
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCytotoxic heteroleptic heptacoordinate salan zirconium(IV)-bis-chelates – synthesis, aqueous stability and X-ray structure analysis†
Fabian
Schneider‡
,
Tiankun
Zhao‡
and
Thomas
Huhn
*
Fachbereich Chemie and Konstanz Research School Chemical Biology, Universität Konstanz, Universitätsstr. 10, D-78457 Konstanz, Germany. E-mail: thomas.huhn@uni-konstanz.de
First published on 21st July 2016
Abstract
Herein we report the synthesis and structural characterization of a series of novel Zr(IV)salan complexes. The initial metalation product [(L1)2Zr] is highly water sensitive while ligand exchanged [L1Zr(dipic)] hydrolyses slowly with a bis-hydroxo Zr(IV) species identified by MS as an intermediate. [L1Zr(dipic)] is cytotoxic in the range of cisplatin against two human carcinoma cell lines.
Stimulated by the discovery of tumor-inhibiting effects of titanocene dichloride1 by Köpf and Köpf-Maier in 1979 other metallocene halides of V,2 Nb,3 Mo,4 W,3 Zr and Hf5 were screened for their anti-tumoral activity in short succession. During the following years research was mainly focused on the development of titanium based drugs while the heavier congener zirconium received considerably less attention. Propelled by the early success of titanocene dichloride other ligand systems like diketonates were investigated and the early development phase of group-IV metal based anti-tumor drugs finally climaxed in clinical trials of budotitane (Ti(bzac)2(OEt)2; bzac = phenylbutane-1,3-dione)6 and titanocene dichloride.7 Further clinical studies were discouraged mainly due to cardiotoxic side effects and formulation problems associated with premature hydrolysis of the complexes under physiological conditions.6b,8
Surprisingly, reports on anti-tumor active zirconium complexes are rather sparse and do not reflect the rich variety of ligand systems available for titanium. With the exception of zirconium 1,3-diketonates9 and an amino functionalized ionic zirconocene dichloride,10 no other structurally well-defined anti-tumor active zirconium complexes are known yet.
Salans are hydrogenated N,N′-bis(salicylidene)ethylenediamine (salen) ligands with [ONNO]-binding motif and variable substitution pattern at nitrogen.11 They are known to form zirconium bis(alkoxides) which are intensely investigated as catalysts in the polymerization of olefins12 and lactide.13 We recently found the functionalization of titanium(IV) salan alkoxides14 with the dianionic tridentate pyridine-2,6-dicarboxylic acid (dipic) is leading to rigid heptacoordinate Ti(IV)-complexes.15 These hetero-bis-chelates are stable in cell-medium, highly toxic in vitro against human carcinoma cell lines and have strong in vivo antitumor efficacy in a mouse cervical-cancer model.15a,16
Based on the above considerations we thus became interested to explore the utilization of our mixed ligand system on zirconium alkoxides to study stability and potential cytotoxicity of the resulting complexes. Herein we present the first synthesis and preliminary bio evaluation of three novel heptacoordinate salan Zr(IV) bis-chelates.
The synthesis of salan zirconium(IV)-bis-chelates [L1–L3Zr(dipic)] is summarized in Scheme 1 and started from readily available ligands H2L1–H2L3.17 Metalation with Zr(OEt)4 in a molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 at 50 °C resulted in the formation of two structurally different zirconium(IV) salan intermediates depending on the type of salan used. While the less bulky H2L1 and H2L2 gave exclusively the homoleptic bis-salan Zr(IV) complexes [(L1)2Zr] and [(L2)2Zr], metalation of H2L3 resulted in the formation of the expected heteroleptic salan–zirconium-bis-alkoxo complex [L3Zr(OEt)2].13a All three were formed in nearly quantitative yield as evident from 1H-NMR spectra recorded from the crude material. The propensity of H2L1,2 to form homoleptic bis-salan complexes might be rationalized by the smaller steric bulk of the methyl group in H2L1,2 compared to the tert-butyl group in H2L3ortho to the phenolic oxygen. Hence, H2L1 and H2L2 permit tighter packing of the ligands in the coordination sphere of the zirconium(IV) ion resulting in exclusive formation of the octacoordinate bis-salans [(L1)2Zr] and [(L2)2Zr], whereas in case of H2L3 instead of a second salan only the small ethoxide is tolerated. Even with an excess of free salan present, [L3Zr(OEt)2] remains the sole reaction product. Ligand exchange to form dipic-stabilized heptacoordinate complexes of type [LZr(dipic)] was initiated by the addition of 1.1 eq. pyridine-2,6-dicarboxylic acid to the corresponding intermediate, i.e. to [(L1)2Zr], [(L2)2Zr] or [L3Zr(OEt)2] dissolved in anhydrous THF was added dipicolinic acid and the reaction mixture stirred at 50 °C until total consumption of starting material was evident by NMR. Corresponding zirconium(IV)-bis-chelates [L1-3Zr(dipic)] were isolated as yellow solids in pure state after filtration from excess dipic. When starting from the homoleptic complexes an additional washing with toluene was necessary to remove liberated salans H2L1 and H2L2, respectively.
