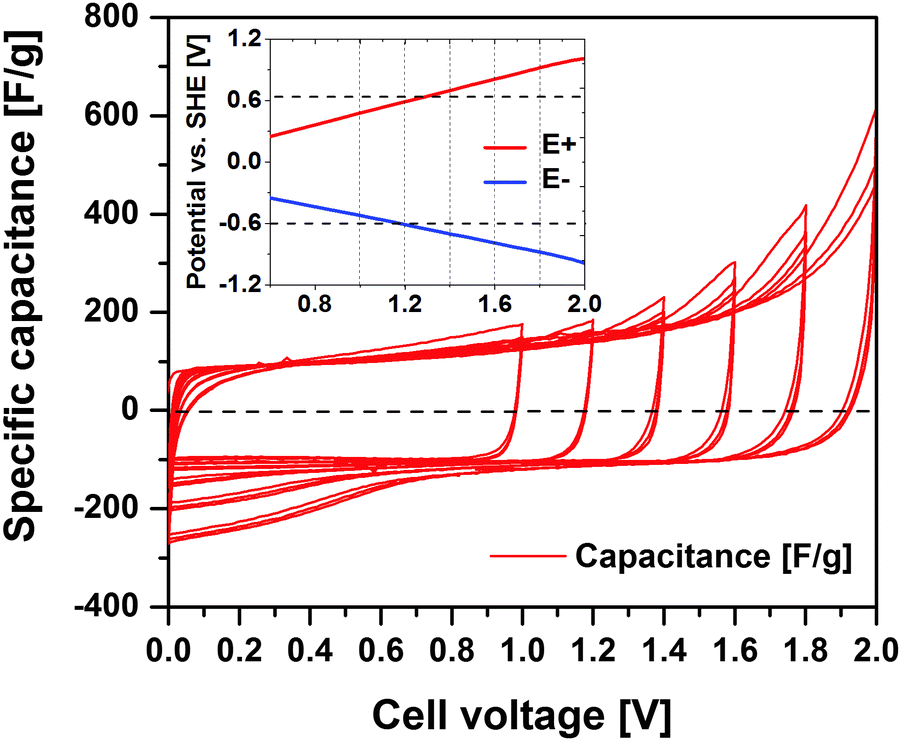 | ||
| Fig. 2 Cyclic voltammogram (1 mV s−1) of the activated carbon based symmetric EDLC cell (1 mol L−1 Li2SO4) with stepwise increasing cell voltage limits. The inset shows the positive (E+) and negative (E−) electrode potentials as a function of cell voltage. The upper and lower horizontal line indicate the thermodynamic limit of O2 evolution and H2 evolution, respectively. | ||
The inset of Fig. 2 shows the positive and negative electrode potentials (labelled as E+ and E−, respectively) for a given voltage applied over the symmetric EDLC cell as given by the reference electrode. The thermodynamic potential limits (represented by horizontal dashed lines) of oxygen evolution reaction (OER) and hydrogen evolution reaction (HER) calculated according to the Nernst equation are 0.63 V (EOER = 1.23–0.059 pH) and −0.60 V (EHER = −0.059 pH), respectively. The positive electrode reaches the thermodynamic OER potential limit when a 1.3 V cell voltage is applied. The practical HER potential is believed to shift towards more negative values due to hydrogen electrosorption on the activated carbon surface and a local pH value increase.10 Consequently, mainly the faradaic processes on the positive electrode are believed to restrict the maximum operating voltage and detrimentally impact on cycle performance of these types of symmetric EDLCs.
In situ internal cell pressure analysis
Fig. 3 shows the voltage, specific current, and internal pressure profiles as well as the specific pressure increase rates of the symmetric EDLC cell during cyclic voltammetry at (a) 10 mV s−1 and (b) 0.1 mV s−1 scan rates as well as during (c) accelerated ageing tests at 1.8 V. The faradaic contribution during CV to the total current is smaller at higher scan rates because the cell remains at higher polarization voltages for shorter periods of time, thus resulting in lower extent of electrochemical side-reactions. The influence of side-reactions is evidenced by comparing the internal cell pressure P (blue solid line in Fig. 3a and b) after the complete CV experiments: a total increase of ΔP ∼ 6 mbar for a 10 mV s−1 sweep rate is smaller than ∼95 mbar for 0.1 mV s−1. The amount of volatile side-products and the long-term cell pressure increase is proportional to the extent of irreversible side-reactions.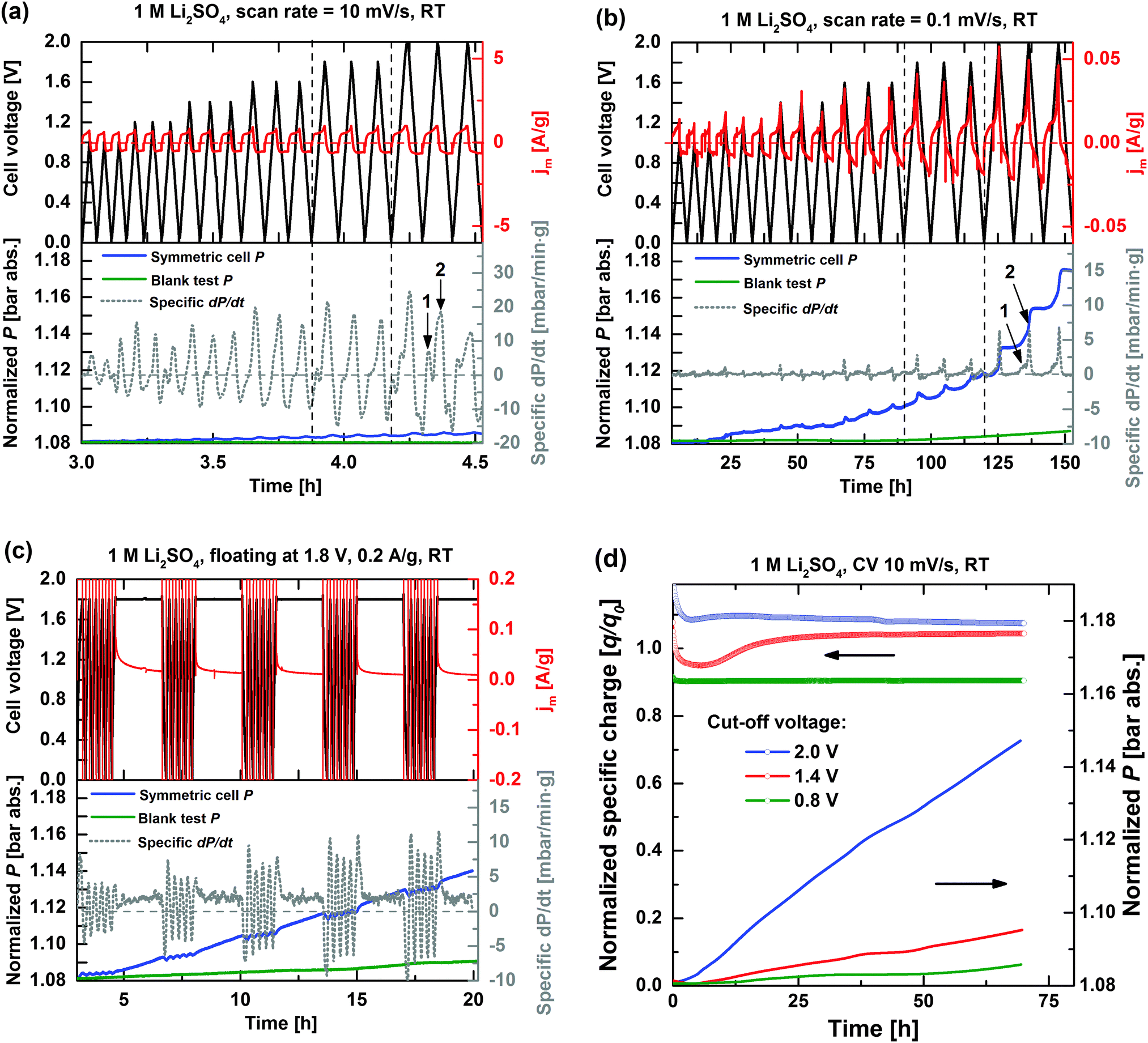 | ||
| Fig. 3 Cell voltage U (black solid lines), specific current jm (red solid lines), internal pressure P (blue solid lines), and calculated specific pressure increase rate dP/dt (grey dashed lines) profiles during CV at scan rates of (a) 10 mV s−1, and (b) 0.1 mV s−1 as well as during the accelerated ageing process floating at a voltage of 1.8 V (c). Internal pressure profiles of blank EDLC cells without activated carbon electrodes (green solid lines) are included. (d) Long-term specific charge retention and internal cell pressure increase during CV cycling at 10 mV s−1 with 0.8 V (green), 1.4 V (red), and 2 V (blue) cut-off voltages. | ||
