
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence35Cl dynamic nuclear polarization solid-state NMR of active pharmaceutical ingredients†
David A.
Hirsh
a,
Aaron J.
Rossini
*bc,
Lyndon
Emsley
d and
Robert W.
Schurko
*a
aDepartment of Chemistry and Biochemistry, University of Windsor, Windsor, ON N9B 3P4, Canada. E-mail: rschurko@uwindsor.ca; Tel: +1-519-253-3000 ext. 3548
bDepartment of Chemistry, Iowa State University, Ames, IA 50011, USA. E-mail: arossini@iastate.edu; Tel: +1-515-294-8952
cUS DOE Ames Laboratory, Ames, Iowa 50011, USA
dInstitut des Sciences et Ingénierie Chimiques, Ecole Polytechnique Fédérale de Lausanne (EPFL), Lausanne, CH-1015, Switzerland
First published on 24th August 2016
Abstract
In this work, we show how to obtain efficient dynamic nuclear polarization (DNP) enhanced 35Cl solid-state NMR (SSNMR) spectra at 9.4 T and demonstrate how they can be used to characterize the molecular-level structure of hydrochloride salts of active pharmaceutical ingredients (APIs) in both bulk and low wt% API dosage forms. 35Cl SSNMR central-transition powder patterns of chloride ions are typically tens to hundreds of kHz in breadth, and most cannot be excited uniformly with high-power rectangular pulses or acquired under conditions of magic-angle spinning (MAS). Herein, we demonstrate the combination of DNP and 1H–35Cl broadband adiabatic inversion cross polarization (BRAIN-CP) experiments for the acquisition of high quality wideline spectra of APIs under static sample conditions, and obtain signals up to 50 times greater than in spectra acquired without the use of DNP at 100 K. We report a new protocol, called spinning-on spinning-off (SOSO) acquisition, where MAS is applied during part of the polarization delay to increase the DNP enhancements and then the MAS rotation is stopped so that a wideline 35Cl NMR powder pattern free from the effects of spinning sidebands can be acquired under static conditions. This method provides an additional two-fold signal enhancement compared to DNP-enhanced SSNMR spectra acquired under purely static conditions. DNP-enhanced 35Cl experiments are used to characterize APIs in bulk and dosage forms with Cl contents as low as 0.45 wt%. These results are compared to DNP-enhanced 1H–13C CP/MAS spectra of APIs in dosage forms, which are often hindered by interfering signals arising from the binders, fillers and other excipient materials.
1. Introduction
The identification of solid forms of active pharmaceutical ingredients (APIs) plays an important role in drug development, both in the discovery of new forms and quality control.1–3 Each polymorph, pseudopolymorph (such as a hydrate or solvate), cocrystal, or salt of an API is uniquely patentable,4,5 and can have substantially different physicochemical properties (stability, solubility, bioavailability etc.).6–11 Undesired phases or impurities in dosage forms are potentially dangerous or costly; hence, new and innovative methods are needed for structurally characterizing APIs, both in the bulk phase and especially within solid dosage forms.Solid APIs are commonly characterized using X-ray diffraction (powder or single-crystal), 1H and 13C solid-state NMR (SSNMR), thermogravimetric methods, and other spectroscopic techniques.12–18 In many cases, these techniques provide adequate characterization of the bulk forms of APIs; however, they are often of limited use for dosage forms (especially those with low weight percentages, wt%, of the API). In particular, both pXRD patterns and 13C SSNMR spectra of dosage forms often display interfering signals from the excipient (e.g., binding ingredients and fillers), which obscure signals arising from the API.
Prior studies by our group19–21 and others22,23 have demonstrated that 35Cl SSNMR is a valuable tool for characterizing the bulk and dosage forms of APIs that have been synthesized as HCl salts (excipients do not contain chloride ions, so there are no interfering signals in 35Cl SSNMR spectra of dosage forms). Since 35Cl is a quadrupolar nucleus (I = 3/2), its spectra are influenced by a combination of anisotropic chemical shift and quadrupolar interactions. The latter are particularly sensitive to small structural differences in the local Cl− anion environments arising from variations in local hydrogen bonding.19,20,24–27 As a result, each solid phase of an API produces a distinct 35Cl SSNMR spectral fingerprint. Given the importance of identifying low concentrations of API phases within dosage forms (including impurities), it is crucial to improve the lower detection limit (LDL) of 35Cl SSNMR experiments.
Our research group has developed pulse sequences that enable the rapid acquisition of broad 35Cl SSNMR patterns (hundreds of kHz or more) with high S/N, even at moderate field strengths (e.g., 9.4 T). Unlike older methods,28–30 these pulse sequences rely on phase-modulated frequency-swept WURST (wideband uniform-rate smooth truncation) pulses31,32 for broadband excitation and polarization transfer. The WURST-CPMG (WCPMG)33,34 and broadband adiabatic inversion cross-polarization (BRAIN-CP)35 pulse sequences are used for direct (35Cl) and indirect (1H–35Cl) broadband excitation of 35Cl SSNMR spectra, respectively. The BRAIN-CP-WCPMG sequence (BCP for short) uses BRAIN-CP to transfer spin polarization from abundant nuclides (e.g., 1H) to dilute nuclides (e.g., 35Cl), and a subsequent WURST-CPMG pulse and windowed acquisition train for further signal enhancement (Fig. S1, ESI†).
Over the past few years, high-field dynamic nuclear polarization (DNP) has become a prominent method for achieving high gains in S/N for SSNMR experiments.36–39 Recent developments in DNP NMR instrumentation (e.g., high-frequency gyrotrons,40,41 low-temperature MAS probes42,43), optimized radical polarizing agents,44–48 and the availability of commercial DNP NMR spectrometers49,50 have enabled signal increases in excess of 300, representing potential time savings by a factor of 90![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000. DNP has enabled SSNMR experiments that were previously considered challenging/impossible, allowing the detailed the study of materials that were previously inaccessible to SSNMR.51–60 Most materials are prepared for DNP experiments by using a simple incipient wetness impregnation procedure to coat the surface of the particles, or fill the porous volume with a radical polarizing agent solution.47,61 Saturation of the EPR transitions of the radicals with microwaves results in enhanced polarization of the nuclei (most often protons) that are in close proximity to the polarizing agents.46 In the case of micro-particulate organic solids (e.g., APIs), DNP-enhanced polarization can be relayed from the surface of the particles into the interiors of the solids by 1H–1H spin diffusion without perturbing their macroscopic structure.62,63 With this technique, DNP-enhanced solid-state NMR can be applied to organic solids,63–66 pure APIs,63,66–69 and low API wt% dosage forms.69 These developments have enabled natural isotopic abundance 13C–13C, 1H–15N and 13C–15N correlation SSNMR experiments, which would be challenging or impossible without DNP.
000. DNP has enabled SSNMR experiments that were previously considered challenging/impossible, allowing the detailed the study of materials that were previously inaccessible to SSNMR.51–60 Most materials are prepared for DNP experiments by using a simple incipient wetness impregnation procedure to coat the surface of the particles, or fill the porous volume with a radical polarizing agent solution.47,61 Saturation of the EPR transitions of the radicals with microwaves results in enhanced polarization of the nuclei (most often protons) that are in close proximity to the polarizing agents.46 In the case of micro-particulate organic solids (e.g., APIs), DNP-enhanced polarization can be relayed from the surface of the particles into the interiors of the solids by 1H–1H spin diffusion without perturbing their macroscopic structure.62,63 With this technique, DNP-enhanced solid-state NMR can be applied to organic solids,63–66 pure APIs,63,66–69 and low API wt% dosage forms.69 These developments have enabled natural isotopic abundance 13C–13C, 1H–15N and 13C–15N correlation SSNMR experiments, which would be challenging or impossible without DNP.
To date, most DNP SSNMR studies have been limited to the characterization of nuclei with fairly narrow powder patterns (i.e., breadths on the order of tens of kHz or less). These reports include the spectral acquisition of a variety of spin-1/2 nuclei as well as quadrupolar nuclei in highly symmetric environments (e.g., 2H, 14N overtone, 17O, 27Al, and 51V).55,56,64,66,70–75 However, the enhancements afforded by DNP also make it an attractive technique for the acquisition of NMR spectra of broader patterns (e.g., 35Cl SSNMR) due to their inherently low S/N that largely results from the distribution of NMR signal across a wide frequency range. During the final preparation of the manuscript describing our work, Kobayashi et al. published one such example, in a report on DNP-enhanced ultra-wideline 195Pt SSNMR.76 One factor that hinders the acquisition of DNP-enhanced wideline spectra from stationary samples is that moderate- and fast-magic angle spinning (MAS) results in substantially higher DNP and sensitivity enhancements.47,50,77–79 MAS rates of ca. 6 to 40 kHz provide DNP enhancements (ε) that are ca. 3 to 5 times higher than those obtained from static (i.e., stationary sample) experiments.47,50,77,78 Unfortunately, MAS experiments are not generally useful for patterns with breadths on the order of 100 kHz or more, especially for quadrupolar nuclides. First, MAS only results in partial averaging of the effects of the second-order quadrupolar interaction, and second, even very fast MAS may not result in the separation of the spinning sidebands from the isotropic centerband.80 Matters are further complicated by the effects of first-order quadrupolar interactions and/or large chemical shift anisotropies. In many cases, MAS spectra of broad patterns are distorted and have low S/N, which prevents the accurate determination of quadrupolar and chemical shift tensor parameters.