:1 at 50 °C resulted in the formation of two structurally different zirconium(IV) salan intermediates depending on the type of salan used. While the less bulky H2L1 and H2L2 gave exclusively the homoleptic bis-salan Zr(IV) complexes [(L1)2Zr] and [(L2)2Zr], metalation of H2L3 resulted in the formation of the expected heteroleptic salan–zirconium-bis-alkoxo complex [L3Zr(OEt)2].13a All three were formed in nearly quantitative yield as evident from 1H-NMR spectra recorded from the crude material. The propensity of H2L1,2 to form homoleptic bis-salan complexes might be rationalized by the smaller steric bulk of the methyl group in H2L1,2 compared to the tert-butyl group in H2L3ortho to the phenolic oxygen. Hence, H2L1 and H2L2 permit tighter packing of the ligands in the coordination sphere of the zirconium(IV) ion resulting in exclusive formation of the octacoordinate bis-salans [(L1)2Zr] and [(L2)2Zr], whereas in case of H2L3 instead of a second salan only the small ethoxide is tolerated. Even with an excess of free salan present, [L3Zr(OEt)2] remains the sole reaction product. Ligand exchange to form dipic-stabilized heptacoordinate complexes of type [LZr(dipic)] was initiated by the addition of 1.1 eq. pyridine-2,6-dicarboxylic acid to the corresponding intermediate, i.e. to [(L1)2Zr], [(L2)2Zr] or [L3Zr(OEt)2] dissolved in anhydrous THF was added dipicolinic acid and the reaction mixture stirred at 50 °C until total consumption of starting material was evident by NMR. Corresponding zirconium(IV)-bis-chelates [L1-3Zr(dipic)] were isolated as yellow solids in pure state after filtration from excess dipic. When starting from the homoleptic complexes an additional washing with toluene was necessary to remove liberated salans H2L1 and H2L2, respectively.
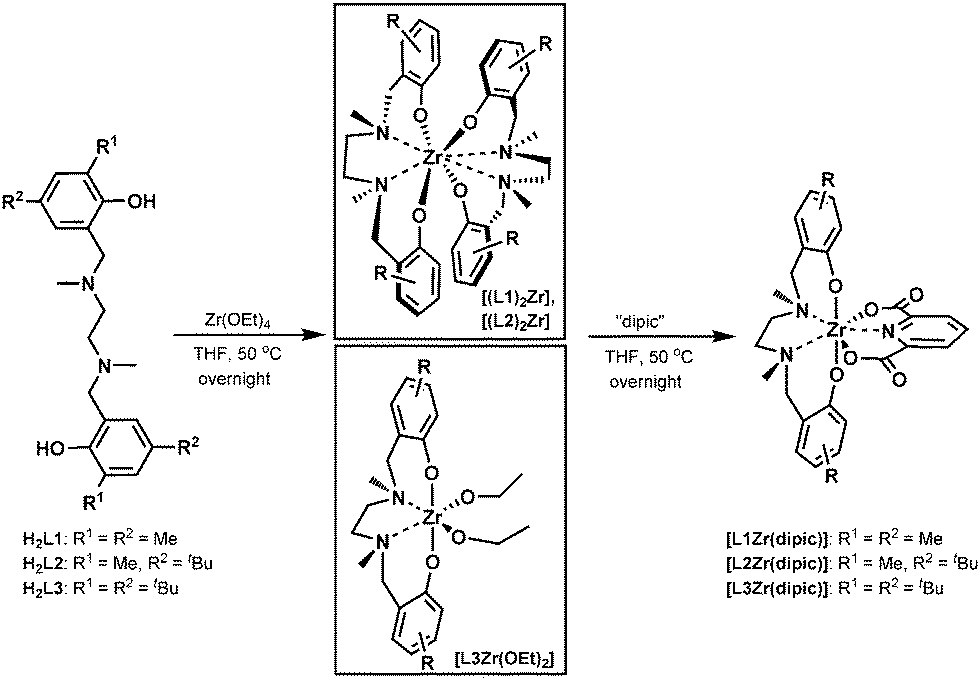 | ||
| Scheme 1 Synthesis of salan Zr(IV) bis-chelates [L1-3Zr(dipic)]. | ||
