
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAllenes, versatile unsaturated motifs in transition-metal-catalysed [2+2+2] cycloaddition reactions
A.
Lledó
,
A.
Pla-Quintana
* and
A.
Roglans
*
Institut de Química Computacional i Catàlisi (IQCC) and Departament de Química, Universitat de Girona (UdG), Facultat de Ciències, Campus de Montilivi, s/n, 17071 Girona, Spain. E-mail: anna.roglans@udg.edu
First published on 3rd February 2016
Abstract
The development of efficient reactions that enable the construction of multiple bonds and/or stereogenic centres in a single synthetic operation is of great interest for greener and step-economical syntheses of complex organic targets. Transition-metal-catalysed [2+2+2] cycloadditions excell in this regard: no fewer than three new sigma bonds and a new ring system are formed from simple unsaturated components. Allenes constitute an important subclass among these. They are more reactive than simple alkenes and generate sp3 centres, with important stereochemical implications. In addition, further manipulation of the resulting cycloadduct through the remaining double bond is possible, increasing molecular complexity. This tutorial review highlights the value that allenes have as reagents in [2+2+2] cycloaddition and their great versatility for the development of methods to generate complex architectures.
 From left to right: A. Lledó, A. Pla-Quintana and A. Roglans | Agustí Lledó (1980, Barcelona) obtained his PhD in chemistry from the University of Barcelona (UB) in 2006. In 2007 he carried out post-doctoral studies with Prof. Julius Rebek, Jr at The Scripps Research Institute (TSRI). In 2010 he returned to Spain and joined the Institute for Research in Biomedicine, Barcelona (IRB Barcelona) as a “Juan de la Cierva” research fellow. In 2014 he transferred to his current position at the University of Girona (UdG), where he works as a “Ramón y Cajal” researcher. His research interests include transition-metal catalysis, organometallic chemistry and supramolecular chemistry. |
Anna Pla-Quintana (1978, Banyoles) obtained her PhD in chemistry from the University of Girona (UdG) in 2004. After post-doctoral studies under the supervision of Dr Jean-Pierre Majoral at the Laboratoire de Chimie de Coordination (LCC) Toulouse (2005–2007), she was promoted to lecturer in chemistry in 2010. Her research focuses on the development of transition-metal-catalyzed processes and the study of the mechanisms involved. |
Anna Roglans (1964, Palafrugell) obtained her PhD in chemistry from the Autonomous University of Barcelona (UAB) in 1994 under the supervision of Prof. M. Moreno-Mañas, before joining the group of Prof. V. Snieckus at the University of Waterloo (Canada) for post-doctoral studies. She returned to the UAB with a post-doctoral contract and in 1998 obtained a position as a lecturer at the University of Girona (UdG). In 2010 she was promoted to full professor in chemistry. She currently leads the Transition Metals in Organic Synthesis Group and her research interests involve the study of transition-metal-catalysis, organometallic chemistry and the elucidation of reaction mechanisms. |
Key learning points(1) The versatility of allenes in transition-metal-catalysed cycloaddition reactions(2) Mechanistic rationale to explain the selectivity of the cycloadditions (3) Recent advances in stereoselective six-membered ring synthesis (4) Effect of the transition-metal on the outcome of the cycloaddition (5) The potential synthetic applications of [2+2+2] cycloadditions involving allenes |
Introduction
The construction of new ring systems is a key step in a multitude of organic syntheses. Among the strategies available to assemble cyclic frameworks, transition-metal-catalysed cycloadditions stand out as one of the most efficient options. A wide range of different six-membered rings can be efficiently built up by the transition-metal-catalysed [2+2+2] cycloaddition reaction,1 a simplified representation of which is shown in Scheme 1. The reaction is remarkable in terms of the increase in molecular complexity, the functional group compatibility, the extremely high atom-efficiency, and the variety of substrates that can be used. On the other hand, the control of the chemo- and regioselectivity is the most critical aspect of this cycloaddition due to the large number of isomers that can be formed.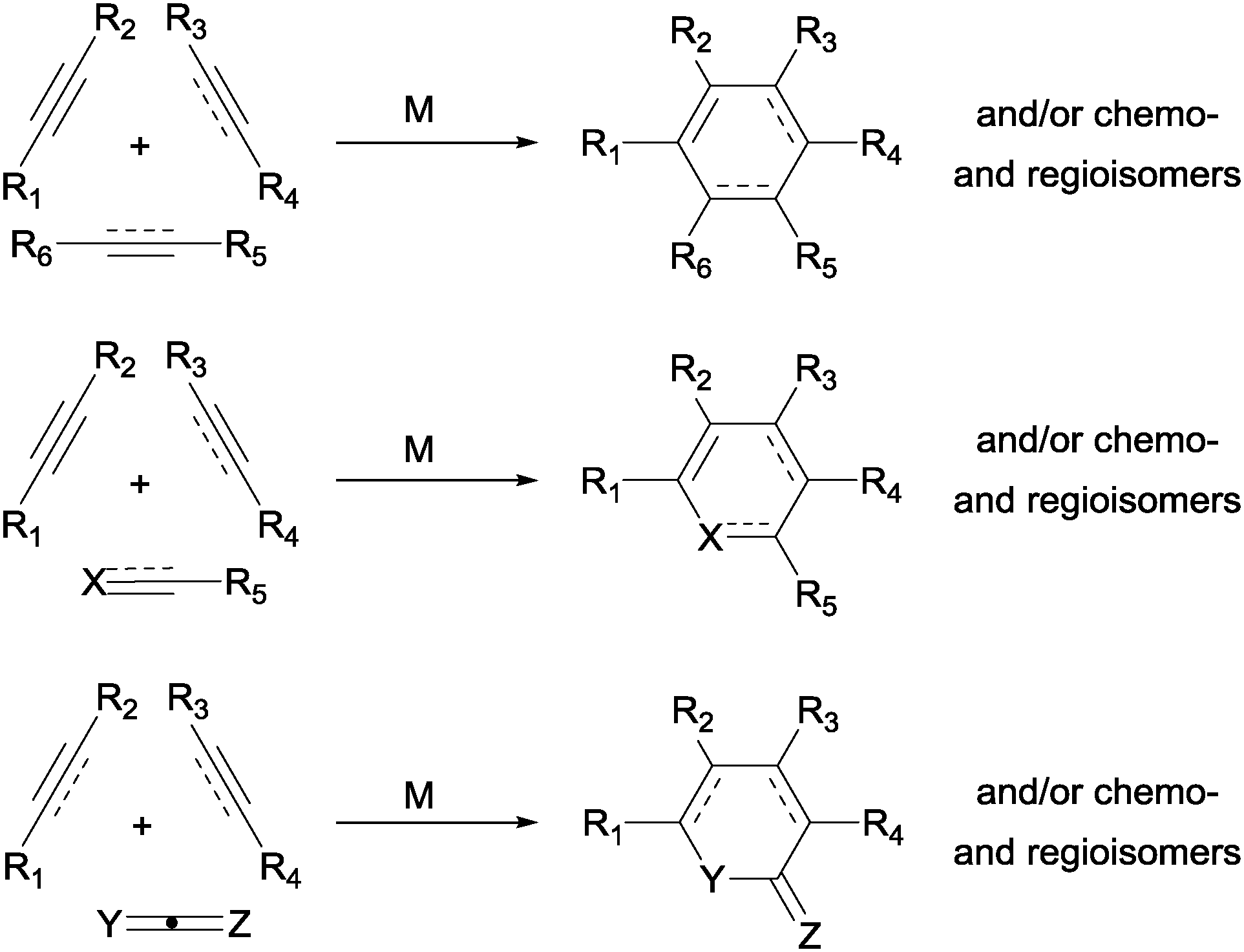 | ||
| Scheme 1 Transition-metal-catalysed [2+2+2] cycloaddition reaction to afford six-membered rings (M = transition-metal complex). | ||
The [2+2+2] cycloaddition reaction can be catalysed by a wide range of early to late transition-metals. Despite the large number of complexes that can catalyse this transformation, the mechanism is postulated to be similar for complexes based on cobalt, rhodium, ruthenium, nickel, and iridium, which are amongst the most commonly used metals in these processes. Taking as an example the [2+2+2] cycloaddition of three alkynes, substrates in which the reaction was first described by Reppe et al.,2 the mechanism can be summarised as outlined in Scheme 2.
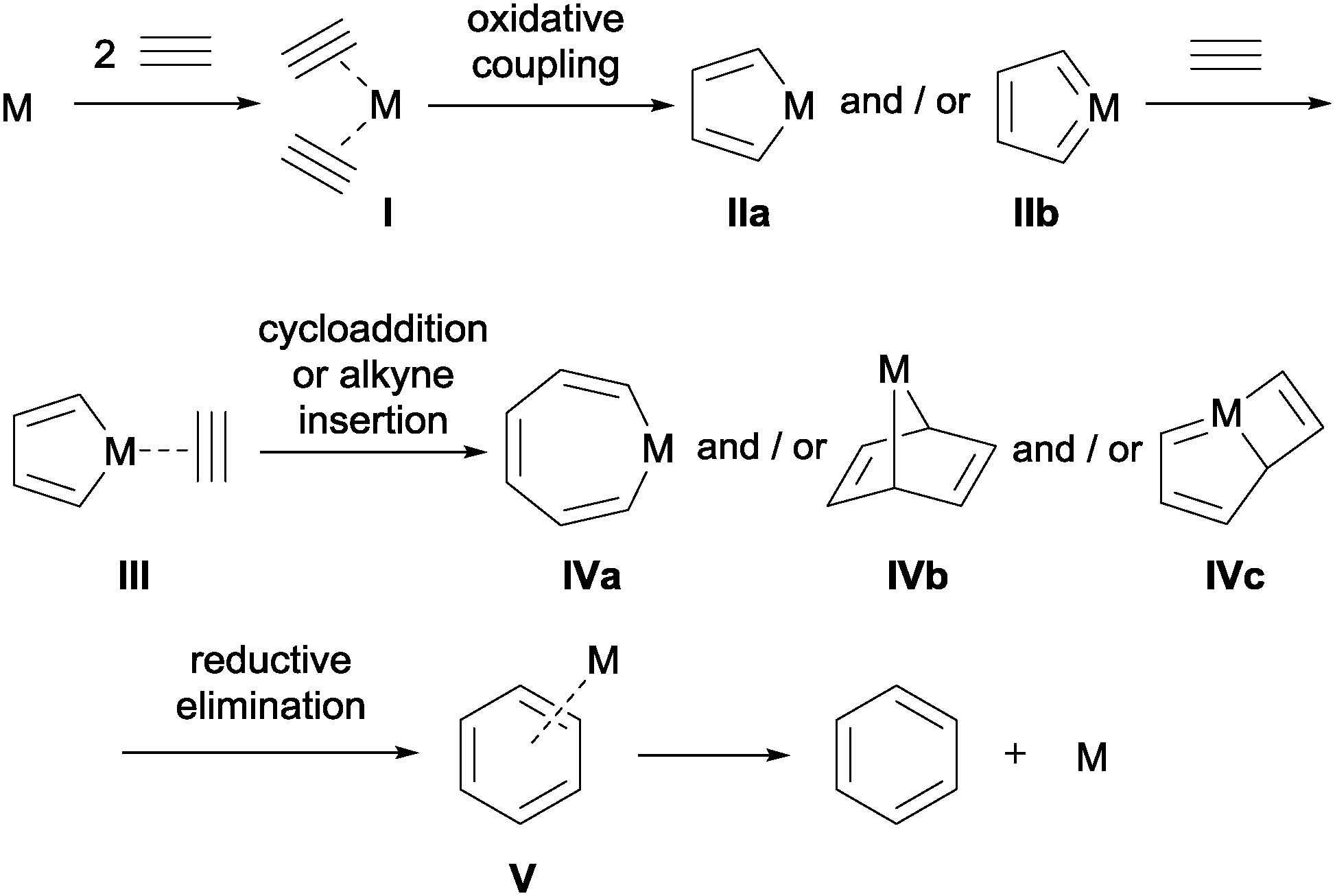 | ||
| Scheme 2 General overview of the mechanistic cycle of the [2+2+2] cycloaddition reaction of alkynes (M = transition-metal complex). | ||
Two alkynes sequentially displace metal-coordinated ligands to form alkyne complex I. Subsequent oxidative coupling results in the formation of either metallacyclopentadiene IIa or metallacyclopentatriene IIb. The coordination of a third alkyne substrate generates reactive species III from which alkyne insertion affords metallacycloheptatriene IVa. Alternative cycloadditions yield 7-metallanorbornadiene complex IVb or metallabicyclo [3.2.0]-heptatriene IVc. Finally, the arene is formed by the reductive elimination of the metal and the catalyst is recovered after decoordination. The mechanistic cycle depicted in Scheme 2 has been validated by theoretical calculations3–5 and by the isolation of metallacyclopentadiene IIa6 and metallacyclopentatriene IIb7 intermediates. More advanced intermediates in the catalytic cycle have also been detected.8 Slight variations in the mechanism (i.e. the intermediates formed) are invoked for different catalytic systems and/or substrates.
The problems of chemo- and regioselectivity that arise when differently substituted alkynes concur in the same reaction can be easily rationalised by the mechanism proposed. On the other hand, when the reaction takes place with at least one substrate that is different from an alkyne, the mechanism operating is assumed to involve analogous key steps and intermediates although it is often not clear at which step the different unsaturations enter the catalytic cycle.
Among the great variety of six-membered rings that can be formed, a large number of the reactions found in the literature describe the formation of aromatic rings, mainly benzene and pyridine (by reaction of three alkynes or two alkynes and a nitrile, respectively). Replacement of sp hybridised substrates by sp2 hybridised ones generally concur with a decrease in reactivity, but has the advantage of allowing for the eventual introduction of stereogenic centres in the newly formed six-membered ring. This is exemplified for carbon-based unsaturations in Scheme 3, where the products that can be formed by the reaction of three alkynes or two alkynes and either an alkene or an allene are outlined. There are advantages and disadvantages in the use of the different carbon-based unsaturations. Alkynes readily participate in the reaction, allowing for the efficient synthesis of benzenes. The use of properly substituted alkenes allows for the synthesis of chiral cyclohexadienes. However, alkenes are more reluctant to react and, therefore, it is generally difficult to control the chemoselectivity given the high reactivity of the alkyne substrates. An excellent alternative to react sp2 hybridised unsaturations, thus generating stereocomplexity, while maintaining the reactivity is the use of allenes.
