
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Identification of genes encoding squalestatin S1 biosynthesis and in vitro production of new squalestatin analogues†
B.
Bonsch
a,
V.
Belt
a,
C.
Bartel
a,
N.
Duensing
a,
M.
Koziol
b,
C. M.
Lazarus
b,
A. M.
Bailey
b,
T. J.
Simpson
c and
R. J.
Cox
*ac
aInstitut für Organische Chemie, Leibniz Universität Hannover, Schneiderberg 1b, 30167 Hannover, Germany. E-mail: russell.cox@oci.uni-hannover.de
bSchool of Biological Sciences, 24 Tyndall Avenue, Bristol BS8 1TQ, UK
cSchool of Chemistry, University of Bristol, Cantock's Close, Bristol, BS8 1TS, UK
First published on 1st April 2016
Abstract
A gene cluster responsible for the biosynthesis of squalestatin S1 (SQS1, 1) was identified by full genome sequencing of two SQS1-producing ascomycetes: Phoma sp. C2932 and unidentified fungus MF5453. A transformation protocol was established and a subsequent knockout of one PKS gene from the cluster led to loss of SQS1 production and enhanced concentration of an SQS1 precursor. An acyltransferase gene from the cluster was expressed in E. coli and the expressed protein MfM4 shown to be responsible for loading acyl groups from CoA onto the squalestatin core as the final step of biosynthesis. MfM4 appears to have a broad substrate selectivity for its acyl CoA substrate, allowing the in vitro synthesis of novel squalestatins.
Squalestatin S1 (SQS1, also known as zaragozic acid A) 1 was independently discovered by Glaxo1 and Merck2 in the early 1990s as a novel lead compound for the treatment of hypercholesterolemia by targeting squalene synthase (SS). SQS1 1 and related compounds are potent pM inhibitors of mammalian and fungal SS.3 SQS1 1 correspondingly shows broad spectrum antifungal properties and lowers the blood cholesterol level of rhesus-monkeys in vivo.4
Numerous different squalestatins have been isolated from more than ten taxa of filamentous fungi, mostly varying in the attached 1-alkyl and 6-O-acyl side chains.5 However the class is distinguished by the highly functionalised 4,8-dioxa-bicyclo[3.2.1]octane core. Feeding experiments using labelled precursors identified the origin of the heavy atoms of 1 (Fig. 1).6,7 The biosynthesis involves the production of two polyketides: a hexaketide initiated by benzoate; and a tetraketide produced by a highly reducing fungal polyketide synthase (hrPKS).8 The remaining carbon atoms are derived from a citric acid cycle intermediate (such as oxaloacetate).
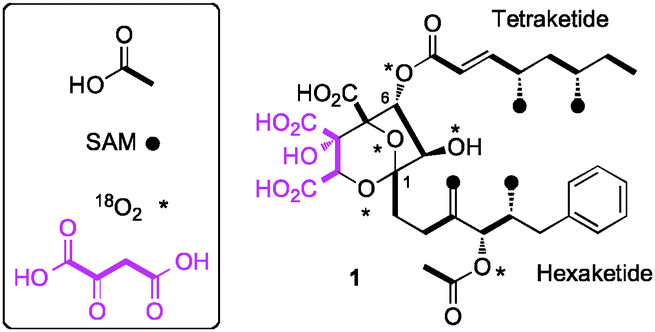 | ||
| Fig. 1 Origin of heavy atoms in SQS1 1. | ||
We previously reported the isolation and sequence of the gene (phpks1) encoding squalestatin tetraketide synthase (SQTKS).9 Expression of phpks1 in Aspergillus oryzae resulted in the isolation of the tetraketide chain of SQS1 from liquid culture.8 Since no other molecular information is available for squalestatin biosynthesis, we undertook to generate more information via full genome sequencing.
Two fungal strains have been previously identified as producers of 1: Phoma sp. C2932 (Glaxo); and unidentified strain MF5453 (Merck). Initial sequencing of internal transcribed spacer (ITS) sequences obtained by PCR showed the two strains to be closely related (see ESI†).
