
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Making the invisible visible: improved electrospray ion formation of metalloporphyrins/-phthalocyanines by attachment of the formate anion (HCOO−)†
Jakob Felix
Hitzenberger
a,
Claudia
Dammann
a,
Nina
Lang
b,
Dominik
Lungerich
b,
Miguel
García-Iglesias
c,
Giovanni
Bottari
 *cd,
Tomás
Torres
*cd,
Norbert
Jux
*b and
Thomas
Drewello
*a
*cd,
Tomás
Torres
*cd,
Norbert
Jux
*b and
Thomas
Drewello
*a
aPhysical Chemistry I, Department of Chemistry and Pharmacy, University of Erlangen-Nuremberg, Egerlandstrasse 3, 91058 Erlangen, Germany. E-mail: thomas.drewello@fau.de
bOrganic Chemistry II, Department of Chemistry and Pharmacy, University of Erlangen-Nuremberg, Henkestraße 42, 91054 Erlangen, Germany. E-mail: norbert.jux@fau.de
cDepartamento de Química Orgánica, Universidad Autónoma de Madrid, Cantoblanco, 28049 Madrid, Spain. E-mail: giovanni.bottari@uam.es; tomas.torres@uam.es
dIMDEA-Nanociencia, c/Faraday 9, Campus de Cantoblanco, 28049 Madrid, Spain
First published on 23rd December 2015
Abstract
A protocol is developed for the coordination of the formate anion (HCOO−) to neutral metalloporphyrins (Pors) and -phthalocyanines (Pcs) containing divalent metals as a means to improve their ion formation in electrospray ionization (ESI). This method is particularly useful when the oxidation of the neutral metallomacrocycle fails. While focusing on Zn(II)Pors and Zn(II)Pcs, we show that formate is also readily attached to Mn(II), Mg(II) and Co(II)Pcs. However, for the Co(II)Pc secondary reactions can be observed. Upon collision-induced dissociation (CID), Zn(II)Por/Pc·formate supramolecular complexes can undergo the loss of CO2 in combination with transfer of a hydride anion (H−) to the zinc metal center. Further dissociation leads to electron transfer and hydrogen atom loss, generating a route to the radical anion of the Zn(II)Por/Pc without the need for electrochemical reduction, although the Zn(II)Por/Pc may have a too low electron affinity to allow electron transfer directly from the formate anion. In addition to single Por molecules, multi Por arrays were successfully analyzed by this method. In this case, multiple addition of formate occurs, giving rise to multiply charged species. In these multi Por arrays, complexation of the formate anion occurs by two surrounding Por units (sandwich). Therefore, the maximum attainment of formate anions in these arrays corresponds to the number of such sandwich complexes rather than the number of porphyrin moieties. The same bonding motif leads to dimers of the composition [(Zn(II)Por/Pc)2·HCOO]−. In these, the formate anion can act as a structural probe, allowing the distinction of isomeric ions with the formate bridging two macrocycles or being attached to a dimer of directly connected macrocycles.
Introduction
Metallo Pors and related metallomacrocycles are of crucial importance in a multitude of areas in natural sciences.1 In biology, iron-containing Pors appear in O2-binding hemoproteins2,3 and heme-based gas sensors.4,5 Structurally related Por derivatives play an important role as magnesium-containing chlorophyll in photosynthesis and as cobalt-containing vitamin B12 with multiple functions on the molecular level in the human body.1 Fossil metallo Pors have been found in crude oil and are used as geochemical markers.6 In medicine, metallo Pors and the structurally related metallo Pcs have been used as photosensitizers in photodynamic therapy.7 The application of metallo Pors and related metallomacrocycles in molecular electronics8 has stimulated intense research across physics, chemistry and materials science, owing to their robustness, the ability to tune electronic properties by variation of the central metal cation and/or derivatization of the macrocycle,9,10 as well as their rich supramolecular chemistry,11 especially in self-assembly systems.12–14The characterization of new functional Por-related materials is closely related to the development of modern mass spectrometry. Nowadays, the two most prominent soft ionization methods, namely matrix-assisted laser desorption/ionization (MALDI)15–17 and electrospray ionization (ESI),18–24 have both been applied intensively to the analysis of Por-related materials. Both methods have their advantages and disadvantages, almost possessing complementary features in their applicability. Since Pors are efficient chromophores, which strongly absorb in the UV region, they have even been analyzed by direct laser desorption/ionization (LDI) without the need for a matrix.15–17 The robustness of these aromatic macrocycles leads to only a little fragmentation. In fact, Pors have been successfully used as matrix materials themselves for the MALDI analysis of other materials.25–29 For MALDI, solubility is not a prerequisite,30,31 which accommodates the often only poorly soluble Pors. ESI, on the other hand, is a softer approach, which is advantageous for the analysis of Pors bearing more fragile substituents, but it requires solubility. In reality, the use of one particular method is often determined by availability rather than performance.
The present investigation focusses on the analysis of divalent metallo Pors and -Pcs by ESI. The ion formation for these molecules constitutes a peculiar case for ESI. In these cases, common electrospray ion formation processes based on acid–base chemistry, such as protonation or deprotonation, are not usually efficient. In M(II)Pors, the two most acidic hydrogen atoms of the free-base Por residing in the inner cavity are substituted by a metal cation, thus deprotonation is not an attractive route. On the other hand, protonation is also problematic, since the addition of acids facilitates M(II)Por demetallation and thus promotes their unwanted conversion to metal-free Pors.32 Interestingly, the most prominent ion formation process has been oxidation.18–20 Under certain circumstances, the ESI source can ionize the molecules in redox reactions, operating as an electrochemical cell.18–20,33–35 A prerequisite for oxidation is, amongst other requirements, a sufficiently low oxidation potential of the analyte (Eox < 1 V). Compound classes susceptible to oxidation by ESI have been reviewed.34,35 M(II)Pors have been used as an indicator of the occurrence of electrochemical oxidation during ESI and employed for mechanistic investigations of this process.18–20 Oxidation of M(II)Pors has also been successfully applied to the generation of adducts with imidazoles, allowing the determination of the bond strengths in these complexes.21,22 ESI oxidation has led to the formation of a stable tricationic dimer of a Zn(II)Por carrying two hexabenzocoronene ligands.23 The analytical use of ESI oxidation in mixtures of Pors has been examined and it was found that if the oxidation potentials of the two Pors differ by more than 0.1 V, discrimination occurs against the Por with the higher oxidation potential.24 Furthermore, oxidation may not be efficient or may fail, if the oxidation potential is too high. Another complication may arise from the required choice of solvents, as oxidation is particularly prominent with aprotic, non-nucleophilic solvents.18–20,34,35 However, these are often not ideal to sustain a stable electrospray. Finally, some of the M(II)Pcs and all multi-M(II)Por arrays of this study (vide infra) could not be oxidized during ESI or only to a minor extent, showing that oxidation is not a viable option for the analysis of these compounds. To enhance the applicability of ESI, a method is presented based on the supramolecular coordination of the formate anion to the macrocycle's metal center of M(II)Pcs, M(II)Pors and arrays of multi-M(II)Pors. The advantage of using the multi-atomic formate anion, as opposed to a mono-atomic charge carrier like Cl, Br and I, will be revealed in MS/MS experiments, allowing the elucidation of the structure of formate-containing dimer ions and providing a way to generate molecular Zn(II)Por/Pc anions without the need for electrochemical reduction in the ESI process.
