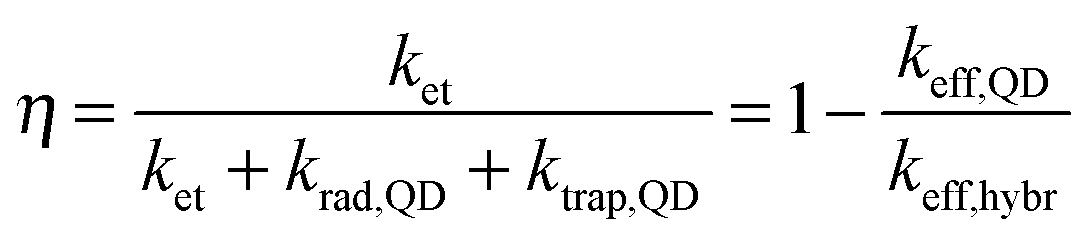Efficient charge transfer in solution-processed PbS quantum dot–reduced graphene oxide hybrid materials†
Received
22nd April 2015
, Accepted 7th June 2015
First published on 8th June 2015
Abstract
Quantum dot–graphene hybrid materials have attracted significant interest due to the unique synergy of the optical properties of colloidal quantum dots (QDs) and the transport properties of graphene. This stimulated the development of low-cost and up-scalable solution-processed strategies for hybrid materials with potential applications in light harvesting and optoelectronic devices. Herein, we report a versatile covalent linking-based approach for the functionalization of reduced graphene oxide (rGO), to prepare a variety of QD–rGO hybrid dispersions with QDs of different sizes and compositions (PbS, PbS–CdS and CdSe QDs), and shapes (CdSe–CdS dot-in-rods). We achieved a well-controlled QD coverage of the rGO sheets by functionalizing the rGO surface with mercapto-silane linkers. A further spectroscopic investigation of near-infrared PbS QD–rGO materials demonstrates efficient electronic coupling between both materials. The QD photoluminescence emission quenching and exciton lifetime shortening of up to 95%, together with subtle graphene Raman G-band shifts upon QD linking, support electron transfer as the dominant relaxation pathway from the QD to the rGO. The use of core–shell PbS–CdS QDs allows tuning of the transfer efficiency from 94% for a 0.2 nm thin CdS shell, down to 30% for a 1.1 nm thick shell.
Introduction
A continuing search for solution-processed materials that are low-cost and can be fabricated and applied on a large scale is paramount to drive the development of future photonic devices, such as solar cells,1–3 photodetectors,3–6 LEDs7 or lasers.8 Efforts are focused on both the optically active materials as well as charge transport layers. In this respect, since the discovery of the extraordinary electronic properties of graphene,9 it has often been incorporated as a nearly transparent and flexible electrode. In solution-based approaches, it is convenient to use graphene derivatives, such as liquid-phase exfoliated graphene and reduced graphene-oxide (rGO), with rGO offering advantages such as a large area coverage as well as tunable optoelectronic properties due to the presence of oxygen functionalities, which also favor chemical interactions.5,10–14 While the light-harvesting properties of graphene and rGO could in principle also be exploited, typically different strategies are followed, for instance, interfacing them with other materials such as colloidal quantum dots (QDs).15–19 These possess a strong and tunable light absorption and emission together with a high chemical and photophysical stability, and therefore, make excellent photoactive materials.20,21 Additionally, combining them with graphene-based materials would at the same time resolve a major shortcoming of nanosized quantum dots as QD thin films are known to suffer from inefficient carrier transport.22,23 Thus, the potential of the resulting hybrid materials clearly lies in the unique synergy of the optical properties of the QDs and the transport properties of the graphene.12,24,25