Genomic DNA from both organisms was sequenced and assembled to give two high quality draft genomes (see ESI†). A putative SQS1 gene cluster was identified in both organisms by comparison of the assembled sequences to the previously characterised phpks1 gene (Fig. 2).10
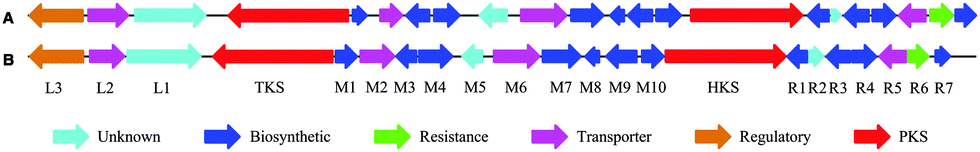 | ||
| Fig. 2 SQS1 genomic region from A, Phoma sp. C2932 and B, MF5453: L, left hand side of pks1; M, between pks1 and pks2; R, right hand side of pks2. | ||
In order to more clearly link the clusters with the biosynthesis of 1, and delineate the boundaries of the cotranscribed genes, MF5453 was grown under producing and non-producing conditions (see ESI†). mRNA prepared under these conditions was used for quantitative reverse transcription polymerase chain reaction (qRT-PCR). The expression level of all genes from mfpks1 to mfR6 was strongly linked to the production of SQS1 1 in the culture medium (Fig. 3), being between 102 and 105 fold more highly expressed under producing conditions. This suggests that the pks1 and R6 genes form the boundaries of a cotranscribed region.
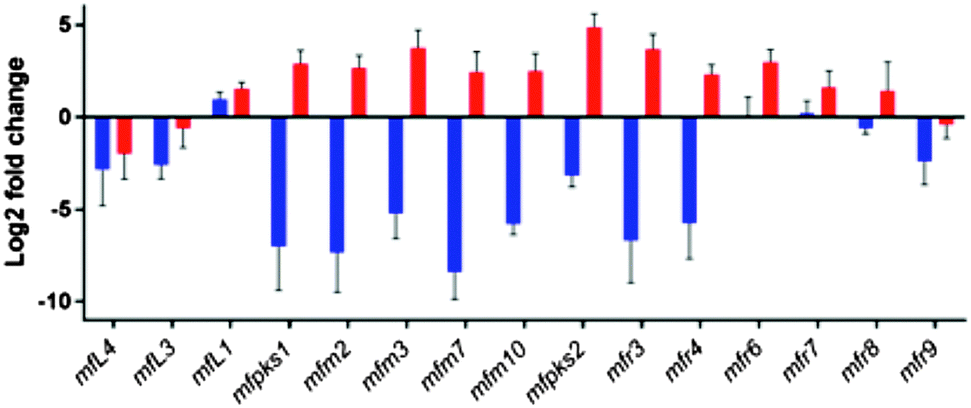 | ||
| Fig. 3 Relative gene expression levels of selected genes in the SQS1 cluster of MF5453 under producing (red) and non-producing (blue) fermentation conditions measured using qRT-PCR, average of four nonproducing and five producing biological replicates. | ||
A comparison of the SQS1 clusters from MF5453 and C2932 shows a highly similar composition of genes and a high identity/similarity (74–93%/82–97%) between proteins with the same predicted activity (see ESI†).11 A search of the NCBI database using sequences from these clusters revealed a third putative squalestatin cluster in Dothistroma septosporum. Comparisons (see ESI†) show that all genes between pks1 and R6 are conserved in all 3 clusters, suggesting these may be the limits of the biosynthetic gene cluster.
Both phenylalanine and benzoic acid are known6 precursors of 1 and so it is unsurprising that the cluster also contains genes potentially involved in benzoate production: phenylalanine ammonia lysase (PAL), encoded by M7, catalyses the first step in the degradation of phenylalanine (Scheme 3). In other microbial and plant systems12 cinnamate is hydrated, oxidised and then subject to a retro-Claisen reaction during the synthesis of benzoyl CoA, and the NADP-dependent dehydrogenase M3 may also be involved in this.
The cluster contains two PKS encoding genes: both PKS belong to the highly reducing class.13 The tetraketide synthase, has already been shown to be responsible for the biosynthesis of the tetraketide sidechain of SQS1 1.8,14 The other is therefore likely to encode squalestatin hexaketide synthase (SQHKS). The hexaketide main chain is initiated by benzoate (Scheme 3, vide infra) which is an unusual starter unit for hrPKS and comparison of the sequences of the putative hexaketide synthase AT domains with AT domain from hrPKS which select acetate as the starter unit, show consistent substitutions in residues adjacent to the AT active site (see ESI†). The cluster also contains a gene encoding a citrate synthase-like protein (encoded by R3) linking the biosynthetic pathway to those recently reported to be involved in maleidride biosynthesis in fungi15,16 and presumably involved in linking the hexaketide to the oxaloacetate moiety (Scheme 3). The biosynthesis of 1 requires several oxidative steps (Scheme 3). Based on sequence comparisons alone (e.g. BLAST) no encoded protein shows significant homology to known oxygenases. However M1 shows modest structural homology (e.g. PHYRE2)17 to copper dependent peptidylglycine monooxygenases and R1 and R2 show some structural homology to non-heme iron dependent enzymes.