Herein, we show that DNP can be used to enhance S/N in static wideline 35Cl SSNMR patterns of APIs acquired with BCP methods. We detail a new protocol, called spinning-on spinning-off (SOSO) acquisition, for enhancing the DNP polarization under MAS and subsequently acquiring a wideline 35Cl pattern under static conditions. These techniques result in DNP enhancements in 35Cl SSNMR spectra of up to a factor of 110 at 100 K and B0 = 9.4 T. We demonstrate the application of 35Cl DNP SSNMR for the characterization of APIs in their bulk forms, as well as in dosage forms with Cl contents of as low as 0.45 wt%. The application of these techniques for polymorph differentiation, impurity identification, and the discovery of new solid phases are also demonstrated.
2. Methods
2.1 Sample preparation
Bulk samples of histidine hydrochloride monohydrate (hist), ambroxol hydrochloride (ambr), isoxsuprine hydrochloride (isox), diphenhydramine hydrochloride (diph), and cetirizine dihydrochloride (ceti), with purities ranging between 98 and 99 wt%, were purchased from Sigma-Aldrich Canada, Ltd and used without further purification (Fig. 1). A 10 mg tablet of isoxsuprine and a 25 mg tablet of Life Brand diphenhydramine obtained at local pharmacies were used as the dosage forms of isox and diph, respectively. The weight percentages of chlorine in the bulk and dosage samples are tabulated in the ESI† (Table S2). To prepare the samples for DNP SSNMR experiments, the bulk powders were ground by hand for several minutes in a mortar and pestle to reduce particle sizes, while the dosage form tablets were only lightly crushed to break the tablets into a fine powder. The samples were then impregnated52,61 with ca. 15 μL of 15 mM TEKpol in 1,1,2,2-tetrachloroethane (TCE)47 (hist, ambr, isox) or 1,3-dibromobutane (DBB) (ceti, diph) and packed into 3.2 mm sapphire rotors. | ||
| Fig. 1 Molecular structures of APIs. | ||
2.2 VT pXRD
Variable-temperature powder X-ray diffraction (pXRD) experiments were conducted using the APEX III software suite and a Bruker Photon 100 CMOS diffractometer with a graphite monochromator with CuKα (λ = 1.5406 Å). Samples were ground, packed into glass capillary tubes, and then cooled with a stream of cold N2 from an Oxford cryostream attached to the diffractometer. pXRD patterns were acquired for all of the samples and the identities of the samples with known crystal structures were confirmed by comparison to simulated patterns (Fig. S13, ESI†).81–842.3 NMR
13C and 35Cl SSNMR DNP experiments were conducted on a 9.4 T (400 MHz)/263 GHz Bruker Avance III solid-state DNP NMR spectrometer50 using a 3.2 mm HXY probe configured for 1H–13C–35Cl experiments located at the DOE Ames Laboratory. Carbon and chlorine chemical shifts were referenced to TMS at 0 ppm, using the unified scale in the IUPAC standard.85 Preliminary DNP NMR experiments were conducted at the EPFL (Lausanne) on a Bruker Avance I solid-state DNP NMR spectrometer equipped with a 3.2 mm HXY probe configured for 1H–13C–35Cl.The full experimental parameters used for the 13C and 35Cl experiments are given in the ESI† (Tables S3–S6). NMR-13C: all 1H–13C CP/MAS experiments used a CP contact time of 2 ms, and a constant 13C spin lock rf field of ca. 74 kHz. An MAS frequency of 8 kHz (or 9 kHz for isox) was used. The 1H spin lock amplitude was linearly ramped86–88 from ca. 75 kHz to 83 kHz. Proton decoupling was applied for each acquisition using an rf field of 100 kHz and the SPINAL-64 pulse sequence.89 NMR-35Cl: 1H–35Cl CP-CPMG and CP-echo spectra of hist were acquired using conventional rectangular pulses with ca. 70 kHz rf on both channels. The 1H–35Cl BRAIN-CP-WURST-CPMG (BCP) pulse sequence35 was used to acquire the spectra of all of the other samples. The sweep direction of the WURST pulse applied during the BCP contact period can result in lopsided powder patterns; to minimize these effects on the 35Cl powder patterns of isox and diph, two sub-spectra were acquired with opposite sweep directions for the BCP contact pulses. These sub-spectra were then co-added together to form the final spectrum (Fig. S11, ESI†). With the exception of the spectra of hist, all of the 35Cl spectra were acquired using WURST-CPMG (WCPMG) refocusing pulses.33,34 To process these spectra, the echoes in each FID were co-added to form a single echo, which was then Fourier transformed and magnitude processed.
Because of the breadth of the 35Cl powder pattern of ceti (vide infra), its 1H–35Cl BCP spectrum was acquired using frequency-stepped acquisition.28,29,90 Four pieces were acquired with the transmitter frequencies separated by 50 kHz increments. These pieces were then co-added together to produce the final spectrum.
The dipolar hetero-nuclear multiple-quantum correlation rotatory-resonance recoupling (D-HMQC-R3) pulse sequence91,92 was used to obtain a 2D dipolar 13C–35Cl correlation spectrum in the hist sample. See Table S4 for the experimental parameters (ESI†).
2.4 Software
All spectra were processed using the TopSpin 3.2 software package. Analytical simulations of the processed 35Cl SSNMR spectra (Fig. S5–S9, ESI†) were generated using the Solid Lineshape Analysis module (v. 2.2.4) within TopSpin. The resulting quadrupolar and chemical shift parameters are listed in Table S7 (ESI†). Simulated 35Cl MAS NMR spectra were obtained using the simulation program for solid-state NMR spectroscopy (SIMPSON).93,943. Results and discussion
3.1 Histidine HCl
Histidine HCl (hist) is an excellent setup standard for DNP experiments due to its long T1(1H), which is ca. 284 s at 100 K. Slow longitudinal relaxation is advantageous for remote DNP, as it allows for increased 1H polarization buildup and greater DNP enhancements.47 The 1H–13C CP/MAS NMR spectra of hist obtained with and without microwaves are shown in Fig. 2a. The corresponding 13C DNP enhancement (εCP(13C) = 260) is the highest measured for all of the compounds in this study. All 13C signals from histidine are easily resolved and differentiated from the broad 1,1,2,2-tetrachloroethane (TCE) solvent peak at ca. 75 ppm. | ||
| Fig. 2 13C and 35Cl SSNMR spectra of finely ground hist impregnated with a 15 mM TEKPol/TCE solution acquired at 100 K and B0 = 9.4 T. (a) 1H–13C CP/MAS spectra acquired with and without microwaves. The top inset shows the two 13C spectra scaled to the same maximum intensity. Asterisks denote spinning sidebands. (b) 1H–35Cl CP-CPMG spectra acquired with microwaves and rotation during part of the recycle delay period (SOSO conditions, red), with microwaves and a stationary sample (blue), and a stationary sample without microwaves (black). The inset shows a vertical expansion of the 35Cl SSNMR spectra. | ||
The 1H–35Cl CP-CPMG NMR spectra of hist acquired with and without DNP are shown in Fig. 2b. Even under completely static sample conditions, a large DNP enhancement is observed when the microwaves are turned on (εCP(35Cl) = 50) (Fig. 2b). However, when the sample is rotated during part of the 30 s recycle/polarization delay, an additional 2.2-fold signal gain over the static DNP experiment is observed (Fig. 2b). We call this technique spinning-on spinning-off (SOSO). To acquire the spectrum with SOSO, we manually controlled the sample spinning at ca. 200 to 2000 Hz during the recycle delay and stopped the sample spinning several seconds before collecting a spectrum under static conditions (see Fig. S1 for a schematic diagram of the pulse sequence timings, ESI†). This procedure was repeated for each of the 4 to 8 scans in the experiment. SOSO allows for a larger 1H polarization build-up due to improved DNP while the sample is spinning and acquisition of a distortion-free wideline spectrum under static conditions. The improved DNP enhancement with spinning is consistent with previous results that show increased DNP enhancements in 1H–13C CP/MAS NMR spectra with increasing sample spinning speeds up to ca. 10–15 kHz.47,50,77,78
In order to examine the effects of slow MAS on DNP efficiency (without the application of SOSO), a 35Cl SSNMR spectrum was acquired with uninterrupted slow MAS at 250 Hz and the CP-echo pulse sequence (Fig. 3a). Slow MAS yields a central transition powder pattern that is very similar in appearance to that obtained from a corresponding experiment on a static sample (Fig. 3a and Fig. S2, ESI†). The signal in the spectrum acquired with continuous slow MAS is ca. 3.1 times greater than that acquired under static conditions (Fig. 3a). Thus, the DNP enhancement under continuous MAS is slightly greater than that obtained with SOSO (2.2 times, vide supra). The 35Cl DNP enhancements in these spectra are lower than the enhancements seen in the 13C NMR spectra, due in part to the slower MAS frequencies used in both the SOSO and slow MAS 35Cl SSNMR experiments.