Compared to non-interacting composite systems, a clear increase of device photo-conversion efficiency and photo-catalytic activity has been demonstrated when a covalent linking is promoted between the optically active and charge transport materials.16–17,26 Among the existing approaches to prepare such hybrid materials, the most common are based on a one-pot in situ synthesis, layer-by-layer electrostatic assembly, or click-chemistry using amino-based functional linkers.17–18,26–28 Especially the latter is advantageous, allowing first the optimization of each constituent individually, followed by coupling with precisely controlled distance. Following this principle, we developed a covalent linking method that efficiently anchors a variety of QDs with different chemical compositions, sizes and shapes to rGO using short-chain silane molecules. By functionalizing rGO with (3-mercaptopropyl) trimethoxysilane (MPTS), we achieved a uniform and density-controlled QD coverage of the rGO sheets. To demonstrate the potential of the QD–rGO coupling approach, we used near infrared (NIR)-emitting PbS QDs to investigate the optoelectronic properties of PbS QD–rGO hybrid materials and the charge/energy transfer at their interface. Several groups have already focused on the understanding of energy and electron transfer processes in different Cd-based QDs and GO- or graphene-based systems.24,29–32 In contrast, PbS QDs are less studied despite their potential to significantly open up a wide spectral range of applications due to their narrow bulk band gap of 0.4 eV. Herein, we show that the proposed coupling leads to a highly efficient carrier transfer between the materials, reaching up to 95% as determined from the PbS QD photoluminescence (PL) quenching and exciton lifetime shortening. Moreover, by using a 0.2–1.1 nm CdS shell synthesized via cation exchange, we were able to carefully tune the charge transfer efficiency. The shell thickness dependence, and shifts in the rGO Raman G-band after functionalization and QD coupling suggest an electron transfer mechanism as the main pathway for carrier relaxation into the rGO, most likely favored by a short (<1 nm) coupling distance in our QD–rGO hybrid materials.
Results and discussion
To build devices such as photodetectors or solar cells, two materials are necessary: an absorber, here QDs, and a transport layer, here chemically reduced graphene oxide (ESI,† Scheme S1 and Fig. S1). The GO reduction has been carried out by caffeic acid, enabling the use of efficient green alternatives and at the same time avoiding residual functionalization by other reducing agents such as hydrazine.29,33 The degree of reduction is determined via X-ray photoelectron spectroscopy (XPS), with an O/C ratio of 0.40 for the initial GO, that is reduced to 0.22 for rGO, comparable to recent results for green reducing agents (0.14), while having the reaction time reduced by three-fold.14