On a short term, cell pressure fluctuations are observed during cycling, which largely is believed to arise from partly reversible gas evolution and consumption processes. For instance, gas bubble formation (e.g. H2 evolution) in the pores would both push the electrolyte out and expand the electrode package. Evolving gas may also leave the electrolyte phase to increase the pressure of the cell headspace. The pressure reduction on depolarization of the cell then corresponds to gas consumption, e.g. H2 oxidation.
Reversible swelling of the electrolyte volume may also occur during cycling due to changes in the space requirement of a given molecule/ion under polarization, which depends on its chemical and sterical environment, and may cause cell pressure increase on the forward sweep and relaxation on the backward sweep.16 A minor ion insertion into the activated carbon electrode or modification of micropore wettability may also cause volume expansion, but both these effects are estimated to have a comparatively minor influence on the internal cell pressure.16,37
In conclusion, two gas evolution mechanisms may therefore be discerned, i.e. a reversible gas evolution/consumption reaction (short-term pressure fluctuations) and an irreversible gas evolution reaction (long-term continuous pressure increase). In order to identify cell currents and pressure increases related to the current collector (e.g. corrosion, over-potential shift, etc.),12 blank tests (green solid line, Fig. 3) with identical polarization conditions, but excluding the two carbon electrodes, were performed and showed significantly lower gas evolutions compared to when the high-surface area activated carbon is included.
The highest cell specific current (∼1 A g−1) also results in the highest specific dP/dt, thus demonstrating the relationship between the overall cell current and the rate of side-reactions. The gas evolution rate inside the closed EDLC cell per gram of the carbon electrode is roughly proportional to the calculated specific dP/dt, which in turn can be further semi-quantitatively analysed by employing the ideal gas law. For instance, ∼25 mbar min−1 g−1 during the first cell polarization cycle to 2 V at 10 mV s−1 (Fig. 3a) roughly corresponds to a gas evolution rate of 2 μmol min−1 g−1 and a total formed gas amount in turn corresponding to an oxidation and reduction of less than 1% of the available electrolyte in the same cycle. The specific dP/dt peak gradually decreases upon repeated cycling, which agrees with the fading of the faradaic current during repeated CV cycles (clearly seen in Fig. 2), again indicating a gradual modification of the electrode surface.
At a lower sweep rate of 0.1 mV s−1 (Fig. 3b), the gas evolution onsets are more easily discernible. The extent of irreversible gas evolution (compare to Fig. 3a) is significantly enhanced when a cell voltage higher than 1.6 V is applied, which is believed to be due to the pronounced influence of water decomposition. The splitted dP/dt peaks (indexed in Fig. 3a and b to 1 and 2 in the 0–2 V range) indicate the presence of at least two different processes of gas evolution between U < 1.6 V and U > 1.6 V, which will be further discussed in detail.
