
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A dimethacrylate cross-linker cleavable under thermolysis or alkaline hydrolysis conditions: synthesis, polymerization, and degradation†
Marios
Elladiou
and
Costas S.
Patrickios
*
Department of Chemistry, University of Cyprus, POB 20537, 1678 Nicosia, Cyprus. E-mail: costasp@ucy.ac.cy
First published on 18th January 2016
Abstract
We develop a new platform based on 2,6-pyridinediethanol diesters for introducing polymer degradability under thermolysis or alkaline hydrolysis conditions, with the latter being rare in polymers. Such labile diesters can be cross-linkers, bifunctional initiators and inimers. We demonstrate the power of this platform through the synthesis of the 2,6-pyridinediethanol dimethacrylate cross-linker, its controlled (co)polymerization, and the thermal and hydrolytic cleavage of its (co)polymers.
Degradable polymers represent an important class of polymeric materials capable of undergoing reduction in their molecular weight and, in certain cases, change in their architecture. These materials are exploited in several important areas, such as in microelectronics1 and in biomedical engineering,2 where they are used as positive photoresists and matrices for tissue engineering or drug delivery, respectively. Degradable polymers comprise at least one degradable component, the monomer, the cross-linker or the initiator. Although degradable monomers are the most common components in degradable polymers, degradable cross-linkers have also been frequently employed, leading to the formation of branched polymers or polymer networks with degradable branching points, whose degradation results in soluble, linear polymers.
To date, a large number of degradable cross-linkers have been developed, (co)polymerized and cleaved. However, the vast majority of these cross-linkers are labile to acidic hydrolysis conditions, with only one example of cross-linker being readily amenable to cleavage under basic conditions. Regarding the former type of cross-linkers, these include several acetal di(meth)acrylates or diacrylamides,3–15 hemiacetal ester dimethacrylates,16,17 ketal di(meth)acrylates, diacrylamides, or di(N-vinylformamides),18–26 silyl ether di(meth)acrylates,27,28 and tertiary ester di(meth)acrylates.29–33 Regarding the latter type, the only example of cross-linker spontaneously degradable under alkaline hydrolysis conditions is, to the best of our knowledge, tetrafluorophenylene diacrylate,34 which is very costly to prepare because of its perfluorinated ring. Other cross-linkers, also sensitive to alkaline hydrolysis conditions, but degrading at much slower rates, include a dilactone dimethacrylate35 and 1,1-ethylene diol (acylal) dimethacrylate.36,37
Thus, there is a need for new, inexpensive divinyl cross-linkers that would be initially stable under acidic hydrolysis conditions (under which most known cross-linker residues would be cleaved), and degrade at a later stage under alkaline hydrolysis conditions. This is exactly the aim of this investigation, where a dimethacrylate cross-linker with these traits is developed. The molecular design in this work is based on the base-labile 2-(pyridin-2-yl)ethyl ester group, which has occasionally been used in organic and peptide synthesis for the protection of the carboxylic acid groups in small molecules,38–44 and was recently imported by our team to polymer chemistry for the protection of methacrylic acid (MAA) before its controlled polymerization, leading to the development of 2-(pyridin-2-yl)ethyl methacrylate (PyEMA).45–47 Our wish to prepare a symmetrical cleavable cross-linker necessitated the presence of two 2-(pyridin-2-yl)ethyl groups. However, rather than synthesizing two such groups and linking them together, we decided, instead, for atom economy, to include only one pyridine ring and introduce two ethyl groups at its 2- and 6-positions.
This design led to 2,6-pyridinediethanol dimethacrylate (PyDMA) whose chemical structure is shown on the left of Scheme 1. The synthesis of PyDMA required the non-commercially available 2,6-pyridinediethanol that was prepared following the procedure of Kelly et al.48 which was based on the original method of Löffler and Thiel.49 Although the procedure possesses low yield, it uses readily available, low-cost and recyclable reagents, with 2,6-lutidine being the main reagent. Thus, 2,6-lutidine was reacted in water with paraformaldehyde in a pressure glass vial at 135 °C for 16 h, leading to three hydroxymethylation products (Fig. S1 in the ESI†): the desired 2,6-pyridinediethanol, its isomeric 2-(6-methylpyridin-2-yl)propane-1,3-diol, and the monosubstituted 2-(6-methylpyridin-2-yl)ethanol. The unreacted 2,6-lutidine was removed by steam distillation, whereas 2-(6-methylpyridin-2-yl)ethanol was removed on the vacuum line at 80 °C. The two diols were separated in a subsequent step using column chromatography and were identified using 1H and 13C NMR spectroscopy (Fig. S2 and S3 in the ESI†); their melting points, determined using differential scanning calorimetry (DSC), were at 40 °C for 2,6-pyridinediethanol and at 68 °C for 2-(6-methylpyridin-2-yl)propane-1,3-diol. Subsequently, the symmetric diol was esterified using methacryloyl chloride to obtain PyDMA which was purified by passage through basic alumina columns. The 1H and 13C NMR spectra of the purified PyDMA cross-linker in d6-DMSO are also given in the ESI† (Fig. S4).