Two putative acyltransferases (AT) are encoded in the cluster. One, encoded by mfM4, shows high sequence homology (20% identity, 37% similarity) to tricothocene-3-O-acetyltransferase (Tri101) from Fusarium species with which it shares the key active site residues H163 and D167.18 Residues involved in CoA binding identified from the crystal structure of Tri101,18 are also conserved in MfM4, including L-282, T-298, D-300, and L-425 (see ESI†). The other AT, encoded by R4, shows low homology to the membrane-spanning acyltransferase-3 superfamily including proteins such as OaC from Shigella flexneri which encodes an O-acetyl transferase involved in serotype development (see ESI†).19
Finally, in support of the identification of the clusters as being responsible for 1 production, all three contain a gene encoding a putative SS (R6), presumably encoding a resistance protein. The genomes of Phoma C2932, MF5453 and D. septosporum also contain housekeeping SS genes. Multiple alignment of these sequences with human SS,20 shows that some identified substrate binding residues of human SS are substituted (e.g. L186V and C299V) in the resistance proteins but not the housekeeping proteins suggesting a likely mechanism for self-resistance (see ESI†).
Neither Phoma sp. C2932 or MF5453 have been reported to undergo genetic transformation. A PEG based transformation protocol using protoplasts for MF5453 and C2932 was therefore established, based on hygromycin selection with an auxiliary egfp marker gene. Establishment of the transformation protocol was confirmed by observation of green fluorescent mycelia of hygromycin-resistant MF5453 transformants (see ESI†).
The phpks1 gene in MF5453 was then knocked out21 which resulted in the creation of ten hygromycin resistant transformants. Three of these were analysed by PCR and shown to have incorporated the hygromycin resistance gene and the expected lesion in phpks1. The LC-MS traces of one MF5453 knockout transformant, grown under 1-producing conditions, were compared to the wild type (WT) strain and different standards (Fig. 4).
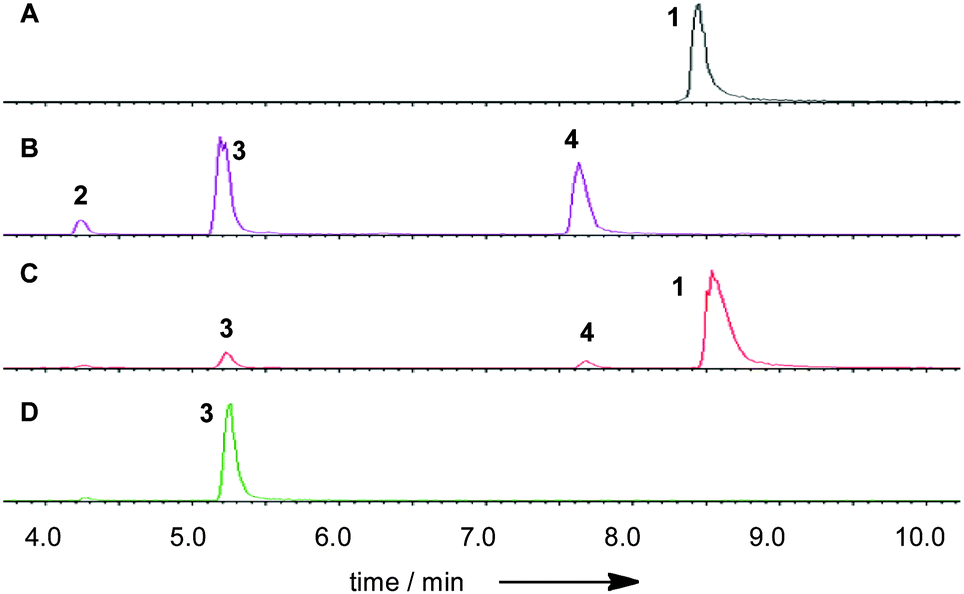 | ||
| Fig. 4 ES− total ion current (TIC) LCMS traces of SQS1 derivatives and MF5453 culture broth extracts: A, SQS1 1 standard; B, hydrolysis products of SQS1 under acidic catalysis; C, wild type extract from a 6 day old MF5453 liquid culture; D, extract of a phpks1-Knockout transformant of MF5453. | ||
Partial hydrolysis of SQS1 1 with aqueous potassium hydroxide gave a mixture of the core metabolite 2, the two mono-acylated cores 3 (loss of 6-O-tetraketide) and 4 (loss of 12-O-acetate) and unreacted starting material 1 (Scheme 1). All compounds were purified by mass-directed reverse-phase chromatography and fully characterised by NMR spectroscopy (see ESI†). Full hydrolysis (acidic conditions) gave the tetraketide 5 in quantitative yield. In the extract of WT MF5453, grown under producing conditions, SQS1 1 is the major product, but traces of compounds 2, 3 and 4 are observable (Fig. 4C). The SQTKS knockout mutant of MF5453 shows no SQS1 1 production but very high titres of 3, in which the tetraketide is missing (Fig. 4D). Compound 4 which would require an active SQTKS was also not evident in the extract.