Fig. 3c presents the internal pressure development during the accelerated ageing test, which contains five galvanostatic cycles (0.2 A g−1, 0 < U < 1.8 V) and a potentiostatic step at 1.8 V for 2 hours (also termed floating step), during which the leakage current is recorded.13 Despite the harsh ageing conditions, the double-layer capacitance of the cells is constant at ∼110 F g−1 regardless of the floating potential, thus indicating that the active electrode surface area is preserved after five ageing cycles. The internal cell pressure fluctuations are also observed for the galvanostatic cycling. However, both the leakage current and the specific dP/dt approach steady state values during floating steps, and show the presence of a single gas evolution process. For higher voltages, e.g. 2 V, the steady state gas evolution rate increases for every floating step (not shown), which points to an increasing reactivity at the activated carbon electrode/electrolyte interface with time at voltages U > 1.8 V.
Furthermore, the specific dP/dt peak amplitude during galvanostatic cycling is generally the highest in the first cycle after each floating step, thereafter it gradually decays. The initial higher peak in specific dP/dt may be related to the formation of reactive species during the floating step, which are slowly consumed during the five subsequent cycles.
The specific dP/dt is considerably higher during galvanostatic cycling compared to the floating steps in the same ageing sequence indicating that the degradation process of EDLCs is harsher during the cycling period than in the potentiostatic period. Such observation is consistent with the result of Avasarala et al., who compared the modification of carbon surface groups under both potential cycling and potential holding conditions by performing XPS characterization, thus further demonstrating that potential cycling significantly enhanced the extent of carbon surface oxidation.36
Fig. 3d shows the long-term specific charge retention and internal cell pressure increase during the CV cycling with cell cut-off voltages (Vc) of 0.8, 1.4, and 2 V. For easy comparison, the specific charge q is normalized with respect to the expected specific charge q0 (=C0·Vc [F V g−1]), simply assuming a double-layer capacitance C0 of 110 F g−1. A higher q reflects the increased influence of faradaic reactions, particularly at higher cell voltages. The initial fluctuations in q are related to the reactions of residual cell impurities and incomplete pore wetting.13 Clearly, higher operational voltages increase the rate of electrode and electrolyte related side-reactions as both cell specific charge loss and internal pressure linearly increase proportionally without any indication of stabilizing. Again, assuming that every mole of formed gas corresponds to a mole of consumed carbon (1 C per gas molecule, e.g. as for CO or CO2), the pressure increase of ∼70 mbar during long-term cycling at 2 V (Fig. 3d) corresponds to a consumption of 5.5 μmol C (∼80 nmol mbar−1, according to the ideal gas law), which is ∼0.4% of the available activated carbon electrode mass. A specific charge loss of 2% can thus only partly be explained by direct carbon corrosion in form of gas, and further effects associated with the electrode morphology and/or the electrolyte have to be considered.
In situ OEMS analysis
Fig. 4a and b show the cell current response as well as the H2, CO, O2, and CO2 mass signal intensities during cyclic voltammetry (0.1 mV s−1) of the symmetric EDLC cell with the positively polarized electrode on the top and bottom, respectively. Higher sweep rates were omitted due to the insufficient time response of the OEMS setup. Compared to the pressure cell, where the formed gases remain inside the cell, the OEMS cell is continuously flushed with a stream of Ar, which removes the gases from the cell once formed (to the inlet of the mass spectrometer) and therefore prevents further reactions. Gases forming on the top electrode have a short diffusion distance before being carried away with the Ar-stream,38 unlike gases forming on the bottom electrode that first have to cross the separator and the top electrode. Therefore, by exchanging the polarity of the cell, gas evolution/interactions associated with positive and negative electrodes may be partly decoupled.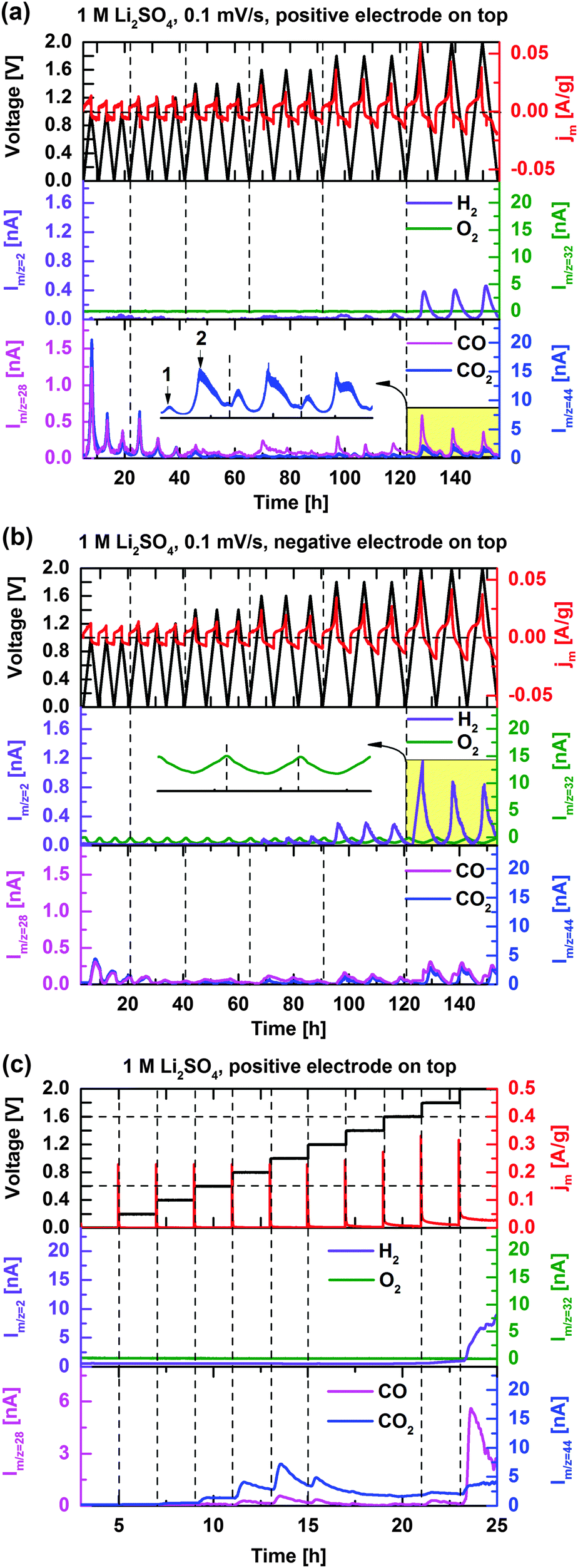 | ||
| Fig. 4 Cell current response and H2, O2, CO, and CO2 mass signal intensities during cyclic voltammetry at 0.1 mV s−1 with (a) the positively polarized carbon electrode on the top, and (b) negatively polarized carbon electrode on the top. Insets: Zoom-in of CO2 and O2 ion-current profiles (yellow labelled region). (c) Cell current response and mass signal intensities during potentiostatic stepping in cell voltage with the positive electrode located at the top. | ||