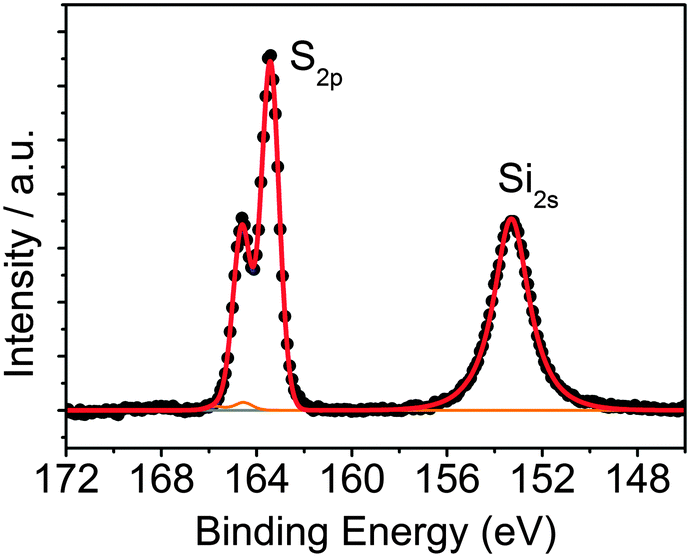
To attach QDs to rGO, common click-chemistry approaches using aminobenzene derivatives, such as 4-aminobenzoic acid (ABA) and 4-aminothiophenol (ATP) have been already proposed.34,35 However, these routes led to a low degree of functionalization and non-controlled QD–rGO assembly (see the ESI,† for further details and results, Fig. S2 and S3). Therefore, here we propose a novel approach using short-chain silanes, more specifically MPTS. This linker can bind to both rGO (via Si–O) as well as QDs (via SH). The coupling results from a simple mixing of MPTS with rGO in EtOH at 60 °C. Evaluating the S/C and Si/C atomic ratios by XPS, we obtained an average ratio of 0.13 (Table 1), significantly increasing the functionalization degree compared to ABA or ATP, where we only achieved an N/C ratio of 0.02 (Table 1, also see the ESI†). This is probably due to the ability of MPTS to bind to the carboxylic acid at the sheet edges, as well as the rGO surface itself via an Si–O–C reaction through the methoxy groups of the silane and the epoxy groups of the rGO,36–40 which are found throughout the carbon network (see ESI†). A more detailed analysis of the S 2p XPS spectrum of silane-functionalized rGO (silane-f-rGO) is shown in Fig. 1. The best fit for the S 2p profiles is obtained by using two components. A minor contribution centered at 164.2 ± 0.2 eV, amounting to 2 ± 2% of the total amount of sulfur in the silane-f-rGO samples can be related to S–S bonds,41 arising from a small amount of coupling between MPTS linkers. However, the main component centered at 163.2 ± 0.2 eV, constituting 98 ± 2%, can be assigned to unbound thiol moieties (–SH).42 This confirms that the chemical interaction between MPTS and rGO is directed by the Si–O–C path, leaving the SH group exposed to bind to the QDs.
Table 1 Atomic ratio of the different elements in GO and rGO and the corresponding functionalized derivatives, as determined by XPS using the amount of carbon as a reference
| Sample |
O/C |
N/C |
S/C |
Si/C |
| GO |
0.40 |
|
— |
— |
| Silane-f-GO |
0.46 |
— |
0.12 |
0.17 |
| rGO |
0.22 |
— |
— |
— |
| ABA-f-rGO |
0.27 |
0.02 |
— |
— |
| ATP-f-rGO |
0.25 |
0.02 |
0.02 |
— |
| Silane-f-rGO |
0.37 |
— |
0.10 |
0.16 |
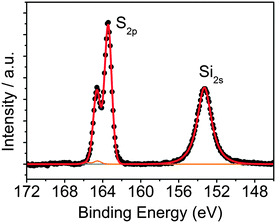 |
| | Fig. 1 XPS core-level S 2p and Si 2s spectra of silane-f-rGO. The Si 2s peak is centered at (153.2 ± 0.2) eV, a typical position for silanes.38 For the S 2p spectrum, the fitted curves correspond to the unbound thiols (blue line) and a small amount of disulfide (orange line). | |
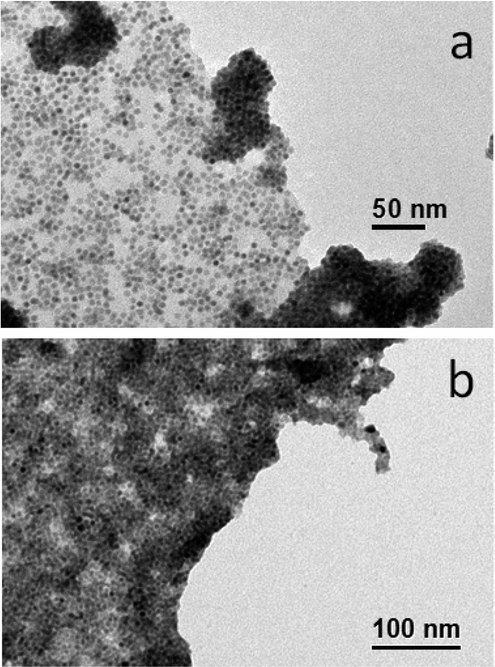