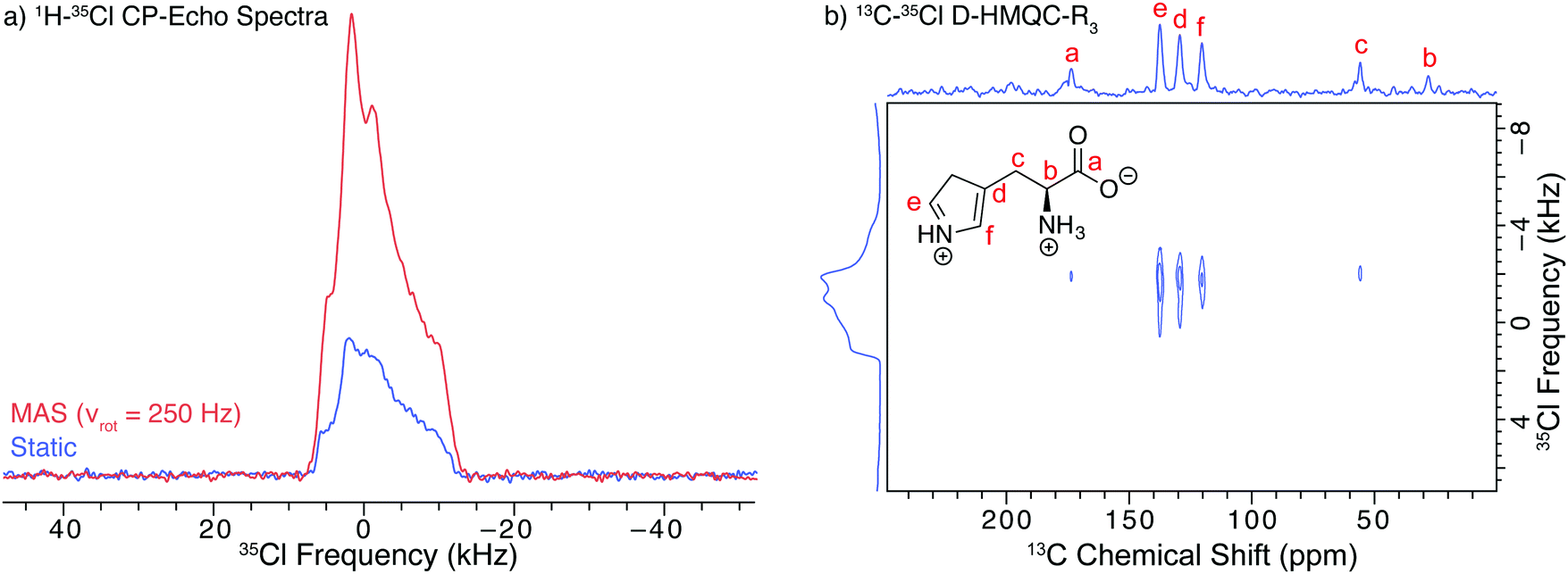 | ||
| Fig. 3 (a) DNP-enhanced 35Cl SSNMR spectra of finely ground hist impregnated with a 15 mM TEKPol/TCE solution acquired with the CP-echo pulse sequence under continuous slow MAS (νrot = 250 Hz) (red) and with the sample stationary at all times (blue). (b) 2D 13C–35Cl D-HMQC-R3 correlation spectrum of hist acquired under MAS (νrot = 8 kHz) with a projection of the 2D data along the direct dimension axis and the 35Cl CPMAS-echo spectrum shown along the indirect dimension axis. | ||
Since hist has a small quadrupolar coupling constant, a high-quality DNP enhanced CP-echo spectrum was also acquired at a faster MAS frequency of 8 kHz (Fig. S3, ESI†), resulting in a powder pattern free of overlapping spinning sidebands. The DNP enhancement of this MAS spectrum (εCP(35Cl) = 230) is comparable to the enhancements seen in the 13C NMR spectra (εCP(13C) = 260, vide supra). While slow or moderate MAS experiments may be useful for acquiring spectra of narrow central transition patterns that can be partially averaged (i.e., static pattern breadths <50 kHz, Fig. S3, ESI†), overlap of the MAS powder patterns with spinning sidebands is problematic for spectra with broader central transition patterns. Such overlap yields spectra without clearly defined discontinuities, which are difficult to analyze (Fig. S4, ESI† and vide infra). Given that the 35Cl SSNMR spectra of anionic chlorides in hydrochloride salts of APIs typically have powder patterns with breadths spanning 100–300 kHz at moderate field strengths,19–21 MAS 35Cl experiments with spinning rates between 5 and 15 kHz are not suitable for the characterization of most APIs. Furthermore, the polarization transfer from 1H to quadrupolar nuclei is often challenging to optimize and inefficient under MAS conditions.95
All of the 35Cl SSNMR spectra of hist can be simulated with the same quadrupolar tensor parameters: CQ = 1.8(1) MHz, ηQ = 0.72(2), δiso = 16(5) ppm (spectral simulations of these and all other 35Cl powder patterns in this work can be found in the ESI:† Fig. S5–S9 and Table S7). While these parameters agree with those reported in a recent study by Pandey et al. (CQ = 1.8 MHz and ηQ = 0.66),22 they do not match those reported by Bryce et al. for the room temperature spectrum of hist (CQ = 4.59 MHz, ηQ = 0.46, δiso = 93 ppm).24 It is possible that the study by Bryce et al. involved a different polymorph of hist.
DNP enhancement also provides access to two-dimensional experiments that would otherwise be challenging or impossible. One such example is a 2D heteronuclear dipolar correlation spectrum of proximate 13C and 35Cl nuclei. As a proof of concept, we obtained a 13C–35Cl dipolar heteronuclear multiple-quantum correlation spectrum (Fig. 3b) with rotatory-resonance recoupling (D-HMQC-R3).91,92 This 2D spectrum shows correlations between the 13C and 35Cl nuclei that are close to each other. Such results may provide valuable distance constraints on the structure of the molecule that may be useful for NMR crystallography.96 With DNP at higher magnetic fields and/or with faster sample spinning rates, it should be possible to acquire 13C–35Cl correlation spectra for APIs with larger values of CQ. 2D 13C–35Cl correlation NMR spectra could enable overlapping 35Cl powder patterns in APIs with multiple 35Cl sites to be resolved by correlation to high resolution 13C resonances.
3.2 Ambroxol HCl
Ambroxol HCl (ambr) is an API that is used to treat a myriad of respiratory diseases by clearing mucus from the respiratory tract. It is sold under a variety of trade names, including Mucosolvan, Mucobrox, Mucol, Lasolvan, Mucoangin, Surbronc, Ambolar, and Lysopain. A substantial DNP-enhancement (εCP(13C) = 92) was measured for the 1H–13C CP/MAS NMR spectra of ambr (Fig. 4a). The decreased enhancement relative to hist could result from less favorable relaxation characteristics; the T1(1H) of ambr at 100 K (ca. 30 s) is far less than that of hist (ca. 284 s), which limits the DNP enhancements of ambr.62,63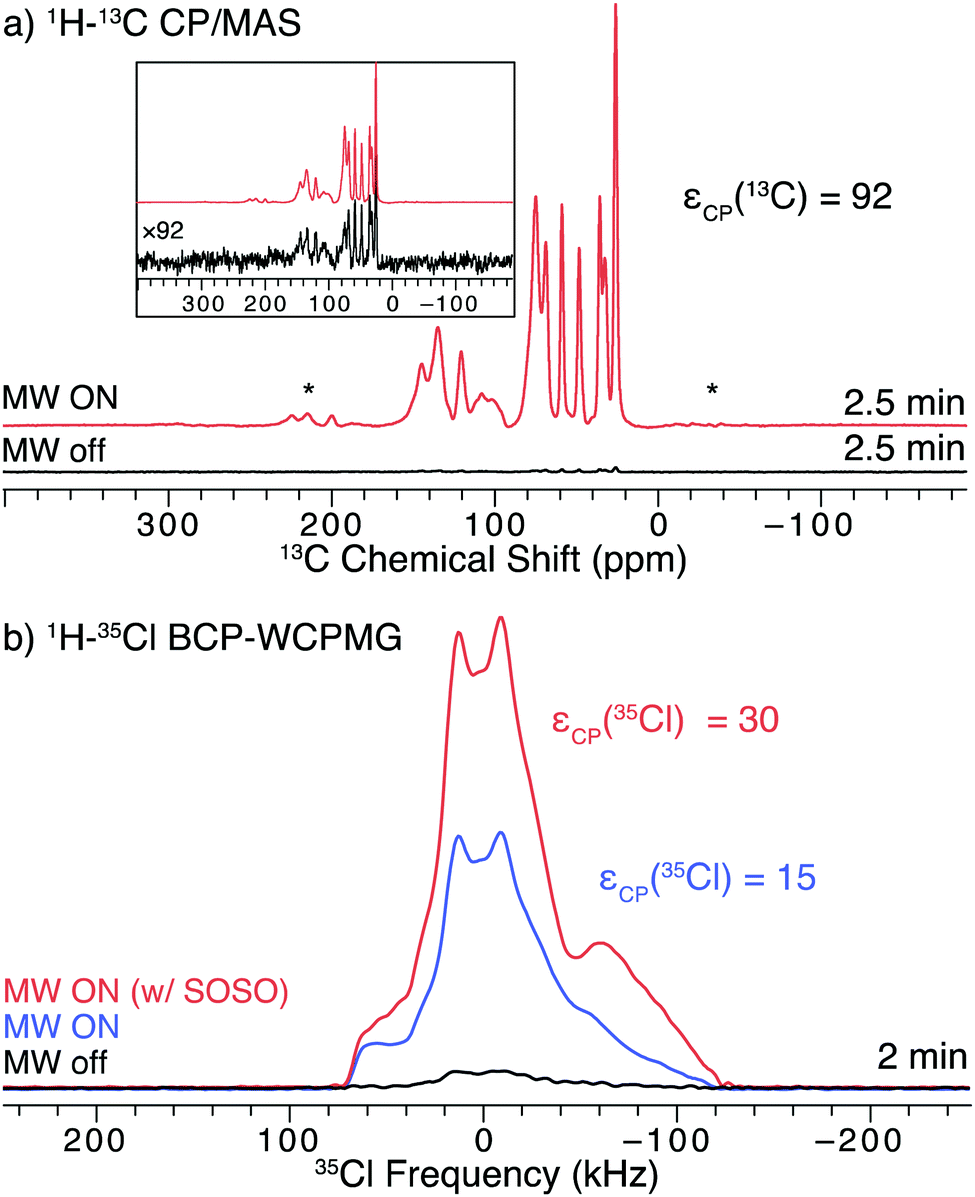 | ||
| Fig. 4 13C and 35Cl SSNMR spectra of finely ground ambr impregnated with 15 mM TEKPol/TCE solution acquired at 100 K and B0 = 9.4 T. (a) 1H–13C CP/MAS spectra acquired with and without microwaves to drive DNP. The top inset shows the two 13C spectra scaled to the same maximum intensity. The asterisks denote spinning sidebands. (b) 1H–35Cl BCP spectra acquired with microwaves and slow MAS rotation during most of recycle delay period (SOSO condition, red), with microwaves and with the sample stationary at all times (blue), and with the sample stationary without microwaves (black). | ||
As with hist, DNP provides a considerable signal enhancement (εCP(35Cl) = 15) in the 1H–35Cl BCP NMR spectra acquired under static conditions (Fig. 4b). If the SOSO procedure is used (i.e., where the sample is spun slowly during most of the recycle delay and then is stationary during the pulse and acquisition periods), the DNP enhancement is further increased by a factor 2 and εCP(35Cl) = 30 is observed (Fig. 4b). The combination of DNP and BCP produces a high S/N spectrum, spanning roughly 200 kHz, (ca. 10 times broader than that of hist).