The highly symmetric disposition of the salan ligands around the zirconium centre of the homoleptic octacoordinate [(L1)2Zr] and [(L2)2Zr] is nicely reflected in their 1H-NMR spectra with a single set of signals demonstrating the magnetic equivalence of both ligands. Besides two pairs of doublets representing the diastereotopic benzylic and bridgehead CH2, respectively, three different methyl groups and two singlets of the Haryl are found. The NMR interpretation was confirmed by X-ray diffraction study, suitable single crystals were grown by slow diffusion of hexane into a saturated solution of [(L1)2Zr] in DCM (Fig. 1 and Table 1). [(L1)2Zr] crystallizes in the monoclinic space group P21/n with an additional molecule of solvent found in the asymmetric unit. The (ONNO)2 binding motif is quite uncommon for group IV complexes and besides two N,N′-bis(salicylidene)alkylenediamine (salen) complexes,18 only two bis(salan) complexes are known.19[(L1)2Zr] differs from these by its methyl-substitution at the nitrogen; while the former are originating from secondary amines (salanH4), the latter is derived from a tertiary amine (salanH2). With an average Zr–N distance of 2.609(2) Å in [(L1)2Zr], the metal protrudes out of the salan binding sphere compared to the two characterized zirconium bis(salan) complexes (Zr–N distance ∼ 2.44–2.45 Å) where both ligands are pulled tighter together. This structural feature becomes more manifest when comparing the O–Zr–O angles of [(L1)2Zr] with above bis(salan) complexes.
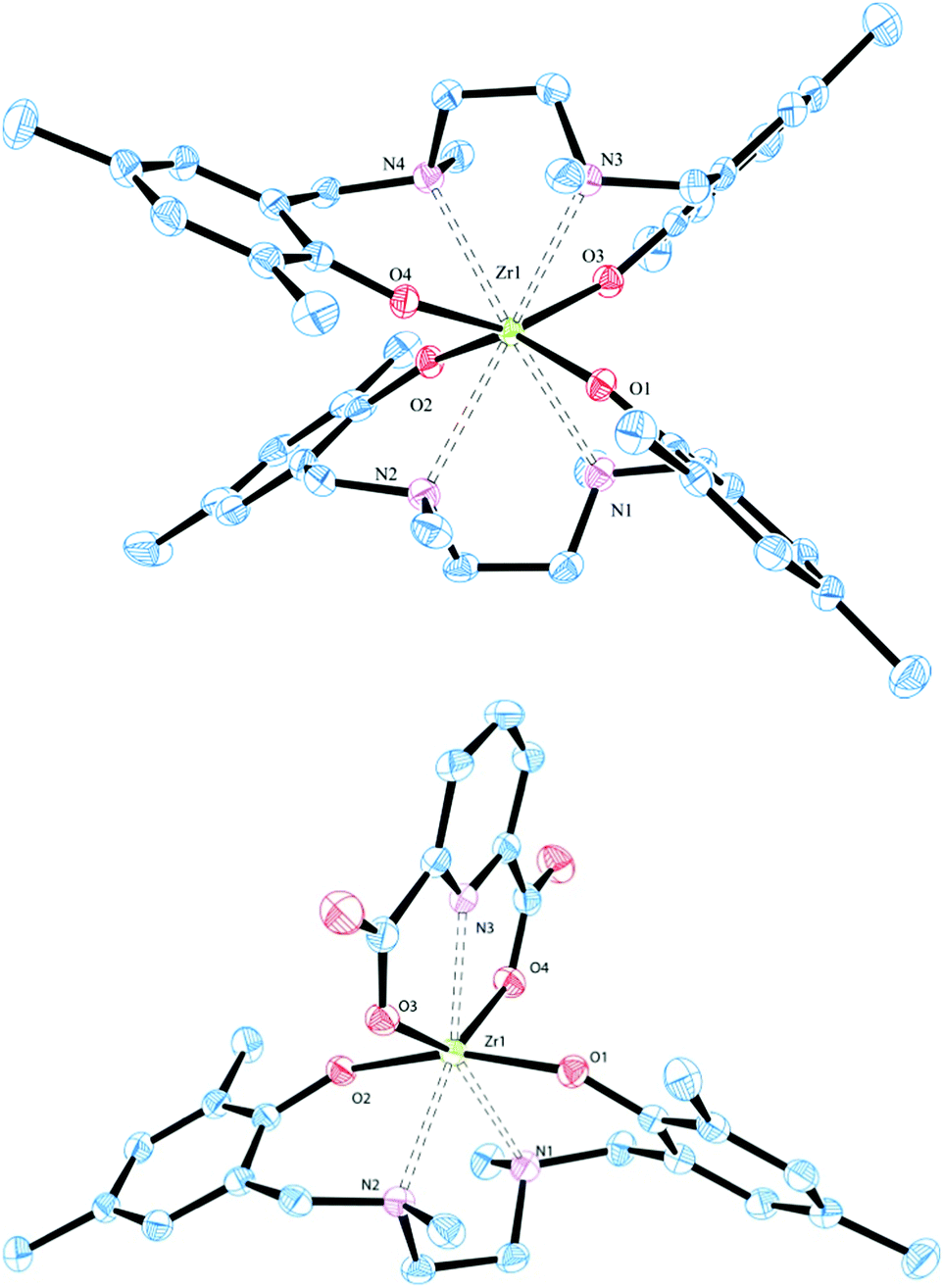 | ||
| Fig. 1 Solid state molecular structures of approximate C2 symmetric octacoordinate homoleptic [(L1)2Zr] (top) and heptacoordinate heteroleptic [L1Zr(dipic)] (bottom). Thermal ellipsoids are drawn at the 50% probability level. Hydrogen atoms are omitted for clarity. | ||
| Complex | HeLa S3a | Hep G2a |
|---|---|---|
| IC50a [μM] | ||
| a 48 h of incubation. | ||
| [L1Zr(dipic)] | 2.7 ± 0.3 | 3.6 ± 0.5 |
| [L1Ti(dipic)] 15a | 4.4 ± 0.4 | 3.4 ± 0.3 |
| [L2Zr(dipic)] | 32.7 ± 6 | 47 ± 9 |