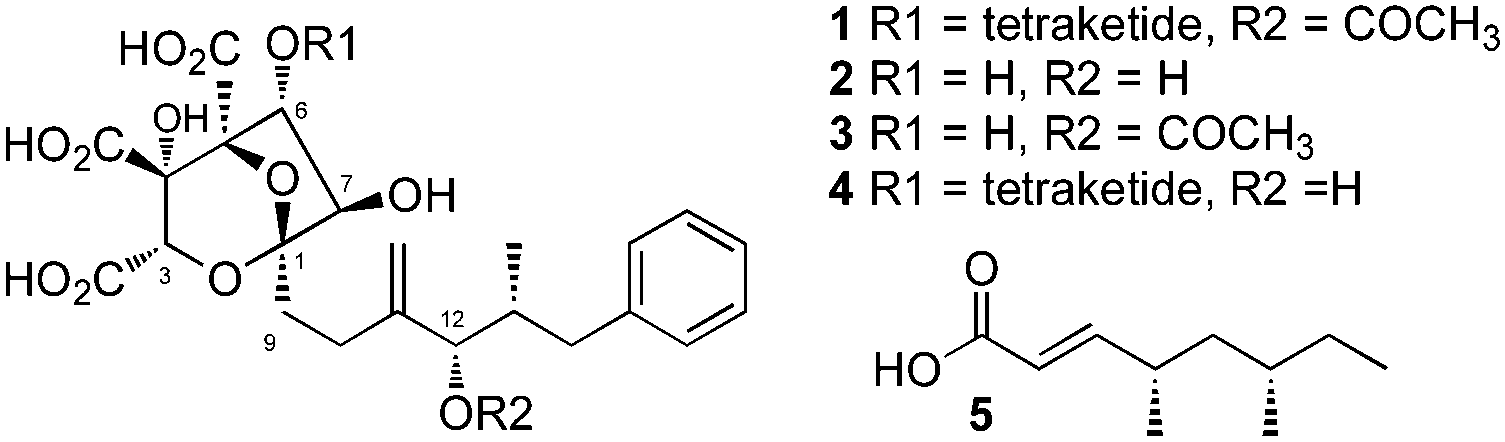 | ||
| Scheme 1 Hydrolysis of SQS1. The main products 2, 3, 4 and 5 were purified by LCMS and identified by NMR spectroscopy. | ||
These results show that transfer of the tetraketide to 3 is likely to be the final step of biosynthesis. As shown above, the gene cluster contains two genes encoding putative ATs (mfM4 and mfR4). The expected intronless sequence of mfM4 was expressed and purified from E. coli (see ESI†). The mfR4 gene was also expressed. However extensive expression trials did not give adequate soluble protein for further study, even using an E. coli optimised sequence.
Squalestatin tetraketide pantetheine 6 and CoA 7 were synthesised by literature procedures and purified by mass-directed HPLC. In vitro assays were set up in which various possible substrates (2, 3 or 4) were incubated in buffer in the presence of MfM4 and various acyl groups (6, 7) and acetyl, hexanoyl and octanoyl CoAs and the reactions were monitored by LCMS (Scheme 2). No evidence could be obtained for the transfer of the tetraketide from pantetheine to any possible acceptor. However, MfM4 does transfer acetate, hexanoate and octanoate from their corresponding CoA thiolesters to 3 (Fig. 5). When the tetraketide CoA 7 was used as a substrate for the reaction with 3 and MfM4, SQS1 1 was synthesised showing that transfer occurs specifically to the 6-hydroxyl of 3. Compounds 8–10 are new squalestatin analogues synthesised in vitro for the first time. Acyl groups were not transferred to 2 or 4 by MfM4 showing that the 12-O-acetate must be attached earlier during biosynthesis, and further confirming that the 6-hydroxyl is the target for acylation.