Given the breadth of the 35Cl powder pattern of ambr at 9.4 T, it is not possible to use conventional MAS (i.e., constant spinning throughout the experiment). At the spinning speeds typically used for DNP-enhanced 13C NMR experiments (i.e., between 8 and 15 kHz), the presence of spinning sidebands distorts the 35Cl pattern and makes analysis of the powder pattern challenging (Fig. S4, ESI†). These issues result because (i) slower MAS rates do not average the effects of the second-order quadrupolar interaction, leading to patterns with many overlapping sidebands that are difficult to simulate, and (ii) spinning speeds exceeding 40 to 50 kHz are necessary to separate the spinning sidebands from the isotropic centerband for typical values of 35Cl CQ at 9.4 T (or hundreds of kHz for other quadrupolar nuclides).80 However, DNP MAS probes with faster spinning rates have recently become available,79 and may enable acquisition of spectra exhibiting undistorted isotropic centerbands for 35Cl sites with larger values of CQ.
There is a slight distortion in the low-frequency shoulder (at ca. −70 kHz) of the pattern acquired with the SOSO method. This distortion may arise from an improperly refocused CPMG echo train, which results from the sample not coming to a complete stop after spinning during the recycle delay – even very slow spinning can be disastrous for these experiments (Fig. S10, ESI†). The starting and stopping of the sample spinning was performed manually for this preliminary set of experiments. In the future, these issues could be addressed with the addition of specialized hardware to precisely control the spinning rate and stop/start timings of the sample. Alternatively, longer recycle delays could be employed, at the expense of a slight reduction in sensitivity.
3.3 Isoxsuprine HCl
The API isoxsuprine HCl (isox), commonly sold as Duvadilan, is a vasodilator used for both human and equine treatments. We have previously characterized isox in its bulk and dosage forms using 35Cl static NMR experiments without DNP.21 The 1H–13C CP/MAS NMR spectra of both the bulk and dosage forms of isox (Fig. 5a and b, respectively) can be acquired in only a few minutes with DNP (εCP(13C) = 86 and 32, respectively). The DNP enhancement of the signal from the API in the dosage form is much less than that from the bulk form of the API (a trend that continues with the other dosage form samples in this study, vide infra). Such lower DNP enhancements for dosage forms are likely due to: (i) differences in the particle sizes of the API in the bulk and dosage forms, (ii) the distribution of the radical solution in the sample, (iii) the presence of the excipient, or (iv) some combination of these factors. First, there are likely differences in the size of the API particles in the bulk and dosage forms due to the processing the API undergoes during tablet manufacturing. Second, the radical solution may be adsorbed preferentially by excipients during the impregnation step, or, if most of the API particles were coated with an excipient (e.g., a polymer) during production of the dosage form, the radical solution may not penetrate the excipient.69 Finally, the API particles may be coated with an excipient phase having an intrinsically short T1(1H), a very high concentration of protons, or unfavorable dielectric properties that reduce local microwave fields. All of these effects would reduce DNP enhancements of the 1H nuclei at the surface of the API particles and subsequently inside the cores of the API particles. DNP enhancements could probably be further improved on a case by case basis by optimizing the solvent used for impregnation (including investigating the use of fully- and partially-deuterated solvents), the concentration of radical in the solution, and the amount of radical solution used for the impregnation step.62,63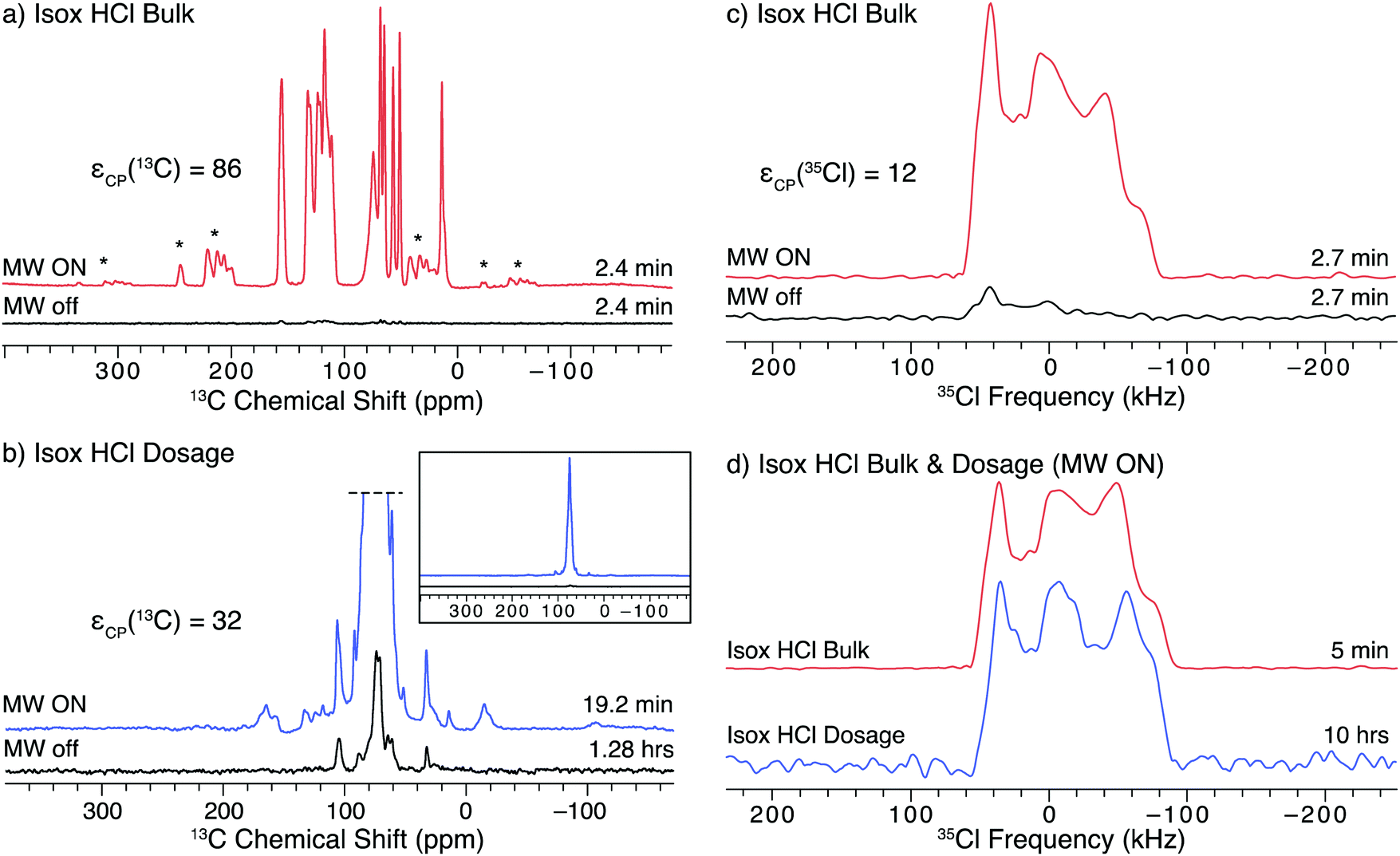 | ||
| Fig. 5 SSNMR spectra of the bulk and dosage forms of isox impregnated with a 15 mM TEKPol/TCE solution acquired at 100 K and B0 = 9.4 T. The left column has 1H–13C CP/MAS spectra of (a) the bulk API and (b) the dosage samples acquired with microwaves on and off. The bottom inset shows the full spectra of the dosage form without vertical clipping. The right column has 1H–35Cl BCP spectra of (c) the bulk API with and without microwaves and (d) the bulk API and dosage samples with DNP enhancement. The lineshapes in (c) are lopsided to the high frequency side because only one sweep direction of the BCP contact pulse was used (see Fig. S10 for more details, ESI†). | ||
Another complication is that the DNP enhancements of signals arising from the API, solvent, and excipient molecules are not the same, as was observed in a prior study of several cetirizine dosage forms using DNP-enhanced 13C SSNMR.69 In the case of isox (Fig. 5b, inset), the strongest signal is observed for a feature at 74.8(2) ppm, which corresponds to the solvent, TCE (εCP(13C) = 104). This intense feature dominates the spectrum of the dosage form and obscures several peaks from the API. The DNP enhancement of the unobscured API signal (e.g., peaks at ca. 110–135 ppm), εCP(13C) = 32, is less than that of the solvent. Finally, there are features that correspond to various types of excipient molecules, including polysaccharides (20–40 ppm), synthetic polymers (60–75 ppm) and stearates (100–110 ppm),69,97,98 which have enhancements ranging from 12 to 50. Overlapping signals from the API and excipient make it challenging to determine the phase of an API in 13C NMR spectra even without the use of DNP;21 however, the differences in the DNP-enhancement such as those observed in the spectra of isox can further complicate the analysis. While the signal from the solvent can be decreased by adding a spin-echo to the pulse sequence99 doing so only marginally improves the resolution of the features from isox (Fig. S14, ESI†).