Experimental
Due to the low solubility of M(II)Pcs 1–5 (Fig. 1),36 these derivatives were sonicated in DMF (N,N-dimethylformamide). The resulting suspension was centrifuged and a small part of the supernatant was diluted further with DMF and, after addition of sodium formate (1.0 × 10−4 M), it was introduced at an unknown concentration into the ESI-MS by direct injection. The Por and Pc derivatives 6–16 (Fig. 1 and Fig. 4) were dissolved in DCM (dichloromethane) at a concentration of 1 mg ml−1, respectively. The obtained stock solutions were diluted further with DMF to a concentration of 5.0 × 10−5 M. After addition of sodium formate (1.0 × 10−4 M) and thorough mixing, the resulting solution was introduced to the ESI-MS by direct injection.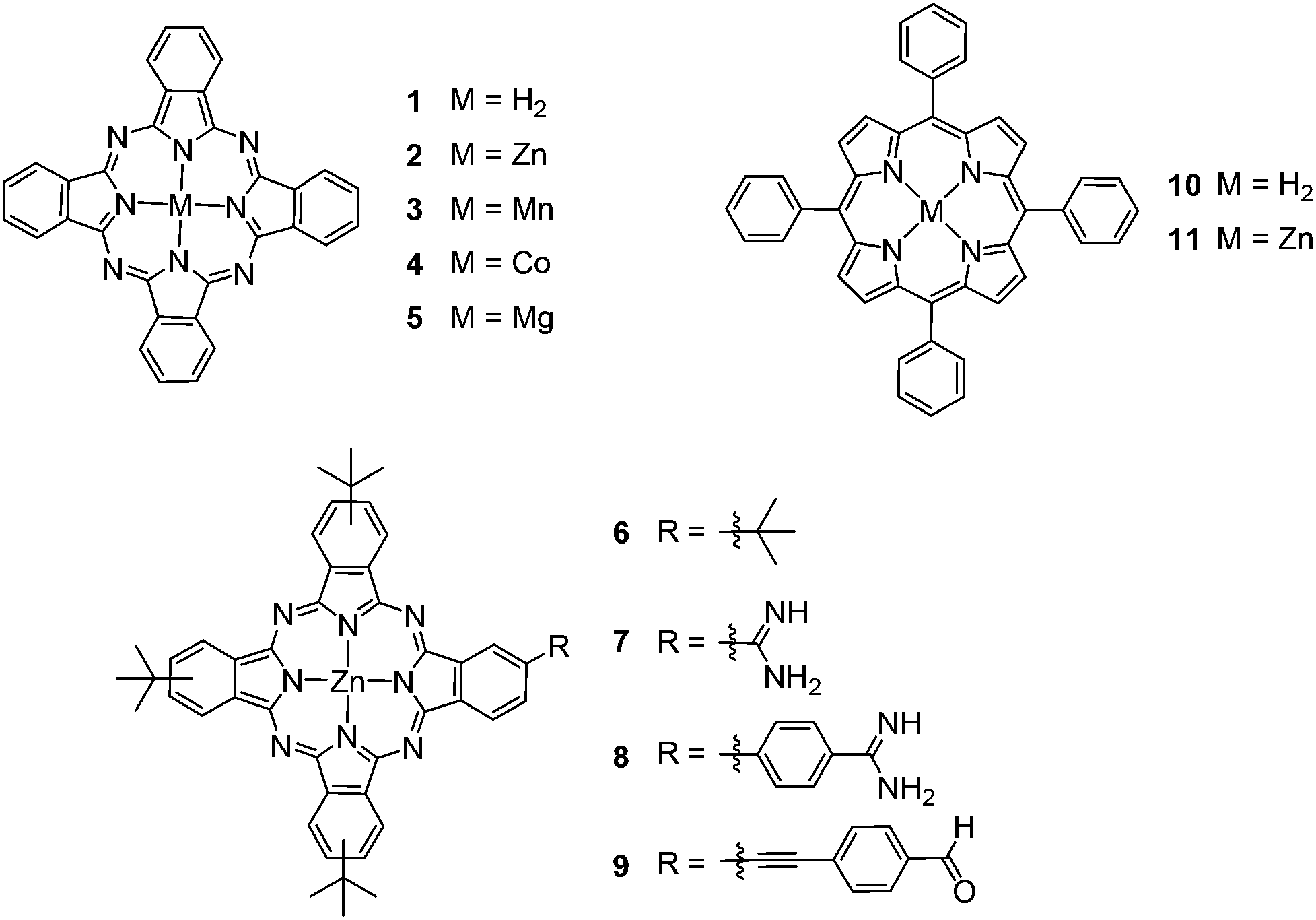 | ||
| Fig. 1 Functionalized and non-functionalized free-base and M(II) Pors/Pcs (M = Zn, Mn, Co, Mg). | ||
Samples 1–6, 10 and 11 were purchased from Sigma-Aldrich. Amidine-substituted Zn(II)Pcs 737 and 8, aldehyde-substituted Zn(II)Pc 9,38,39 and Zn(II)Por dimer, -trimers, -pentamer and -hexamer 12–1640 were synthesized by using published procedures. All solvents used were of HPLC grade purity. DMF-d7 (C3D7NO) was purchased from Deutero GmbH, Germany and DCOONa from Sigma-Aldrich.
The ESI experiments were conducted with an ESI-quadrupole time-of-flight (qToF) mass spectrometer (micrOTOF-Q II, Bruker, Bremen, Germany) and an ESI-quadrupole ion trap instrument (esquire6000, Bruker, Bremen, Germany). Both instruments have been employed in earlier Por related studies.23,41–43 Further details can be found in the ESI.†
Results and discussion
Metal–ligand axial coordination of the formate anion to Zn(II)Pors/Pcs
Fig. 2a shows the negative-ion ESI mass spectrum of aldehyde-substituted Zn(II)Pc 9 from DMF/DCM solution upon addition of HCOONa. The spectrum is characterized by one major signal at m/z 917.6, which corresponds to the mass of derivative 9 (872.3 Da) incorporating one formate anion (45 Da). Comparison of measured and simulated isotope patterns confirms the addition of the formate anion to 9. Further confirmation is obtained in two labelling experiments. Firstly, the experiment was performed using DMF-d7, which led to exactly the same outcome as obtained in Fig. 2a, excluding any incorporation of the solvent entities in the resulting ion. Secondly, HCOONa was replaced by DCOONa, which led to a shift of the signal by 1 Da to m/z 918.7 (Fig. 2b), in line with the addition of DCOO− to compound 9. The formate adduct anion 9·HCOO− shows an under collision-induced dissociation (CID) loss of CO2 accompanied by H− transfer to Zn(II)Pc 9, resulting in the species 9·H− at m/z 873.2 (Fig. 2c). The transfer of hydride anions to zinc (and other metals) is generally well-documented and known in the literature.44,45 But upon further collisional activation, the ion 9·H− at m/z 873.2 dissociates additionally by H˙ loss into the radical anion of Zn(II)Pc 9˙− at m/z 872.1 (MS3, Fig. 2e). In this way, the true molecular anion of Pc 9 is generated, without the need for 9 to be reduced in the ESI process. Interestingly, the formate radical, i.e. the carrier of the extra electron in the formate anion, has a higher electron affinity (EA) (3.5 