| [L3Zr(dipic)] | Non toxic | Non toxic |
| [(L1)2Zr] | Non toxic | Non toxic |
| [(L2)2Zr] | Non toxic | Non toxic |
| [L3Zr(OEt)2] | 62.3 ± 6 | 60.2 ± 6 |
| Cisplatin | 3.3 ± 0.2 | 6.0 ± 1.1 |
The O–Zr–O angle (average 147.12(8)°) of [(L1)2Zr] is much wider than in the two comparable complexes (average 139.9(1)°) while the N–Zr–N angle (average 67.48(7)°) of [(L1)2Zr] is more acute than in above bis(salan) complexes (average 70.4(1)°).19 This situation results from the steric repulsion of the tertiary amines in [(L1)2Zr] which forces both salan ligands in a zig–zag like conformation. This contrasts with the binding geometry in bis(salan) and bis(salen) complexes where the disposition of the ligands resembles two interpenetrating planes.18,19
The structures of [L1-3Zr(dipic)] were confirmed by 1H-NMR analysis and resembles closely those found for their titanium congeners with two pairs of doublets, each representing a pair of diastereotopic benzylic and bridgehead protons.15,16b Except for a slight upfield shift for the benzylic protons, the spectra of [L1Zr(dipic)] and [L1Ti(dipic)] are nearly indistinguishable (see Fig. S23, ESI†). The structure was further proven by X-ray diffraction study of suitable single crystals of [L1Zr(dipic)] grown by slow diffusion of hexane in a saturated solution of complex in DCM. [L1Zr(dipic)] crystallizes in the monoclinic space group C2/c with one molecule in the asymmetric unit. With respect to the spatial arrangement around the zirconium centre, [L1Zr(dipic)] compares extremely well with its titanium congener.15a Dipicolinic acid acts as bisanionic tridentate coordinating to the zirconium–salan fragment via both carboxylates (average Zr–O 2.151 Å) and the pyridine N (Zr–N3 2.293(1) Å). The nitrogen atoms N1, N2 (average Zr–N 2.449 Å) of the salan complement the base of the pentagonal bipyramidal coordination sphere. The phenolic oxygen atoms O1 and O2 are occupying the axial positions in an average distance of Zr–O 1.991 Å.