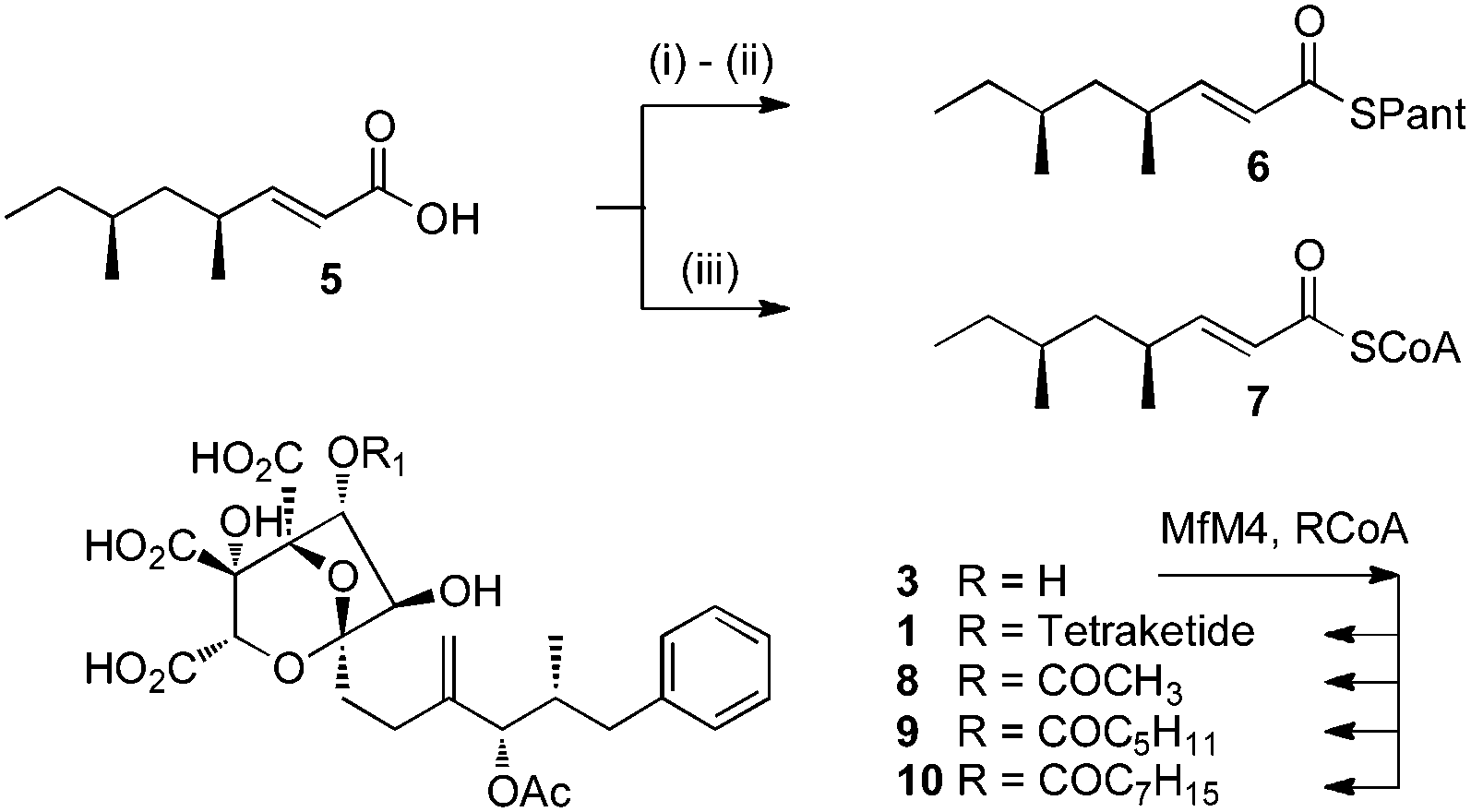 | ||
| Scheme 2 In vitro preparation of squalestatin S1 and analogues. (i) Pantetheine dimethylacetal, DMAP, EDCI, CH2Cl2; (ii) 10% TFA in CH2Cl2, 20 min; (iii) CDI, CoASH. | ||
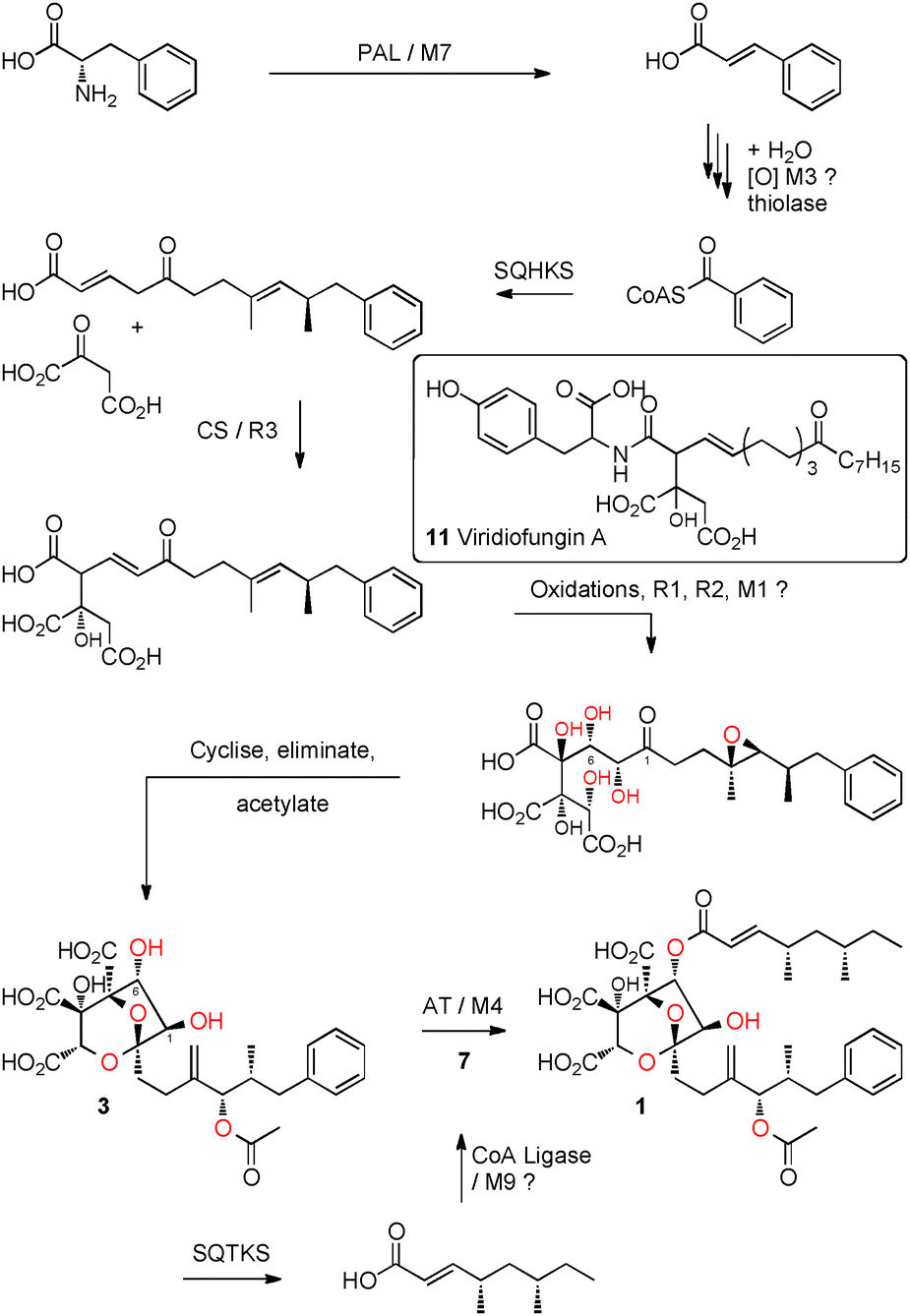 | ||
| Scheme 3 Proposed biosynthesis of SQS1 1. | ||
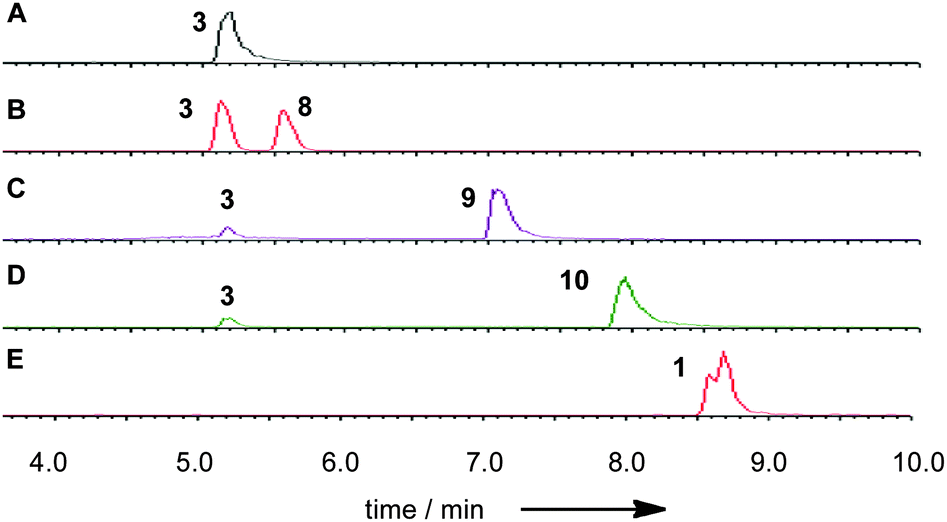 | ||
| Fig. 5 ES− LCMS traces showing the transfer of acyl groups from CoA to 3: A, standard 3 from hydrolysis of SQS1 1 (TIC); in vitro reaction product of AT1 with B, acetyl CoA + 3 (TIC); C, hexanoyl CoA + 3 (TIC); D, octanoyl CoA + 3 (TIC); E, tetraketide CoA 7 + 3 (EIC). | ||
Thus our results reveal the biosynthetic gene cluster for the squalestatins for the first time. The cluster encodes two highly-reducing PKS, similar to those known for lovastatin biosynthesis. In the case of the lovastatins a diketide is transferred to a nonaketide as an ester-bound sidechain. This occurs via a specific acyl transferase LovD,22 which appears to unload the complete diketide from its PKS onto itself before then passing it directly to the hexaketide hydroxyl so there is no enzyme-free diketide intermediate.