eV)46 than Zn(II)Pc 9, thus direct electron transfer from the formate anion to 9 is prevented. The EA of Zn(II)Pc 9 is unknown, but we assume that it is similar to the EA values established for other divalent M(II)Pors (EA = 1.5–2.1 eV).47–49 The same scenario is observed for the addition of common monoatomic anions like Cl− (EA = 3.6 eV), Br− (EA = 3.4 eV) and I− (EA = 3.0 eV).50 In this case, CID would result in the loss of the charge carrier without any observable analyte ions. Only the CO2 loss from the formate anion enables H− transfer to the Zn(II)Pc. The EA of the hydrogen atom amounts to only 0.76 eV,51 so that electron transfer to the Zn(II)Por/Zn(II)Pc (EA = 1–2 eV) now becomes feasible. In comparison, the chloride and bromide adduct of Zn(II)Pc 7 and 9 only show disappearance of the precursor ion with increasing CID energy since the only fragment ions, Cl− and Br−, lie below the detection limit of both instruments used in this study.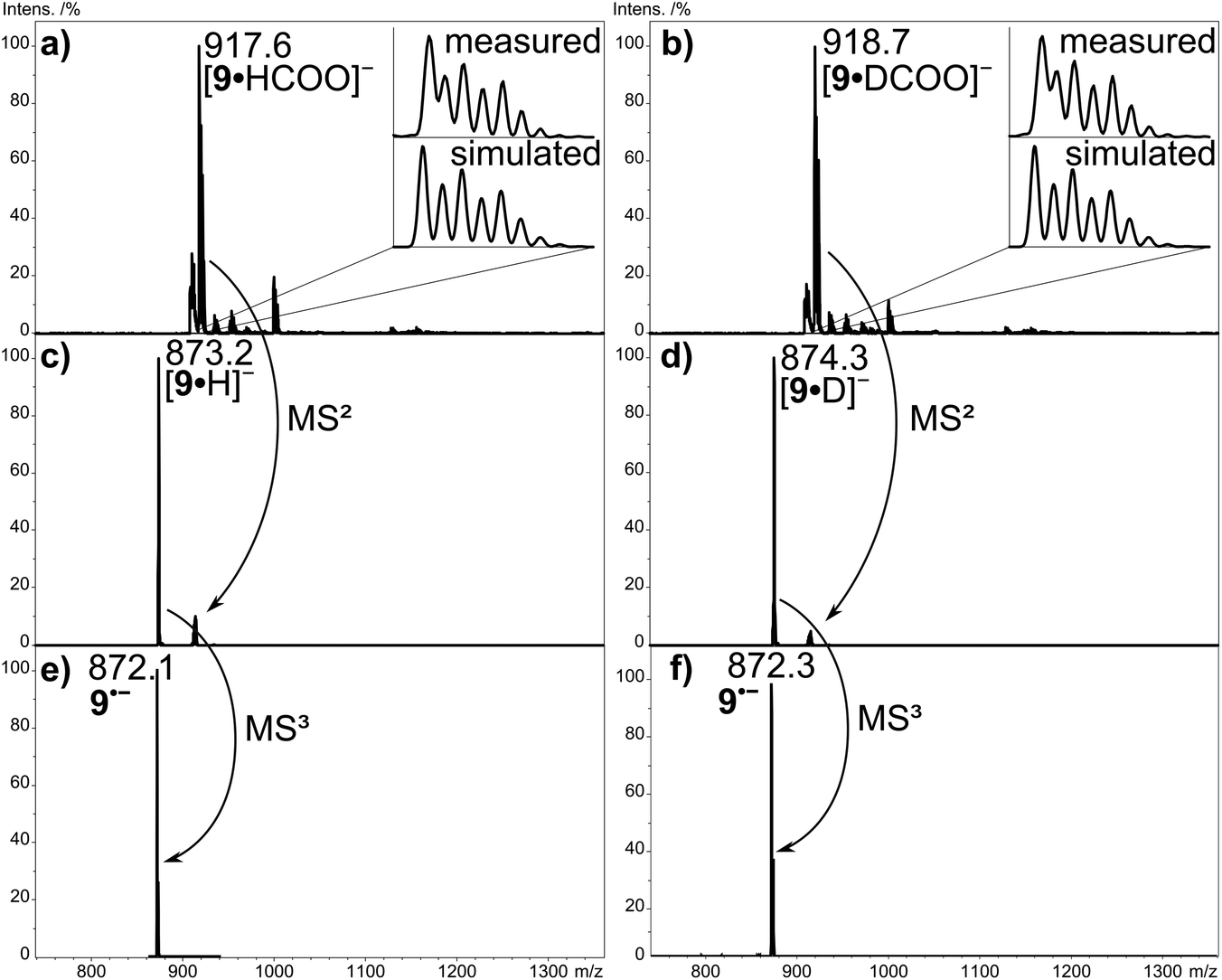 | ||
| Fig. 2 Negative-ion ESI ion trap mass spectra of aldehyde-substituted Zn(II)Pc 9 with (a) sodium formate (NaOOCH) and (b) deuterated sodium formate (NaOOCD). CID (MS2) of (c) the [9·HCOO]− and (d) [9·DCOO]− species. (e) CID (MS3) of the [9·H]− and (f) [9·D]− adduct. | ||
To confirm that the formate anion coordination to the M(II)Por/M(II)Pc occurs exclusively at the metal center, free-base Pc 1 (Fig. 3c), tetra(tert-butyl) Zn(II)Pc 6 (Fig. 3d), free-base tetraphenylPor 10 (Fig. 3a), and Zn(II)tetraphenylPor 11 (Fig. 3b), were electrosprayed in the presence of formate. These experiments revealed that only the M(II)macrocycles showed formate addition, while the free-base macrocycles did not, thus suggesting that the interaction of the formate anion occurs through coordination to the zinc metal center.
 | ||
| Fig. 3 Negative-ion ESI qTOF mass spectra of (a) tetraphenylPor 10, (b) tetraphenylZn(II)Por 11, (c) free-base Pc 1 and (d) tetra(tert-butyl)Zn(II)Pc 6 with sodium formate in DMF. The intensities of each pair (a,b and c,d) were normalized for better comparability. | ||
All Zn(II)macrocycles displayed in Fig. 1 and Fig. 4 readily undergo the coordination of the formate anion in the negative-ion mode. In light of the pronounced formate addition in the negative-ion mode, the question arises to which extent the established positive-ion mode oxidation18–24,33–35 would occur in these samples. Therefore, the positive-ion mode formation of radical cations was investigated for all Zn(II)Pors and Zn(II)Pcs by electrospraying from acetonitrile (ACN) solutions as required for oxidation. ACN has been established as a solvent promoting ESI oxidation.24 While Zn(II)tetraphenylPor 11 is readily oxidized (Fig. S4, ESI†), in the case of unsubstituted Zn(II)Pc 2, a radical cation species was observed only in low abundance (Fig. S5, ESI†). For the other metallomacrocycles (Zn(II)Pors and Zn(II)Pcs), no formation of radical cations could be detected (Fig. S6–S8 ESI†). In summary, the oxidation process in the positive-ion mode is rather inefficient with most Zn(II)Pors and Zn(II)Pcs under investigation, while the ion formation by formate addition in the negative-ion mode is established as an abundantly occurring process.