When the positive electrode is located at the top (Fig. 4a), H2 evolution is detected in the cell voltage ranges of 0–1.8 V and 0–2 V, as the negative electrode potential is beyond the thermodynamic limit of HER (−0.6 V vs. SHE, Fig. 2). When the negative electrode is located at the top, the H2 evolution onset is found earlier (at a cell voltage of 1.6 V, Fig. 4b) and at significantly higher H2 evolution rates. H2 evolution is thus confirmed to follow on the negative electrode exceeding thermodynamic HER potential, leading to pronounced H2 trapping and/or oxidation in the presence of the positive electrode on top. Even though the positive electrode is polarized beyond the onset potential of OER (0.63 V vs. SHE, Fig. 2) at cell voltages U > 1.2 V, no O2 evolution was observed at any potential. However, drops in the O2 background signal (inset of Fig. 4b) are noticed during cell polarization and related to electrochemical reduction of trace O2 present in the Ar stream (∼100 ppm O2) of the OEMS (e.g. from leakage). Because of the high sensitivity of the mass spectrometer, such small variations in O2 partial pressure due to O2 reduction on the negative electrode are readily observed,39 and provides a clue to the reversible gas pressure variations in the closed pressure cell configuration above. However, traces of O2 are found at very low concentrations and do not significantly influence cell chemistry. The absence of O2 evolution is probably due to carbon corrosion, consuming the reactive oxygen once formed. However, the absence of O2 may also be explained by the increased over-potentials for OER due to the high H2O de-solvation energy of the electrolyte salt. The strong ion solvation of Li+ and SO42− ions has been proposed as the origin of the enhanced capacitance and voltage in 1 mol L−1 Li2SO4 electrolytic media.6
The ion-current signals of CO and CO2 appearing on m/z = 28 and 44, respectively, present nearly identical evolution profiles when the symmetric EDLC cell is cycled. The overlap is partly explained by the minor contribution of CO2 to m/z = 28 due to induced fragmentation by the electron impact ionization of the MS.40 The CO evolution can however be anticipated by comparing the m/z = 28 and 44 evolution profiles. The peak intensities of CO/CO2 in each CV cycle are detected at the maximum applied voltages. The evolution at relatively low cell voltages U < 1.2 V are assigned to the decomposition of carbon surface functionalities, which is supported by the gradually decreasing signal intensities until the cell voltage reaches 1.8 V (Fig. 4a and b). In order to more precisely determine the voltage onset for the carbon surface modification, Fig. 4c shows gas evolution profiles of the EDLC cell when the cell voltage is stepwise increased from the open circuit voltage to 2 V. Apparently, CO2 evolution starts already at a cell voltage of 0.6 V and a maximum ion-current intensity is observed at 1 V under the present conditions, the pristine carbon surface functional groups are assumed to be continuously decomposed at cell voltages U > 0.6 V with the electrochemical potential as the driving force. Once cell voltages higher than 1 V are reached, the surface groups approach depletion and the CO2 evolution declines.
In agreement with the specific dP/dt pattern in Fig. 3a and b, two CO2 evolution peaks in voltage range of 0–2 V can be clearly distinguished (inset of Fig. 4a). The first CO2 evolution peak (0 < U < 1.6 V) corresponds to the decomposition of carbon surface groups while carbon corrosion is believed to account for the second CO2 evolution peak at cell voltages higher than 1.6 V. The ion-current intensity related to CO (m/z = 28) increases to a larger extent at U > 1.6 V than for CO2, indicating that CO gas evolution is higher during the carbon corrosion process (Fig. 4a–c). CO and CO2 evolution intensities significantly decrease after moving the negative polarized electrode to the top (Fig. 4b), which shows that these gases derive from the positive electrode. Besides, chemical absorption of CO2 into the electrolyte may also have influence on CO2 intensity. Accelerated ageing tests were also performed with the OEMS configuration and confirm the above findings in the cell pressure characterization section.
Mechanism discussion
In situ pressure cell and OEMS analysis provide complementary data on gas evolution in EDLC cells. The type, onset, and quantity of gas evolution are correlated with the potential of the activated carbon electrodes. H2, CO and CO2 are the detected volatile species, and the involved reaction processes may presumably be classified into four general categories:1. Carbon surface functional group decomposition.
2. Water splitting (HER and OER).
3. Carbon corrosion.
4. Chemical/electrochemical gas consumption.
The proposed gas evolution mechanisms for an activated carbon based EDLC cell containing 1 mol L−1 Li2SO4 electrolyte are illustrated in Scheme 1, and further details of the potential dependence and reaction mechanisms are given in Table 1.