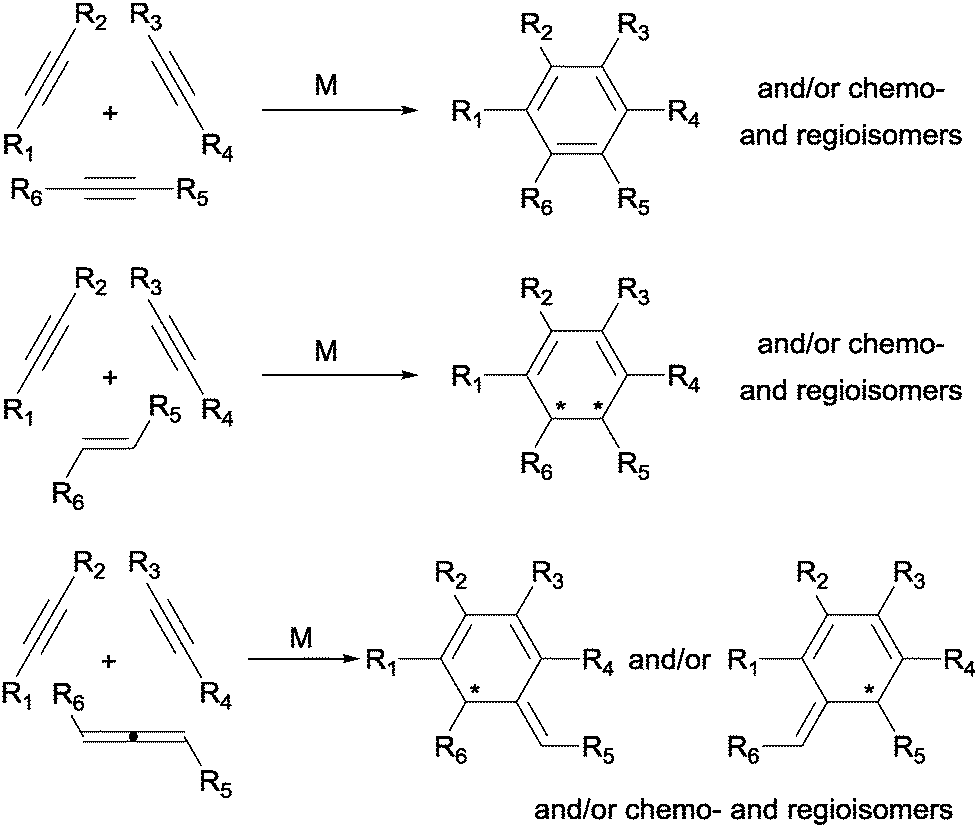 | ||
| Scheme 3 General scaffolds that can be obtained by [2+2+2] cycloaddition of two alkynes and either an alkyne, an alkene or an allene (M = transition-metal complex). | ||
Allenes, which are cumulated systems with two contiguous carbon–carbon double bonds, are increasingly being used as substrates in many reactions.9 The development of efficient methodologies for the synthesis of a broad range of allenes has fostered their use as substrates.10 There are several particularities that make allenes especially appealing carbon-based unsaturated substrates in cycloaddition reactions. They have higher reactivity compared to alkenes and, when properly substituted, have inherent axial chirality. Both the electronic density and the reactivity of each carbon atom can be modulated by their substitution pattern. Furthermore, when allenes are reacted as two-carbon synthons, they deliver cycloadducts with an exocyclic double bond that can subsequently be manipulated. On the other hand, the use of allenes implies increased selectivity problems compared both to alkynes and alkenes. Apart from the chemoselectivity towards other unsaturations, and the issues of regio- and stereoselectivity, the chemoselectivity as to which of its two double bonds reacts also needs to be controlled.11
Scope, limitation and organisation of the review
This review will cover all the transition-metal-catalysed or mediated [2+2+2] cycloaddition reactions involving allenes published up until October 2015. To be more concise, we have considered all works in which three new bonds are formed between an equal number of unsaturations to yield a six-membered ring, independently of whether the mechanism postulated follows the general scheme proposed in the introduction.The review is organised by the type of unsaturations participating in the cycloaddition. Special attention has been paid to rationalise the selectivity of the reaction.
In all of the schemes in this review, specific case studies have been drawn, highlighting the point of diversity in red. The number of examples is given together with the range of yield.
Seminal studies
Pioneering studies into the use of allenes in metal catalysed cyclisations were first reported by Benson and Lyndsey12 at DuPont in 1958 (Scheme 4). The authors found that the parent allene cyclotrimerised in the presence of nickel(0) catalysts in a moderate yield but with good selectivity versus cyclotetramerisation. The corresponding trimethylenecyclohexanes were obtained as a quasi-statistic mixture of 1,2,4 and 1,3,5 isomers (1a–b). Alternatively, the introduction of substoichiometric amounts of an alkyne in the mixture and the use of nickel(II) acetylacetonate gave dimethylenecyclohexenes 3 with good chemoselectivity, considering the large number of possible products.13 The experimental settings used (>50 g scale, low boiling point of substrates and products) do not make these compounds particularly interesting for modern laboratory-scale research, but the chemoselectivities observed did anticipate the potential of allenes in cyclotrimerisation reactions. Notwithstanding, 35 years would go by before new examples of [2+2+2] cycloadditions involving allenes were reported by Malacria (vide infra).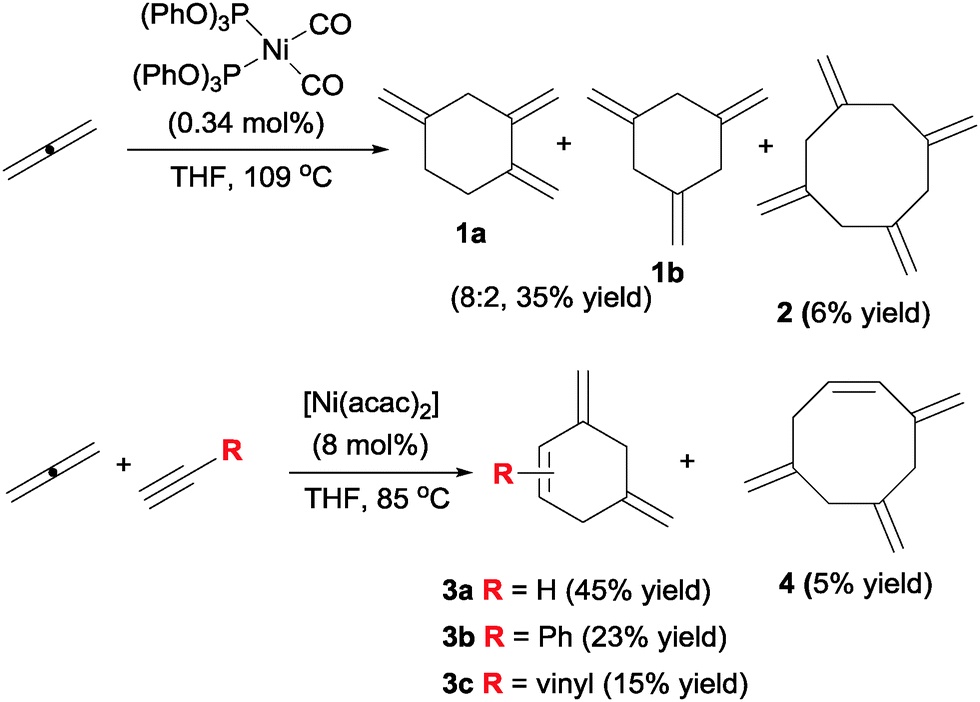 | ||
| Scheme 4 Pioneering studies of nickel catalysed cycloadditions with allenes. | ||
[2+2+2] Cycloaddition reactions of three allenes
The cyclisation of three allenes was reinvestigated by Ma et al.14,15 in 2005, this time under Rh(I) catalysis and in a partially intermolecular setting, which constitutes an efficient entry into steroidal structures. In the presence of [RhCl(CO)(PPh3)2] as a catalyst, two molecules of a bis-allene 5 engaged in a cycloaddition to yield a cross-conjugated diene, which spontaneously underwent Diels–Alder cycloaddition with the remaining allene to yield tetracyclic compound 6 (Scheme 5). Remarkably, the [2+2+2] cycloaddition occurred with total chemoselectivity: the initial intramolecular oxidative addition step occurred through one terminal and one internal bond, while the second bis-allene incorporated reacted through the terminal position. Ma et al.16 later implemented this reaction in a combinatorial approach to these steroidal compounds, employing mixtures of bis-allenes analogous to 5 with different tethers (C(SO2Ph)2 in 5). Not surprisingly, however, this strategy furnished mixtures of the four possible products with little selectivity and a preference for homocoupling products, if any.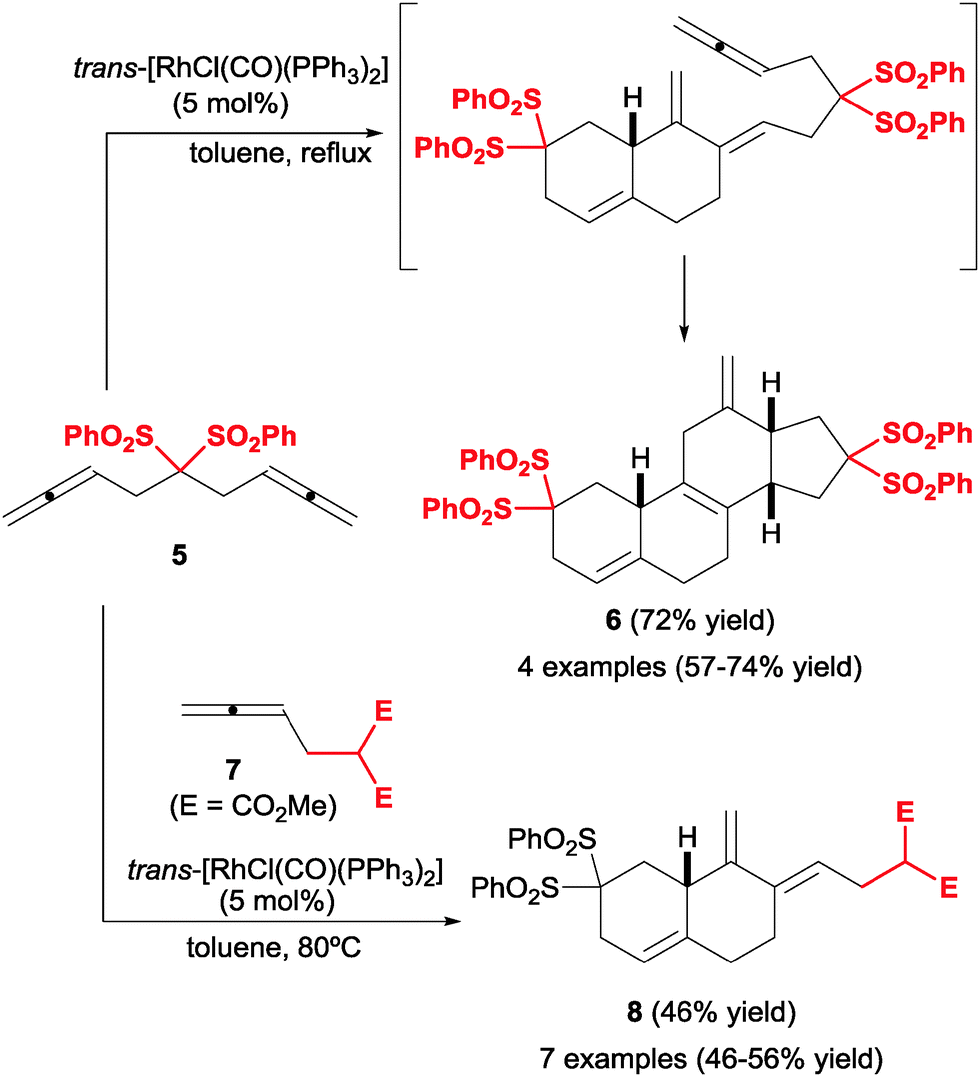 | ||
| Scheme 5 Cyclisation of three allene unsaturations catalysed by Rh(I). | ||
Notwithstanding, the overall process was still completely chemo- and stereoselective, delivering a rapid increase in molecular complexity. A follow-up approach to this process consisted in combining a bis-allene and a mono-allene (Scheme 5).17 Under the right conditions (slow addition of bis-allene to excess mono-allene) selective cyclisation was achieved, delivering versatile diene products (8), which can be utilised in subsequent Diels–Alder reactions with a variety of dienophiles. The yields of this transformation were moderate but the reaction was relatively insensitive to variations in the structure of the allene 7. Only one tether was reported, often a critical point of variation in these transformations. Overall, this tandem cycloaddition/Diels–Alder scheme provides an efficient and modular entry into tetracyclic steroid-like compounds.