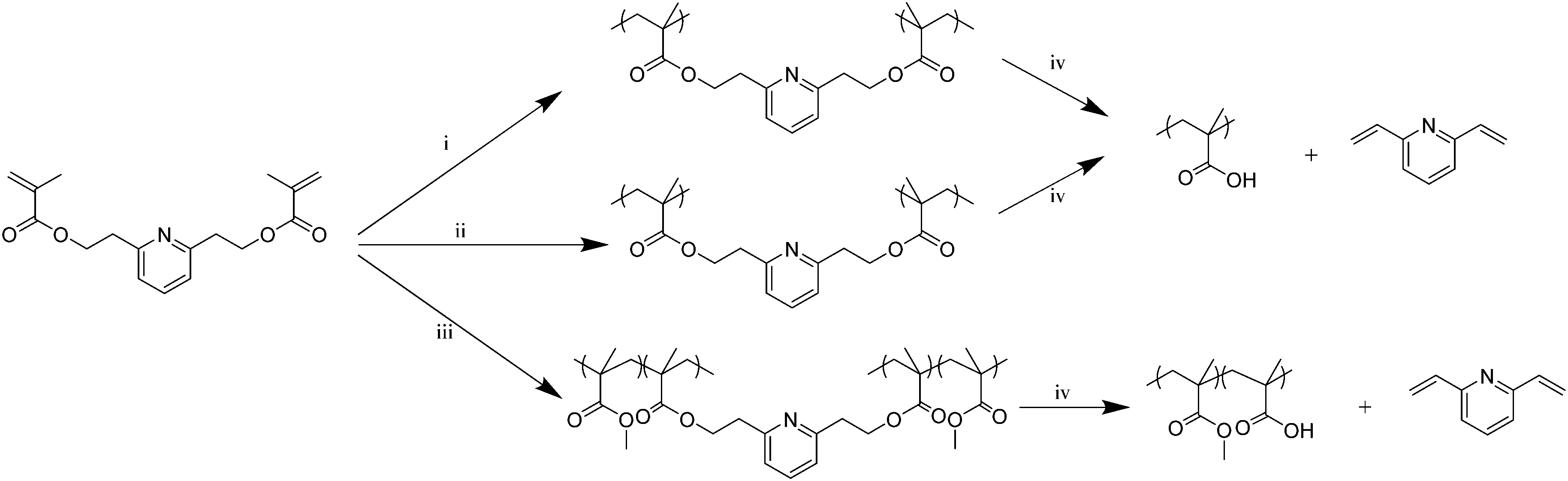 | ||
| Scheme 1 RAFT (co)polymerization of the PyDMA cross-linker, and the thermal and alkaline hydrolysis treatments of the resulting polymers. (i) RAFT homopolymerization of PyDMA (2.50 M); (ii) RAFT homopolymerization of PyDMA (0.66 M); (iii) RAFT copolymerization of MMA plus PyDMA (total concentration 3.00 M); (iv) NaOH (aq) in DMSO at RT overnight or 130 °C in a vacuum oven for 6 h. | ||
Following its synthesis and purification, PyDMA was polymerized in a controlled way. In particular, reversible addition–fragmentation chain transfer (RAFT) polymerization was employed to homopolymerize PyDMA at two different concentrations, and also to copolymerize it with methyl methacrylate (MMA), as illustrated in Scheme 1. Scheme 1 also illustrates the cleavage of the PyDMA units in the (co)polymers thermally or via alkaline hydrolysis at room temperature. Upon its cleavage, each polymerized PyDMA unit was converted to two MAA units and 2,6-divinylpyridine. In all cases, 2-cyanoprop-2-yl dithiobenzoate (2-CPDB), 2,2′-azobis(isobutyronitrile) (AIBN) and 1,4-dioxane served as the chain transfer agent (CTA), the radical source and the solvent for the polymerizations, respectively. PyDMA concentrations of 0.66 and 2.50 M were used for the homopolymerizations (at a PyDMA to CTA molar ratio, corresponding to the nominal degree of polymerization, DP, equal to 10, for both cases), leading to a hyperbranched polymer and a polymer network, respectively. Additionally, a randomly cross-linked PyDMA–MMA copolymer network was also synthesized. Table S1 in the ESI† summarizes the main characteristics of the three PyDMA-containing polymers.