35Cl SSNMR can selectively probe the API in the dosage form without interfering signals from the excipients, since chloride ions are only found in the API and the 35Cl signal from covalently-bound chlorines would be extremely broad and of too low intensity to be detected.100–102 A comparison of the DNP-enhanced 1H–35Cl BCP NMR spectra of bulk and dosage forms of isox is shown in Fig. 5d. The DNP enhancement observed for isox under static conditions (Fig. 5c, εCP(35Cl) = 12) is comparable to that of ambr (cf.Fig. 4b). Both the 35Cl SSNMR spectra of the bulk and dosage forms of isox are consistent with spectra acquired without DNP19 (see Fig. S7 for the spectral simulation and associated quadrupolar parameters, ESI†). The fact that the powder pattern of the tablet matches that of the bulk compound confirms that both samples contain the same polymorph of isox. There are additional features in the centers of both patterns (with centers of gravity at ca. 0 ppm). While the origin of this feature is still under study, it may result from Cl− anions coordinated to H2O (e.g., as a result of disproportionation of the HCl salt), or some other chemical or physical alteration of the sample (see VT-pXRD patterns in Fig. S12, ESI†).
A primary advantage of DNP experiments is that dosage forms of APIs can be studied even if the wt% of the API is very low, as is the case for isox (4.95 wt% API, 0.52 wt% Cl). Here, the combined acquisition time of the 35Cl spectra of the pure and dosage forms of isox with DNP was just over 10 hours, roughly half the experiment time necessary to acquire a comparable set of spectra at room temperature.21 Such time savings are critical in the high-throughput screening of dosage forms with low wt% APIs. Given the long acquisition times required to obtain sufficient 35Cl signal without microwaves, we have not attempted to acquire these spectra, and cannot report an enhancement in the spectrum of dosage isox (or the other dosage forms, vide infra).
SOSO experiments were not conducted on isox because of its short T1(1H) of 15 s. The short T1(1H) limits the amount of time available for 1H polarization build up before the polarization is lost to longitudinal relaxation. Roughly 25 s were required to start the sample spinning and completely stop rotation, which was not fast enough to fit within the optimal polarization time (20 s) for the experiments with isox. Of course, a longer polarization time could be applied to perform the SOSO method; however, the gains from increased DNP enhancement would be partially offset from reduced sensitivity arising from use of a recycle delay longer than 1.3 × T1. As with ambr (vide supra), the 35Cl powder pattern is too broad for conventional MAS to be used.
3.4 Diphenhydramine HCl
Diphenhydramine HCl (diph) is a widely used antihistamine, most commonly sold under the trade name Benadryl. The 1H–13C CP/MAS NMR spectra of both bulk diph (Fig. 6a) and the dosage form (Fig. 6b) show fair DNP enhancement (εCP(13C) = 25 and 16, respectively) and can be acquired in 2 minutes or less. The most intense feature in both of these spectra is a broad feature at ca. 50(2) ppm, which corresponds to the solvent, 1,3-dibromobutane (DBB). DBB was used for these experiments because diph was found to be soluble in TCE. Distinct features corresponding to the API (e.g., at 128(1) ppm) can be distinguished from those of the solvent and excipient in the spectrum of the dosage form (Fig. 6b). | ||
| Fig. 6 13C and 35Cl SSNMR spectra of the bulk and dosage forms of diph impregnated with a 15 mM TEKPol/1,3-dibromobutane solution acquired at 100 K and B0 = 9.4 T. The left column has 1H–13C CP/MAS spectra of (a) the bulk API and (b) the dosage samples acquired with microwaves on and off. Asterisks denote spinning sidebands. The right column has 1H–35Cl BCP spectra of (c) the bulk API with and without microwaves and (d) the bulk API and dosage samples with DNP enhancement. The lineshape in (c) is lopsided to the high frequency side because only one sweep direction of the BCP contact pulse was used (see Fig. S10 for more details, ESI†). | ||
The DNP enhancement observed in these spectra is not as high as those observed in the spectra of the other samples (Fig. 2, 4, and 5). One contributing factor is that diph, like isox, has a relatively short T1(1H) (ca. 18 s), which limits the build-up of enhanced polarization in the microcrystalline solid. The choice of solvent for the radical also plays an important role, as prior studies have reported decreased enhancements when using DBB rather than TCE, however, DBB was required for solubility reasons.47,48,103 Another disadvantage of the use of DBB is that it produces a broad solvent peak in the 13C SSNMR spectra, which can obscure features from the API and excipient and make confirmation of the phase and purity even more difficult. Clearly, the identification of other compatible solvents is an important future step for the further optimization of DNP experiments on APIs.
Unlike the 13C spectra, the 1H–35Cl BCP NMR spectra of diph (Fig. 6d) are free from signal interference from the excipient and solvent. The breadths of the powder patterns and locations of the discontinuities are identical, which confirm that the same phase of diph is present in both the bulk and dosage forms. These features are also consistent with previous work done at room temperature.21 As with the spectra of isox, there is an additional low-intensity feature at ca. −4(2) ppm that may result from disproportionation of the API. It is difficult to measure the DNP enhancement of these spectra due to the low S/N in the spectrum acquired without microwaves (Fig. 6c). The estimated minimum value of εCP(35Cl) = 7 was obtained by comparing the spectra after applying a Fourier transform directly to the CPMG echo train (see Fig. S15, ESI†).
Prior 35Cl SSNMR studies of this compound have relied on direct excitation experiments (e.g., WCPMG)21 because attempts to use CP at room temperature were unsuccessful due to poor CP efficiency. While CP experiments are still a challenge at low temperature, they are possible with the use of DNP. Unfortunately, the variation of CP efficiency with temperature when using the BCP pulse sequence is not well understood. DNP experiments, such as those reported here, could provide opportunities for further understanding the CP dynamics, and lead to improvements to the BCP sequence and related experiments under DNP conditions.
3.5 Cetirizine HCl
SSNMR is extremely useful for the study of APIs that form solid amorphous phases.104,105 One such API is cetirizine dihydrochloride (ceti), an antihistamine that is commonly sold under the trade names Zyrtec and Reactine.The 1H–13C CP/MAS NMR spectra of the bulk and dosage samples of ceti are shown in Fig. 7a and b, respectively, and are consistent with previous studies of this compound using DNP-enhanced 13C SSNMR.69 Several distinct features from the API are apparent in the spectrum of bulk ceti (e.g., peaks at 60–70 ppm and 130–200 ppm). However, only the strongest signal from the API (at ca. 130 ppm) can be distinguished in the spectrum of the dosage form due to interference from the excipients. In both spectra of ceti, there is less interference from the solvent than was observed in the spectra of diph (cf.Fig. 6a and b), because the DNP enhancement of the solvent peak is not as large (ε(DBB, ceti) = 24, ε(DBB, diph) = 50). The enhancement of the solvent varies from sample to sample, which may depend on the types and concentrations of different excipients, the quality of the glass formation when the radical-containing solution freezes, the amount of dissolved O2(g), the dielectric properties of the sample that affect the microwave field,106 or the other factors discussed above.
 | ||
| Fig. 7 13C and 35Cl SSNMR spectra of the bulk and dosage forms of ceti impregnated with 15 mM TEKPol/1,3-dibromobutane solution acquired at 100 K and B0 = 9.4 T. The left column has 1H–13C CP/MAS spectra of (a) the bulk API and (b) the dosage samples acquired with and without microwaves. Asterisks denote spinning sidebands. The right column shows the 1H–35Cl BCP spectra of (c) the bulk API with and without microwaves and (d) the bulk API and dosage samples with DNP enhancement. | ||
The enhancement observed in the spectrum of bulk ceti (εCP(13C) = 20) is close to what was previously reported for this compound (εCP(13C) = 31).69 However, for ceti in the dosage form we measured εCP(13C) = 8, which is lower than the 55-fold enhancement previously reported. We attribute these decreased enhancements to the use of DBB as the radical solvent. In the previous DNP SSNMR study, ceti was found to be sparingly soluble in TCE.69 We chose to use DBB (in which ceti is insoluble) to maximize the amount of solid sample in the rotor and to better maintain the structure of the API in the dosage form. Given that the discrepancy in enhancements is particularly apparent in the spectra of the dosage form, it is possible that DBB does not penetrate the excipients as well as TCE. Further optimization of the sample preparation could likely yield improved enhancements.