[2+2+2] Cycloaddition reactions of allenes with carbon–carbon π-unsaturations
Intramolecular cycloadditions of allenes with alkynes and alkenes
In 1994, Malacria et al.18,19 started a series of studies employing allenes which prompted a revamped interest in the utility of these substrates in [2+2+2] cycloadditions. A number of linear allene–diyne substrates were prepared and tested for Co(I)-mediated intramolecular cycloaddition (Scheme 6). Upon exposure to equimolar amounts of [CpCo(CO)2] and under ultraviolet light activation, polyunsaturated yne–allene–yne substrates bearing tetrasubstituted allene moieties (9/10) cleanly underwent cycloaddition to yield tricyclic conjugated trienes, which were isolated as their corresponding η4 cobalt complexes 11/12 and 13/14. No reaction progress was observed when catalytic amounts of the cobalt complex were used.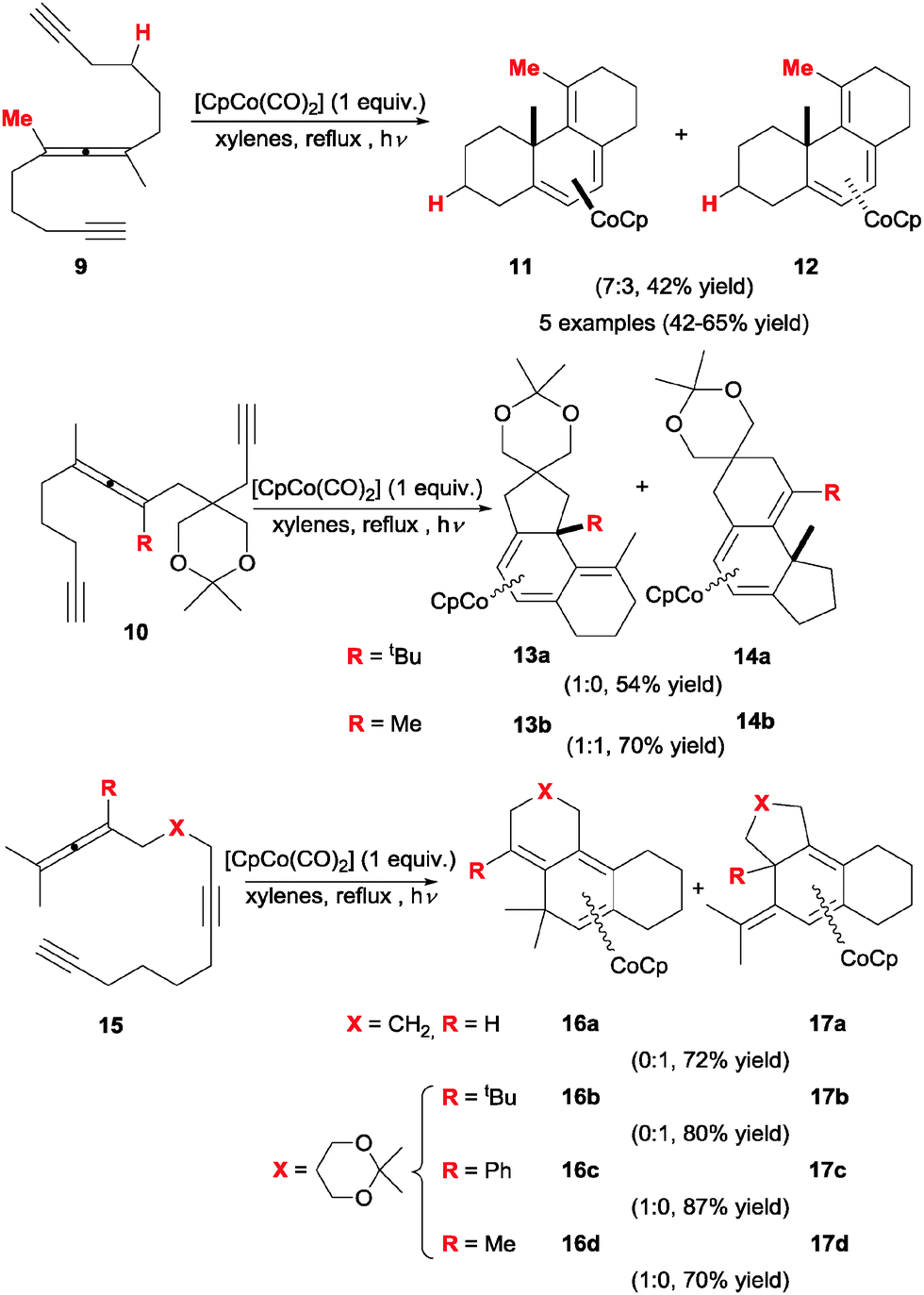 | ||
| Scheme 6 Co(I)-mediated cycloaddition of allene–diyne substrates. | ||
Further efforts in this direction showed that allene substitution is key in this type of transformations, greatly affecting the outcome of the cycloaddition and its chemoselectivity. For instance, the cyclisation of analogous substrates bearing tri- or di- substituted allene portions (not shown) only met with failure under these conditions, most likely due to the instability of the resulting cyclisation products and intermediates. Analogous allene–diyne substrates 15 were also subjected to cycloaddition under the same conditions. Although the chemoselectivity is complete, it is quite erratic and difficult to predict beforehand. This transformation illustrates the strong effect of the allene substitution pattern on the reaction. With this particular arrangement of unsaturations, a trisubstituted allene exceptionally underwent clean cyclisation to 17a. Generally speaking, complexes 11–14/16–17 are not particularly robust, and the reported silica promoted decomplexation to release the free triene products was low yielding and accompanied by alkene isomerisation in most cases, somewhat diminishing the synthetic utility of this approach.
An interesting feature of these cyclisations is that they occur with effective axial to central chirality transfer, as demonstrated by Malacria et al.20 in a separate study. When homochiral allene 18 (obtained from an enantiopure propargylic substrate) was subjected to the Co(I)-mediated cyclisation, the corresponding triene-η4-cobalt complex 19 was obtained as a single diastereoisomer without any erosion in enantiomeric excess (Scheme 7). It is worth noting that the introduction of a phosphine oxide group at the alkyne terminus was found to be beneficial for reaction yields. It was reasoned that this group increases the stability of intermediate reaction complexes. This function and similar ones (sulphones) have since been incorporated as a common structural motif of cycloaddition substrates in many related studies.
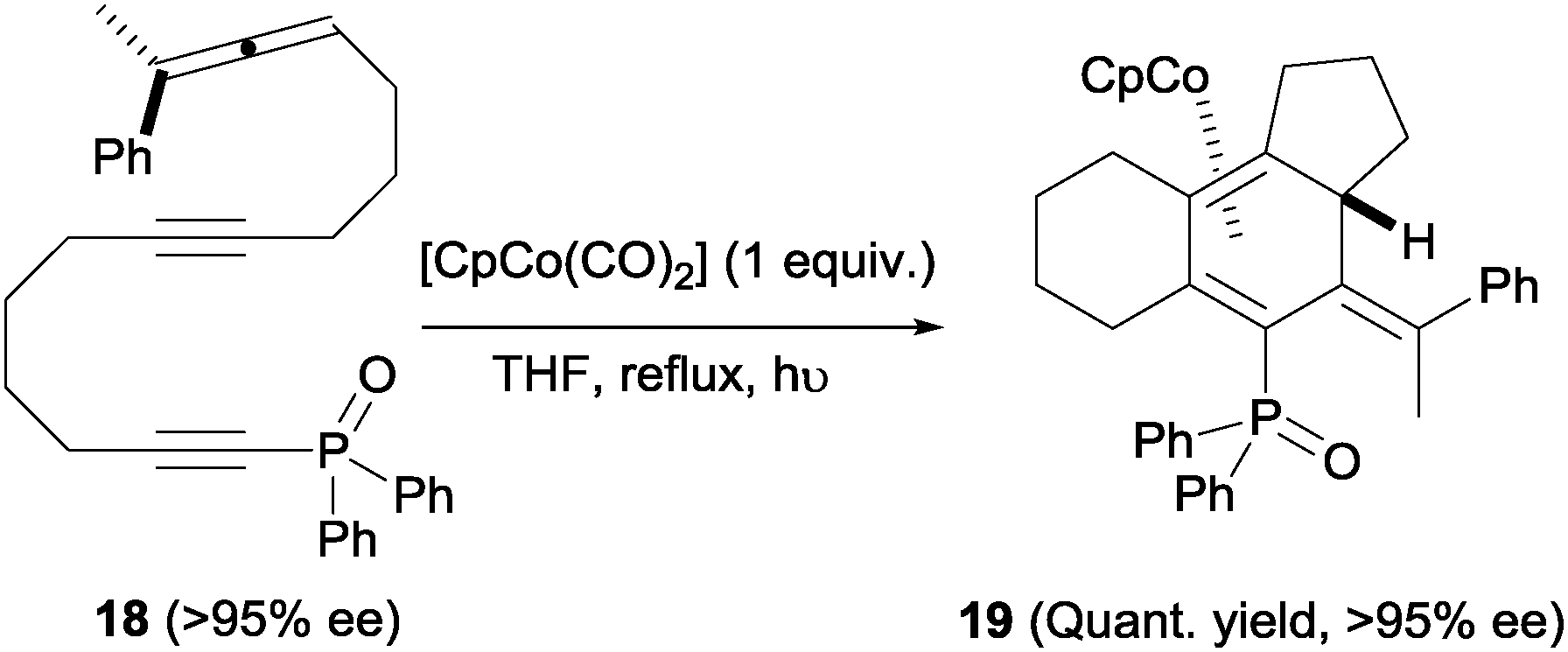 | ||
| Scheme 7 Axial to central chirality transfer in a Co(I)-mediated allene–diyne cycloaddition. | ||
Malacria et al.21,22 later exploited the high chemo- and diastereoselectivity observed in these processes to access steroidal scaffolds in a straightforward manner. Based on the previous exploratory studies, the synthesis of a suitable precursor to the complete ABCD steroid ring system was addressed. Eventually, it was possible to obtain diastereomerically pure yne–allene–yne substrate 20. Upon cyclisation, followed by silica-promoted decomplexation, steroid analogue 21 was obtained in moderate yield but with complete diastereoselectivity (Scheme 8). This approach allows a rapid increase in molecular complexity, but is hampered by difficulties in finding and accessing the appropriate cyclisation partner. In the case of other stereoisomers and close analogues of 20, it was either not possible to isolate them in pure form or they did not give [2+2+2] cyclisation products.
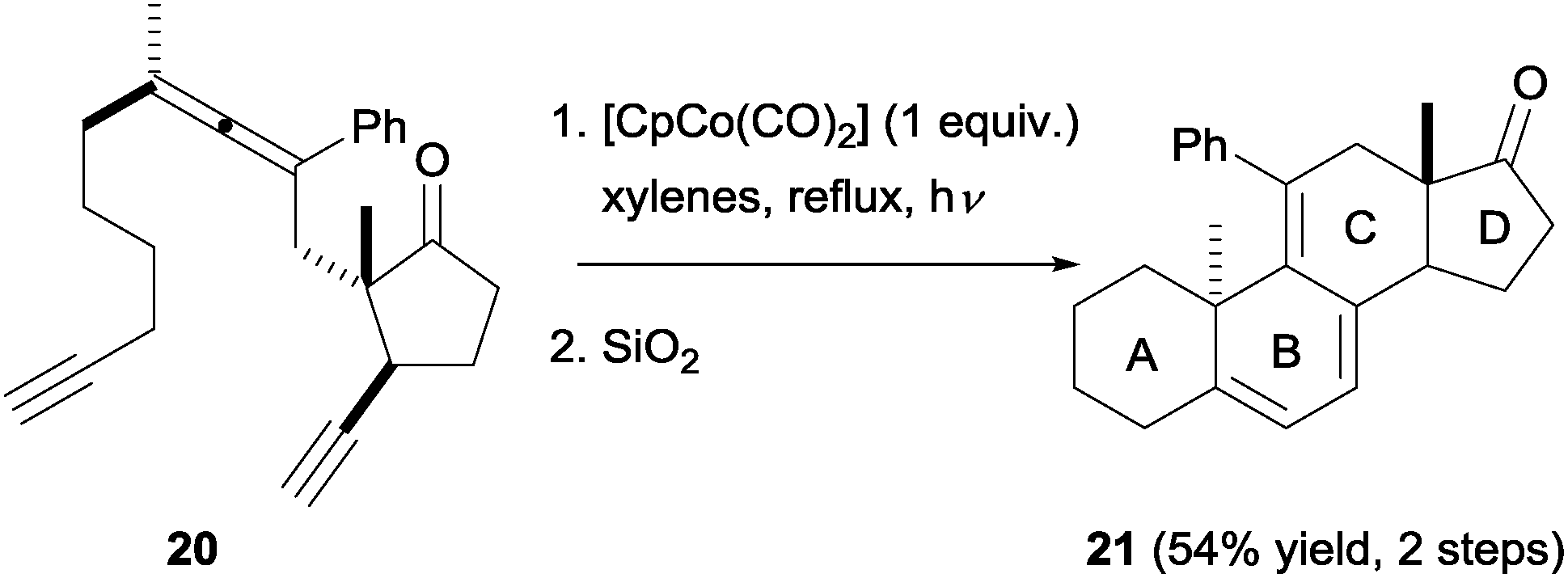 | ||
| Scheme 8 Diastereoselective synthesis of a steroid-like compound from a diastereomerically pure yne–allene–yne precursor. | ||
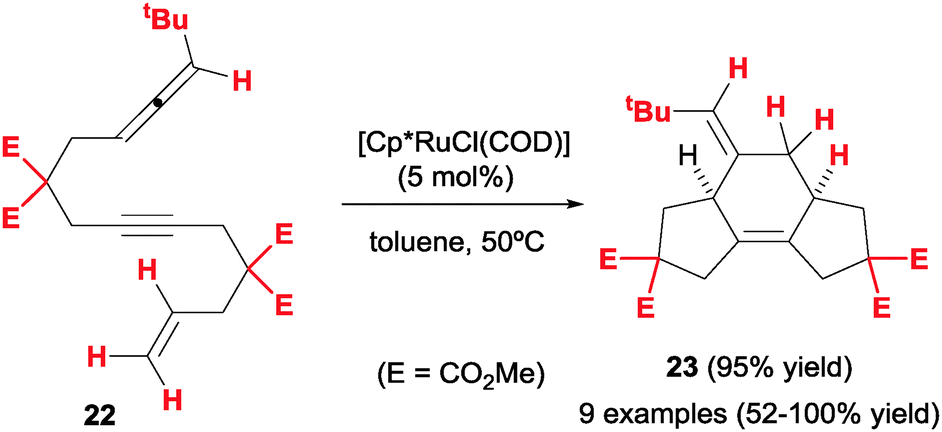
In contrast to what is seen with allene–diyne cycloaddition precursors, the combination of an allene, an alkyne and an alkene appears to be more robust for intramolecular [2+2+2] cycloadditions. Saito, Sato et al.23 first described the preparation of a series of allene–yne–ene substrates (22) and their cyclisation under ruthenium(II) catalysis (Scheme 9). Employing three-membered tethers between unsaturations, skipped diene products with a 5/6/5 ring system (23) were obtained with complete diastereoselectivity by selective reaction at the internal double bond of the allene. Moderate to good yields were obtained for most substrates with the exception of those bearing oxygen tethers. The process can be thus considered quite general and relatively insensitive to substitution at the reactive sp2 positions, unlike previously discussed examples.
 | ||
| Scheme 9 Ru(II)-catalysed cycloaddition of allene–yne–enes. | ||
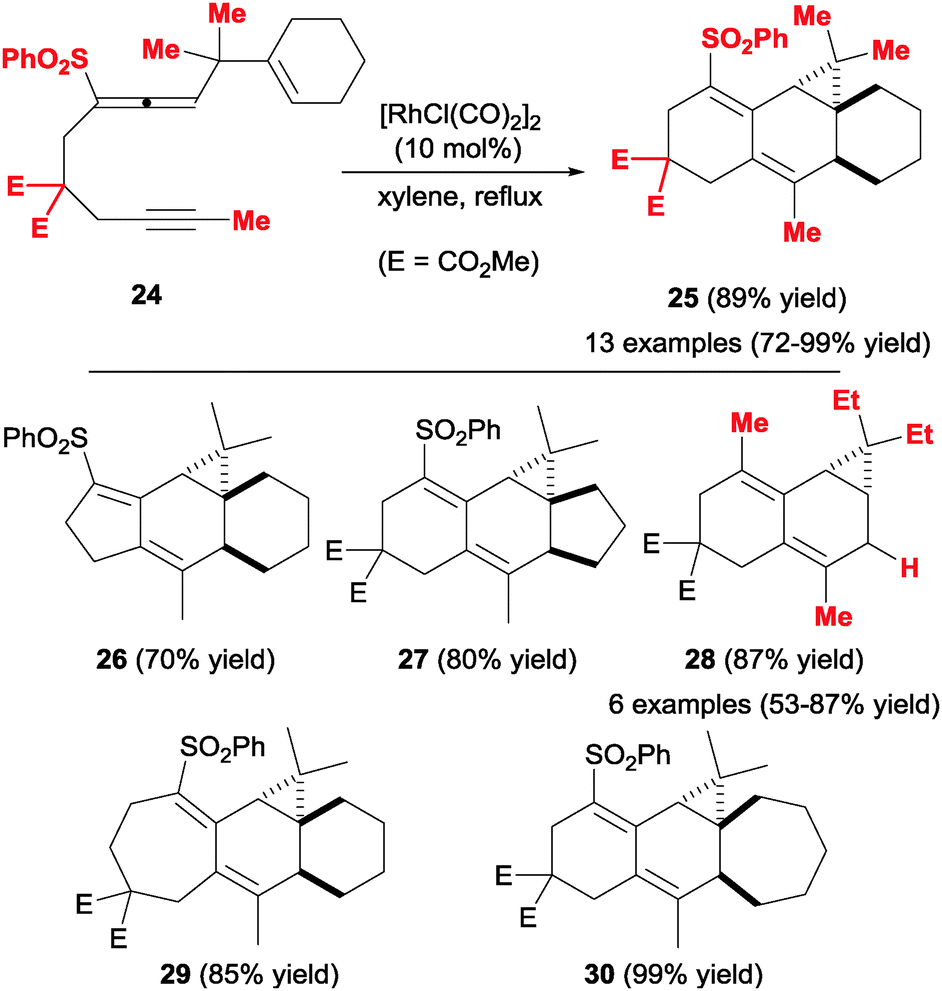
A similar study by Mukai et al.24 inverted the order of the unsaturations in the cyclisation precursor, placing the allene moiety in the middle of the chain (24, Scheme 10). This arrangement proved to be extremely versatile in terms of structural variability and allowed the formation of complex tri- and tetracyclic scaffolds 25–30, again in a totally diastereoselective fashion. In these cases, the authors employed a Rh(I) catalyst and obtained the corresponding products in good to excellent yields. The flexibility of this methodology is illustrated by the introduction of tethers of different lengths in the precursor structure, successfully leading to a variety of fused polycyclic systems of different sizes (compounds 26–30, Scheme 10). An interesting feature of this approach is the possibility of forming a cyclopropane-fused polycyclic structure. The sole drawback of this strategy is the requirement for a gem-dialkyl substituted tether between the allene and the alkene moieties, which prevents undesired side reactions occurring through β-elimination processes of intermediate Rh(I) complexes.