The hyperbranched PyDMA homopolymer was successfully hydrolyzed in a 0.4 M NaOD solution in d6-DMSO at room temperature within 2 h, as shown using 1H NMR spectroscopy (Fig. S5 in the ESI†). A similar sample in DCl solution was completely stable for at least 24 h (also Fig. S5, ESI†). Upon alkaline hydrolysis, a two-phase system was formed, comprising a d6-DMSO solution top phase, and a precipitate phase soluble in D2O. 1H NMR spectroscopy analyses of the two phases in the respective deuterated solvents indicated that the bottom phase consisted of poly(sodium methacrylate) and 2,6-divinylpyridine, whereas the top phase primarily contained 2,6-divinylpyridine. These 1H NMR spectra are displayed in Fig. 1, together with that of the starting hyperbranched homopolymer in d6-DMSO before the addition of NaOD. The weak sharp signals at 8.0 ppm in the spectra corresponded to the aromatic protons of the 2-CPDB residue. The hyperbranched PyDMA homopolymer was also thermolyzed in d6-DMSO solution (without the addition of NaOD) at 130 °C for 8 h. The 1H NMR spectrum of the thermolysis product (in d6-DMSO) is also shown in Fig. 1, and it displayed broad polymeric peaks due to the polyMAA units (including the carboxylic acid protons at 12.5 ppm) and sharp peaks due to the newly-formed 2,6-divinylpyridine.
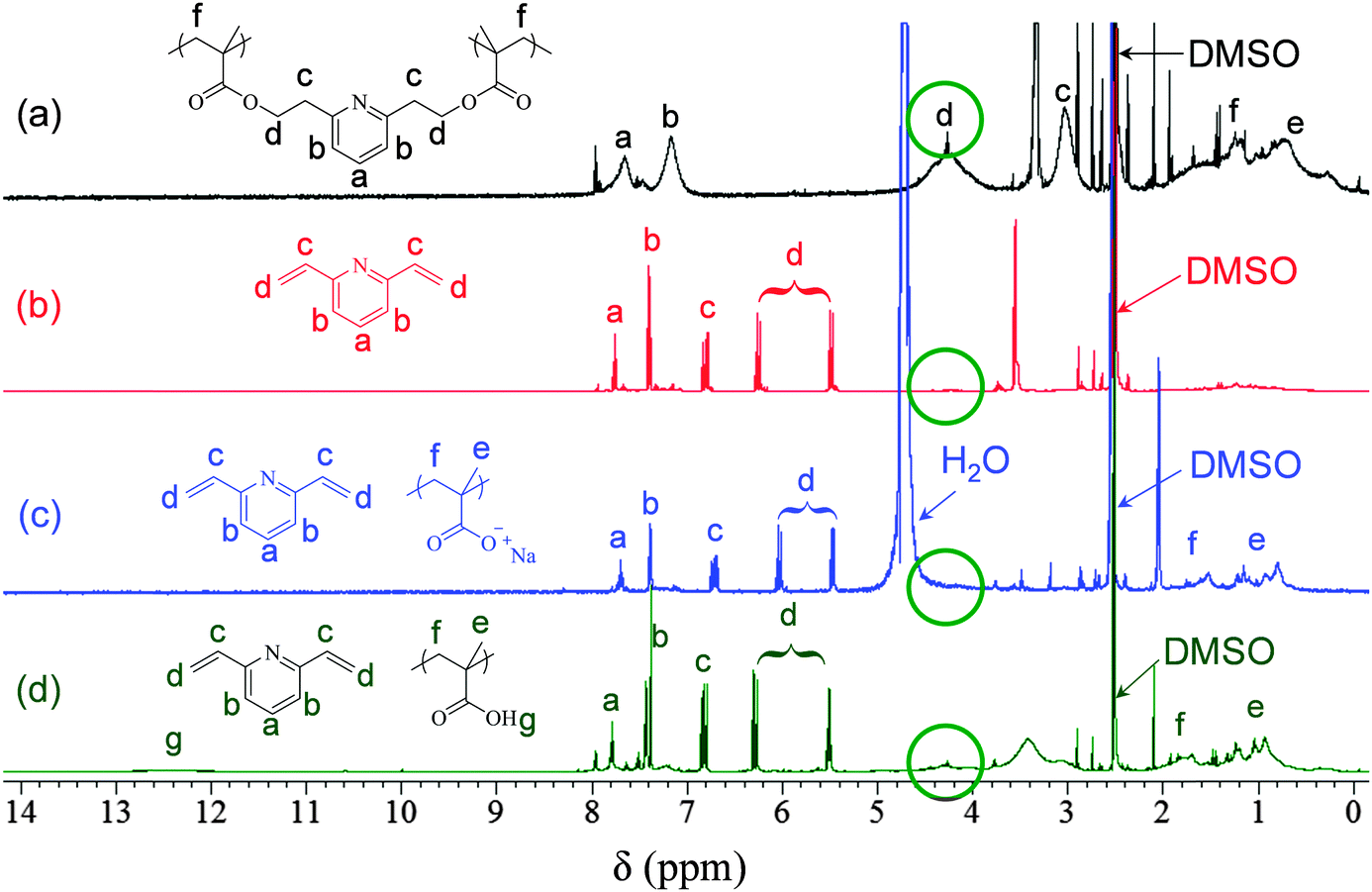 | ||
| Fig. 1 1H NMR spectra of the hyperbranched PyDMA homopolymer (a) before any treatment in d6-DMSO (black), (b) after alkaline hydrolysis using NaOD in d6-DMSO (red), (c) after alkaline hydrolysis using NaOD in D2O (blue), and (d) after thermolysis in a vacuum oven at 130 °C for 6 h in d6-DMSO (green). | ||
The PyDMA homopolymer network was also successfully cleaved both by solution thermolysis and by alkaline hydrolysis at room temperature. Here, the cleavage was most dramatically demonstrated by the conversion of the insoluble network to a polymer solution. Furthermore, the 1H NMR spectra of the network cleavage products (via thermolysis or hydrolysis) in d6-DMSO indicated the presence of the olefinic protons of 2,6-divinylpyridine and the disappearance of the polymeric oxyethylene protons. Fig. S6 in the ESI† shows the 1H NMR spectra of the cleavage products after subjecting the PyDMA homopolymer network to alkaline hydrolysis or to thermolysis. In the 1H NMR spectrum of the thermolysis product, a weak signal due to the polyMAA carboxylic acid protons was also visible.