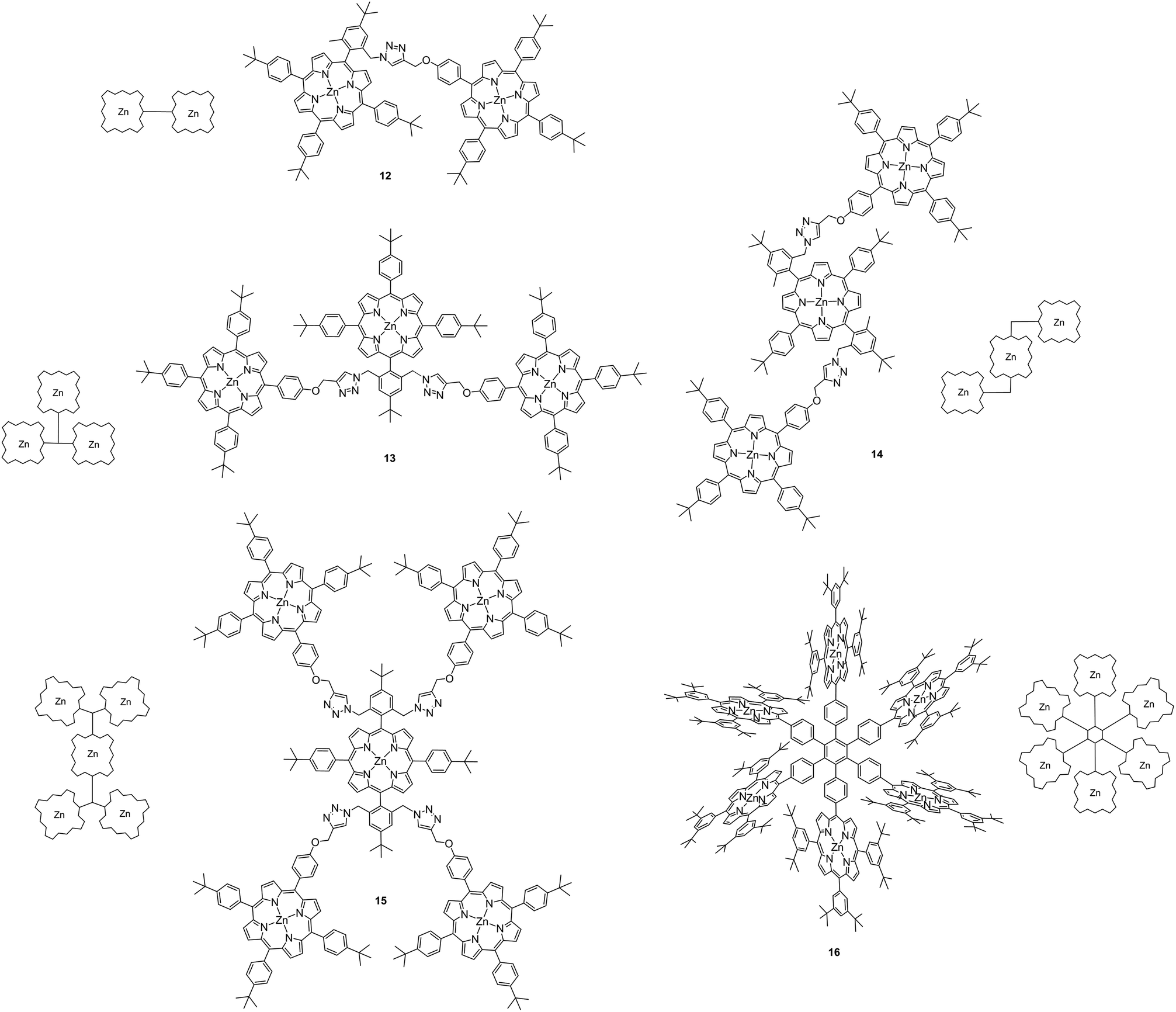 | ||
| Fig. 4 Covalently-bound porphyrin arrays with multiple zinc centers. | ||
Metal–ligand axial coordination of the formate anion to metallo(II)Pcs other than Zn(II)
The coordination of the formate anion to other central metals has also been studied by electrospraying metallo Pcs with Mn(II) (3), Mg(II) (5) and Co(II) (4) in the presence of HCOONa. All three M(II)Pcs show the addition of the formate anion. However, some important differences were observed compared to the mass spectra of the Zn(II)macrocycles. Mg(II)Pc 5 shows, similarly to the Zn(II)macrocycles, the abundant formation of a HCOO−-coordinated species, namely 5·HCOO− and to a lower extent the formation of a formate-bridged dimer ion [2 × 5·HCOO]−. Dimeric ions of this type will be discussed in detail below. For Mn(II)Pc 3 the formate addition is also abundantly observed. Additionally, a singly charged monomer with two formate anions can be detected (Fig. S2a, ESI†). Due to the two negative charges of the formate anions, the manganese ion has to be in a III+ oxidation state, resulting in [Mn(III)Pc·2 × (HCOO)]−. Differently from the Zn(II)macrocycles case, the formation of a Pc dimeric species incorporating a formate anion and an extra oxygen atom was detected. However, Mn(II)Pors are known to form μ-oxo dimers in the ESI experiment,52 so that the structure of the dimeric ion would correspond most likely to an oxygen-bridged dimer of Pc 3 with external formate anion addition, [3-O-3·HCOO]−, with Mn in a III+ oxidation state.53,54 The structure of this ion was confirmed by its CID fragmentation pattern and will be addressed below within the discussion of dimeric ions. Co(II)Pc 4 shows the intact formate adduct ion only to a minor amount. Instead, radical anion [4]˙− and hydride transfer [4·H]− are more prominently observed (Fig. S2b, ESI†). The presence of [4·Cl]− and [2 × 4·Cl]− adducts was also observed due to the presence of Cl− traces. Furthermore, [4·HCOONa]˙− was observed.From these data, it is clear that the addition of the formate anion to metallo(II)Pcs is not limited to zinc species. While Mg(II) and Mn(II)Pcs show a similar behavior to zinc as the central metal, there are more pronounced side reactions with Co(II)Pcs, which suggests that the efficiency of the formate addition to this metal should be checked with appropriate model compounds prior to the analytical application.
Metal–ligand axial coordination of the formate anion to metallo(II)Pors containing multiple metal centers
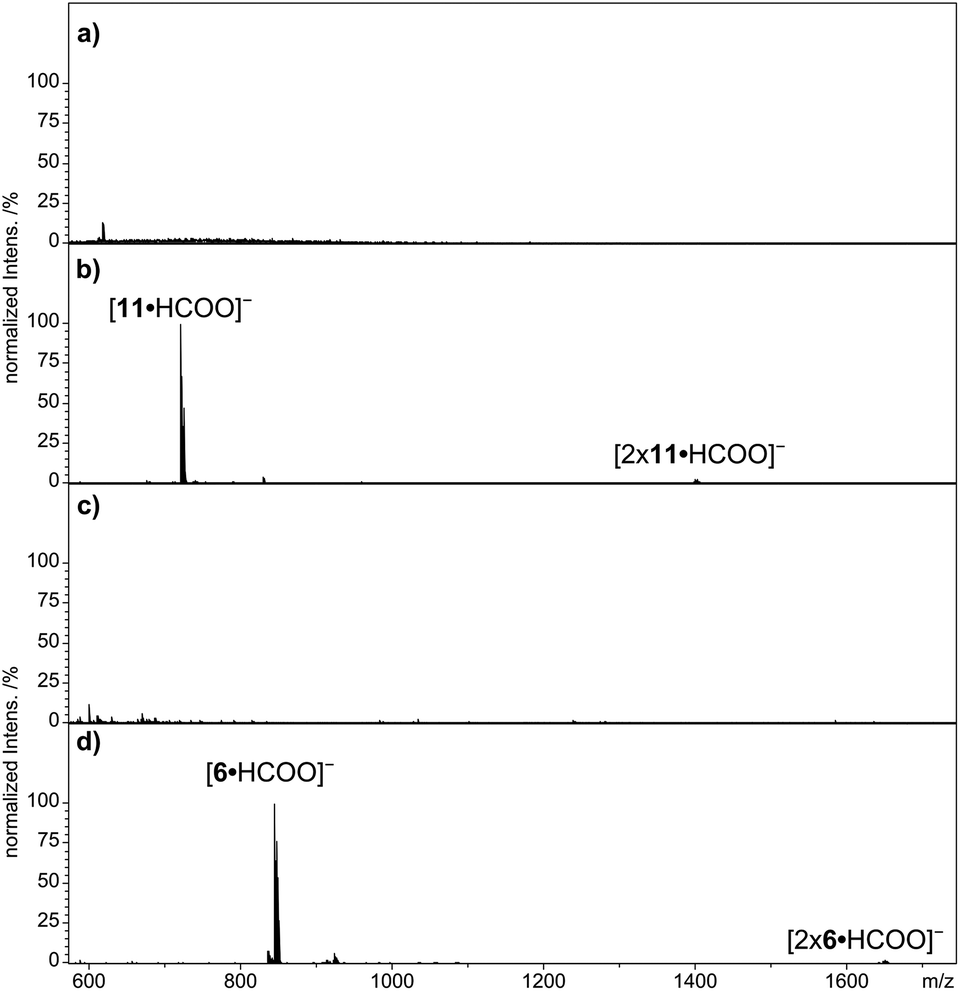
The multi Por arrays 12–16 (Fig. 4), which incorporate two (12), three (13 and 14), five (15) or six (16) zinc(II) centers have been electrosprayed in the presence of sodium formate. Fig. 5 shows the resulting negative-ion ESI mass spectra. For none of the multi Por derivatives the formation of radical cations or higher oxidized ions were observed in the positive-ion mode under the conditions that are known to favor electrochemical oxidation.24 In contrast, all multi Por arrays readily show the addition of the formate anion (Fig. 5). However, the most interesting finding is that despite the occurrence of