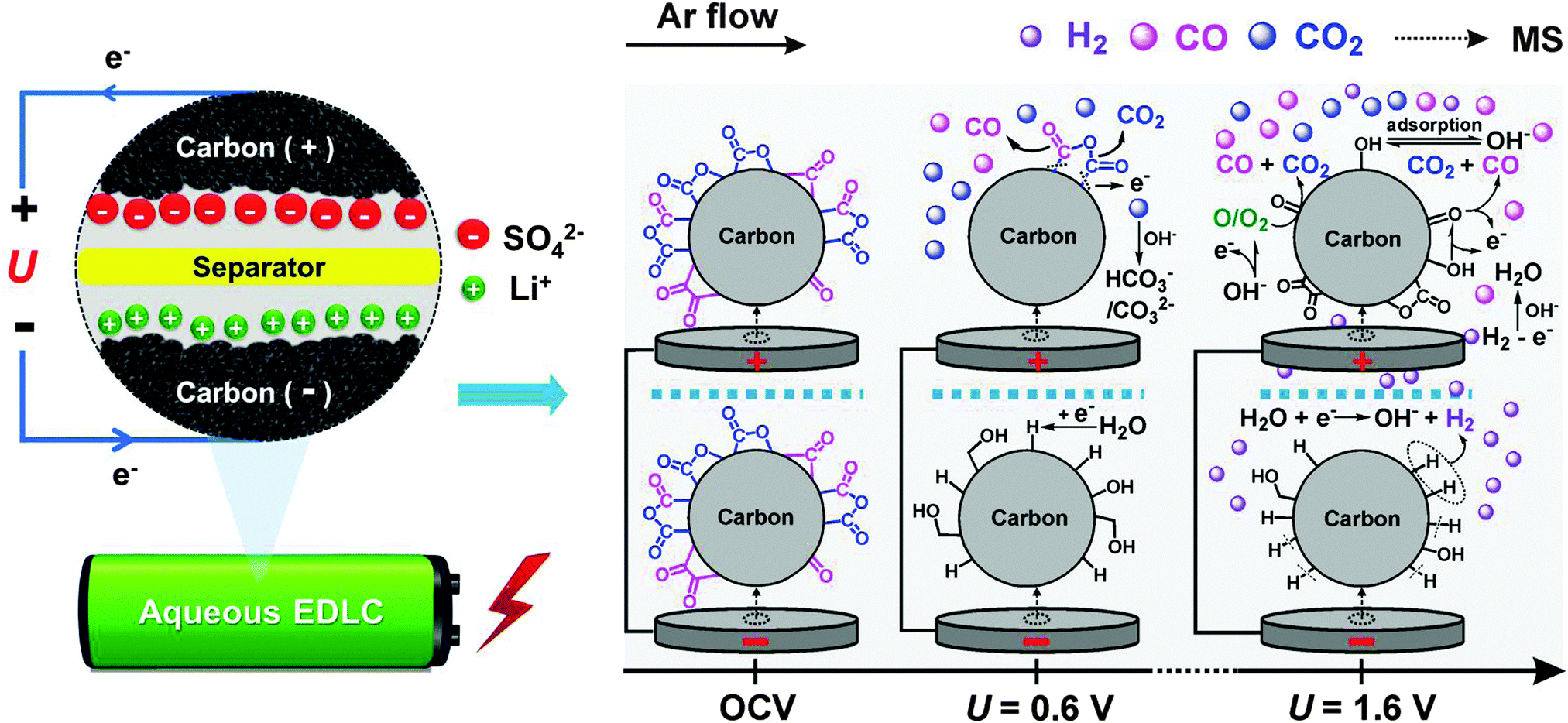 | ||
| Scheme 1 Gas evolution and electrochemical processes occurring on the activated carbon surface at different cell voltages in EDLCs with 1 mol L−1 Li2SO4 electrolyte (positively polarized electrode on the top). | ||
| Process | U [V] | E [V vs. SHE] | Reactions | # | Ref. |
|---|---|---|---|---|---|
| Carbon surface group oxidation | ∼0.6 | 0.25 (∼0.6, pH = 1) | e.g. (RCO) 2 O − 4e − → 2R + CO↑ + CO 2 ↑ | 1 | 32, 36 and 41 |
| R 1 COCOR 2 − 4e−→ R1+ R2+ 2CO↑ | 2 | ||||
| Carbon surface group reduction | n.a. | n.a. (∼−0.1, pH = 10) | e.g., R 1 COR 2 + 2e − + 2H + → R 1 CHOHR 2 | 3 | 36, 42 and 43 |
| R 1 COCOR 2 + 4e − + 4H + → R 1 (CHOH) 2 R 2 | 4 | ||||
| H2 chemisorption | n.a. | n.a. (<−0.35, pH = 7) | AC + xH2O + xe− → ACHx + xOH− | 5 | 7, 10, 44 and 45 |
| H2 evolution | >1.6 | <−0.79 | CHad + H2O + e− → H2↑ + OH− + C | 6 | 46 |
| CHad + CHad → H2↑ + 2C | 7 | ||||
| O2 evolution | None | None | 2H2O–4e− → O2↑ + 4H+ | 8 | 47 |
| Carbon corrosion | >1.6 | >0.81 | H2O − ne− → OH˙ + H+ (n = 1), O˙ + 2H+ (n = 2) | 9 | 36 and 48–51 |
C + O˙/OH˙ → C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/i_char_e001.gif) O, C–OHads → CO (+H+)↑ O, C–OHads → CO (+H+)↑
|
10 | ||||
| C + 2H2O − 4e−→ CO/CO2↑ + 4H+ | 11 | ||||
| Oxidation of hydrogen | n.a. | n.a. (∼0.2, pH = 0) | 2CHad–2e− → 2C + 2H+ | 12 | 52 and 53 |
| H2–2e− → 2H+ | 13 | ||||
| Trace O2 reduction | ∼0.4 | ∼0.1 | O2 + H2O + 2e− → HO2− + OH− | 14 | 39 |
| O2 + 4H2O + 4e− → 4OH− | 15 | ||||
| CO oxidation | n.a. | n.a. | CO + O/O2 → CO2↑ | 16 | 54 |
| CO2 chemical absorption | n.a. | n.a. | CO2 + 2OH− → H2O + CO32− | 17 | 55 and 56 |
| 2Li+ + CO32−→ Li2CO3↓ | 18 | ||||
The simultaneous CO and CO2 evolution observed at cell voltages U > 0.6 V (Scheme 1) are assumed to be initiated by the electrochemical oxidation of the existing pristine surface groups of the activated carbon electrode (eqn (1) and (2), Table 1).36,57 TPD indicates the existence of phenol, anhydride, lactone, quinone and carbonyl groups etc. Anhydride and lactone groups may electro-oxidize to form CO2 at an electrode potential of 0.25 V, whereas oxidation of quinone and carbonyl groups preferentially yields CO evolution at identical onsets, thus possibly explaining the apparent parallel evolution of those species in our experiments.32,48 Despite a stable cycling in a low voltage regime (<0.8 V), the carbon surface functional groups appear to be continuously oxidized and reformed, thus explaining the very minor long-term internal pressure increase (Fig. 3d). On the negatively polarized electrode, surface functional groups containing sp-hybridized oxygen, e.g. quinone and carbonyl groups, can be reduced at the same voltages to yield alcohol-like species (eqn (3) and (4)).