Ceti is a dihydrochloride, and its 35Cl SSNMR spectra should show two distinct 35Cl patterns corresponding to structurally unique Cl− anion sites. With the use of DNP, it is possible to identify two overlapping powder patterns in the 35Cl spectrum of the bulk form in just 5 minutes (εCP(35Cl) = 5.8, Fig. 7c). Due to hardware limitations, it is challenging to uniformly excite the entire breadth of the two 35Cl patterns of ceti, even with BCP. As such, we used frequency-stepped acquisition28,29 and combined 4 sub-spectra at evenly spaced transmitter frequencies to obtain the full pattern (which is ca. 250 kHz broad, Fig. S9, ESI†). The two overlapping 35Cl powder patterns with distinct quadrupolar parameters can be readily distinguished using analytical simulations. Nonetheless, the most important discontinuities in the two patterns can be observed in the central sub-spectrum (as in Fig. 7c and d).
Acquiring a 35Cl SSNMR spectrum of the dosage form with similar signal-to-noise ratio takes more than 11 hours, due to the extremely low wt% Cl in this sample (5.78 wt% API, 0.45 wt% Cl), which is the lowest discussed herein. Comparison of the 35Cl spectra of the bulk and dosage forms (Fig. 7d) shows that the dosage form likely contains two 35Cl environments that are similar to those of the bulk form. However, the poorer resolution of the discontinuities in the 35Cl spectrum of the dosage form (most evident at the center of the pattern) is consistent with the lower crystallinity of the API within the formulation. This API was previously confirmed to exist as an amorphous form in all formulations using DNP-enhanced 13C and 15N SSNMR in the previous study.69
4. Conclusions
We have demonstrated the acquisition of high-quality static wideline 35Cl SSNMR spectra of APIs using the 1H–35Cl BRAIN-CP-WURST-CPMG pulse sequence under DNP conditions. DNP has been observed to enhance the 35Cl SSNMR signal by as much as 110 times for stationary samples. This enhancement is achieved by using a new spinning-on spinning-off (SOSO) protocol, in which the sample is spun during the recycle delay and halted shortly before the pulse/acquisition periods. The use of SOSO results in a build-up in 1H polarization under MAS conditions and allows for the acquisition of a wideline 35Cl spectrum free of spinning sidebands under static conditions. This method provides an additional two-fold signal enhancement over the spectra acquired with DNP under purely static conditions. The use of DNP dramatically decreases the lower detection limit for 35Cl SSNMR spectra of dosage forms; we report successful characterization of APIs in bulk and dosage forms with Cl contents as low as 0.45 wt%. These 35Cl NMR spectra are particularly useful for the identification of the API within the dosage form because they are not affected by interfering signals from excipient molecules in the pill. In this respect, the DNP-enhanced 1H–13C CP/MAS spectra of the dosage forms are limited, despite having higher signal enhancements than the corresponding 35Cl NMR spectra. For all of the systems in this study, we observed lower DNP enhancements in the spectra of the dosage forms than in those of the bulk API, possibly due to the presence of excipients that reduce DNP efficiency via a number of different mechanisms, or due to differences in the particle size of the API in the bulk and dosage samples. These techniques show potential for investigating the sizes of micro- and nanoparticles of APIs in dosage forms.69 Finally, we have demonstrated the use of DNP signal enhancement for the acquisition of a two-dimensional 13C–35Cl correlation NMR spectrum of histidine HCl monohydrate. The increasing availability of DNP MAS probes with faster spinning rates79 will allow for the acquisition of 1D and 2D 35Cl MAS NMR spectra of most chloride salts of APIs and other organic molecules, with CQ values as high as 7 to 8 MHz at 9.4 T.The techniques we have reported in this study will help expand the use of DNP to the study of other wideline and ultra-wideline (breadths > 250 kHz) powder patterns. While the spectra reported herein are dominated by the second-order quadrupolar interaction, the techniques described should would work equally well for patterns that are broadened by the first-order quadrupolar interaction, chemical shift anisotropy, or combinations thereof. These developments make DNP useful for the study of a wide range of materials whose NMR spectra suffer from inherently low S/N largely due to the wide breadth of the signal.
Acknowledgements
R. W. S. acknowledges the Natural Science and Engineering Research Council (NSERC, Canada) for support in the form of Discovery, Accelerator, and Research Tools and Instruments (RTI) grants. He also acknowledges the University of Windsor for a 50th Golden Jubilee Research Chair. We are also grateful to the Canadian Foundation for Innovation (CFI), the Ontario Innovation Trust (OIT), and the University of Windsor for support of the Laboratories for Solid-State Characterization. The DNP solid-state NMR spectrometer at the Ames Laboratory was funded by US Department of Energy (DOE), Office of Science, Basic Energy Sciences, Division of Materials Science and Engineering and Division of Chemical Sciences, Geosciences, and Biosciences. The Ames Laboratory is operated for the US DOE by Iowa State University under contract no. DE-AC02-07CH11358. A. J. R. thanks the Ames Laboratory (Royalty Account) and Iowa State University for support. L. E. acknowledges the support of ERC Advanced Grant No. 320860. We thank Professors Holger Eichhorn and Jeremy Rawson (Windsor) for their assistance with the VT-pXRD measurements. We are grateful to Prof. P. Tordo, Dr O. Ouari and Dr G. Casano (Aix-Marseille Université, France) for providing the TEKPol biradical used in the DNP experiments.References
- F. Kesisoglou, S. Panmai and Y. Wu, Adv. Drug Delivery Rev., 2007, 59, 631–644 CrossRef CAS PubMed.
- J. Aaltonen, M. Alleso, S. Mirza, V. Koradia, K. Gordon and J. Rantanen, Eur. J. Pharm. Biopharm., 2009, 71, 23–37 CrossRef CAS PubMed.
- A. Newman, G. Knipp and G. Zografi, J. Pharm. Sci., 2012, 101, 1355–1377 CrossRef CAS PubMed.
- F. G. Vogt, eMagRes, John Wiley & Sons, Ltd, Chichester, UK, 2015, vol. 4, pp. 181–188 Search PubMed.
- A. V. Trask, Mol. Pharmaceutics, 2007, 4, 301–309 CrossRef CAS PubMed.
- S. R. Chemburkar, J. Bauer, K. Deming, H. Spiwek, K. Patel, J. Morris, R. Henry, S. Spanton, W. Dziki, W. Porter, J. Quick, P. Bauer, J. Donaubauer, B. A. Narayanan, M. Soldani, D. Riley and K. McFarland, Org. Process Res. Dev., 2000, 4, 413–417 CrossRef CAS.
- M. R. Caira, Design of Organic Solids, 1998, 198, 163–208 CAS.
- J. Bauer, J. Morley, S. Spanton, F. J. J. Leusen, R. Henry, S. Hollis, W. Heitmann, A. Mannino, J. Quick and W. Dziki, J. Pharm. Sci., 2006, 95, 917–928 CrossRef CAS PubMed.
- D. Hörter and J. B. Dressman, Adv. Drug Delivery Rev., 1997, 25, 3–14 CrossRef.
- Z. A. Langham, J. Booth, L. P. Hughes, G. K. Reynolds and S. A. C. Wren, J. Pharm. Sci., 2012, 101, 2798–2810 CrossRef CAS PubMed.
- L.-F. Huang and W.-Q. Tong, Adv. Drug Delivery Rev., 2004, 56, 321–334 CrossRef CAS PubMed.
- D. E. Bugay, Adv. Drug Delivery Rev., 2001, 48, 43–65 CrossRef CAS PubMed.
- S. Datta and D. J. W. Grant, Nat. Rev. Drug Discovery, 2004, 3, 42–57 CrossRef CAS PubMed.
- R. K. Harris, Analyst, 2006, 131, 351–373 RSC.
- R. K. Harris, J. Pharm. Pharmacol., 2007, 59, 225–239 CrossRef CAS PubMed.
- F. G. Vogt, New Applications of NMR in Drug Discovery and Development, 2013, pp. 43–100 Search PubMed.
- G. A. Monti, A. K. Chattah and Y. G. Linck, Annu. Rep. NMR Spectrosc., Elsevier Ltd, 2014, vol. 83, pp. 221–269 Search PubMed.