The cleavage of the PyDMA–MMA copolymer network was also performed both thermally and hydrolytically, and the results are shown in Fig. S7 in the ESI,† where the 1H NMR spectra were consistent with the expected degradation products, which were a linear MAA–MMA random copolymer plus 2,6-divinylpyridine. Importantly, the cleavage also became evident from the transformation of the insoluble copolymer network to a soluble copolymer.
The bulk thermolysis of the PyDMA-containing polymers was investigated using DSC and thermogravimetric analysis (TGA), and the results are shown in Fig. 2 and Fig. S8 (ESI†), respectively. Fig. 2 displays an overlay of the DSC traces of the three PyDMA-bearing polymeric materials, all presenting an endotherm at around 200 °C, corresponding to the cleavage of the 2-(pyridin-2-yl)ethyl ester group.45 The highest cleavage temperature, at 210 °C was exhibited by the PyDMA–MMA copolymer network, possibly a result of the protection offered to the PyDMA units by the MMA units. Note that the cleavage temperature of the same ester group in (linear) polyPyEMA, determined also using DSC, was much lower, at 160 °C.45 The higher, by 50 °C, cleavage temperatures of the 2-(pyridin-2-yl)ethyl ester groups in the PyDMA units might be due to electronic effects arising from the presence of two such groups in the same pyridine ring. The cleavage molar (per PyDMA unit) enthalpies were also calculated from the DSC traces and were found to be 52.1, 45.2 and 45.9 kJ mol−1 for the PyDMA hyperbranched homopolymer, the PyDMA homopolymer network and the PyDMA–MMA copolymer network, respectively. These values were close to each other, as expected, but lower than those for the PyEMA units in their homopolymers of ∼70 kJ mol−1. This difference can be understood considering that each PyDMA unit bears two 2-(pyridin-2-yl)ethyl ester groups.