multiple formate anion attachment, the maximum number of formate anions added does not correspond to the number of Por units within each array. For instance, covalently-bridged Zn(II)Por dimer 12 attaches only one formate anion (Fig. 5a). A signal for the doubly negatively charged ion [12·2 × HCOO]2− could not be observed. Derivatives 13 and 14 with three Zn(II)Pors show the attachment of one and two formate anions, with the doubly charged ion being even more abundantly formed than the single formate addition (Fig. 5b). Again a maximum of three formate anions, matching the number of Por units, could not be detected. In the case of derivatives 15 and 16, having five and six Por units, respectively, species corresponding to the attachment of three formate anions were observed (Fig. 5c and d). For these Pors, the attachment of a single formate anion is only detected in low abundance. For 15, the most intense peak corresponds to the attachment of two, and for 16, of three formate anions. Evidently, addition of three formate anions to hexa Zn(II)Por 16 is more favorable than in the case of Zn(II)Por pentamer 15. Moreover, the experiment was carried out in a reaction mixture containing Zn(II)Por 16 and three structurally related hexameric Zn(II)Por derivatives in the presence of the formate anion (Scheme S13, ESI†). The data revealed the presence of peaks corresponding to the coordination of three formate anions to each of the four hexameric Zn(II)Pors (Fig. 12, ESI†).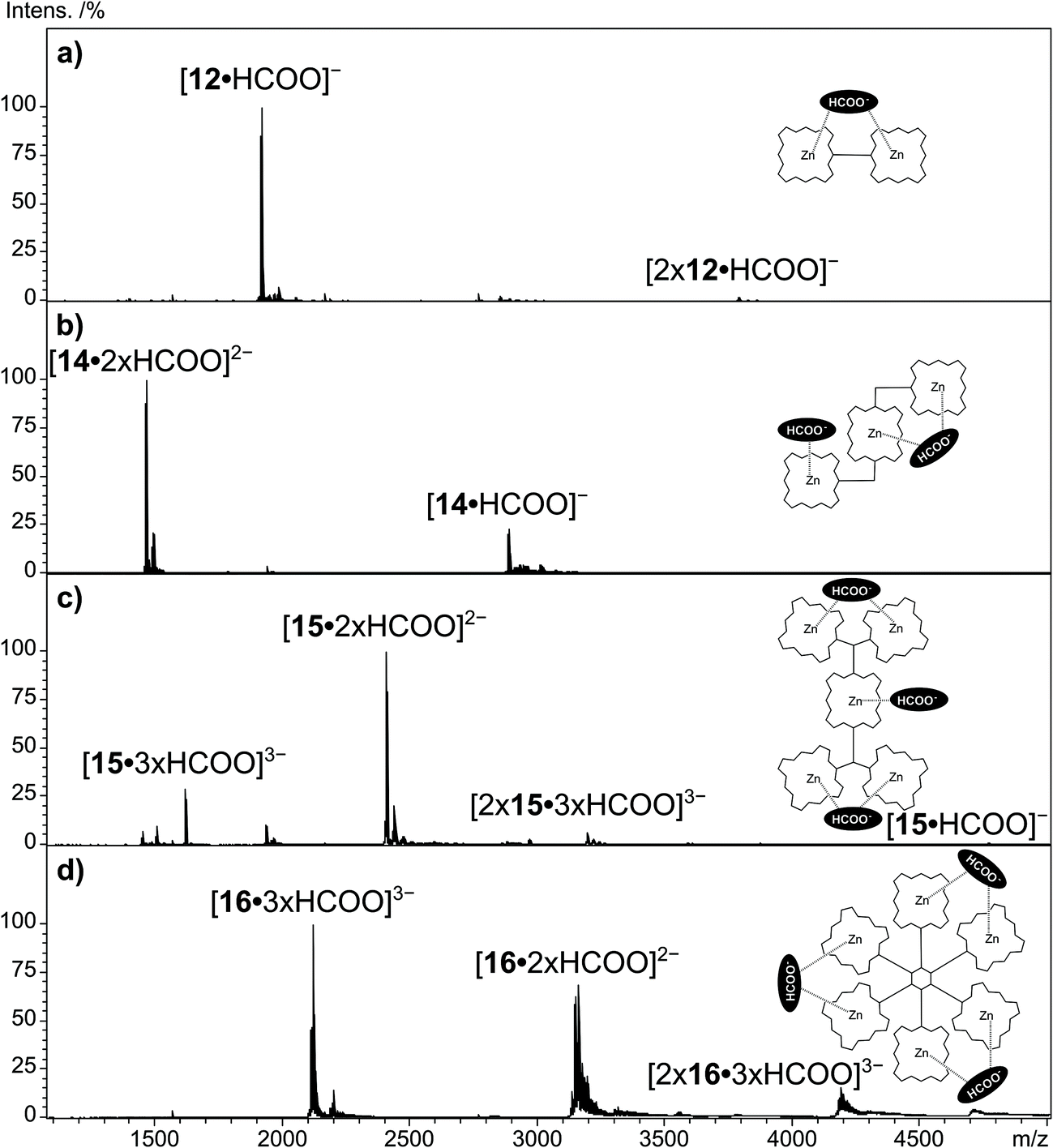 | ||
| Fig. 5 Negative-ion ESI qTOF mass spectra of multi Por zinc derivatives (a) 12, (b) 14, (c) 15 and (d) 16 with addition of sodium formate in DMF. | ||
In order to explain the number of formate anions attached to the multi Zn(II)Por derivatives, a mechanism is postulated involving the chelation of a formate anion by two zinc metal centers within the same multi Por array, in a sandwich-type arrangement. As a result, a maximum of one and three formate anion attachments was observed in the case of a Zn(II)Por dimer and a hexamer, respectively. On the other hand, when using multi Zn(II)Por arrays containing an uneven number of Por units, such as in the case of derivatives 13–15, then the “unpaired” macrocycle not involved in the sandwich-type chelation of the formate anion, can individually bind a HCOO− anion. Careful inspection of the mass spectra in Figure 5 reveals that also small amounts of multiply charged Por dimers are formed. These come in the form of [2xZn(II)Por·3xHCOO]3− trianionic species and are seen for the trimeric Pors and larger arrays. In these ions, two Por arrays are most likely kept together by formate anions that function as supramolecular bridges between them. Considering the bonding motif of the suggested sandwich structure in which one formate connects with two Pors, it is not surprising, that for all mononuclear Zn(II)macrocycles in this study, dimers of the type [2x(Zn(II)Por/Zn(II)Pc)·HCOO]− could be found. However, CID experiments conducted on these dimeric ions reveal that the sandwich is not the only bonding motif of these dimeric species and CID experiments enable the distinction between the isomeric ions.