While the Zr–N distance of 2.293 Å observed for [L1Zr(dipic)] is in good agreement with 2.180–2.186 Å found in zirconium dipicolinates of the bis(cyclopentadienyl)20 and bis(indenyl)20a type, the Zr–O distance in [L1Zr(dipic)] is slightly shorter compared to values observed for above metallocenes (2.180–2.186 Å). As a consequence the pyridine is tilted by 18.40° out of the plane defined by the carboxylate O atoms and Zr. Comparison of the salans bond distances to Zr in [L1Zr(dipic)] and its precursor [(L1)2Zr] reveal slightly longer Zr–O distances of 2.040–2.054 Å of the homoleptic complex over the heteroleptic one with Zr–O distances of 1.989–1.992 Å. A similar trend was observed for the Zr–N distances with 2.592–2.627 Å for [(L1)2Zr] and 2.439–2.459 Å for [L1Zr(dipic)]. A side by side comparison of bond lengths and angles of [(L1)2Zr] and [L1Zr(dipic)] is given in Table S15 (ESI†).
The kinetics of hydrolysis of selected complexes was studied by time resolved 1H-NMR16b and time resolved UV-spectra for selected dipic-complexes. Upon addition of 1.000 eq. of D2O to 15 μmol of the bulky bis-tert-butyl-substituted [L3Zr(OEt)2] in [D8]-THF, ethanol is liberated within seconds accompanied by the precipitation of presumably polynuclear species. Upon longer incubation time signals of free salan become apparent indicating a slow release of salan from the precipitate. [(L1)2Zr], the less hindered of the homoleptic complexes shows a rather similar behavior. Upon addition of D2O the complex decomposes within seconds with concomitant precipitation and slow release of H2L1.
In contrast to the rather sensitive complexes above, stability of the dipic-stabilized heptacoordinate [L1Zr(dipic)] could be assessed by UV-vis spectroscopy in the concentration range of 10−5 mol l−1 utilizing larger amounts of water. Consequently, UV-vis spectra were recorded after the addition of 100.000 eq. of water in intervals of 1 hour. During the measurement period of 140 h a slow decrease of λmax at 250 nm with an overall decrease of absorption over the whole spectral range was observed. The decay followed a rate law of higher order which prevented the exact determination of t1/2. Nevertheless, when exposed to such a large excess of water, the half-life of [L1Zr(dipic)] is still in the range of several hours and outperforms the other Zr-complexes under study by several orders of magnitude (Fig. S24, ESI†). To explore the possible endpoint of hydrolysis, we exposed [L1Zr(dipic)] to neat water for prolonged time. To facilitate reaction of the suspension, the mixture was subjected to ultrasound irradiation every 24 hours for 30 min during the first 4 days. Under these conditions a 50:50 mixture of [L1Zr(dipic)] and free salan was detected after 24 h. Longer exposure led to complete liberation of the salan ligand with only minute quantities of intact [L1Zr(dipic)] left.