 | ||
| Scheme 10 Rh(I)-catalysed cycloaddition of ene–allene–ynes. | ||
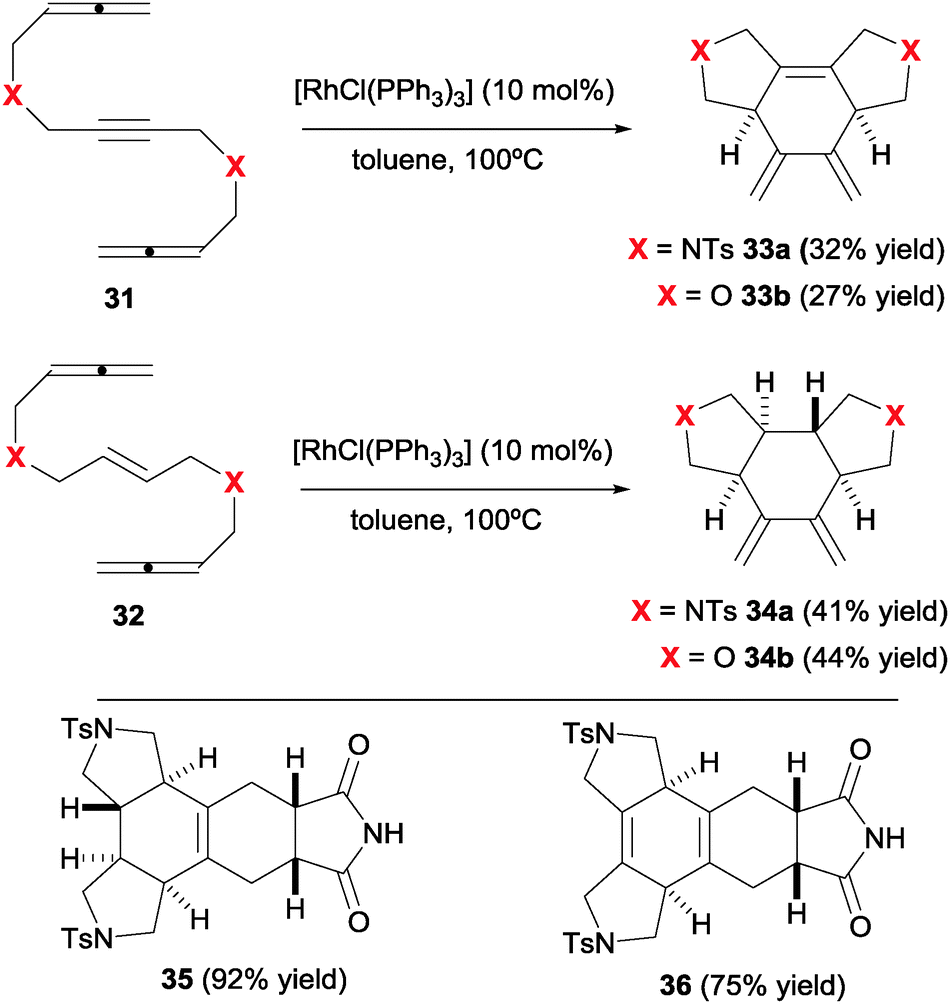
The combination in a linear substrate of two allenes and a third different unsaturation is an interesting approach to polycyclic 1,3-diene products reported by our group (Scheme 11).25 These cycloadducts have additional potential for increasing molecular complexity through successive cycloadditions. The Rh(I) catalysed cyclisation of allene–yne–allene and allene–ene–allene substrates (31–32) delivered cross-conjugated dienes 33–34 with complete chemo- and diastereoselectivity. In this case, the Wilkinson's catalyst, a convenient and practical source of Rh(I), was used.
 | ||
| Scheme 11 Rh(I)-catalysed cyclisation of allene–yne/ene–allene substrates and the Diels–Alder adducts obtained. | ||
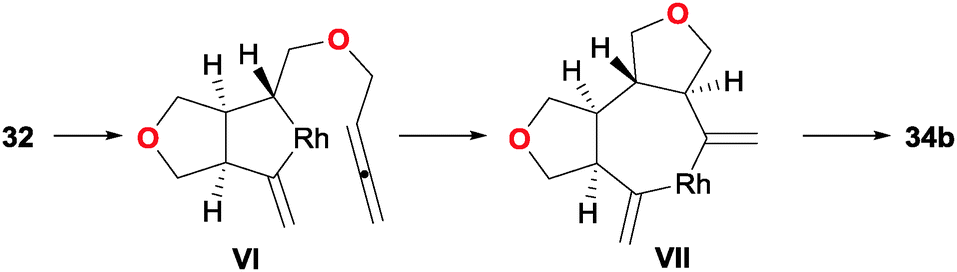
Additionally, detailed DFT calculations were carried to rationalise the observed diastereoselectivity and to propose a mechanistic cycle. After a detailed analysis of the many possibilities for the key reaction intermediates, oxidative addition was postulated as taking place between the central alkene and the internal double bond of one of the allenes, leading to a cis-fused bicyclic intermediate VI. The remaining allene is then inserted into the Rh–Csp3 bond of the rhodacyclopentane in VI to give a trans fused ring junction in intermediate VII (Scheme 12). The preference for cyclisation at the internal double bond of the allene was also analysed. This concurred with a higher energy of the occupied π molecular orbital localised at this double bond. It is worth mentioning that this is the first and only example in which a mechanistic hypothesis has been validated by rigorous computational techniques. Finally, the products obtained easily engaged in Diels–Alder reactions with maleimide, delivering complex pentacyclic products 35/36 in excellent yields and with total stereoselectivity.
 | ||
| Scheme 12 Key intermediates proposed on the basis of DFT calculations in the Rh(I) catalysed cyclisation of allene–ene–allene substrates. | ||
Two additional examples of intramolecular cyclisation are found in the literature, although they are of lesser synthetic interest. In the context of a study on rhodium-catalysed carbonylative cycloaddition reactions, Ojima et al.26 found that allene–diyne 37 underwent regular [2+2+2] cycloaddition (without CO insertion) and concomitant isomerisation to yield the corresponding benzo-fused tricyclic product 38 (Scheme 13). A similar result was observed by our research group.27 Allene–diyne 39 bearing a longer tether gave, in the presence of Wilkinson's complex, tricyclic benzene scaffold 40. Although both reactions proceed selectively at the outer double bond of the allene, its utility is somewhat limited: in a completely intramolecular setting, the same structures can be easily accessed employing analogous triyne precursors.
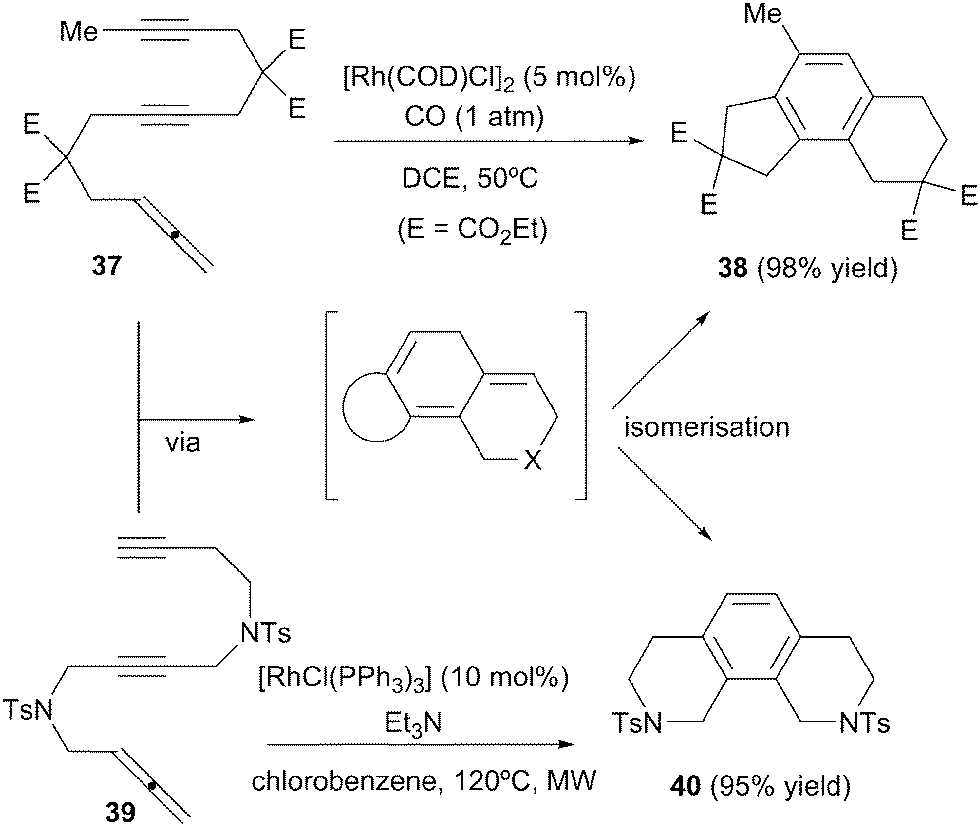 | ||
| Scheme 13 Rh(I)-catalysed cyclisation/isomerisation of allene–diynes to obtain benzo-fused tricyclic compounds. | ||
Intermolecular and partially intermolecular cycloadditions of allenes with alkynes
The viability of the intermolecular [2+2+2] cycloaddition of allenes with alkynes was first established in two papers by Dieck et al.28,29 While studying the synthesis of naphthalene derivatives they found that a diazadiene stabilised palladacyclopentadiene was able to catalyse the [2+2+2] cycloaddition of an allene with two equivalents of dimethyl acetylenedicarboxylate 41 but only in the specific case in which phenoxyallene 42 was used (Scheme 14a).28 When increasing the temperature to 60 °C and increasing the catalyst loading, the benzene derivative was obtained quantitatively.29 The authors postulate that the benzene derivative is formed after an isomerisation process of the initially formed 1,3-cyclohexadiene derivative.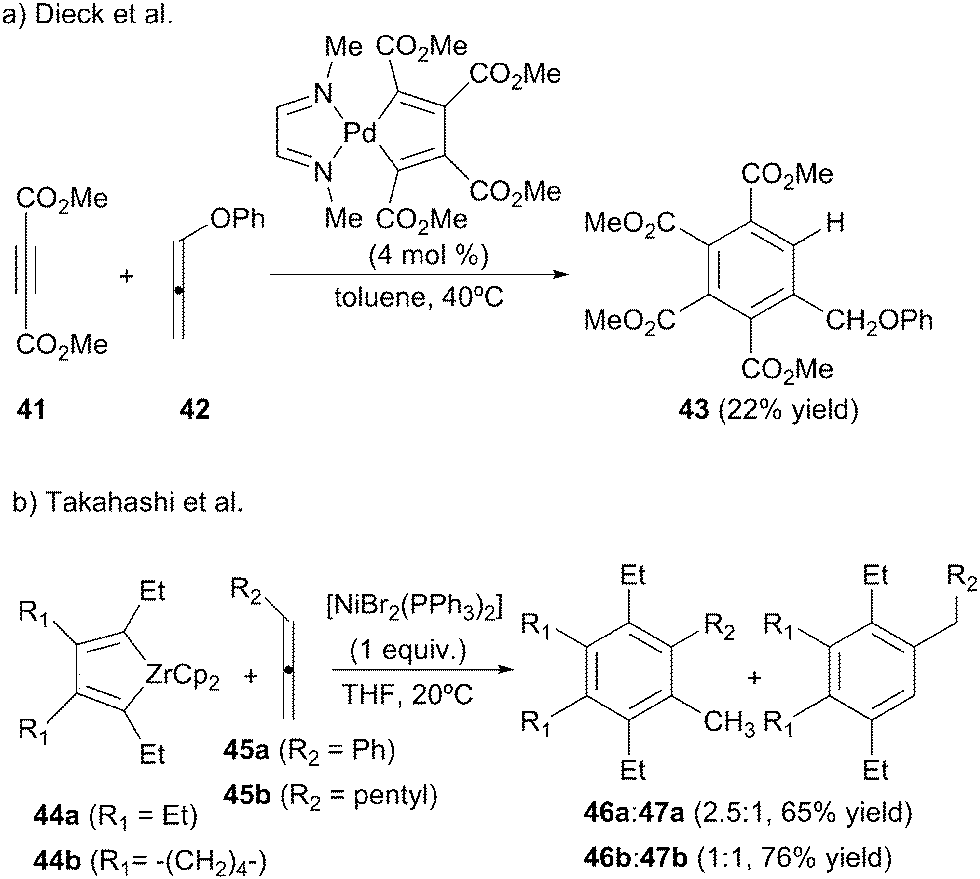 | ||
| Scheme 14 [2+2+2] Cycloadditions between monoalkynes and diynes with allenes. | ||
Takahashi et al.30 also used a metallacyclopentadiene for the [2+2+2] cycloaddition reaction although in this case as a substrate. In the framework of a study into nickel-mediated cycloadditions of zirconacyclopentadienes 44 with alkynes, two allenes were tested in the process. The reaction was not chemoselective as they obtained a mixture of isomers 46 and 47 from the addition of each of the two double bonds of the allene (Scheme 14b). The authors proposed a mechanism consisting of a transmetallation step from the zirconacyclopentadiene to form the corresponding nickelacyclopentadiene followed by the insertion of the allene and, finally, reductive elimination and isomerisation.