Structure and fragmentation behavior of Zn(II)Por/Zn(II)Pc supramolecular dimers complexing one formate anion
Fig. 6 shows the CID daughter ion mass spectra of selected [2 × (Zn(II)Por/Zn(II)Pc)·HCOO]− precursor ions. The Zn(II)-tetraphenylPor 11 is an example of the formation of a formate-bridged dimer, [11·HCOO·11]−, featuring the loss of one neutral metallo Por while the other Zn(II)Por retains the formate anion as the charge carrier (Fig. 6a). In this case, the loss of CO2, detected in the case of the monomers, forming [112·H]− is not observed. Evidently, the sandwiched formate anion does not show CO2 loss. The same fragmentation pattern is also observed for Zn(II)Pc derivative 6 (Fig. S9, ESI†). However, the fragmentation pattern changes when the Zn(II)Pc presents a heteroatom at its peripheral position (not the case for 6 and 11), which could coordinate to a neighboring zinc center and thus connecting two metallomacrocycles directly.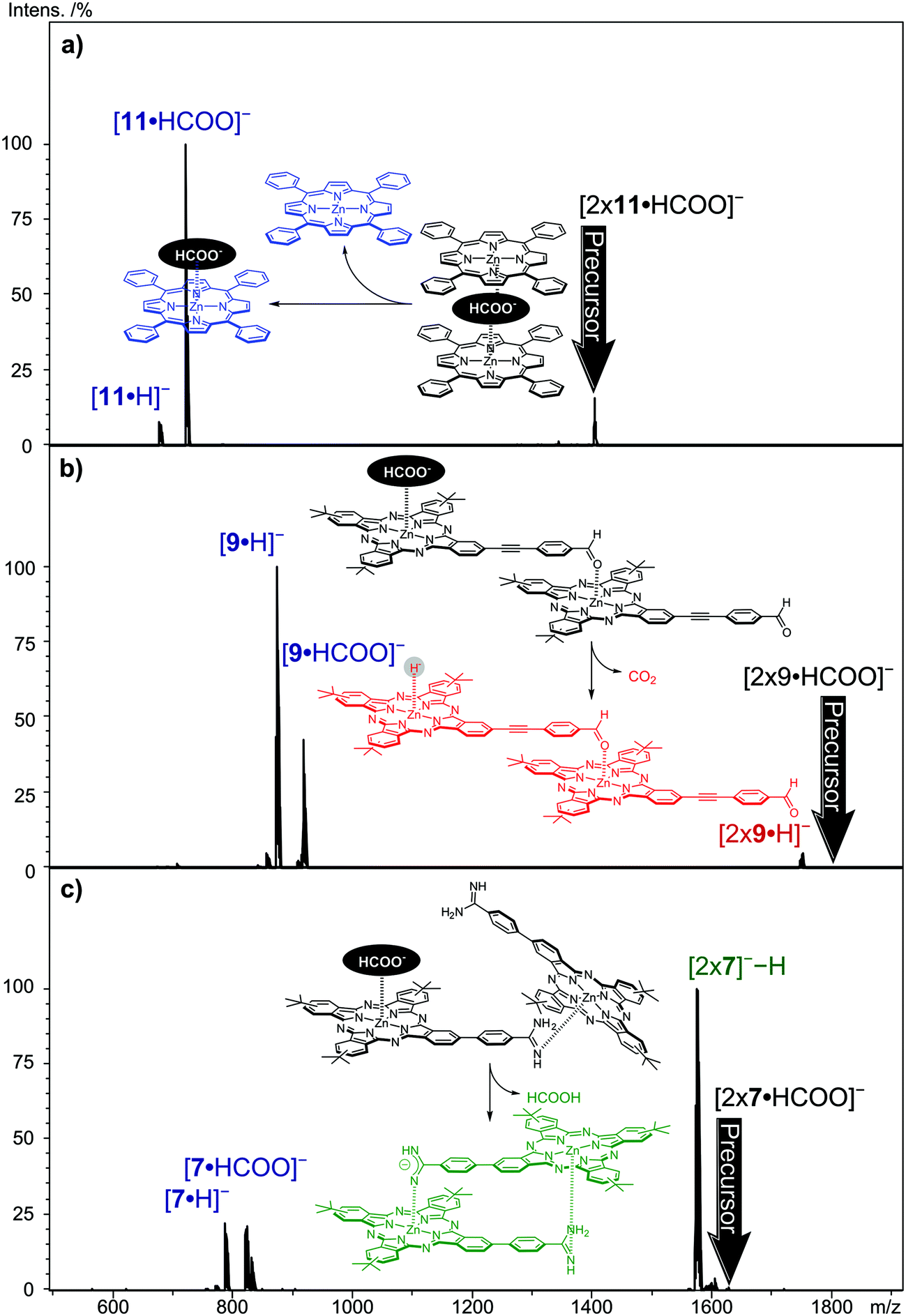 | ||
| Fig. 6 Negative-ion CID (MS2) of the formate-containing dimers (a) [2 × 11·HCOO]−, (b) [2 × 9·HCOO]−, and (c) [2 × 7·HCOO]−. | ||
CID (MS2) on aldehyde-substituted Zn(II)Pc 9, by selecting the ion corresponding to [2 × 9·HCOO]−, shows, besides the fragmentation pattern of a formate-bridged species, one additional signal resulting from the CO2 loss from the dimeric ions, leading to [92·H]−, representing a Zn(II)Pc2 dimer with a hydride ion (Fig. 6b). It is most likely to assume that the CO2 loss has occurred from a “non-bridging” formate anion attached to only one zinc center, while the two Zn(II)Pcs are directly linked together. The subsequent decay, the loss of neutral hydrogen, is not observed for the dimer, probably the competing dissociation of the dimer by the loss of one Zn(II)Pc unit is more feasible. Support for the “outside” coordination of HCOO− to this dimer comes from the dissociation behavior of the HCOO−-coordinated dimer of Mn(II)Pc 3, featuring the extra oxygen atom. This ion most likely possesses the [3-O-3-HCOO]− structure in which the Pc units are covalently connected and which shows abundantly the loss of CO2 in the CID experiment, perfectly in line with its assumed structure (Fig. S11, ESI†).
The CID experiment with Zn(II)Pc 7 points even more clearly to the presence of a directly connected dimer with “outside” attachment of formate (Fig. 6c). In this case, however, it is not CO2 loss, but the loss of formic acid (HCOOH) which is observed, leading to the [72]−−H ion. The formation of the [72]−−H ion provides evidence of the non-anion-bridged, directly connected Pc-dimer. Despite the high basicity of amidine,55,56 the formate anion seems to be able to abstract a proton, leaving the amidine negatively charged. However, the formation of the amidine anion must receive considerable support from interactions with the adjacent, neighboring zinc center. We emphasize that the monomeric formate adduct, 7·HCOO−, is not showing formic acid but CO2 loss, thus, deprotonation without the second Zn(II)Pc molecule is not observed. It is known that amidine-substituted Zn(II)Pc 7 forms dimers in solution with high association constants.37 In the dimer, each amidine moiety axially coordinates to a zinc metal center of the other Zn(II)Pc in a head-to-tail fashion. The addition of carboxylic acid in solution leads to strong hydrogen bonding between the amidine group and carboxylic acid. In the gas-phase dissociations of [72·HCOO]−, the formate anion may interact with the amidine group and an additional stabilizing interaction with the zinc center of the neighboring molecule may facilitate the formation and subsequent loss of formic acid. Amidine-substituted Zn(II)Pc 8 and 7 show similar behavior, but the formation of [82]−−H species is less abundant. We assume that the longer linker between the zinc center and the amidine group in derivative 8 results in a more loosely bound dimer. For both [2 × 7·HCOO]− and [2 × 8·HCOO]− species, the loss of formic acid in CID MS2 experiments clearly indicates a direct linkage of the two monomer units with the formate anion coordinated to only one zinc metal center and not acting as a bridging unit.