- F. G. Vogt, eMagRes, 2015, 4, 255–268 CAS.
- M. P. Hildebrand, H. Hamaed, A. M. Namespetra, J. M. Donohue, R. Fu, I. Hung, Z. Gan and R. W. Schurko, CrystEngComm, 2014, 16, 7334–7356 RSC.
- H. Hamaed, J. M. Pawlowski, B. F. T. Cooper, R. Fu, S. H. Eichhorn and R. W. Schurko, J. Am. Chem. Soc., 2008, 130, 11056–11065 CrossRef CAS PubMed.
- A. M. Namespetra, D. A. Hirsh, M. P. Hildebrand, A. R. Sandre, H. Hamaed, J. M. Rawson and R. W. Schurko, CrystEngComm, 2016, 18, 6213–6232 RSC.
- M. K. Pandey, H. Kato, Y. Ishii and Y. Nishiyama, Phys. Chem. Chem. Phys., 2016, 18, 6209–6216 RSC.
- F. G. Vogt, G. R. Williams, M. Strohmeier, M. N. Johnson and R. C. B. Copley, J. Phys. Chem. B, 2014, 118, 10266–10284 CrossRef CAS PubMed.
- D. L. Bryce and G. D. Sward, J. Phys. Chem. B, 2006, 110, 26461–26470 CrossRef CAS PubMed.
- B. J. Butler, J. M. Hook and J. B. Harper, Recent Advances in the NMR Spectroscopy of Chlorine, Bromine and Iodine, Elsevier Ltd, 2011, vol. 73 Search PubMed.
- C. M. Widdifield, R. P. Chapman and D. L. Bryce, Annu. Rep. NMR Spectrosc., Elsevier Ltd, 2009, vol. 66, pp. 195–326 Search PubMed.
- D. L. Bryce and E. B. Bultz, Chem. – Eur. J., 2007, 13, 4786–4796 CrossRef CAS PubMed.
- W. G. Clark, M. E. Hanson, F. Lefloch and P. Segransan, Rev. Sci. Instrum., 1995, 66, 2453–2464 CrossRef CAS.
- D. Massiot, I. Farnan, N. Gautier, D. Trumeau, A. Trokiner and J. P. Coutures, Solid State Nucl. Magn. Reson., 1995, 4, 241–248 CrossRef CAS PubMed.
- R. W. Schurko, Acc. Chem. Res., 2013, 46, 1985–1995 CrossRef CAS PubMed.
- E. Kupce and R. Freeman, J. Magn. Reson., 1995, 115, 273–276 CrossRef CAS.
- R. Bhattacharyya and L. Frydman, J. Chem. Phys., 2007, 127, 194503 CrossRef PubMed.
- L. A. O'Dell, A. J. Rossini and R. W. Schurko, Chem. Phys. Lett., 2009, 468, 330–335 CrossRef.
- L. A. O'Dell and R. W. Schurko, Chem. Phys. Lett., 2008, 464, 97–102 CrossRef.
- K. J. Harris, A. Lupulescu, B. E. G. Lucier, L. Frydman and R. W. Schurko, J. Magn. Reson., 2012, 224, 38–47 CrossRef CAS PubMed.
- D. A. Hall, D. C. Maus, G. J. Gerfen, S. J. Inati, L. R. Becerra, F. W. Dahlquist and R. G. Griffin, Science, 1997, 276, 930–932 CrossRef CAS PubMed.
- R. G. Griffin and T. F. Prisner, Phys. Chem. Chem. Phys., 2010, 12, 5737–5740 RSC.
- T. Maly, G. T. Debelouchina, V. S. Bajaj, K.-N. Hu, C.-G. Joo, M. L. Mak-Jurkauskas, J. R. Sirigiri, P. C. A van der Wel, J. Herzfeld, R. J. Temkin and R. G. Griffin, J. Chem. Phys., 2008, 128, 052211 CrossRef PubMed.
- L. R. Becerra, G. J. Gerfen, R. J. Temkin, D. J. Singel and R. G. Griffin, Phys. Rev. Lett., 1993, 71, 3561–3564 CrossRef CAS PubMed.
- L. R. Becerra, G. J. Gerfen, B. F. Bellew, J. A. Bryant, D. A. Hall, S. J. Inati, R. T. Weber, S. Un, T. F. Prisner, A. E. McDermott, K. W. Fishbein, K. E. Kreischer, R. J. Temkin, D. J. Singel and R. G. Griffin, J. Magn. Reson., 1995, 117, 28–40 CrossRef CAS.
- C. D. Joye, R. G. Griffin, M. K. Hornstein, K.-N. Hu, K. E. Kreischer, M. Rosay, M. A. Shapiro, J. R. Sirigiri, R. J. Temkin and P. P. Woskov, IEEE Trans. Plasma Sci., 2006, 34, 518–523 CrossRef PubMed.
- A. B. Barnes, M. L. Mak-Jurkauskas, Y. Matsuki, V. S. Bajaj, P. C. A. van der Wel, R. DeRocher, J. Bryant, J. R. Sirigiri, R. J. Temkin, J. Lugtenburg, J. Herzfeld and R. G. Griffin, J. Magn. Reson., 2009, 198, 261–270 CrossRef CAS PubMed.
- M. Rosay, J. C. Lansing, K. C. Haddad, W. W. Bachovchin, J. Herzfeld, R. J. Temkin and R. G. Griffin, J. Am. Chem. Soc., 2003, 125, 13626–13627 CrossRef CAS PubMed.
- C. Song, K.-N. Hu, C.-G. Joo, T. M. Swager and R. G. Griffin, J. Am. Chem. Soc., 2006, 128, 11385–11390 CrossRef CAS PubMed.
- C. Sauvée, M. Rosay, G. Casano, F. Aussenac, R. T. Weber, O. Ouari and P. Tordo, Angew. Chem., Int. Ed., 2013, 52, 10858–10861 CrossRef PubMed.
- K.-N. Hu, H. Yu, T. M. Swager and R. G. Griffin, J. Am. Chem. Soc., 2004, 126, 10844–10845 CrossRef CAS PubMed.
- A. Zagdoun, G. Casano, O. Ouari, M. Schwarzwälder, A. J. Rossini, F. Aussenac, M. Yulikov, G. Jeschke, C. Copéret, A. Lesage, P. Tordo and L. Emsley, J. Am. Chem. Soc., 2013, 135, 12790–12797 CrossRef CAS PubMed.
- D. J. Kubicki, G. Casano, M. Schwarzwälder, S. Abel, C. Sauvée, K. Ganesan, M. Yulikov, A. J. Rossini, G. Jeschke, C. Copéret, A. Lesage, P. Tordo, O. Ouari and L. Emsley, Chem. Sci., 2016, 7, 550–558 RSC.
- M. Rosay, M. Blank and F. Engelke, J. Magn. Reson., 2016, 264, 88–98 CrossRef CAS PubMed.
- M. Rosay, L. Tometich, S. Pawsey, R. Bader, R. Schauwecker, M. Blank, P. M. Borchard, S. R. Cauffman, K. L. Felch, R. T. Weber, R. J. Temkin, R. G. Griffin and W. E. Maas, Phys. Chem. Chem. Phys., 2010, 12, 5850–5860 RSC.
- T. Kobayashi, F. A. Perras, I. I. Slowing, A. D. Sadow and M. Pruski, ACS Catal., 2015, 5, 7055–7062 CrossRef CAS.
- A. J. Rossini, A. Zagdoun, M. Lelli, A. Lesage, C. Copéret and L. Emsley, Acc. Chem. Res., 2013, 46, 1942–1951 CrossRef CAS PubMed.
- O. Lafon, A. S. L. Thankamony, T. Kobayashi, D. Carnevale, V. Vitzthum, I. I. Slowing, K. Kandel, H. Vezin, J.-P. Amoureux, G. Bodenhausen and M. Pruski, J. Phys. Chem. C, 2013, 117, 1375–1382 CAS.
- W. R. Gunther, V. K. Michaelis, M. A. Caporini, R. G. Griffin and Y. Román-Leshkov, J. Am. Chem. Soc., 2014, 136, 6219–6222 CrossRef CAS PubMed.
- F. A. Perras, T. Kobayashi and M. Pruski, J. Am. Chem. Soc., 2015, 137, 8336–8339 CrossRef CAS PubMed.
- F. Blanc, L. Sperrin, D. A. Jefferson, S. Pawsey, M. Rosay and C. P. Grey, J. Am. Chem. Soc., 2013, 135, 2975–2978 CrossRef CAS PubMed.
- A. Lund, M.-F. Hsieh, T.-A. Siaw and S. Han, Phys. Chem. Chem. Phys., 2015, 17, 25449–25454 RSC.
- H. Takahashi, D. Lee, L. Dubois, M. Bardet, S. Hediger and G. De Paëpe, Angew. Chem., Int. Ed., 2012, 51, 11766–11769 CrossRef CAS PubMed.
- D. Lee, G. Monin, N. T. Duong, I. Z. Lopez, M. Bardet, V. Mareau, L. Gonon and G. De Paëpe, J. Am. Chem. Soc., 2014, 136, 13781–13788 CrossRef CAS PubMed.
- Z. Guo, T. Kobayashi, L.-L. Wang, T. W. Goh, C. Xiao, M. A. Caporini, M. Rosay, D. D. Johnson, M. Pruski and W. Huang, Chem. – Eur. J., 2014, 20, 16308–16313 CrossRef CAS PubMed.