Cheng et al.31–33 made a substantial contribution to the topic by systematically studying the intermolecular cycloaddition of allenes with alkynes and diynes (Scheme 15). In these studies, the less substituted double bonds of the allenes were found to participate in the cycloaddition. Presumably, the first compound obtained in these processes was the 1,3-cyclohexadiene derivative, which was followed by an isomerisation/aromatisation process to afford the corresponding benzene compound. Therefore, the allenes acted here as synthetic equivalents of monosubstituted alkynes. The first study by Cheng et al.31 was based on a completely intermolecular reaction between two molecules of alkyne and one molecule of allene under nickel catalysis, to afford polysubstituted benzene derivatives in a highly regio- and chemoselective fashion (Scheme 15a). The best catalytic system found was [Ni(dppe)Br2] with an excess of metallic Zn as the reductant of Ni(II) to Ni(0), which was the actual catalytic species. With a ratio of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 of starting materials 49:48 no cyclotrimerisation of the alkyne was observed. The presence of an ester group in the alkyne was necessary for the success of the reaction. Remarkably, the reaction was highly selective and only isomer 50 was observed. Curiously, when the propiolate had an aryl group attached, products with different regiochemistry were obtained.
:1 of starting materials 49:48 no cyclotrimerisation of the alkyne was observed. The presence of an ester group in the alkyne was necessary for the success of the reaction. Remarkably, the reaction was highly selective and only isomer 50 was observed. Curiously, when the propiolate had an aryl group attached, products with different regiochemistry were obtained.
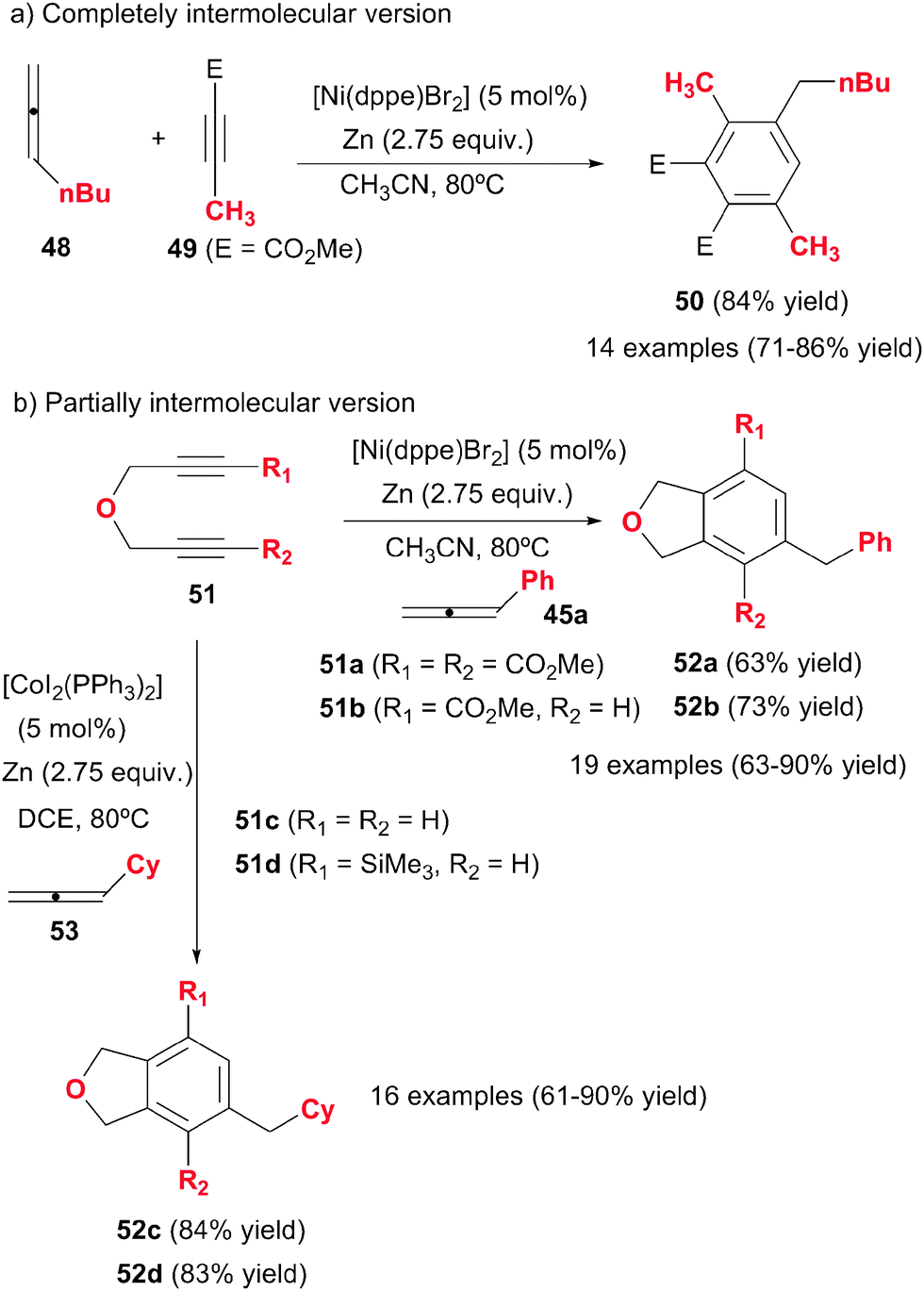 | ||
| Scheme 15 [2+2+2] Cycloadditions between monoalkynes and diynes with terminal allenes. | ||
This first study was later on extended to the cycloaddition of diynes with allenes.32,33 The partially intermolecular version is often more advantageous in the control of the regioselectivity of the cycloaddition. The same Ni-based catalytic system as before was used (Scheme 15b). Homocoupling of the diyne was not observed even when a stoichiometric ratio of starting materials was used. When the diyne was not symmetrical (51b), the cycloaddition was highly regioselective, giving the meta-isomer 52b as the predominant compound. The presence of an ester group in the diyne was again necessary. The use of [CoI2(PPh3)2]/Zn system, instead of nickel complexes, improved the reaction broadening the cycloaddition scope to electronically neutral diynes (51c–d). The cycloaddition also resulted in high chemo- and regio- selectivity (Scheme 15b). The authors demonstrate the advantages of allenes in comparison with analogous alkynes by performing the reaction with 1-heptyne instead of its isomeric derivative, n-butylallene (48), obtaining, in all cases, a mixture of compounds and much lower yields of the desired product.
The mechanistic proposal for these cases follows the general scheme commented in the introduction, with the allene entering in the last step of the catalytic cycle. The meta-regioselectivity is justified by steric hindrance of the intermediate leading to the ortho-isomer.
Cheng et al.34 made a further contribution using the previously mentioned nickel catalytic system to trigger the [2+2+2] cycloaddition between an allene and two arynes (Scheme 16a). While arynes have been reacted with a variety of alkynes, diynes, allyl derivatives and CO under palladium catalysis to afford [2+2+2] cycloadducts,35 this was the first time that they were reacted with allenes. The arynes were generated in situ by fluoride-induced β-elimination of ortho-(trimethylsilyl)benzene triflates. An allene was reacted with two equivalents of the in situ generated benzynes to provide 10-methylene-9,10-dihydrophenanthrene derivatives 55 in an efficient and selective manner (Scheme 16a). In contrast to the previous studies by the same group, the monosubstituted allenes reacted through the internal double bond, giving a highly chemoselective reaction. However, when the allenes were 1,1-disubstituted a mixture of the two isomers was formed, which the authors attributed to steric effects. Another difference of this study in comparison with earlier ones is that the exocyclic double bond in the cyclohexadiene initially generated did not undergo isomerisation to give the more stable aromatic derivative. This exocyclic double bond is maintained in the final cycloadduct 55. Phenylallene failed to react, and only alkyl substituents were tolerated on the allene and only terminal allenes were tested. Small amounts of the homotrimerisation product of benzyne were observed in all of the reactions. These studies would seem to show that both the chemoselectivity of the process and the isomerisation step are quite unpredictable.
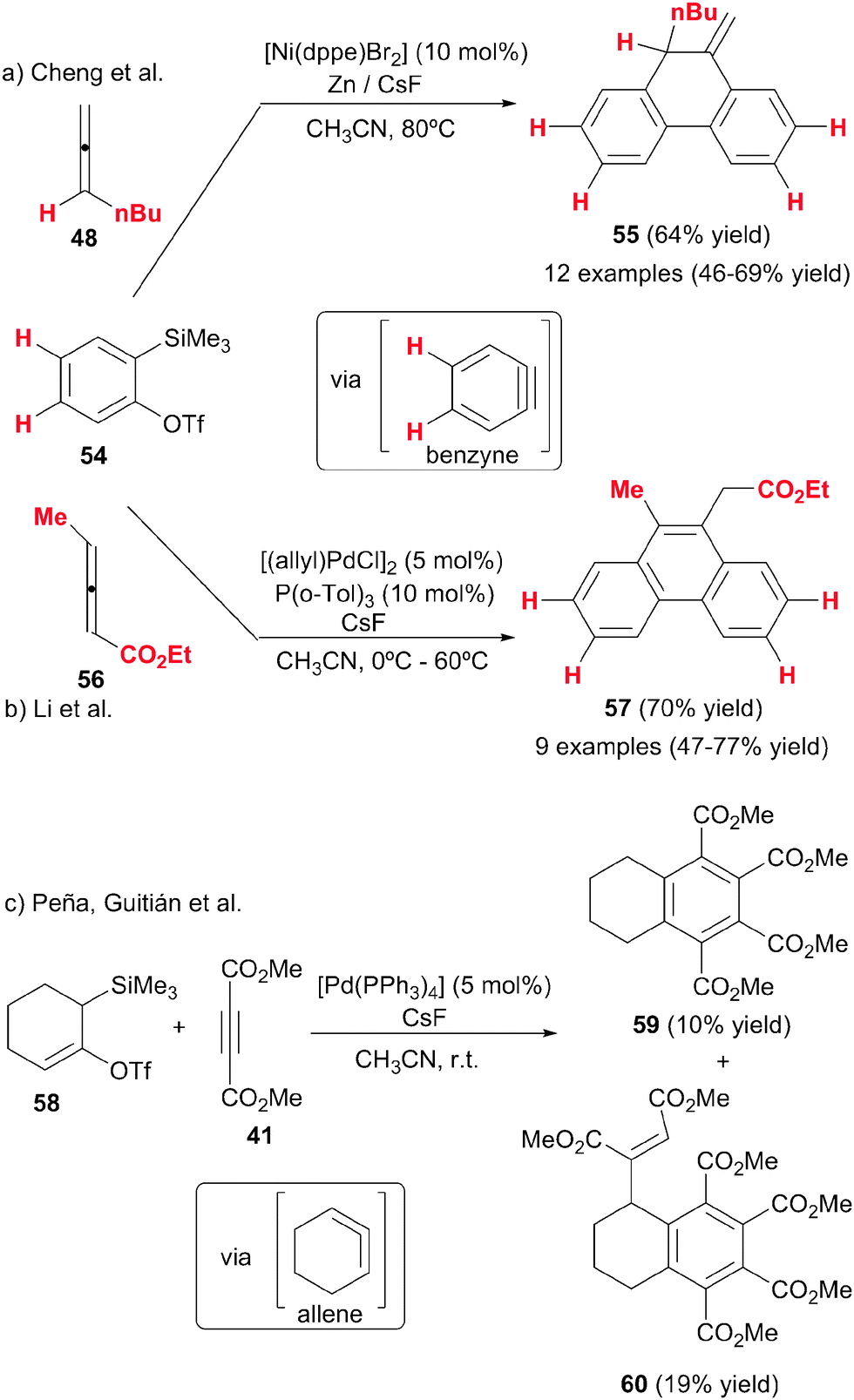 | ||
| Scheme 16 [2+2+2] Cycloadditions involving benzynes and 1,2-cyclohexadienes. | ||
Later Liang, Li et al.,36 following on from Cheng, studied the synthesis of phenanthrene derivatives by the cycloaddition of allenes with two molecules of arynes but under palladium catalysis (Scheme 16b). After optimisation, the [(allyl)PdCl]2/P(o-Tol)3 catalytic system was found to be the most efficient. When they tested 1,3-disubstituted allenes 56 with an electron-withdrawing group, the final product was the benzene derivative 57 obtained after the isomerisation step. The reaction was highly chemoselective as it took place with the double bond not linked to the electron-withdrawing group. Using trisubstituted allenes and alkyl terminal allenes, the authors obtained the same tricyclic derivatives 55 as Cheng without aromatisation. Here again it seems to be rather difficult to predict the final product in these processes and probably both electronic and steric effects play an important role in the process as a whole.
Fluoride-induced β-elimination of ortho-triflate 58, was also used by Peña, Guitián et al.37 for the in situ preparation of 1,2-cyclohexadiene, a highly reactive and strained bent allene. In order to study the reactivity of this cyclic allene, precursor 58 and dimethyl acetylenedicarboxylate were treated with catalytic amounts of [Pd(PPh3)4] in the presence of CsF, affording a mixture of benzene derivatives 59 and 60 in 10 and 19% yields, respectively (Scheme 16c). The authors proposed a palladium-catalysed [2+2+2] cycloaddition reaction between the cyclic allene and two molecules of alkynes followed by a fluoride-induced deprotonation/aromatisation to give derivatives 59 and 60.
Another example by Ma et al.38 of [2+2+2] cycloaddition reactions involving alkynes and allenes is the dimeric cycloaddition of propargylic 2,3-dienoates 61 catalysed by a modified Wilkinson complex (Schemes 17 and 18). A [2+2+2] cycloaddition took place between the propargylic triple bond, the remote double bond of the allene moiety and the triple bond of a second molecule of the propargylic dienoate 61. The 1,3-cyclohexadiene 62 obtained in a first stage, underwent an intramolecular Diels–Alder reaction to afford tricyclic derivative 63 in good yields. Although 63 has four stereogenic centres, only two diastereoisomers were formed, which could be separated by column chromatography (Scheme 17).