The present experiments do not allow the establishment of the relative amount of the two isomeric dimers, that is formate anion-bridged [Zn(II)Pc·HCOO·Zn(II)Pc]−versus directly connected (Zn(II)Pc)2 with the “externally” attached formate anion [(Zn(II)Pc)2·HCOO]−. The presence of the directly connected [(Zn(II)Pc)2·HCOO]− dimer with “externally” coordinated formate anions is indicated by the diagnostic dimeric fragment ion caused by the loss of CO2 or HCOOH. However, the quantification of this isomer in relation to the formate anion-bridged [Zn(II)Pc·HCOO·Zn(II)Pc]− isomer is prevented by the fact that both isomers can fragment into the same daughter ion [Zn(II)Pc·HCOO]−.
Finally, it should be stressed that only the use of the formate anion as a charge carrier allows the distinction of the isomeric dimer ions. A mono-atomic anion as the charge carrier would not necessarily induce different fragmentations for anion-bridged vs. dimers with direct monomer-to-monomer bonding. To test the validity of this assumption, the chloride anion was used instead of the formate as the charge carrying anion with the dimer of amidine-substituted Zn(II)Pc 7. The resulting fragmentation pattern is shown in Fig. S3 (ESI†). The dimer fragments exclusively into the chloride-carrying monomer. Assuming that directly connected dimer ion species are formed in equal amounts in the experiment with chloride, there is no indication that would allow the distinction of the isomeric dimer ions.
Conclusion
In this work, we propose the axial coordination of the formate anion to different metallated Pors, Pcs and multi Por species containing divalent metal centers (Zn, Mn, Mg, Co) as an effective and superior strategy for the detection of these macrocycles in ESI experiments. From these studies, a general rule for the coordination of the formate anion to the metallated macrocycles can be revealed. While in the case of mononuclear Zn(II)Por/Zn(II)Pc species, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and 2:1 macrocycle-to-formate complexes were observed, in the case of the multinuclear Zn(II)Por systems, complexes with distinct macrocycle-to-formate ion stoichiometry were found. In particular, the maximum formate coordination amounts to “n” for multi Por arrays with “2n” zinc centers and to “n + 1” for “2n + 1” zinc centers. The adducts of Zn(II)Por/Zn(II)Pc with HCOO− can produce the molecular radical anion of the Zn(II)Por/Zn(II)Pc, without the need for electrochemical reduction to occur even though the formate radical has a larger electron affinity than the Zn(II)Por/Zn(II)Pc. For these systems, the electron transfer proceeds via collision-induced CO2 loss in conjunction with hydride transfer to Zn(II)Por/Zn(II)Pc, followed in a second step by hydrogen atom loss and electron transfer to the Zn(II)Por/Zn(II)Pc. Interestingly, in the case of Zn(II)Pc species bearing a peripheral heteroatom, the ESI experiments showed the formation of dimeric species through coordination of the heteroatom on one macrocycle to the zinc metal of a nearby macrocycle accompanied by the axial coordination of one formate ion. The CID pattern of their formate adducts is different and shows additional CO2/HCOOH losses involving the formate moiety. Monoatomic anions as charge carriers would not allow the distinction of these dimeric ions. In future, we want to evaluate the utilization of further organic bases like acetate and propionate, which did not show the same ease of addition as formate under similar conditions and, therefore, were not included in the present study.
:1 and 2:1 macrocycle-to-formate complexes were observed, in the case of the multinuclear Zn(II)Por systems, complexes with distinct macrocycle-to-formate ion stoichiometry were found. In particular, the maximum formate coordination amounts to “n” for multi Por arrays with “2n” zinc centers and to “n + 1” for “2n + 1” zinc centers. The adducts of Zn(II)Por/Zn(II)Pc with HCOO− can produce the molecular radical anion of the Zn(II)Por/Zn(II)Pc, without the need for electrochemical reduction to occur even though the formate radical has a larger electron affinity than the Zn(II)Por/Zn(II)Pc. For these systems, the electron transfer proceeds via collision-induced CO2 loss in conjunction with hydride transfer to Zn(II)Por/Zn(II)Pc, followed in a second step by hydrogen atom loss and electron transfer to the Zn(II)Por/Zn(II)Pc. Interestingly, in the case of Zn(II)Pc species bearing a peripheral heteroatom, the ESI experiments showed the formation of dimeric species through coordination of the heteroatom on one macrocycle to the zinc metal of a nearby macrocycle accompanied by the axial coordination of one formate ion. The CID pattern of their formate adducts is different and shows additional CO2/HCOOH losses involving the formate moiety. Monoatomic anions as charge carriers would not allow the distinction of these dimeric ions. In future, we want to evaluate the utilization of further organic bases like acetate and propionate, which did not show the same ease of addition as formate under similar conditions and, therefore, were not included in the present study.
Acknowledgements
Financial support from the Deutsche Forschungsgemeinschaft (DFG)-SFB 953 “Synthetic Carbon Allotropes”, the MINECO, Spain (CTQ-2014-52869-P) and the Comunidad de Madrid (S2013/MIT-2841 FOTOCARBON, T.T.) is acknowledged.References
-
K. M. Kadish, K. M. Smith and R. Guilard, The Porphyrin Handbook, Academic Press, Amsterdam, 2003 Search PubMed
.