To identify possible intermediates of the hydrolysis, a defined amount of [L1Zr(dipic)] was incubated with a range of water concentrations spanning three orders of magnitude. Products were analyzed by means of 1H-NMR and LDI-TOF mass spectroscopy. To this end, 30 mg of complex were dissolved in 30 ml THF and 5, 10, 100 and 1.000 eq. water were added. After continuous stirring at r.t. for 6 days the solvent was removed and the remainder taken up in CDCl3. While the lowest water concentrations (5–100 eq.) only led to limited hydrolysis, the experiment with 1.000 eq. of water showed considerable formation of hydrolysis products. Besides the starting complex and free salan ligand, an apparent highly symmetric new species showing the typical 1H-NMR pattern of a salan-complex was identified. The three constituents were found in a 1:1.4:2 ratio (Fig. S25, ESI†). The newly formed complex was identified directly from the NMR-sample as the monomeric bishydroxo zirconium salan [L1Zr(OH)2] by LDI-TOF mass spectroscopy (Fig. S26, ESI†). Similar mono-molecular Zr-hydroxo-species were previously isolated and characterized.21 Apparently, the bishydroxo species is a transient intermediate in the hydrolysis of [L1Zr(dipic)] since it was not detected in experiments applying higher water concentrations as described above.
All Zr(IV) complexes were preliminary tested for their cytotoxic activity utilizing an AlamarBlue-based proliferation assay (details see ESI†).22 The cytotoxicity was studied in two different cell lines: the human cervix carcinoma cell line HeLa S3 and the Hep G2 cell line, a human hepatocarcinoma cell line with epithelial morphology. Cisplatin was used as a reference (IC50 HeLa S3: 3.3 ± 0.2 μM; Hep G2: 6.0 ± 1.1 μM). Results are summarized in Table 1. [L3Zr(OEt)2] is the only octahedral complex found cytotoxic, albeit with low activity. Because its titanium homologue is known to be non-active in the same cell lines, as is the free ligand, it might act via a different pathway.14d From the heptacoordinate complexes [L1Zr(dipic)] showed the best bioactivity with IC50 values of 2.7 ± 0.3 μM against HeLa S3 and 3.6 ± 0.5 μM against Hep G2 cell lines. This is comparable to the values determined for cisplatin in both cell lines (Table 1, entries 1 and 8). Even though [L1Zr(dipic)] is less robust in aqueous media compared with its titanium homologue [L1Ti(dipic)] its cytotoxicity is on par (Table 1, entry 2).15a Likewise, a heptacoordinate Hf(IV) tris β-diketonato chloro complex was recently found to be cytotoxic against HT-29 and MCF-7 in the range of cisplatin and of higher activity than similar Ti and Zr-complexes.9b This might indicate an efficient cellular uptake mechanism for the heavier group IV-complexes.
The sterically more demanding [L2Zr(dipic)] showed about 10 times lower cytotoxicity than [L1Zr(dipic)] (entry 2). Enhancing the sterical demand even further like in [L3Zr(dipic)] with 2,4-di-tert-butyl substitution on the salan backbone, led to a complete loss of cytotoxicity in both cell lines. A systematic decrease in cytotoxic activity with increased sterical demand was previously reported for octahedral salan Ti(IV) bisalkoxides.14d
In summary, we have synthesized three members of a new type of heptacoordinate salan dipic Zr(IV)-bis-chelate. Their structures were elucidated by NMR and X-ray data analysis. Preliminary in vitro biological evaluation against two human cell lines reveals that heptacoordinate [L1Zr(dipic)] exhibits excellent anticancer activity in the range of cisplatin. The lack of labile ligands suggests a different mode of action to that of previously reported titanium salan bisalkoxides. In contrast to the non-active complexes, [L1Zr(dipic)] exhibits improved aqueous stability with a t1/2 in the range of several hours. However, during long term exposure of [L1Zr(dipic)] to varying amounts of water a bishydroxo Zr(IV) complex was identified by mass and 1H-NMR spectroscopy as the major intermediate of hydrolysis. Further studies towards the identification of other stabilizing ligand systems and their application in the development of zirconium based antitumoral drugs are ongoing work.
We thank the Konstanz Research School Chemical Biology (KoRS-CB) for financial and scientific support. T. Z. is grateful to the KoRS-CB for providing a personal scholarship. The authors would also like to thank Dipl.-chem. Malin Bein for help with the biological assays, Dr Inigo Göttker-Schnetmann and MSc Fabian Geist for assistance with the X-ray measurements. The support by Umicore through a generous gift of cisplatin is gratefully acknowledged.