Electrochemical decomposition of water, i.e. hydrogen/oxygen evolution reaction (HER/OER), is expected at higher cell voltages (U > 1.2 V). On the negative electrode, the reduction process initiates with hydrogen chemisorption (eqn (5)) and storage. In situ Raman spectroscopy has demonstrated that considerable amounts of hydrogen can be reversibly stored in activated carbons on HER.52 The H+/C–Had redox couple offers additional pseudo-capacitance for activated carbon based aqueous EDLCs as hydrogen may strongly adsorb into the activated carbon micropores45 and can later be reversibly oxidized under anodic conditions (eqn (12)). At cell voltages U > 1.6 V, recombination of nascent chemisorbed hydrogen (Scheme 1) is clearly observed to form H2 gas (eqn (6) and (7)) on the negative electrode.46
On the positive electrode, OER would be the expected counter reaction (eqn (8)) as the electrode is above the thermodynamic potential for water splitting (Fig. 2). However, no O2 is observed under any investigated conditions partly due to high overpotential for OER, which in turn is assumed to be related to the higher de-solvation energy of SO42−–H2O and Li+–H2O bonds in the 1 mol L−1 Li2SO4 aqueous electrolyte system.6 However, instead of O2, water oxidation most probably yields reactive atomic oxygen O˙ or OH˙ (eqn (9)) that rapidly react with the activated carbon to preferentially form carbonyl and phenol (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, C–OH) surface groups,36,58 which in turn immediately oxidize to form predominantly CO (eqn (10)), as outlined by Figueiredo et al.32 Such carbon corrosion processes appear to be the governing gas evolution process at cell voltages U > 1.6 V (Scheme 1) and lead to significantly higher specific charge loss and pressure increase rates (∼2 V, Fig. 3d) compared to intermediate (∼1.4 V) or lower cell voltage cut-offs (∼0.8 V).
O, C–OH) surface groups,36,58 which in turn immediately oxidize to form predominantly CO (eqn (10)), as outlined by Figueiredo et al.32 Such carbon corrosion processes appear to be the governing gas evolution process at cell voltages U > 1.6 V (Scheme 1) and lead to significantly higher specific charge loss and pressure increase rates (∼2 V, Fig. 3d) compared to intermediate (∼1.4 V) or lower cell voltage cut-offs (∼0.8 V).
Carbon corrosion is generally complex, and sensitively dependent on electrolyte pH and electrode potential. Several reaction mechanisms have been proposed for neutral and alkaline solutions, in which the activated carbon is either indirectly oxidized via the formation of surface groups (eqn (9) and (10)) or directly oxidized (eqn (11)).59 Yang et al. proposed that at lower anodic potential (i.e. E < 1.44 V vs. SHE in 0.5 mol L−1 H3PO4) surface oxide species were first transformed to quinone-like and carboxylic-like species, while at higher anodic potentials (i.e., E > 1.89 V vs. SHE in 0.5 mol L−1 H3PO4) atomic oxygen from the water oxidation was assumed to cause direct carbon corrosion (eqn (9)).48 Several well documented carbon degradation mechanisms occurring in fuel cell applications could also be considered (eqn (11)).50,51
In addition to the above-described gas evolution processes, H2, O2, CO and CO2 gases may be chemically or electrochemically consumed. The competition between gas evolution and consumption processes may at least partially explain the breathing-like pressure fluctuation inside EDLC cells. H2 gas forming on the negative electrode may oxidize on the backward potential sweep and/or diffuse to and oxidize on the positive electrode (eqn (13)) during cycling. As observed in the OEMS experiment of Fig. 4b, trace oxygen in Ar stream was readily reduced on the negative electrode (eqn (14) and (15)). Even though the ion-current on m/z = 28 partly belongs to CO2, the relative ratio between m/z = 28 and 44 changes at higher potentials (Fig. 4a and b) indicating the increasing formation of CO. However, the low amounts of CO may be explained by its further oxidation to CO2 before leaving the cell (eqn (16)). Such a reaction would however not contribute to the cell pressure build-up. Alkaline solutions are furthermore well known to form an equilibrium with gaseous CO2 to form insoluble carbonate precipitates (eqn (17) and (18)). However, neither a significant pH value change of the electrolyte nor the formation of any precipitate was observed after the cycling experiments. On the other hand, local pH changes in the electrode bulk should be considered.