- Y. T. Li, Y. L. Hsieh, J. D. Henion and B. Ganem, J. Am. Soc. Mass Spectrom., 1993, 4, 631–637 CrossRef CAS PubMed
- Y.-L. P. Ow, D. R. Green, Z. Hao and T. W. Mak, Nat. Rev. Mol. Cell Biol., 2008, 9, 532–542 CrossRef CAS PubMed
- M.-A. Gilles-Gonzalez and G. Gonzalez, J. Appl. Physiol., 2004, 96, 774–783 CrossRef CAS PubMed
- T. Shimizu, D. Huang, F. Yan, M. Stranava, M. Bartosova, V. Fojtíková and M. Martínková, Chem. Rev., 2015, 115, 6491–6533 CrossRef CAS PubMed
- M. F. Ali, H. Perzanowski, A. Bukhari and A. A. Al-Haji, Energy Fuels, 1993, 7, 179–184 CrossRef CAS
-
R. Bonnett, Chemical Aspects of Photodynamic Therapy, Gordon and Breach Science Publishers, Amsterdam, 2000 Search PubMed
- M. Jurow, A. E. Schuckman, J. D. Batteas and C. M. Drain, Coord. Chem. Rev., 2010, 254, 2297–2310 CrossRef CAS PubMed
- D. M. Guldi, Chem. Soc. Rev., 2002, 31, 22–36 RSC
- G. Bottari, G. de la Torre, D. M. Guldi and T. Torres, Chem. Rev., 2010, 110, 6768–6816 CrossRef CAS PubMed
- I. Beletskaya, V. S. Tyurin, A. Y. Tsivadze, R. Guilard and C. Stern, Chem. Rev., 2009, 109, 1659–1713 CrossRef CAS PubMed
- A. D'Urso, M. E. Fragala and R. Purrello, Chem. Commun., 2012, 48, 8165–8176 RSC
- M. Trapani, M. R. Plutino, G. Sabatino, I. Occhiuto, A. Borriello, G. De Luca and L. M. Scolaro, Chem. Commun., 2012, 48, 5136–5138 RSC
- E. Alessio, M. Casanova, E. Zangrando and E. Iengo, Chem. Commun., 2012, 48, 5112–5114 RSC
- R. S. Brown and C. L. Wilkins, Anal. Chem., 1986, 58, 3196–3199 CrossRef CAS PubMed
- N. Srinivasan, C. A. Haney, J. S. Lindsey, W. Zhang and B. T. Chait, J. Porphyrins Phthalocyanines, 1999, 03, 283–291 CrossRef CAS
- E. Stulz, C. C. Mak and J. K. M. Sanders, J. Chem. Soc., Dalton Trans., 2001, 604–613 RSC
- G. J. Van Berkel and F. Zhou, Anal. Chem., 1994, 66, 3408–3415 CrossRef CAS
- G. Van Berkel and F. Zhou, J. Am. Soc. Mass Spectrom., 1996, 7, 157–162 CrossRef CAS PubMed
- G. Van Berkel and V. Kertesz, Anal. Bioanal. Chem., 2012, 403, 335–343 CrossRef CAS PubMed
- E. Mishra, J. Worlinsky, T. Gilbert, C. Brückner and V. Ryzhov, J. Am. Soc. Mass Spectrom., 2012, 23, 1428–1439 CrossRef CAS PubMed
- E. Mishra, J. Worlinsky, T. Gilbert, C. Brückner and V. Ryzhov, J. Am. Soc. Mass Spectrom., 2012, 23, 1135–1146 CrossRef CAS PubMed
- D. Lungerich, J. F. Hitzenberger, M. Marcia, F. Hampel, T. Drewello and N. Jux, Angew. Chem., Int. Ed., 2014, 53, 12231–12235 CrossRef CAS PubMed
- V. E. Vandell and P. A. Limbach, J. Mass Spectrom., 1998, 33, 212–220 CrossRef CAS
- F. O. Ayorinde, D. Z. Bezabeh and I. G. Delves, Rapid Commun. Mass Spectrom., 2003, 17, 1735–1742 CrossRef CAS PubMed
- F. O. Ayorinde, P. Hambright, T. N. Porter and Q. L. Keith, Rapid Commun. Mass Spectrom., 1999, 13, 2474–2479 CrossRef CAS PubMed
- C. Hlongwane, I. G. Delves, L. W. Wan and F. O. Ayorinde, Rapid Commun. Mass Spectrom., 2001, 15, 2027–2034 CrossRef CAS PubMed
- J. J. A. van Kampen, P. C. Burgers, R. de Groot and T. M. Luider, Anal. Chem., 2006, 78, 5403–5411 CrossRef CAS PubMed
- J. J. A. van Kampen, T. M. Luider, P. J. A. Ruttink and P. C. Burgers, J. Mass Spectrom., 2009, 44, 1556–1564 CrossRef CAS PubMed
- S. Trimpin, S. Keune, H. J. Räder and K. Müllen, J. Am. Soc. Mass Spectrom., 2006, 17, 661–671 CrossRef CAS PubMed
- S. G. Kotsiris, Y. V. Vasil'ev, A. V. Streletskii, M. Han, L. P. Mark, O. V. Boltalina, N. Chronakis, M. Orfanopoulos, H. Hungerbühler and T. Drewello, Eur. J. Mass Spectrom., 2006, 12, 397–408 CrossRef CAS PubMed
- G. J. Van Berkel, S. A. McLuckey and G. L. Glish, Anal. Chem., 1991, 63, 1098–1109 CrossRef CAS
- T. D. McCarley, M. W. Lufaso, L. S. Curtin and R. L. McCarley, J. Phys. Chem. B, 1998, 102, 10078–10086 CrossRef CAS
- M. Schäfer, M. Drayß, A. Springer, P. Zacharias and K. Meerholz, Eur. J. Org. Chem., 2007, 5162–5174 CrossRef
- A. E. M. C. R. Vessecchi, T. Guaratini, P. Colepicolo, S. E. Galembeck and N. P. Lopes, Mini-Rev. Org. Chem., 2007, 4, 75–87 CrossRef
- F. Ghani, J. Kristen and H. Riegler, J. Chem. Eng. Data, 2012, 57, 439–449 CrossRef CAS
- M. García-Iglesias, K. Peuntinger, A. Kahnt, J. Krausmann, P. Vázquez, D. González-Rodríguez, D. M. Guldi and T. Torres, J. Am. Chem. Soc., 2013, 135, 19311–19318 CrossRef PubMed
- M. Ince, M. V. Martinez-Diaz, J. Barbera and T. Torres, J. Mater. Chem., 2011, 21, 1531–1536 RSC
- G. Bottari, D. Olea, C. Gómez-Navarro, F. Zamora, J. Gómez-Herrero and T. Torres, Angew. Chem., Int. Ed., 2008, 47, 2026–2031 CrossRef CAS PubMed
-
N. Lang, Ph. D. thesis, Friedrich-Alexander-University Erlangen-Nürnberg, 2010
- J. Li, K. Duerr, M. S. von Gernler, N. Jux, I. Ivanović-Burmazović and T. Drewello, Int. J. Mass spectrom., 2013, 354–355, 406–413 CrossRef CAS
- K. Duerr, J. Olah, R. Davydov, M. Kleimann, J. Li, N. Lang, R. Puchta, E. Hubner, T. Drewello, J. N. Harvey, N. Jux and I. Ivanovic-Burmazovic, Dalton Trans., 2010, 39, 2049–2056 RSC
- K. Duerr, O. Troeppner, J. Olah, J. Li, A. Zahl, T. Drewello, N. Jux, J. N. Harvey and I. Ivanovic-Burmazovic, Dalton Trans., 2012, 41, 546–557 RSC
- G. N. Khairallah, C. C. L. Thum, D. Lesage, J.-C. Tabet and R. A. J. O'Hair, Organometallics, 2013, 32, 2319–2328 CrossRef CAS
- G. Rodríguez-Blanco, K. J. Jobst, T. M. Luider, J. K. Terlouw and P. C. Burgers, ChemPlusChem, 2013, 78, 1184–1189 CrossRef
- E. H. Kim, S. E. Bradforth, D. W. Arnold, R. B. Metz and D. M. Neumark, J. Chem. Phys., 1995, 103, 7801–7814 CrossRef CAS
- H. L. Chen, P. E. Ellis, T. Wijesekera, T. E. Hagan, S. E. Groh, J. E. Lyons and D. P. Ridge, J. Am. Chem. Soc., 1994, 116, 1086–1089 CrossRef CAS
- R. S. Ruoff, K. M. Kadish, P. Boulas and E. C. M. Chen, J. Phys. Chem., 1995, 99, 8843–8850 CrossRef CAS
- M.-S. Liao and S. Scheiner, J. Chem. Phys., 2002, 117, 205–219 CrossRef CAS
- R. S. Berry and C. W. Reimann, J. Chem. Phys., 1963, 38, 1540–1543 CrossRef
- J. C. Rienstra-Kiracofe, G. S. Tschumper, H. F. Schaefer, S. Nandi and G. B. Ellison, Chem. Rev., 2002, 102, 231–282 CrossRef CAS PubMed
- T. Gozet, L. Huynh and D. K. Bohme, J. Mass Spectrom., 2010, 45, 35–42 CAS
- O. V. Dolotova, N. I. Bundina, O. L. Kaliya and E. A. Lukyanets, J. Porphyrins Phthalocyanines, 1997, 1, 355–366 CrossRef CAS
- L. H. Vogt, A. Zalkin and D. H. Templeton, Science, 1966, 151, 569–570 CAS
- E. P. L. Hunter and S. G. Lias, J. Phys. Chem. Ref. Data, 1998, 27, 413–656 CrossRef CAS
- E. D. Raczyńska, M. Decouzon, J. F. Gal, P. C. Maria, G. Gelbard and F. Vielfaure-Joly, J. Phys. Org. Chem., 2001, 14, 25–34 CrossRef
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5an02148k |
| This journal is © The Royal Society of Chemistry 2016 |