In the case of squalestatins the mechanism is different: the tetraketide synthase appears to transfer its product to CoA as an enzyme-free intermediate. The AT MfM4 then transfers the acyl group selectively to the 6-OH of 3. MfM4 has a broad substrate selectivity in terms of its CoA substrate, transferring chains from 2 to 10 carbons. This explains the wide range of known squalestatins with various acyl groups at O-6, which presumably arise by use of the prevailing CoA thiolester pool in various host organisms.5 Formation of the tetraketide CoA from 5 may be catalysed by the M9 CoA ligase, but the mechanism of release of 5 and the hexaketide from their respective PKS remains unknown, although the cluster encodes a potential esterase (M8) and a possible hydrolase (M10) which could be involved in these processes.
Our experiments open up new opportunities for further investigating squalestatin biosynthesis. For example formation of the 4,8-dioxa-bicyclo[3.2.1]octane core involves linkage of the hexaketide to oxaloacetate, presumably by the R3 citrate synthase, but the timing and precise substrates of this step are unknown. However, comparison to the fungal metabolite viridiofungin A 1123 suggests this step occurs early during biosynthesis. The hexaketide and oxaloacetate require several oxidative steps, but the cluster encodes no proteins with strong sequence homology to known oxygenases. However three of the encoded proteins show some structural homology to known oxygenase proteins (i.e. M1, R1 and R2) and the activity of M5 is unknown, so it is possible that these may be new types of multifunctional oxidases similar to LovA which catalyses two oxidations during lovasatin biosynthesis.24 SQS1 1 is one of very few fungal polyketides with a benzoate starter unit and further investigations are required to determine the origin of this selectivity.
Our present work thus focusses on in vitro and in vivo explorations of the cluster to address these and other questions. Furthermore, the results presented here show that new squalestatins can be engineered, and further work to alter the skeleton and substitution pattern of 1 is currently underway.
We thank EPSRC (EP/F066104/1), Leibniz Universität Hannover and DFG (INST 187/621) for funding LCMS instruments. CB Thanks the MINAS programme of Lower Saxony for funding. We thank the University of Bristol Genomics facility for Illumina genome sequencing. Ms Luxi Qiao is thanked for technical assistance. Researchers at Glaxo, notably Dr Brian Rudd, Dr Barrie Wilkinson, Dr Frank van Middlesworth and Dr Mike Dawson are thanked for helpful discussions and the gift of standard 1.
Notes and references
- P. J. Sidebottom, R. M. Highcock, S. J. Lane, P. A. Procopiou and N. S. Watson, J. Antibiot., 1992, 45, 648–658 CrossRef CAS PubMed.
- J. D. Bergstrom, M. M. Kurtz, D. J. Rew, A. M. Amend, J. D. Karkas, R. G. Bostedor, V. S. Bansal, C. Dufresne, F. L. VanMiddlesworth and O. D. Hensens, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 80–84 CrossRef CAS.
- K. Hasumi, K. Tachikawa and K. Sakai, J. Antibiot., 1993, 46, 689–691 CrossRef CAS PubMed.
- R. J. Cannell, M. J. Dawson, R. S. Hale, R. M. Hall, D. Noble, S. Lynn and N. L. Taylor, J. Antibiot., 1993, 46, 1381–1389 CrossRef CAS PubMed; A. Baxter, B. J. Fitzgerald, J. L. Hutson, A. D. McCarthy, J. M. Motteram, B. C. Ross, M. Sapra, M. A. Snowden, N. S. Watson, R. J. Williams and C. Wright, J. Biol. Chem., 1992, 267, 11705–11708 Search PubMed.
- J. D. Bergstrom, C. Dufresne and G. F. Bills, Annu. Rev. Microbiol., 1995, 49, 607–639 CrossRef CAS PubMed.