Conclusions
Online electrochemical mass spectrometry, combined with in situ pressure cell analysis, was for the first time employed to study the performance-determining side-reactions in high-voltage supercapacitors based on an aqueous electrolyte and activated carbon. Rapid short-term cycling leads to breathing-like and partly reversible cell pressure fluctuation, which mainly is believed to be related to reversible gas formation/consumption reactions. Although these side-reactions provide additional pseudo-capacitance at higher voltages, both the long-term performance and the safety are compromised due to the irreversible formation of volatile side-reaction products. Gas evolution may further increase the overall cell impedance. For instance, the formation of gas bubbles at the current collector and/or inside the electrode pores introduce both electronic and ionic resistance by inducing electric contact loss and/or obstructing ionic transport, respectively.Processes, such as carbon surface functional group decomposition, water splitting, and carbon corrosion, were shown to be distinguishable according to their potential dependent onsets, type and amount of gas evolved. The activated carbon electrode with a high surface area and significant amounts of phenol, anhydride, lactone, quinone and carbonyl functional groups, apparently oxidize early to form mainly CO2 gas at cell voltage U > 0.6 V until depletion from the electrode surface. H2 evolution starts close to the thermodynamic HER potentials (U > 1.2 V), but because of carbon surface group modification and hydrogen storage, significant H2 gas evolution occurs first at a cell voltage of U > 1.6 V. No O2 gas evolution was observed at any potential as the formed oxygen species are believed to directly cause carbon corrosion at high cell voltages U > 1.6 V. Direct carbon corrosion on the positive electrode is believed to yield mainly CO and C–OH surface groups due to the strong reactivity of the carbon towards oxidized H2O in the form of oxygen radicals, thus explaining the predominant CO evolution at high voltages. Repeated cycling in a wide electrochemical window, especially in the mild current/scan rate regime supplying more charge for parasitic reactions (gas evolution) appears to be harsher on the chemical integrity of the cell compared to the potential holding steps.
In summary, in situ gas analysis is shown to provide unique tools in the identification, characterization and fundamental understanding of aqueous supercapacitor side-reactions. These investigations constitute the first critical steps to mitigate cell ageing issues and design the next-generation high-energy aqueous based supercapacitors.
Acknowledgements
Financial support from the Polish-Swiss Research Programme (INGEC PSPB-107/2010) is gratefully acknowledged.References
- P. Simon and Y. Gogotsi, Nat. Mater., 2008, 7, 845–854 CrossRef CAS PubMed.
- M. Winter, Chem. Rev., 2004, 104, 4245 CrossRef CAS PubMed.
- F. Béguin, V. Presser, A. Balducci and E. Frackowiak, Adv. Mater., 2014, 26, 2219–2251 CrossRef PubMed.
- V. Aravindan, J. Gnanaraj, Y.-S. Lee and S. Madhavi, Chem. Rev., 2014, 114, 11619–11635 CrossRef CAS PubMed.
- D. Qu, J. Appl. Electrochem., 2009, 39, 867–871 CrossRef CAS.
- K. Fic, G. Lota, M. Meller and E. Frackowiak, Energy Environ. Sci., 2012, 5, 5842–5850 CAS.
- E. Frackowiak, Q. Abbas and F. Béguin, J. Energy Chem., 2013, 22, 226–240 CrossRef CAS.
- K. Fic, M. Meller and E. Frackowiak, Electrochim. Acta, 2014, 128, 210–217 CrossRef CAS.
- L. Demarconnay, E. Raymundo-Piñero and F. Béguin, Electrochem. Commun., 2010, 12, 1275–1278 CrossRef CAS.
- Q. Gao, L. Demarconnay, E. Raymundo-Pinero and F. Beguin, Energy Environ. Sci., 2012, 5, 9611–9617 CAS.
- Q. Abbas, D. Pajak, E. Frackowiak and F. Béguin, Electrochim. Acta, 2014, 140, 132–138 CrossRef CAS.
- P. Ratajczak, K. Jurewicz, P. Skowron, Q. Abbas and F. Béguin, Electrochim. Acta, 2014, 130, 344–350 CrossRef CAS.
- P. Ratajczak, K. Jurewicz and F. Béguin, J. Appl. Electrochem., 2014, 44, 475–480 CrossRef CAS.
- M. Nakamura, M. Nakanishi and K. Yamamoto, J. Power Sources, 1996, 60, 225–231 CrossRef CAS.
- T. Brousse, M. Toupin and D. Bélanger, J. Electrochem. Soc., 2004, 151, A614–A622 CrossRef CAS.
- M. Hahn, R. Kötz, R. Gallay and A. Siggel, Electrochim. Acta, 2006, 52, 1709–1712 CrossRef CAS.
- R. Kötz, M. Hahn, P. Ruch and R. Gallay, Electrochem. Commun., 2008, 10, 359–362 CrossRef.
- M. Hahn, A. Würsig, R. Gallay, P. Novák and R. Kötz, Electrochem. Commun., 2005, 7, 925–930 CrossRef CAS.
- R. Imhof and P. Novák, J. Electrochem. Soc., 1998, 145, 1081–1087 CrossRef CAS.
- Z. Jusys and R. J. Behm, J. Phys. Chem. B, 2001, 105, 10874–10883 CrossRef CAS.
- O. Wolter and J. Heitbaum, Ber. Bunsen–Ges., 1984, 88, 2–6 CrossRef CAS.
- P. Novák, F. Joho, R. Imhof, J.-C. Panitz and O. Haas, J. Power Sources, 1999, 81–82, 212–216 CrossRef.
- R. Imhof and P. Novák, J. Electrochem. Soc., 1999, 146, 1702–1706 CrossRef CAS.