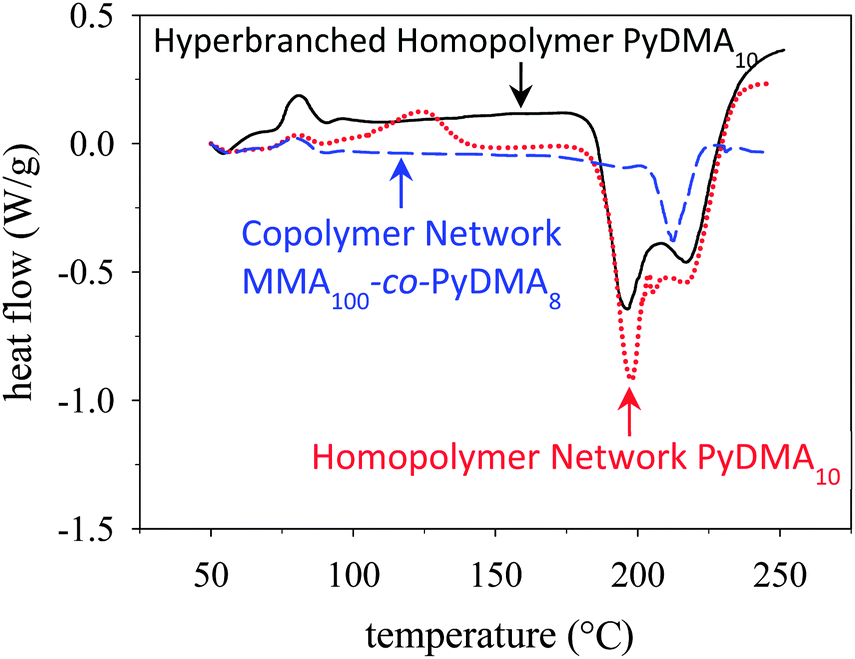 | ||
| Fig. 2 DSC thermograms of the PyDMA hyperbranched homopolymer (black, continuous line), the PyDMA homopolymer network (red, dotted line) and the PyDMA–MMA copolymer network (blue, dashed line). | ||
The weak exotherm in the DSC trace of the PyDMA homopolymer network at 120 °C was due to a small percentage of unreacted methacrylate groups, identified in the ATR-FTIR spectrum of the dried sample as a weak signal at 1630 cm−1 (Fig. S9 in the ESI†). Such a signal was absent from the ATR-FTIR spectrum of dried PyDMA–MMA copolymer network. Similarly, complete vinyl group conversion was also accomplished in the PyDMA hyperbranched homopolymer, as no olefinic signal was present in its 1H NMR spectrum (Fig. 1(a)).
In conclusion, a new labile dimethacrylate cross-linker based on pyridine was designed, prepared, (co)polymerized by controlled radical polymerization, and its residues were cleaved in the resulting polymers under relatively mild conditions, hydrolytically in base at room temperature or thermally. Thermal degradation occurred instantaneously at ∼200 °C (in DSC and TGA), or within a few hours, both in the bulk and in solution, at 130 °C. Hydrolytic degradation took place at room temperature and, selectively, under alkaline conditions, a rare occasion among cleavable dimethacrylate cross-linkers. The availability of this newly-developed base-labile cross-linker will be important for cases in which initial stability under acidic conditions is desired (where most other cross-linkers would degrade), and subsequent degradation is planned. Finally, the syntheses of similarly degrading bifunctional initiators and inimers based on 2,6-pyridinediethanol are currently under way.
The authors wish to thank the European Regional Development Fund and the Republic of Cyprus for cofunding this work (project NEA YPODOMH/NEKYP/0311/27) through the Cyprus Research Promotion Foundation. The same organizations are also thanked for funding the purchase of the 500 MHz NMR spectrometer used in this work (project NEA YPODOMH/NEKYP/0308/02). Finally, we thank the following colleagues at the University of Cyprus for their assistance in various aspects of this work: Dr A. Kalogirou, Dr S. I. Mirallai and Prof. P. A. Koutentis.