To promote the coupling between QDs and silane-f-rGO, we prepared the hybrid material in a mixed medium of toluene and EtOH at room temperature. We added OA-capped PbS QDs, dispersible in non-polar solvents (here toluene with a polarity of 0.09943), to the silane-f-rGO dispersible in polar solvents (here EtOH with a polarity of 0.65443), leading to the formation of aggregates over the course of 5 min. The resulting hybrid material then is precipitated by centrifugation and finally redispersed in DMF, a solvent with intermediate polarity (0.38643), in which a stable dispersion can be formed (for at least one month). Fig. 2a–c shows the TEM images of a typical hybrid material formed using 5.0 nm PbS QDs. They reveal that the PbS QDs cover exclusively the rGO sheets, as no unbound PbS QDs are observed. Additionally, Fig. 2a shows how the PbS QD-decorated rGO sheets can even fold or overlap without affecting the QD–rGO coupling. Our approach also allows controlling the QD surface density by changing the relative PbS QD–rGO concentration in the starting solution (Fig. 3a and b). By increasing the QD–rGO ratio from 0.2 to 0.8 nmol QDs per μg of rGO it is possible to increase the density of QDs on the sheets by a factor of 1.7 (see the ESI,† Fig. S4 and Table S1 for quantitative assessment). Interestingly, the degree of reduction also plays a role. Control experiments with GO, following the same MPTS functionalization and hybrid preparation procedure (see the ESI,† for XPS, Fig. S4, TEM images, Fig. S5, and QDs density analysis, Fig. S7 and Table S1), show that despite a similar degree of functionalization obtained for the MPTS linkers (Table 1), rGO sheets typically show a 3-fold higher density of coupled PbS QDs. Possibly, this is due to the reduced surface polarization of rGO facilitating the binding of non-polar OA-capped PbS QDs.
 |
| | Fig. 2 Representative TEM images of core PbS QD (5.0 nm diameter)–silane-f-rGO. | |
 |
| | Fig. 3 Representative TEM images of PbS QD (5.0 nm diameter)–rGO hybrid materials prepared from silane-f-rGO for the comparison of the QD coverage achieved varying the QD–rGO material ratio from 0.2 (a) to 0.8 (b) nmol QDs per μg rGO. | |
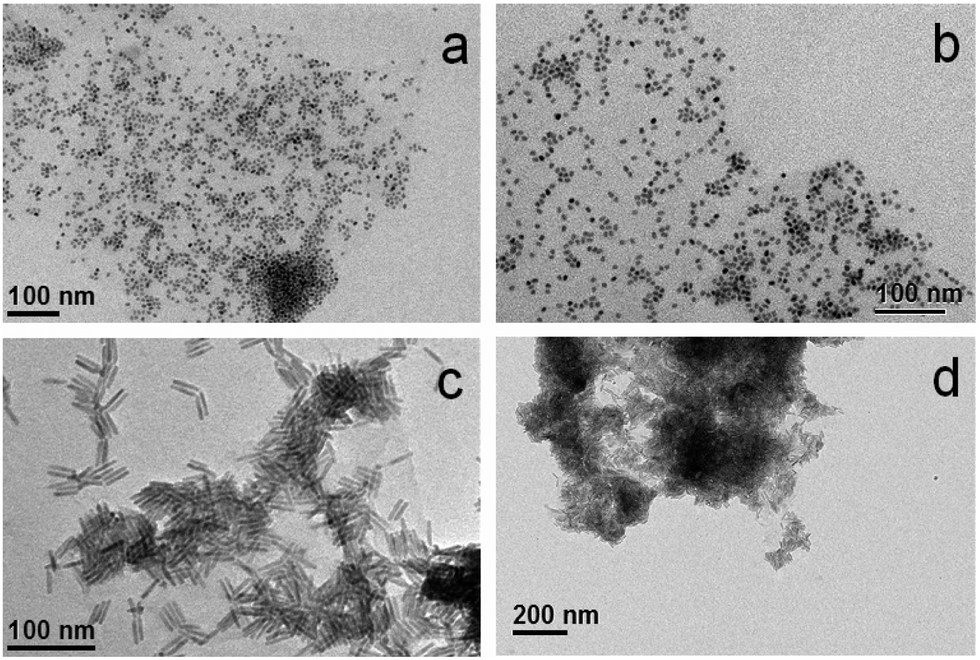
The methodology to prepare hybrid materials from silane-f-rGO is also applicable to QDs of different compositions and shapes. As in the case of PbS QDs, spherical core CdSe QDs (Fig. 4a and b), and CdSe–CdS core–shell dot-in-rods (DIRs) (Fig. 4c and d) were found to exclusively cover the rGO, and were well dispersed over the sheets. Due to their elongated shape, the latter were linked along the DIR long axis, enabling a larger interfacial contact between the materials. This implies a high versatility of the proposed functionalization, since depending on the QDs one can target applications covering a wide spectral range from the visible to the NIR, while the QD–rGO interactions can be carefully tuned by adjusting the QD–rGO contact area in shape-controlled heterostructures.