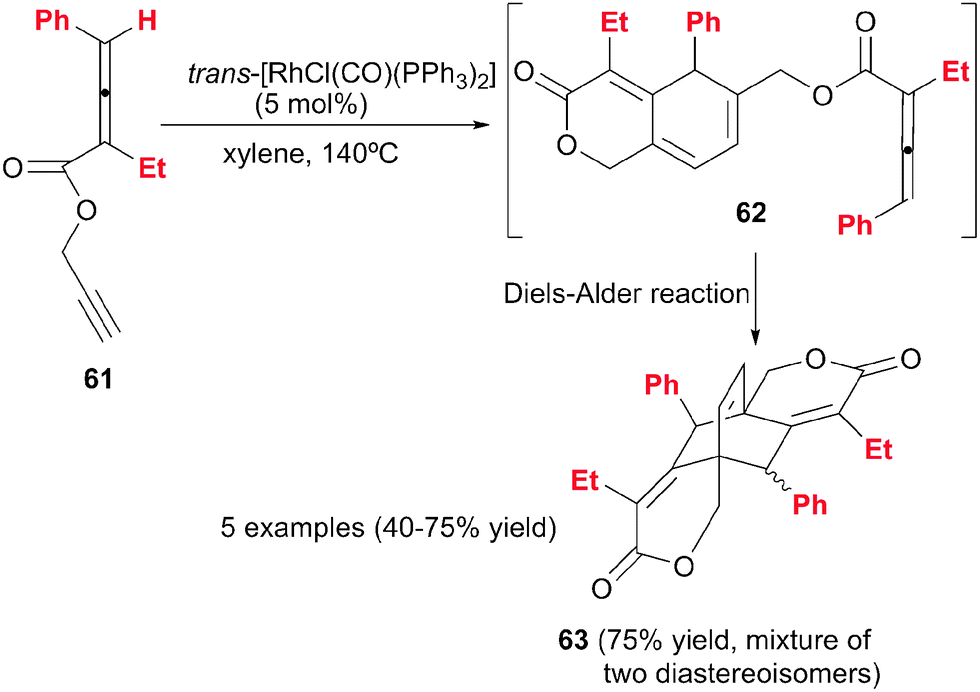 | ||
| Scheme 17 Rh(I)-catalysed bimolecular cyclisation of propargylic allenoates. | ||
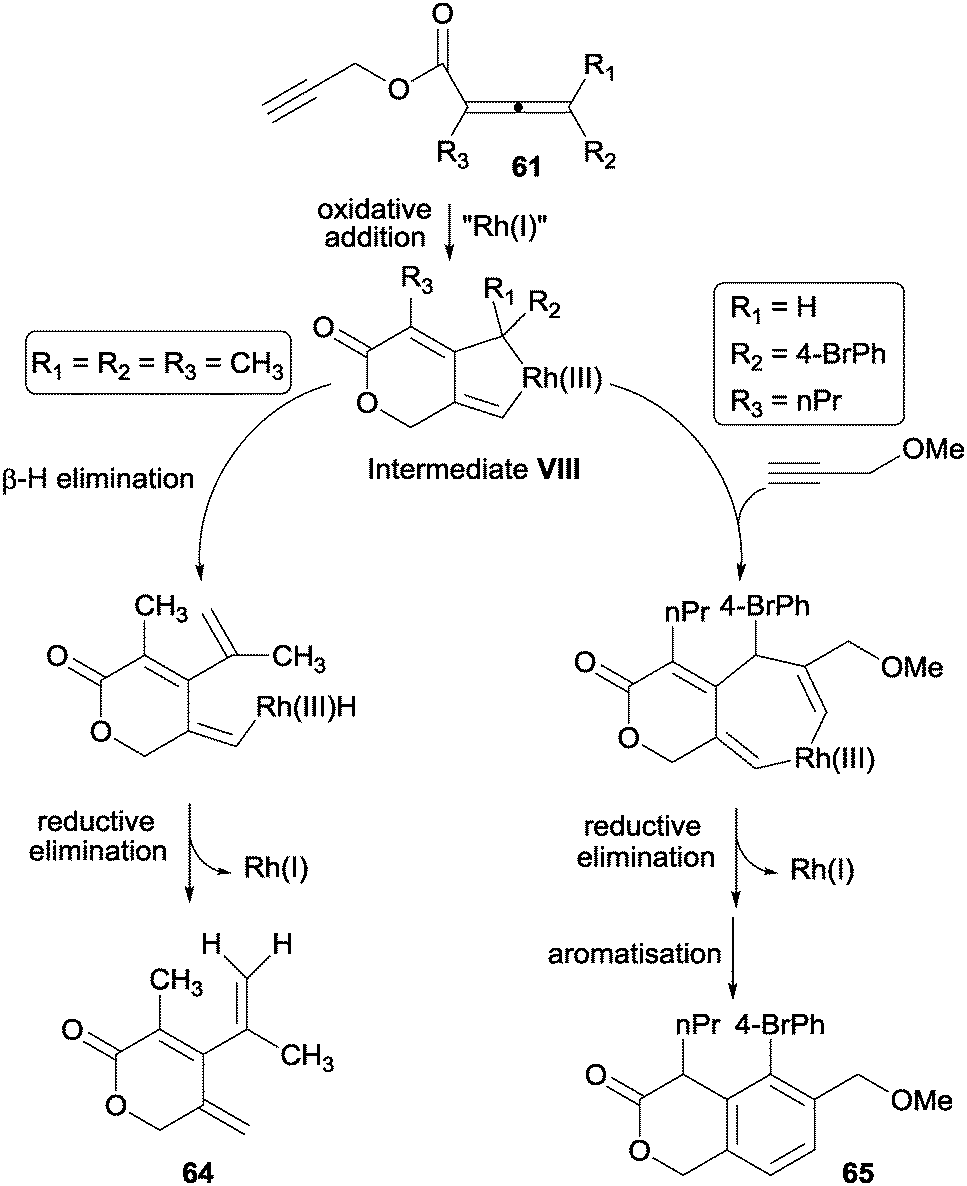 | ||
| Scheme 18 Mechanistic proposal for the Rh(I)-catalysed unimolecular and bimolecular cyclisations of propargylic allenoates. | ||
Certain specific substrates delivered, under the optimised reaction conditions, monocyclic dihydropyranone derivatives rather than the expected cycloadducts. This prompted the authors to study the process mechanistically. The substituents at the 4-position of the starting allenoate moiety (R1 and R2) were found to control the reaction course (Scheme 18). The rhodacyclopentene formed after the oxidative addition between the alkyne and the remote double bond of the allene (intermediate VIII in Scheme 18) can evolve through two different pathways depending on the substituents on the allene: (i) a β-H elimination to afford monocyclic dihydropyranone derivatives 64; or (ii) an alkyne insertion to deliver the [2+2+2] cycloadduct when the substituent at the 4-position did not have a β-H atom. The mechanistic proposal for both pathways was confirmed by trapping intermediate VIII with 3-methoxyprop-1-yne to afford derivative 65.
Partially intermolecular cycloadditions of allenes with alkenes
Allenes were also reacted with alkenes. In this respect, Alexanian et al.39–41 studied the partially intermolecular rhodium-catalysed [2+2+2] cycloadditions of ene-allenes with allenes and alkenes, respectively, to afford carbocycles with up to four contiguous stereogenic centres, including quaternary stereocentres.In their first study,39 the cycloaddition between ene–allenes substrates 66 and monosubstituted allenes 67 afforded regio- and diastereoselectively trans-fused carbocycles 68 with up to four stereogenic centres (Scheme 19). The substituted double bonds of the two allene moieties were involved chemoselectively in the cycloaddition, affording an exo-hexacyclic diene 68. After some experimentation the optimal catalytic system was found to be a rhodium chloride dimer with H8-BINAP ligand treated with silver salts in order to generate in situ a cationic rhodium complex. Several tethers in 66 were tolerated, as well as different substituents in the alkene moiety. The use of trisubstituted alkenes (66b) produced cycloadducts containing a quaternary stereocentre (68b). No substituents were introduced on the tethered allene and the scope of the second allene was limited to ethyl allenoate and phenyl allene. The mechanistic proposal was based on the initial oxidative addition between the two substituted double bonds of the allene moieties and when the alkene is inserted in the corresponding rhodacyclopentane, the trans ring fusion was established. An extension of this study was to investigate the enantioselectivity of the cycloaddition.40 Therefore, with the same substrates 66 and monosubstituted allenes, the reaction was studied using (R)-H8-BINAP as the chiral ligand with the same rhodium-based catalytic system. On comparing this ligand with 39 chiral BINAP-type ligands they found that (R)-H8-BINAP was the most efficient. The enantiomeric excess of the cycloadducts obtained ranged from 48 to 74%.
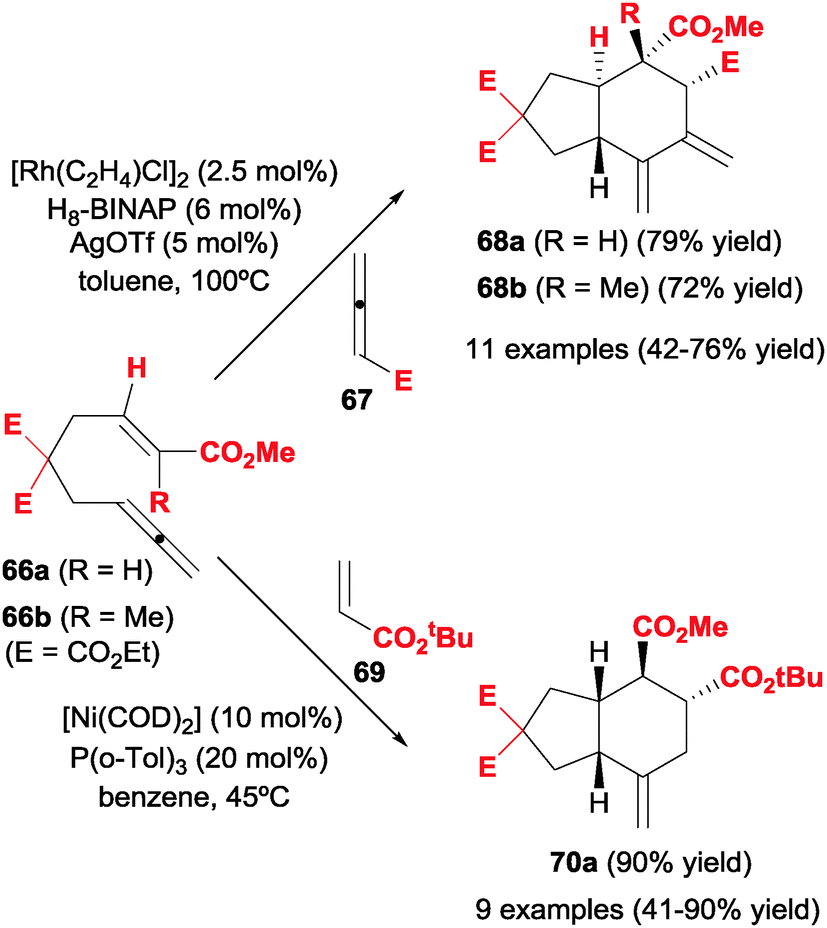 | ||
| Scheme 19 [2+2+2] Cycloaddition reactions between ene–llenes and allenes or alkenes. | ||
The cycloaddition of ene–allenes 66 with electron-poor alkenes was also studied by the same research group (Scheme 19).41 The optimal rhodium-based catalytic system used in the two previous studies did not catalyse the [2+2+2] process in this case. Therefore, the authors turned to nickel complexes and found that the combination of [Ni(COD)2] with monophosphine P(o-Tol)3 was the best catalytic system to afford regio- and diastereoselectively cis-fused carbocycles 70 in good yields. An excess of alkene and a slow addition of the ene–allene to the reaction mixture improved the yield. Different tethers in 66 were also tolerated. No substituents were introduced on the tethered allene, which also reacted with the inner double bond, and the alkenes were always electron-poor. Interestingly, the process seems to be highly sensitive to the nature of the phosphine used as a ligand. When P(tBu)3 was used instead of P(o-Tol)3, the reaction led to an alkenylative carbocyclisation process and when a bidentate phosphine such as XANTPHOS was used, an ene–allene [2+2] cycloaddition took place. In neither case was [2+2+2] cycloadduct observed.
[2+2+2] Cycloaddition reactions of allenes with carbon-heteroatom π-unsaturations
One of the most interesting advances in the field of [2+2+2] cycloadditions is the involvement of heterounsaturations to the generation of different types of heterocycles. Murakami et al.42 studied the reaction of allenes with heterocumulenes such as isocyanates. In a completely intermolecular version, the authors described the cycloaddition of one molecule of allene with two molecules of isocyanates to afford dihydropyrimidine-2,4-dione derivatives (73/74) under nickel(0) catalysis (Scheme 20). This is not an easy process given that it is necessary to control both the chemoselectivity, which was achieved using an excess of the isocyanate, as well as the double bond of the allene involved in the reaction. In the case that the main isomer was derivative 73, a stereogenic centre would be created. When the (S,S)-iPr-Foxap chiral ligand, an unusual ligand for enantioselective [2+2+2] cycloaddition reactions, was used, regioisomer 73 was predominantly formed (73:74 > 20:1) in high enantiomeric excess. The use of bidentate ligands was crucial for the success of the reaction. Only alkyl terminal allenes and aryl isocyanates were tested to cover the scope of the process. Remarkably, this was the first reported example of enantioselective [2+2+2] cycloaddition involving allenes.
 | ||
| Scheme 20 Ni(0)/(S,S)-iPr-Foxap catalysed enantioselective [2+2+2] cycloadditions between allenes and isocyanates. | ||
Another heterounsaturation that has been involved in cycloadditions with allenes is the carbonyl group of aldehydes and ketones to afford compounds with a pyran ring in their structure. Oonishi, Sato et al.43 constructed tricyclic pyran derivatives 76 in a stereoselective manner from acyclic allene–yne aldehydes 75 (Scheme 21).
 | ||
| Scheme 21 Rhodium-catalysed intramolecular [2+2+2] cycloaddition reactions of allenes involving aldehydes. | ||
Given that one of the competing pathways is the cyclopropanation/cyclisation of the allenyne moiety44 to afford derivatives 77, screening of the reaction conditions was undertaken. Curiously, the use of phosphine and NHC-carbene ligands were detrimental to the process as the best combination was [Rh(COD)Cl]2/AgBF4 in DCE at room temperature. However, the process was highly sensitive to the nature of lineal substrate 75. When there were four methylene units between the triple bond and the aldehyde, the competing pathway opened up to form 77. When there were two methylene units between the allene and the tether, a rearrangement of the 5-alkynal moiety in the substrate took place. The process was chemoselective to the double bond of the allene with the tBu substituent. The formation of the syn-isomer was justified mechanistically to avoid steric repulsion between the substituent of the allene and the α-CH2 of the carbonyl group when the aldehyde was inserted into the rhodacyclopentene intermediate.
The same authors45,46 studied the rhodium-catalysed cyclisation of analogous acyclic derivatives 78 and 79 in which the allene was placed in the middle of the chain, and the heterounsaturation was a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group (78) or an imine (79) (Scheme 22). In both cases, when the alleneyne compounds did not have a hydrogen atom at the β-position of the carbonyl or sulphinylimine moieties, pyran and piperidine derivatives containing fused five and seven-membered rings 80 and 81 were obtained respectively in good yields. In the case of 78, alkyl, aryl and silyl ketones covered the scope of the process. In the case of sulfinylimines 79 the scope was tested with different substituents of the alkyne such as hydrogen, alkyls, esters, a phenyl and a TMS group. In both cases, when the starting lineal substrates had a hydrogen atom at the β-position of the carbonyl or sulfinylimine moieties, the rhodacycle intermediate suffered a β-hydride elimination giving 5,7-fused cyclic alcohols 82 and amides 83, respectively (Scheme 22).