- C. A. Jones, P. J. Sidebottom, R. J. Cannell, D. Noble and B. A. Rudd, J. Antibiot., 1992, 45, 1492–1498 CrossRef CAS PubMed.
- K. M. Byrne, B. H. Arison, M. Nallinomstead and L. Kaplan, J. Org. Chem., 1993, 58, 1019–1024 CrossRef CAS.
- R. J. Cox, F. Glod, D. Hurley, C. M. Lazarus, T. P. Nicholson, B. A. M. Rudd, T. J. Simpson, B. Wilkinson and Y. Zhang, Chem. Commun., 2004, 2260–2261 RSC.
- T. P. Nicholson, B. A. Rudd, M. Dawson, C. M. Lazarus, T. J. Simpson and R. J. Cox, Chem. Biol., 2000, 8, 157–178 CrossRef.
- K. Blin, M. H. Medema, D. Kazempour, M. A. Fischbach, R. Breitling, E. Takano and T. Weber, Nucleic Acids Res., 2013, 41, W204–W212 CrossRef PubMed.
- Gene nomenclature – the genes have been named by their relative position to the two PKS genes: i.e.mfL1 is the first gene to the left of the tetraketide synthase (phpks1) in the MF5453 cluster; phL4 is the fourth gene to the left of the hexaketide synthase in Phoma species; mfM3 is the third gene between the two PKS genes etc. A full list of gene and protein names is included in the ESI.† The MF5453 cluster has been deposited at genbank with the accession number KU946987.
- A. V. Qualley, J. R. Widhalm, F. Adebesin, C. M. Kish and N. Dudareva, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 16383–16388 CrossRef CAS PubMed.
- R. J. Cox, Org. Biomol. Chem., 2007, 5, 2010–2026 CAS.
- E. J. Skellam, D. Hurley, J. Davison, C. M. Lazarus, T. J. Simpson and R. J. Cox, Mol. BioSyst., 2010, 6, 680–682 RSC.
- R. Fujii, Y. Matsu, A. Minami, S. Nagamine, I. Takeuchi, K. Gomi and H. Oikawa, Org. Lett., 2015, 17, 5658–5661 CrossRef CAS PubMed.
- K. Williams, A. J. Szwalbe, N. P. Mulholland, J. L. Vincent, A. M. Bailey, C. L. Willis, T. J. Simpson and R. J. Cox, Angew. Chem., 2016 DOI:10.1002/anie.201511882; A. J. Szwalbe, K. Williams, D. E. O'Flynn, A. M. Bailey, N. P. Mulholland, J. L. Vincent, C. L. Willis, R. J. Cox and T. J. Simpson, Chem. Commun., 2015, 51, 17088–17091 RSC.
- L. A. Kelley, S. Mezulis, C. M. Yates, M. N. Wass and M. J. E. Sternberg, Nat. Protoc., 2015, 10, 845–858 CrossRef CAS PubMed.
- G. S. Garvey, S. P. McCormick and I. Rayment, J. Biol. Chem., 2008, 283, 1660–1669 CrossRef CAS PubMed.
- F. Thanweer and N. K. Verma, BMC Biochem., 2012, 13, 13 CrossRef CAS PubMed.
- J. Pandit, D. E. Danley, G. K. Schulte, S. Mazzalupo, T. A. Pauly, C. M. Hayward, E. S. Hamanaka, J. F. Thompson and H. J. Harwood, J. Biol. Chem., 2000, 275, 30610–30617 CrossRef CAS PubMed.
- M. Nielsen, L. Albertsen, G. Lettier, J. Nielsen and U. Mortensen, Fungal Genet. Biol., 2006, 43, 54–64 CrossRef CAS PubMed.
- X. Xie, K. Watanabe, W. A. Wojcicki, C. C. C. Wang and Y. Tang, Chem. Biol., 2006, 13, 1161–1169 CrossRef CAS PubMed.
- S. M. Mandala, R. A. Thornton, B. R. Frommer, S. Dreikorn and M. B. Kurtz, J. Antibiot., 1997, 50, 339–343 CrossRef CAS PubMed.
- J. Barriuso, D. T. Nguyen, J. W. H. Li, J. N. Roberts, G. MacNevin, J. L. Chaytor, S. L. Marcus, J. C. Vederas and D.-K. Ro, J. Am. Chem. Soc., 2011, 133, 8078–8081 CrossRef CAS PubMed; R. V. K. Cochrane and J. C. Vederas, Acc. Chem. Res., 2014, 47, 3148–3161 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6cc02130a |
| This journal is © The Royal Society of Chemistry 2016 |