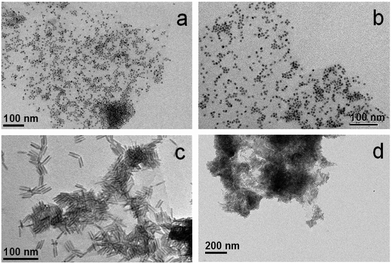 |
| | Fig. 4 Representative TEM images of hybrid materials prepared from silane-f-rGO using different materials: (a, b) CdSe QDs (5.8 nm), and (c, d) CdSe–CdS DIRs (28.3 nm length, 5.1 nm diameter). | |
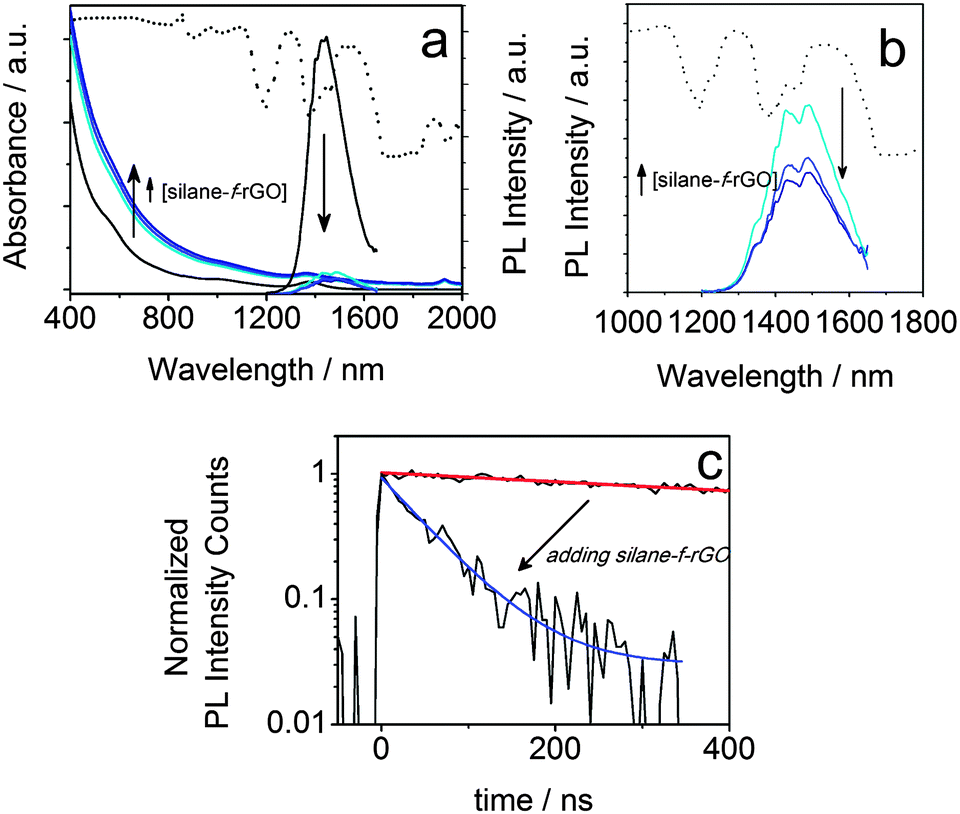
To highlight the relevance of our solution-processed QD–rGO hybrid materials for photonic applications, we investigated the resulting optical properties. We focused our attention on PbS QDs, considering their importance in NIR solar cells, photodetectors, and LEDs,1,15,23,44–46 all of which strongly rely on efficient photon–electron conversion. In thin-film configuration, they have already been combined showing an efficient energy or charge transport depending on the PbS QD–graphene distance.15,44,47 Here, in our solution-based PbS QD–rGO hybrid material we also observed a significant PL quenching of up to 92% after coupling with silane-f-rGO (Fig. 5). This is accompanied by a PL lifetime reduction from 1.4 μs (PbS QDs in TCE) to merely 66 ns (95% reduction, among the highest values reported for QD–(r)GO systems29,30). Possible effects of the preparation method on the PL quenching were discarded by various control experiments. For instance, the PL quenching is not simply due to the use of EtOH during the hybrid preparation, or the phase transfer of the QDs to DMF. Both were evaluated independently (see ESI,† Fig. S8 and S9 and Table S2). EtOH washing only induced a severe PL quenching after three additional precipitation steps and left the PL decay unmodified