O group (78) or an imine (79) (Scheme 22). In both cases, when the alleneyne compounds did not have a hydrogen atom at the β-position of the carbonyl or sulphinylimine moieties, pyran and piperidine derivatives containing fused five and seven-membered rings 80 and 81 were obtained respectively in good yields. In the case of 78, alkyl, aryl and silyl ketones covered the scope of the process. In the case of sulfinylimines 79 the scope was tested with different substituents of the alkyne such as hydrogen, alkyls, esters, a phenyl and a TMS group. In both cases, when the starting lineal substrates had a hydrogen atom at the β-position of the carbonyl or sulfinylimine moieties, the rhodacycle intermediate suffered a β-hydride elimination giving 5,7-fused cyclic alcohols 82 and amides 83, respectively (Scheme 22).
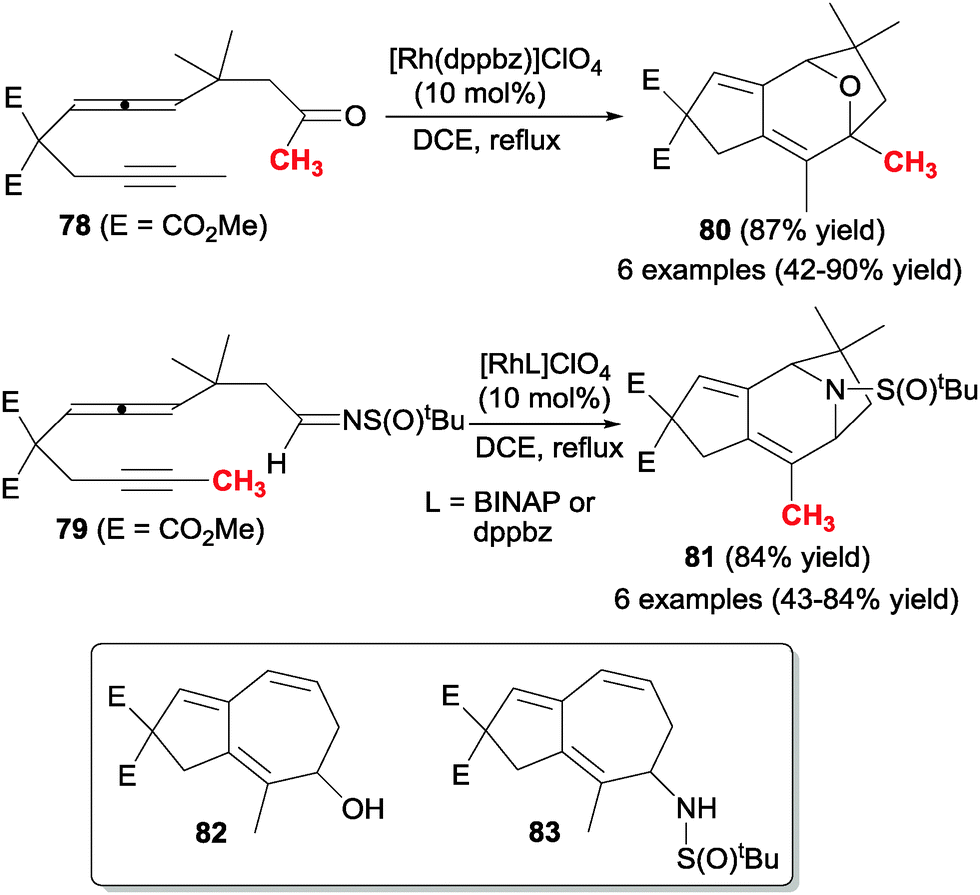 | ||
| Scheme 22 Rhodium-catalysed intramolecular [2+2+2] cycloaddition reactions of allenes involving aldehydes and imines. | ||
If we analyse the different outcome of allenynes 75 in comparison with 78/79, we can deduce that the selective insertion of the carbonyl bond to the Rh–Csp2 bond of the rhodacyclopentene intermediate determines the nature of the final product. In the case of 78 and 79, the reaction proceeds through a strained transition state XI to afford intermediate XII (Scheme 23).
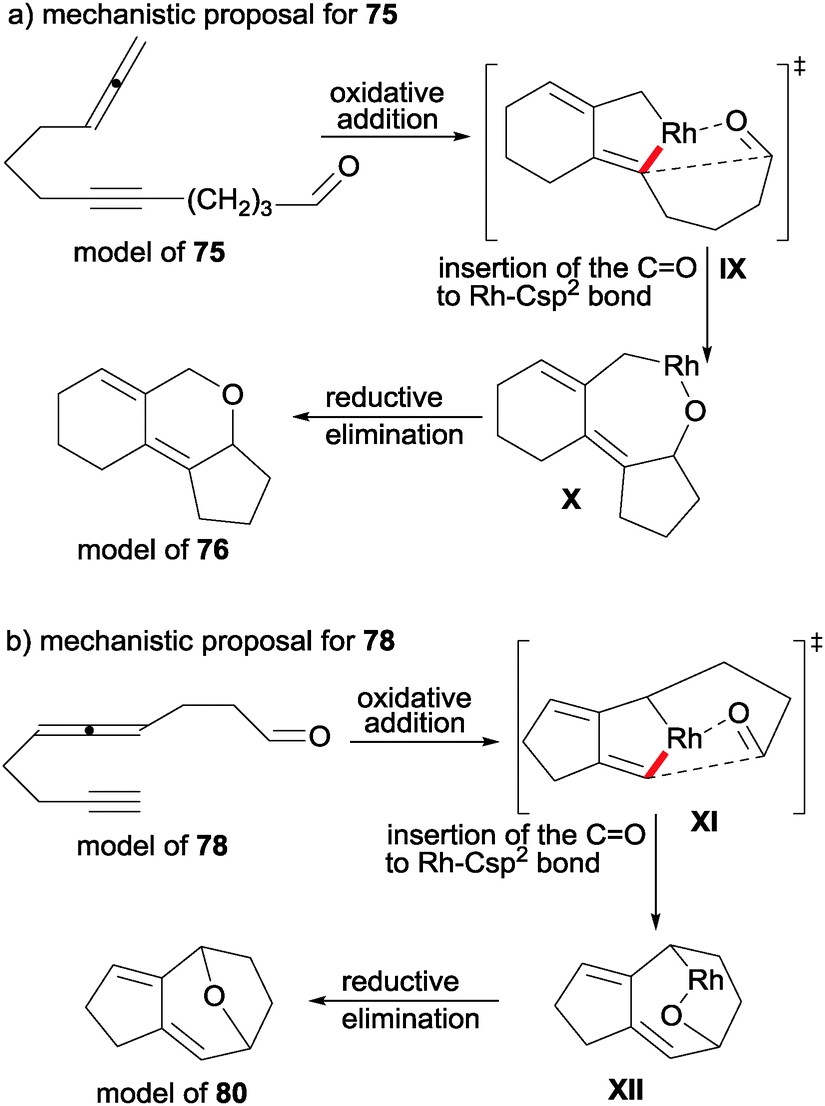 | ||
| Scheme 23 Mechanistic proposal for intramolecular [2+2+2] cycloaddition reactions of alleneynes and aldehydes. | ||
Pursuing the same idea as Oonishi and Sato, Tanaka et al.47 designed a partially intermolecular enantioselective version of tosylamide-linked 5-allenal 84 and 5-allenone with internal alkynes 85 under rhodium catalysis to afford 3,4-dihydropyran derivatives 86 (Scheme 24). In this case, the best catalytic system was found to be a combination of 20% molar of cationic complex [Rh(COD)2]BF4 with the chiral ligand (S)-BINAP. The scope of the process was quite limited since the alkynes had to be non-terminal and a tosyl linker was necessary in the starting material. Surprisingly, the regioselectivity of the process seems to be erratic although the enantioselectivity was good.
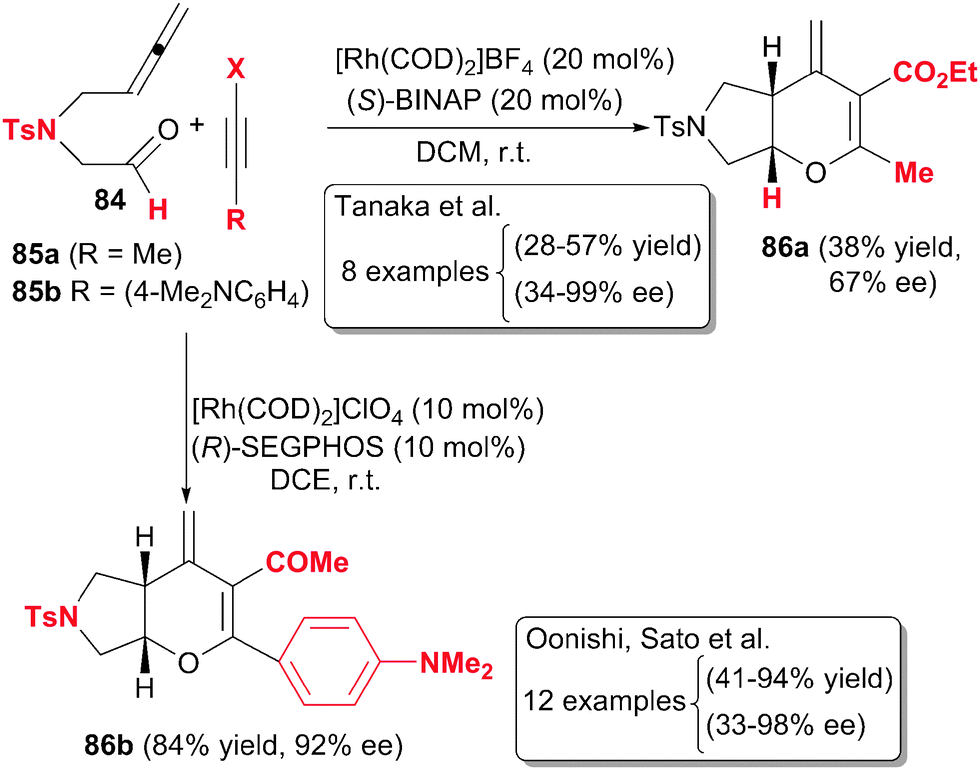 | ||
| Scheme 24 Rhodium-catalysed partially intermolecular [2+2+2] cycloaddition reactions of allenes involving aldehydes. | ||
Oonishi, Sato et al.48 later studied the same reaction, the cycloaddition of allenyl aldehydes 84 with alkynes under rhodium catalysis, broadening the scope of the allenal derivative: other tethers were used instead of the tosyl group, the carbon chain between the allene and the aldehyde moieties was elongated and an aromatic ring was added to the tether. Internal alkynes with an electron-rich aromatic ring and an electron-withdrawing group (85b) were efficient although symmetrical alkynes did not react. The process was regio- and enantioselective using (R)-SEGPHOS as the chiral biphosphine. As in the study by Tanaka, terminal alkynes did not work and, in this case, dienyl ketones were obtained instead of cycloaddition products. The authors proposed an initial oxidative addition between the internal double bond of the allene and the carbonyl group of 84 generating an oxarhodacyclopentane followed by the insertion of the alkyne. The regioselectivity observed in the insertion step was justified by electronic effects of the alkyne.
One contribution of our research group was to involve nitriles in [2+2+2] cycloadditions with allenes to generate 2,6-naphthyridine scaffolds 89 for the first time (Scheme 25).27 The use of Wilkinson's catalyst allowed the chemoselective reaction of allenes through their external double bond to give unsaturated pyridine-containing scaffolds 88 after a dehydrogenative step. After some experimentation, it was found that it was necessary to use a catalytic amount of Et3N and microwave rather than conventional heating in order for the reaction to proceed successfully. Removal of the tosyl group allowed entry into 2,6-naphthyridine nuclei, as is seen in derivative 89.
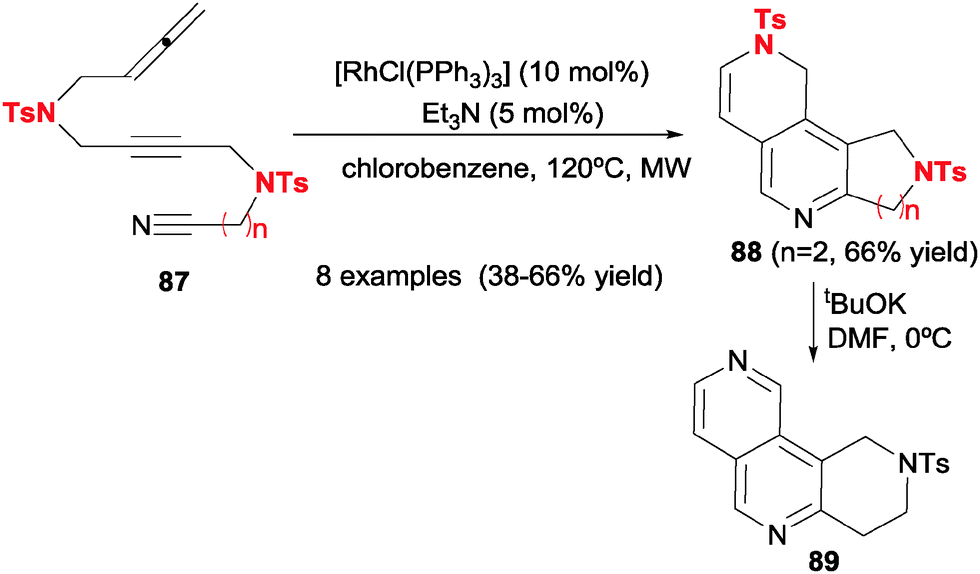 | ||
| Scheme 25 Wilkinson's complex-catalysed [2+2+2] cycloadditions of acyclic allene–yne–cyano derivatives. | ||
Miscellaneous cycloadditions
In this section we will comment on studies in which products are reported that present structures (six-membered carbo- or heterocycles) which come from formal [2+2+2] reactions of three unsaturations, but which mechanistically display different patterns.Ma et al.49 disclosed a palladium-catalysed cyclisation of diyne carbonates 90 and allenoic acids 91 (Scheme 26). The reaction is applicable to both secondary and tertiary carbonates with different tethers between the alkynes, and the alkyne in a remote position with respect to the carbonate may be terminal or non-terminal. When a stereogenic centre was introduced in the propargylic position, the chirality was not erased in the course of the reaction. With respect to the allenoic acids, aryl, alkyl and allyl substituents were tolerated. When 4-monosubstituted allenoic acids were used, the initial cycloadduct suffered an isomerisation to deliver aromatised compounds.
 | ||
| Scheme 26 Palladium-catalysed cyclisation of diyne carbonates and allenoic acids. | ||
The authors postulate a mechanism for the transformation (Scheme 27) consisting of initial oxidative addition and cyclic carbopalladation of the diyne carbonate to form a 1,3,4-trienyl methoxide palladium intermediate XIII. A subsequent trans-oxopalladation generates a palladacyclopentadiene XIV in which the ester has already been formed. From this point, the mechanism is analogous to that which is presented in the introduction, i.e. insertion of the electron-rich double bond of the allene, followed by reductive elimination.