- A. Lesage, M. Lelli, D. Gajan, M. A. Caporini, V. Vitzthum, P. Miéville, J. G. Alauzun, A. Roussey, C. Thieuleux, A. Mehdi, G. Bodenhausen, C. Coperet and L. Emsley, J. Am. Chem. Soc., 2010, 132, 15459–15461 CrossRef CAS PubMed.
- P. C. A. van der Wel, K.-N. Hu, J. Lewandowski and R. G. Griffin, J. Am. Chem. Soc., 2006, 128, 10840–10846 CrossRef CAS PubMed.
- A. J. Rossini, A. Zagdoun, F. Hegner, M. Schwarzwälder, D. Gajan, C. Copéret, A. Lesage and L. Emsley, J. Am. Chem. Soc., 2012, 134, 16899–16908 CrossRef CAS PubMed.
- A. J. Rossini, L. Emsley and L. A. O'Dell, Phys. Chem. Chem. Phys., 2014, 16, 12890–12899 RSC.
- K. Märker, M. Pingret, J. M. Mouesca, D. Gasparutto, S. Hediger and G. De Paëpe, J. Am. Chem. Soc., 2015, 137, 13796–13799 CrossRef PubMed.
- A. J. Rossini, J. Schlagnitweit, A. Lesage and L. Emsley, J. Magn. Reson., 2015, 259, 192–198 CrossRef CAS PubMed.
- G. Mollica, M. Dekhil, F. Ziarelli, P. Thureau and S. Viel, Angew. Chem., Int. Ed., 2015, 54, 6028–6031 CrossRef CAS PubMed.
- A. C. Pinon, A. J. Rossini, C. M. Widdifield, D. Gajan and L. Emsley, Mol. Pharmaceutics, 2015, 12, 4146–4153 CrossRef CAS PubMed.
- A. J. Rossini, C. M. Widdifield, A. Zagdoun, M. Lelli, M. Schwarzwälder, C. Copéret, A. Lesage and L. Emsley, J. Am. Chem. Soc., 2014, 136, 2324–2334 CrossRef CAS PubMed.
- T. Maly, L. B. Andreas, A. A. Smith and R. G. Griffin, Phys. Chem. Chem. Phys., 2010, 12, 5872–5878 RSC.
- V. K. Michaelis, E. Markhasin, E. Daviso, J. Herzfeld and R. G. Griffin, J. Phys. Chem. Lett., 2012, 3, 2030–2034 CrossRef CAS PubMed.
- A. J. Perez Linde, D. Carnevale, P. Miéville, A. Sienkiewicz and G. Bodenhausen, Magn. Reson. Chem., 2015, 53, 88–92 CrossRef CAS PubMed.
- D. Lee, H. Takahashi, A. S. L. Thankamony, J.-P. Dacquin, M. Bardet, O. Lafon and G. De Paëpe, J. Am. Chem. Soc., 2012, 134, 18491–18494 CrossRef CAS PubMed.
- M. Valla, A. J. Rossini, M. Caillot, C. Chizallet, P. Raybaud, M. Digne, A. Chaumonnot, A. Lesage, L. Emsley, J. A. van Bokhoven and C. Copéret, J. Am. Chem. Soc., 2015, 137, 10710–10719 CrossRef CAS PubMed.
- V. Vitzthum, P. Miéville, D. Carnevale, M. A. Caporini, D. Gajan, C. Copéret, M. Lelli, A. Zagdoun, A. J. Rossini, A. Lesage, L. Emsley and G. Bodenhausen, Chem. Commun., 2012, 48, 1988–1990 RSC.
- T. Kobayashi, F. A. Perras, T. W. Goh, T. L. Metz, W. Huang and M. Pruski, J. Phys. Chem. Lett., 2016, 7, 2322–2327 CrossRef CAS PubMed.
- K. R. Thurber and R. Tycko, J. Chem. Phys., 2012, 137, 084508 CrossRef PubMed.
- F. Mentink-Vigier, Ü. Akbey, Y. Hovav, S. Vega, H. Oschkinat and A. Feintuch, J. Magn. Reson., 2012, 224, 13–21 CrossRef CAS PubMed.
- S. R. Chaudhari, P. Berruyer, D. Gajan, C. Reiter, F. Engelke, D. L. Silverio, C. Copéret, M. Lelli, A. Lesage and L. Emsley, Phys. Chem. Chem. Phys., 2016, 18, 10616–10622 RSC.
- A. Jerschow, Prog. Nucl. Magn. Reson. Spectrosc., 2005, 46, 63–78 CrossRef CAS.
- H. Fuess, D. Hohlwein and S. A. Mason, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1977, 33, 654–659 CrossRef.
- D. Schollmeyer and M. Henk, CCDC 189162: Private communication to the Cambridge Structural Database, 2002, DOI:10.5517/cc6bv0c.
- H. S. Yathirajan, B. Nagaraj, R. S. Narasegowda, P. Nagaraja and M. Bolte, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2004, 60, o2228–o2229 CAS.
- R. Glaser and K. Maartmann-Moe, J. Chem. Soc., Perkin Trans. 1, 1990, 1205–1210 RSC.
- R. K. Harris, E. D. Becker, S. M. Cabral de Menezes, R. Goodfellow and P. Granger, Pure Appl. Chem., 2001, 73, 1795–1818 CrossRef CAS.
- O. Peersen, X. Wu and S. Smith, J. Magn. Reson., 1994, 106, 127–131 CrossRef CAS.
- O. Peersen, X. Wu, I. Kustanovich and S. O. Smith, J. Magn. Reson., 1993, 104, 334–339 CrossRef CAS.
- G. Metz, X. L. Wu and S. O. Smith, J. Magn. Reson., 1994, 110, 219–227 CrossRef CAS.
- B. M. Fung, A. K. Khitrin and K. Ermolaev, J. Magn. Reson., 2000, 142, 97–101 CrossRef CAS PubMed.
- J. A. Tang, J. D. Masuda, T. J. Boyle and R. W. Schurko, ChemPhysChem, 2006, 7, 117–130 CrossRef CAS PubMed.
- Z. Gan, J.-P. Amoureux and J. Trébosc, Chem. Phys. Lett., 2007, 435, 163–169 CrossRef CAS.
- B. Hu, J.-P. Amoureux, J. Trébosc and S. Hafner, J. Magn. Reson., 2008, 192, 8–16 CrossRef CAS PubMed.
- M. Bak, J. T. Rasmussen and N. C. Nielsen, J. Magn. Reson., 2000, 147, 296–330 CrossRef CAS PubMed.
- Z. Tošner, R. Andersen, B. Stevensson, M. Edén, N. C. Nielsen and T. Vosegaard, J. Magn. Reson., 2014, 246, 79–93 CrossRef PubMed.
- A. J. Vega, J. Magn. Reson., 1992, 96, 50–68 CAS.
- C. Martineau, J. Senker and F. Taulelle, Annu. Rep. NMR Spectrosc., 2014, vol. 82, pp. 1–57 Search PubMed.
- D. M. Pisklak, M. A. Zielińska-Pisklak, Ł. Szeleszczuk and I. Wawer, J. Pharm. Biomed. Anal., 2016, 122, 81–89 CrossRef CAS PubMed.
- D. M. Sperger and E. J. Munson, AAPS PharmSciTech, 2011, 12, 821–833 CrossRef CAS PubMed.
- W. R. Grüning, A. J. Rossini, A. Zagdoun, D. Gajan, A. Lesage, L. Emsley and C. Copéret, Phys. Chem. Chem. Phys., 2013, 15, 13270 RSC.
- D. L. Bryce and G. D. Sward, Magn. Reson. Chem., 2006, 44, 409–450 CrossRef CAS PubMed.
- K. E. Johnston, C. A. O'Keefe, R. M. Gauvin, J. Trébosc, L. Delevoye, J. P. Amoureux, N. Popoff, M. Taoufik, K. Oudatchin and R. W. Schurko, Chem. – Eur. J., 2013, 19, 12396–12414 CrossRef CAS PubMed.
- C. A. O’Keefe, K. E. Johnston, K. Sutter, J. Autschbach, L. Delevoye, N. Popo, M. Taou, K. Oudatchin and R. W. Schurko, Inorg. Chem., 2014, 53, 9581–9597 CrossRef PubMed.
- A. Zagdoun, A. J. Rossini, D. Gajan, A. Bourdolle, O. Ouari, M. Rosay, W. E. Maas, P. Tordo, M. Lelli, L. Emsley, A. Lesage and C. Coperet, Chem. Commun., 2012, 48, 654–656 RSC.
- M. Geppi, G. Mollica, S. Borsacchi and C. A. Veracini, Appl. Spectrosc. Rev., 2008, 43, 202–302 CrossRef CAS.
- R. T. Berendt, D. M. Sperger, E. J. Munson and P. K. Isbester, Trends Anal. Chem., 2006, 25, 977–984 CrossRef CAS.
- D. J. Kubicki, A. J. Rossini, A. Purea, A. Zagdoun, O. Ouari, P. Tordo, F. Engelke, A. Lesage and L. Emsley, J. Am. Chem. Soc., 2014, 136, 15711–15718 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6cp04353d |
| This journal is © the Owner Societies 2016 |