- M. Lanz and P. Novák, J. Power Sources, 2001, 102, 277–282 CrossRef CAS.
- W. Siriwatcharapiboon, Y. Kwon, J. Yang, R. L. Chantry, Z. Li, S. L. Horswell and M. T. M. Koper, ChemElectroChem, 2014, 1, 172–179 CrossRef.
- H. Liu, Q. Xu and C. Yan, Electrochem. Commun., 2013, 28, 58–62 CrossRef.
- S. Meini, N. Tsiouvaras, K. U. Schwenke, M. Piana, H. Beyer, L. Lange and H. A. Gasteiger, Phys. Chem. Chem. Phys., 2013, 15, 11478–11493 RSC.
- J. Jagiello and J. P. Olivier, Carbon, 2013, 55, 70–80 CrossRef CAS.
- M. He, E. Castel, A. Laumann, G. Nuspl, P. Novák and E. J. Berg, J. Electrochem. Soc., 2015, 162, A870–A876 CrossRef CAS.
- P. Novák, W. Scheifele, F. Joho and O. Haas, J. Electrochem. Soc., 1995, 142, 2544–2550 CrossRef.
- E. Castel, E. J. Berg, M. El Kazzi, P. Novák and C. Villevieille, Chem. Mater., 2014, 26, 5051–5057 CrossRef CAS.
- J. L. Figueiredo, M. F. R. Pereira, M. M. A. Freitas and J. J. M. Órfão, Carbon, 1999, 37, 1379–1389 CrossRef CAS.
- K. H. Kangasniemi, D. A. Condit and T. D. Jarvi, J. Electrochem. Soc., 2004, 151, E125–E132 CrossRef CAS.
- M. J. Bleda-Martínez, D. Lozano-Castelló, E. Morallón, D. Cazorla-Amorós and A. Linares-Solano, Carbon, 2006, 44, 2642–2651 CrossRef.
- C. Moreno-Castilla, M. V. López-Ramón and F. Carrasco-Marín, Carbon, 2000, 38, 1995–2001 CrossRef CAS.
- B. Avasarala, R. Moore and P. Haldar, Electrochim. Acta, 2010, 55, 4765–4771 CrossRef CAS.
- M. Hahn, O. Barbieri, R. Gallay and R. Kötz, Carbon, 2006, 44, 2523–2533 CrossRef CAS.
- E. Berg and P. Novák, Annual Report 2012, Electrochemistry Laboratory, Paul Scherrer Institut, Switzerland, 47.
- S. G. Bratsch, J. Phys. Chem. Ref. Data, 1989, 18, 1–21 CrossRef CAS.
- NIST Mass Spectrometry Data Center, NIST MS Number 69, NIST Chemistry WebBook, http://webbook.nist.gov, 2015.
- P. E. Fanning and M. A. Vannice, Carbon, 1993, 31, 721–730 CrossRef CAS.
- H. P. Boehm and M. Voll, Carbon, 1970, 8, 227–240 CrossRef CAS.
- B. D. Epstein, E. Dalle-Molle and J. S. Mattson, Carbon, 1971, 9, 609–615 CrossRef CAS.
- S. Leyva-García, E. Morallón, D. Cazorla-Amorós, F. Béguin and D. Lozano-Castelló, Carbon, 2014, 69, 401–408 CrossRef.
- A. Wesełucha-Birczyńska, K. Babeł and K. Jurewicz, Vib. Spectrosc., 2012, 60, 206–211 CrossRef.
- K. Jurewicz, E. Frackowiak and F. Béguin, Appl. Phys. A: Mater. Sci. Process., 2004, 78, 981–987 CrossRef CAS.
- P. K. Dubey, A. S. K. Sinha, S. Talapatra, N. Koratkar, P. M. Ajayan and O. N. Srivastava, Int. J. Hydrogen Energy, 2010, 35, 3945–3950 CrossRef CAS.
- Y. Yang and Z. G. Lin, J. Appl. Electrochem., 1995, 25, 259–266 CrossRef CAS.
- X. Jin, W. Zhou, S. Zhang and G. Z. Chen, Small, 2007, 3, 1513–1517 CrossRef CAS PubMed.
- H. Tang, Z. Qi, M. Ramani and J. F. Elter, J. Power Sources, 2006, 158, 1306–1312 CrossRef CAS.
- Y. Yu, Z. Tu, H. Zhang, Z. Zhan and M. Pan, J. Power Sources, 2011, 196, 5077–5083 CrossRef CAS.
- K. Fic, M. Meller and E. Frackowiak, J. Electrochem. Soc., 2015, 162, A5140–A5147 CrossRef CAS.
- R. M. Q. Mello and E. A. Ticianelli, Electrochim. Acta, 1997, 42, 1031–1039 CrossRef CAS.
- S. Huang, K. Hara and A. Fukuoka, Energy Environ. Sci., 2009, 2, 1060–1068 CAS.
- G. Astarita, Ind. Eng. Chem. Fundam., 1963, 2, 294–297 Search PubMed.
- D. Roberts and P. V. Danckwerts, Chem. Eng. Sci., 1962, 17, 961–969 CrossRef CAS.
- M. Zhu, C. J. Weber, Y. Yang, M. Konuma, U. Starke, K. Kern and A. M. Bittner, Carbon, 2008, 46, 1829–1840 CrossRef CAS.
- A. Pandy, Z. Yang, M. Gummalla, V. V. Atrazhev, N. Y. Kuzminyh, V. I. Sultanov and S. Burlatsky, J. Electrochem. Soc., 2013, 160, F972–F979 CrossRef CAS.
- K. Kinoshita, Carbon: Electrochemical and Physicochemical Properties, John Wiley & Sons, New York, 1988 Search PubMed.
| This journal is © The Royal Society of Chemistry 2016 |