Notes and references
- (a) B. D. Gates, Q. Xu, M. Stewart, D. Ryan, C. G. Willson and G. M. Whitesides, Chem. Rev., 2005, 105, 1171 CrossRef CAS PubMed; (b) in Polymers for Microelectronics and Nanoelectronics, ACS Symposium Series, ed. Q. Lin, R. A. Pearson and J. C. Hedrick, American Chemical Society, Washington, DC, 2004, vol. 874 Search PubMed; (c) M. Geoghegan and G. Hadziioannou, Polymer Electronics, Oxford University Press, Oxford, 2013 Search PubMed.
- (a) V. Delplace and J. Nicolas, Nat. Chem., 2015, 7, 771 CrossRef PubMed; (b) B. Guo, L. Glavas and A.-C. Albertsson, Prog. Polym. Sci., 2013, 38, 1263 CrossRef CAS; (c) Nanocomposites with Biodegradable Polymers: Synthesis, Properties, and Future Perspectives, ed. V. Mittal, Oxford University Press, Oxford, 2011 Search PubMed.
- E. Themistou and C. S. Patrickios, Macromolecules, 2006, 39, 73 CrossRef CAS.
- E. Themistou and C. S. Patrickios, Macromolecules, 2007, 40, 5231 CrossRef CAS.
- E. Themistou and C. S. Patrickios, Macromol. Chem. Phys., 2008, 209, 1021 CrossRef CAS.
- N. Murthy, Y. X. Thng, S. Schuck, M. C. Xu and J. M. J. Fréchet, J. Am. Chem. Soc., 2002, 124, 12398 CrossRef CAS PubMed.
- M. Murthy, M. Xu, S. Schuck, J. Kunisawa, N. Shastri and J. M. J. Fréchet, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 4995 CrossRef PubMed.
- S. M. Standley, Y. J. Kwon, N. Murthy, J. Kunisawa, N. Shastri, S. J. Guillaudeu, L. Lau and J. M. J. Fréchet, Bioconjugate Chem., 2004, 15, 1281 CrossRef CAS PubMed.
- Y. J. Kwon, S. M. Standley, S. L. Goh and J. M. J. Fréchet, J. Controlled Release, 2005, 105, 199 CrossRef CAS PubMed.
- Y. Chan, V. Bulmus, M. H. Zareie, F. L. Byrne, L. Barner and M. Kavallaris, J. Controlled Release, 2006, 115, 197 CrossRef CAS PubMed.
- V. Bulmus, Y. Chan, Q. Nguyen and H. L. Tran, Macromol. Biosci., 2007, 7, 446 CrossRef CAS PubMed.
- Y. Chan, T. Wong, F. Byrne, M. Kavallaris and V. Bulmus, Biomacromolecules, 2008, 9, 1826 CrossRef CAS PubMed.
- V. Knorr, V. Russ, L. Allmendinger, M. Ogris and E. Wagner, Bioconjugate Chem., 2008, 19, 1625 CrossRef CAS PubMed.
- R. Jain, S. M. Standley and J. M. J. Fréchet, Macromolecules, 2007, 40, 452 CrossRef CAS.
- S. Kaihara, S. Matsumura and J. P. Fisher, Macromolecules, 2007, 40, 7625 CrossRef CAS.
- Y. Shi, Z. F. Fu and X. C. Sui, Chin. Chem. Lett., 2011, 22, 374 CrossRef.
- E. Ruckenstein and H. Zhang, Macromolecules, 1999, 32, 3979 CrossRef CAS.
- W. H. Heath, F. Palmieri, J. R. Adams, B. K. Long, J. Chute, T. W. Holcombe, S. Zieren, M. J. Truit, J. L. White and C. G. Willson, Macromolecules, 2008, 41, 719 CrossRef CAS.
- K. Heyden, K. Babooram, M. Ahmed and R. Narain, Eur. Polym. J., 2009, 45, 1689 CrossRef.
- Y. J. Kwon, S. M. Standley, A. P. Goodwin, E. R. Gillies and J. M. J. Fréchet, Mol. Pharmacol., 2004, 2, 83 CrossRef PubMed.