Notes and references
- H. Köpf and P. Köpf-Maier, Angew. Chem., Int. Ed. Engl., 1979, 18, 477–478 CrossRef PubMed.
- P. Köpf-Maier and H. Köpf, Z. Naturforsch., B: Chem. Sci., 1979, 34B, 805–807 Search PubMed.
- P. Köpf-Maier, M. Leitner and H. Köpf, J. Inorg. Nucl. Chem., 1980, 42, 1789–1791 CrossRef.
- P. Köpf-Maier, M. Leitner, R. Voigtländer and H. Köpf, Z. Naturforsch., C: Biosci., 1979, 34C, 1174–1176 Search PubMed.
- P. Köpf-Maier, B. Hesse and H. Köpf, J. Cancer Res. Clin. Oncol., 1980, 96, 43–51 CrossRef.
- (a) H. Bischoff, M. R. Berger, B. K. Keppler and D. Schmähl, J. Cancer Res. Clin. Oncol., 1987, 113, 446–450 CrossRef CAS PubMed; (b) T. Schilling, K. B. Keppler, M. E. Heim, G. Niebch, H. Dietzfelbinger, J. Rastetter and A. R. Hanauske, Invest. New Drugs, 1995, 13, 327–332 CrossRef CAS.
- (a) C. V. Christodoulou, D. R. Ferry, D. W. Fyfe, A. Young, J. Doran, T. M. Sheehan, A. Eliopoulos, K. Hale, J. Baumgart, G. Sass and D. J. Kerr, J. Clin. Oncol., 1998, 16, 2761–2769 CAS; (b) A. Korfel, M. E. Scheulen, H. J. Schmoll, O. Gründel, A. Harstrick, M. Knoche, L. M. Fels, M. Skorzec, F. Bach, J. Baumgart, G. Sass, S. Seeber, E. Thiel and W. E. Berdel, Clin. Cancer Res., 1998, 4, 2701–2708 CAS; (c) G. Lümmen, H. Sperling, H. Luboldt, T. Otto and H. Rübben, Cancer Chemother. Pharmacol., 1998, 42, 415–417 CrossRef PubMed; (d) N. Kröger, U. R. Kleeberg, K. Mross, L. Edler and D. K. Hossfeld, Onkologie, 2000, 23, 60–62 Search PubMed.
- (a) J. H. Toney and T. J. Marks, J. Am. Chem. Soc., 1985, 107, 947–953 CrossRef CAS; (b) X. Chen and L. Zhou, J. Mol. Struct., 2010, 940, 45–49 CrossRef CAS.
- (a) B. K. Keppler and K. Michels, Arzneim. Forsch., 1985, 35, 1837–1839 CAS; (b) R. M. Lord, J. J. Mannion, A. J. Hebden, A. E. Nako, B. D. Crossley, M. W. McMullon, F. D. Janeway, R. M. Phillips and P. C. McGowan, ChemMedChem, 2014, 9, 1136–1139 CrossRef CAS PubMed.
- O. R. Allen, R. J. Knox and P. C. McGowan, Dalton Trans., 2008, 5293–5295 RSC.
- D. A. Atwood, Coord. Chem. Rev., 1997, 165, 267–296 CrossRef CAS.
- (a) A. Cohen, A. Yeori, J. Kopilov, I. Goldberg and M. Kol, Chem. Commun., 2008, 2149–2151 RSC; (b) A. Cohen, J. Kopilov, I. Goldberg and M. Kol, Organometallics, 2009, 28, 1391–1405 CrossRef CAS; (c) E. Sergeeva, J. Kopilov, I. Goldberg and M. Kol, Chem. Commun., 2009, 3053–3055 RSC.
- (a) S. Gendler, S. Segal, I. Goldberg, Z. Goldschmidt and M. Kol, Inorg. Chem., 2006, 45, 4783–4790 CrossRef CAS PubMed; (b) M. D. Jones, S. L. Hancock, P. McKeown, P. M. Schafer, A. Buchard, L. H. Thomas, M. F. Mahon and J. P. Lowe, Chem. Commun., 2014, 50, 15967–15970 RSC; (c) S. L. Hancock, M. F. Mahon, G. Kociok-Kőhn and M. D. Jones, Eur. J. Inorg. Chem., 2011, 4596–4602 CrossRef CAS.