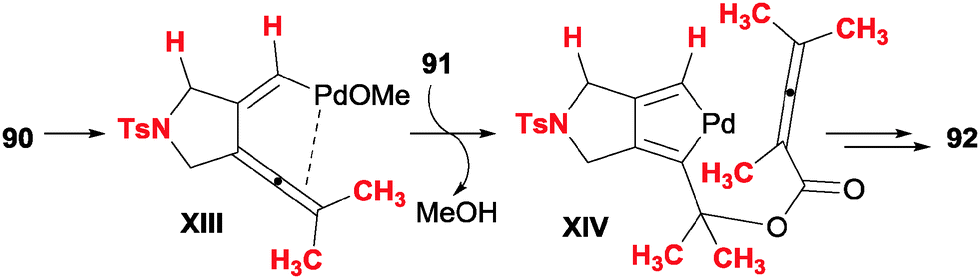 | ||
| Scheme 27 Key intermediates proposed in the Pd-catalysed cyclisation of diyne carbonates and allenoic acids. | ||
Another study by Mascareñas, López et al.50 describes the fully intermolecular [2+2+2] cycloaddition of three different components such as allenamides 93, alkenes 94 and aldehydes 95 to afford tetrahydropyrans 96 under gold catalysis (Scheme 28). The authors found that a phosphite gold complex catalysed the cycloaddition at very low temperatures (−45 °C or −78 °C) in a high regio- and 2,6-cis diastereoselective fashion. The scope of the process is extremely broad: alkenes, styrenes, cyclic alkenes, and electron-rich alkenes were used as alkenes; for aldehydes, benzaldehyde and alkylaldehydes; for allenamides, γ-methyl-substituted allenamides and N-tosylphenyl allenamides.
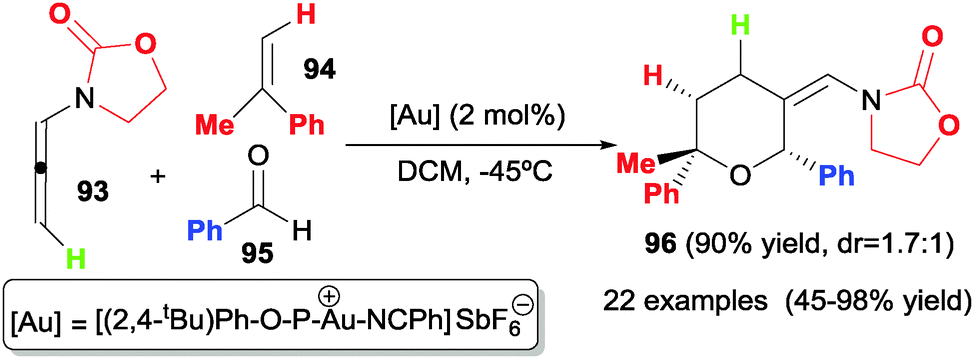 | ||
| Scheme 28 Au-catalysed [2+2+2] cycloaddition reactions of allenamides, alkenes and aldehydes. | ||
The authors proposed an initial π-activation of the allene by the gold complex to form the conjugated acyliminium intermediate XV, which undergoes 1,4-addition by the alkene, giving intermediate XVI. The nucleophilic attack of the carbonyl group of the aldehyde led to intermediate XVII and finally to cycloadduct 96 (Scheme 29).
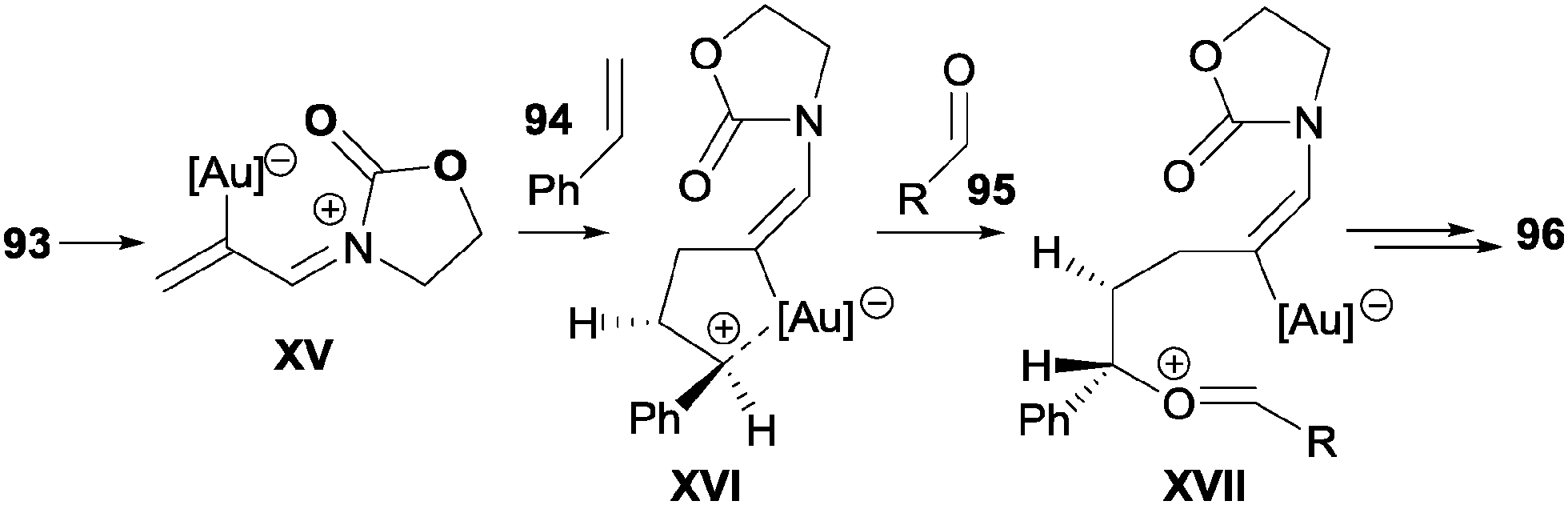 | ||
| Scheme 29 Key intermediates proposed for the Au-catalysed [2+2+2] cycloaddition reactions of allenamides, alkenes and aldehydes. | ||
Conclusions
Allenes are versatile reaction partners in transition-metal-catalysed [2+2+2] cycloadditions for a number of reasons. They display a good reactivity profile, rivalling that of alkynes and far superior to the sluggish reactivity of alkenes. Moreover, cycloadditions involving allenes proceed with good stereoselectivities and much improved chemo- and regioselectivities with respect to all-alkyne cycloadditions. The latter is advantageous for the implementation of intermolecular multicomponent reactions. However, the nature of the chemo- and regioselectivity is highly dependent on the allene structural environment and is difficult to predict. Special care must be taken in the choice of substrates and the design of polyunsaturated precursors, in order to avoid detrimental β-elimination processes. The analysis of the potential mechanistic scenarios by computational techniques may be a solution to these shortcomings. As we progress in the understanding of such intricacies, [2+2+2] cycloadditions involving allenes should gradually find use in target-oriented applications.Acknowledgements
We gratefully acknowledge the financial support of our own research in this area by the Spanish Ministry of Education and Science (MINECO) (Project CTQ2014-54306-P and a RyC contract to A. L.) and the DIUE of the Generalitat de Catalunya (Project 2014-SGR-931).Notes and references
- Transition-Metal-Mediated Aromatic Ring Construction, ed. K. Tanaka, Wiley, Hoboken, 2013 Search PubMed.
- W. Reppe and W. J. Schweckendiek, Justus Liebigs Ann. Chem., 1948, 560, 104 CrossRef CAS.
- N. Agenet, V. Gandon, K. P. C. Vollhardt, M. Malacria and C. Aubert, J. Am. Chem. Soc., 2007, 129, 8860 CrossRef CAS PubMed.
- A. Dachs, S. Osuna, A. Roglans and M. Solà, Organometallics, 2010, 29, 562 CrossRef CAS.
- C.-H. Guo, H.-S. Wu, M. Hapke and H. Jiao, J. Organomet. Chem., 2013, 748, 29 CrossRef CAS.
- H. Nishiyama, E. Niwa, T. Inoue, Y. Ishima and K. Aoki, Organometallics, 2002, 21, 2572 CrossRef CAS.
- B. Dutta, B. F. E. Curchod, P. Campomanes, E. Solari, R. Scopelliti, U. Rothlisberger and K. Severin, Chem. – Eur. J., 2010, 16, 8400 CrossRef CAS PubMed.
- M. Parera, A. Dachs, M. Solà, A. Pla-Quintana and A. Roglans, Chem. – Eur. J., 2012, 18, 13097 CrossRef CAS PubMed.
- Modern Allene Chemistry, ed. N. Krause and A. S. K. Hashmi, Wiley-VCH, Weinheim, 2004 Search PubMed.
- S. Yu and S. Ma, Chem. Commun., 2011, 47, 5384 RSC.
- A. S. K. Hashmi, Angew. Chem., Int. Ed., 2000, 39, 3590 CrossRef CAS.
- R. E. Benson and R. V. Lindsey, J. Am. Chem. Soc., 1959, 81, 4247 CrossRef CAS.
- R. E. Benson and R. V. Lindsey, J. Am. Chem. Soc., 1959, 81, 4250 CrossRef CAS.
- S. Ma, P. Lu, L. Lu, H. Hou, J. Wei, Q. He, Z. Gu, X. Jiang and X. Jin, Angew. Chem., Int. Ed., 2005, 44, 5275 CrossRef CAS PubMed.
- G. Chen, X. Jiang, C. Fu and S. Ma, Chem. Lett., 2010, 39, 78 CrossRef CAS.
- S. Ma and L. Lu, Chem. – Asian J., 2007, 2, 199 CrossRef CAS PubMed.
- P. Lu and S. Ma, Org. Lett., 2007, 9, 5319 CrossRef CAS PubMed.
- C. Aubert, D. Llerena and M. Malacria, Tetrahedron Lett., 1994, 35, 2341 CrossRef CAS.
- D. Llerena, O. Buisine, C. Aubert and M. Malacria, Tetrahedron, 1998, 54, 9373 CrossRef CAS.
- O. Buisine, C. Aubert and M. Malacria, Synthesis, 2000, 985 CrossRef CAS.
- M. Petit, C. Aubert and M. Malacria, Org. Lett., 2004, 6, 3937 CrossRef CAS PubMed.
- M. Petit, C. Aubert and M. Malacria, Tetrahedron, 2006, 62, 10582 CrossRef CAS.
- N. Saito, T. Ichimaru and Y. Sato, Chem. – Asian J., 2012, 7, 1521 CrossRef CAS PubMed.
- Y. Ohta, S. Yasuda, Y. Yokogawa, K. Kurokawa and C. Mukai, Angew. Chem., Int. Ed., 2015, 54, 1240 CrossRef CAS PubMed.
- E. Haraburda, Ò. Torres, T. Parella, M. Solà and A. Pla-Quintana, Chem. – Eur. J., 2014, 20, 5034 CrossRef CAS PubMed.
- B. Bennacer, M. Fujiwara, S.-Y. Lee and I. Ojima, J. Am. Chem. Soc., 2005, 127, 17756 CrossRef CAS PubMed.
- E. Haraburda, A. Lledó. A. Roglans and A. Pla-Quintana, Org. Lett., 2015, 17, 2882 CrossRef CAS PubMed.
- C. Munz, C. Stephan and H. t. Dieck, J. Organomet. Chem., 1990, 395, C42 CrossRef CAS.
- C. Stephan, C. Munz and H. t. Dieck, J. Organomet. Chem., 1994, 468, 273 CrossRef CAS.
- T. Takahashi, F.-Y. Tsai, Y. Li, K. Nakajima and M. Kotora, J. Am. Chem. Soc., 1999, 121, 11093 CrossRef CAS.
- M. Shanmugasundaram, M.-S. Wu and C.-H. Cheng, Org. Lett., 2001, 3, 4233 CrossRef CAS PubMed.
- M. Shanmugasundaram, M.-S. Wu, M. Jeganmohan, C.-W. Huang and C.-H. Cheng, J. Org. Chem., 2002, 67, 7724 CrossRef CAS PubMed.
- M. S. Wu, M. Shanmugasundaram and C.-H. Cheng, Chem. Commun., 2003, 718 RSC.
- J.-C. Hsieh, D. Kumar Rayabarapu and C.-H. Cheng, Chem. Commun., 2004, 532 RSC.
- For a review on Pd-catalysed cycloaddition reactions of arynes, see: E. Guitián, D. Pérez and D. Peña, Top. Organomet. Chem., 2005, 14, 109 Search PubMed.
- Y.-L. Liu, Y. Liang, S.-F. Pi, X.-C. Huang and J.-H. Li, J. Org. Chem., 2009, 74, 3199 CrossRef CAS PubMed.
- I. Quintana, D. Peña, D. Pérez and E. Guitián, Eur. J. Org. Chem., 2009, 5519 CrossRef CAS.
- X. Jiang and S. Ma, J. Am. Chem. Soc., 2007, 129, 11600 CrossRef CAS PubMed.
- A. T. Brusoe and E. J. Alexanian, Angew. Chem., Int. Ed., 2011, 50, 6596 CrossRef CAS PubMed.
- A. T. Brusoe, R. V. Edwankar and E. J. Alexanian, Org. Lett., 2012, 14, 6096 CrossRef CAS PubMed.
- N. N. Noucti and E. J. Alexanian, Angew. Chem., Int. Ed., 2013, 52, 8424 CrossRef CAS PubMed.
- T. Miura, M. Morimoto and M. Murakami, J. Am. Chem. Soc., 2010, 132, 15836 CrossRef CAS PubMed.
- Y. Oonishi, Y. Kitano and Y. Sato, Tetrahedron, 2013, 69, 7713 CrossRef CAS.
- Y. Oonishi, Y. Kitano and Y. Sato, Angew. Chem., Int. Ed., 2012, 51, 7305 CrossRef CAS PubMed.
- Y. Oonishi, T. Yokoe, A. Hosotani and Y. Sato, Angew. Chem., Int. Ed., 2014, 53, 1135 CrossRef CAS PubMed.
- Y. Oonishi, Y. Hato and Y. Sato, Adv. Synth. Catal., 2015, 357, 3033 CrossRef CAS.
- K. Sakashita, K. Masutomi, K. Noguchi and K. Tanaka, Chem. Lett., 2014, 43, 1260 CrossRef CAS.
- Y. Oonishi, A. Saito and Y. Sato, Asian J. Org. Chem., 2015, 4, 81 CrossRef CAS.
- X. Lian and S. Ma, Angew. Chem., Int. Ed., 2008, 47, 8255 CrossRef CAS PubMed.
- H. Faustino, I. Varela, J. L. Mascareñas and F. López, Chem. Sci., 2015, 6, 2903 RSC.
| This journal is © The Royal Society of Chemistry 2016 |