- L. Shi and C. Berkland, Adv. Mater., 2006, 18, 2315 CrossRef CAS.
- L. Shi and C. Berkland, Macromolecules, 2007, 40, 4635 CrossRef CAS PubMed.
- L. Shi, S. Khondee, T. H. Linz and C. Berkland, Macromolecules, 2008, 41, 6546 CrossRef CAS.
- N. Bhuchar, R. Sunasee, K. Ishihara, T. Thundat and R. Narain, Bioconjugate Chem., 2012, 23, 75 CrossRef CAS PubMed.
- X. Hu, T. Yang, R. Gu, Y. Cui, C. Yuan, H. Ge, W. Wu, W. Li and Y. Chen, J. Mater. Chem. C, 2014, 2, 1836 RSC.
- D. L. Roberts, Y. Ma, S. E. Bowles, C. M. Janczak, J. Pyun, S. S. Saavedra and C. A. Aspinwall, Langmuir, 2009, 25, 1908 CrossRef CAS PubMed.
- E. Themistou and C. S. Patrickios, Macromolecules, 2004, 37, 6734 CrossRef CAS.
- M. C. Parrott, J. C. Luft, J. D. Byrne, J. H. Fain, M. E. Napier and J. M. DeSimone, J. Am. Chem. Soc., 2010, 132, 17928 CrossRef CAS PubMed.
- M. F. Montague and C. J. Hawker, Chem. Mater., 2007, 19, 526 CrossRef CAS.
- B. D. Mather, S. R. Williams and T. E. Long, Macromol. Chem. Phys., 2007, 208, 1949 CrossRef CAS.
- D. Kafouris, E. Themistou and C. S. Patrickios, Chem. Mater., 2005, 18, 85 CrossRef.
- K. Ogino, J. Chen and C. K. Ober, Chem. Mater., 1998, 10, 3833 CrossRef CAS.
- L. Kilian, Z. Wang and T. E. Long, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 3083 CrossRef CAS.
- N. Metz and P. Theato, Macromolecules, 2009, 42, 37 CrossRef CAS.
- J. J. Gallagher, M. A. Hillmyer and T. M. Reineke, Macromolecules, 2014, 47, 498 CrossRef CAS.
- E. Themistou, A. Kanari and C. S. Patrickios, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 5811 CrossRef CAS.
- E. Themistou and C. S. Patrickios, J. Polym. Sci., Part A: Polym. Chem., 2009, 75, 5853 CrossRef.
- A. R. Katritzky, G. R. Khan and O. A. Schwarz, Tetrahedron Lett., 1984, 25, 1223 CrossRef CAS.
- H. Kunz and S. Birnbach, Tetrahedron Lett., 1984, 25, 3567 CrossRef CAS.
- H. Kunz and M. Kneip, Angew. Chem., Int. Ed. Engl., 1984, 23, 716 CrossRef.
- H. Kessler, G. Becker, H. Kogler and M. Wolff, Tetrahedron Lett., 1984, 25, 3971 CrossRef CAS.
- H. Kessler, G. Becker, H. Kogler, J. Friese and R. Kerssebaum, Int. J. Pept. Protein Res., 1986, 28, 342 CrossRef CAS.
- C.-Y. Yu and O. Meth-Cohn, Tetrahedron Lett., 1999, 40, 6665 CrossRef CAS.
- B. W. Lund, A. E. Knapp, F. Piu, N. K. Gauthier, M. Begtrup, U. Hacksell and R. Olsson, J. Med. Chem., 2009, 52, 1540 CrossRef CAS PubMed.
- M. Elladiou and C. S. Patrickios, Polym. Chem., 2012, 3, 3228 RSC.
- K. S. Pafiti, M. Elladiou and C. S. Patrickios, Macromolecules, 2014, 47, 1819 CrossRef CAS.
- M. Elladiou and C. S. Patrickios, Macromolecules, 2015, 48, 7503 CrossRef CAS.
- W. S. J. Kelly, G. H. Ford and S. M. Nelson, J. Chem. Soc. A, 1971, 388 RSC.
- K. Löffler and L. Thiel, Chem. Ber., 1909, 42, 132 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and NMR spectra. See DOI: 10.1039/c5cc09445c |
| This journal is © The Royal Society of Chemistry 2016 |