- (a) M. Shavit, D. Peri, C. M. Manna, J. S. Alexander and E. Y. Tshuva, J. Am. Chem. Soc., 2007, 129, 12098–12099 CrossRef CAS PubMed; (b) D. Peri, S. Meker, M. Shavit and E. Y. Tshuva, Chem. – Eur. J., 2009, 15, 2403–2415 CrossRef CAS PubMed; (c) H. Glasner and E. Y. Tshuva, Inorg. Chem., 2014, 53, 3170–3176 CrossRef CAS PubMed; (d) T. A. Immel, U. Groth and T. Huhn, Chem. – Eur. J., 2010, 16, 2775–2789 CrossRef CAS PubMed.
- (a) T. A. Immel, M. Grützke, A. K. Späte, U. Groth, P. Öhlschläger and T. Huhn, Chem. Commun., 2012, 48, 5790–5792 RSC; (b) M. Grützke, T. Zhao, T. A. Immel and T. Huhn, Inorg. Chem., 2015, 54, 6697–6706 CrossRef PubMed.
- (a) T. A. Immel, U. Groth, T. Huhn and P. Öhlschläger, PLoS One, 2011, 6, e17869 CAS; (b) T. Zhao, M. Grützke, K. H. Götz, T. Druzhenko and T. Huhn, Dalton Trans., 2015, 44, 16475–16485 RSC.
- (a) E. Y. Tshuva, N. Gendeziuk and M. Kol, Tetrahedron Lett., 2001, 42, 6405–6407 CrossRef CAS; (b) S. H. Kim, J. Lee, D. J. Kim, J. H. Moon, S. Yoon, H. J. Oh, Y. Do, Y. S. Ko, J.-H. Yim and Y. Kim, J. Organomet. Chem., 2009, 694, 3409–3417 CrossRef CAS; (c) J. Balsells, P. J. Carroll and P. J. Walsh, Inorg. Chem., 2001, 40, 5568–5574 CrossRef CAS PubMed.
- (a) M. L. Illingsworth, B. P. Cleary, A. J. Jensen, L. J. Schwartz and A. L. Rheingold, Inorg. Chim. Acta, 1993, 207, 147–163 CrossRef CAS; (b) H.-J. Zhu, M. Wang, K. Jin, D. Dai, L.-C. Sun and C.-N. Chen, Transition Met. Chem., 2005, 30, 517–522 CrossRef CAS.
- (a) T. Shalumova and J. M. Tanski, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2008, 64, m1029–m1030 CAS; (b) M. D. Jones, M. G. Davidson and G. Kociok-Kohn, Polyhedron, 2010, 29, 697–700 CrossRef CAS.
- (a) U. Niemann, J. Diebold, C. Troll, U. Rief and H.-H. Brintzinger, J. Organomet. Chem., 1993, 456, 195–204 CrossRef CAS; (b) T. C. Stamatatos, S. P. Perlepes, C. P. Raptopoulou, V. Psycharis and N. Klouras, Polyhedron, 2011, 30, 451–457 CrossRef CAS.
- (a) R. Bortolin, V. Patel, I. Munday, N. J. Taylor and A. J. Carty, J. Chem. Soc., Chem. Commun., 1985, 456–458 RSC; (b) M. Kessler, S. Hansen, C. Godemann, A. Spannenberg and T. Beweries, Chem. – Eur. J., 2013, 19, 6350–6357 CrossRef CAS PubMed; (c) P.-M. Pellny, V. V. Burlakov, W. Baumann, A. Spannenberg and U. Rosenthal, Z. Anorg. Allg. Chem., 1999, 625, 910–918 CrossRef CAS.
- R. Hamid, Y. Rotshteyn, L. Rabadi, R. Parikh and P. Bullock, Toxicol. In Vitro, 2004, 18, 703–710 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details of synthesis, cell viability assays and hydrolysis study as well as NMR, UV and MS spectra. CCDC 1439012 and 1439014. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6cc05359a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2016 |