otherwise. Additionally, we observed a somewhat reduced PL lifetime of 0.6 μs after transfer of 5.0 nm PbS QDs to DMF using 11-mercaptoundecanoic acid (MUA) ligands, however the retention of at least 40% of the PL decay time implies that in the QD–rGO hybrid materials, charge/energy transfer to rGO is the dominant pathway for the PL quenching. To quantify the effect of the coupling to rGO, we evaluated the transfer efficiency, η, via the effective decay rates of PbS QDs before, keff,QD, and after hybridization, keff,hybr, using OA-capped PbS QDs as a reference (Table 2).
| | 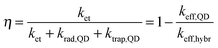 | (1) |
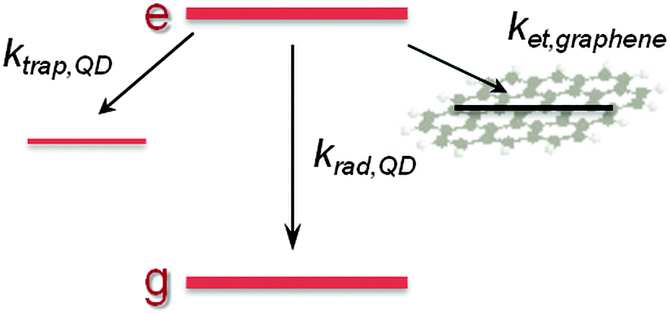
krad,QD equals the QD radiative decay rate. This competes with two additional channels:
ktrap,QD, the carrier trapping rate due to QD surface defects, and in the case of QD–rGO hybrids,
ket, which represents the energy/charge transfer rate from the QDs to rGO (
Scheme 1). Although the transfer process and hybridization with rGO imply some uncertainties in
ktrap,QD and thus
keff,QD, control experiments with MUA-capped PbS QDs in DMF suggest that these are minor (see ESI,
† Fig. S8 and
Table 2).
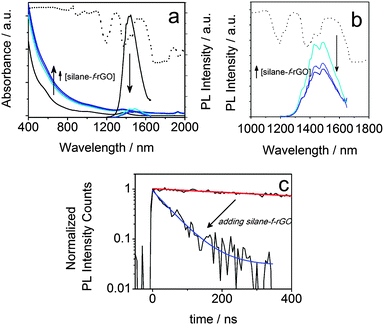 |
| | Fig. 5 Absorbance (a) and PL (a, b) spectra of PbS QD–silane-f-rGO hybrid materials dispersed in DMF with varying relative QD concentrations (ratios 0.2, 0.4 and 0.8 nmol QDs per μg rGO). The data from the reference, OA-capped PbS QDs in TCE are shown as black lines. (c) The corresponding PL decay traces and fits for pure QDs (red line) and rGO hybrid materials (blue line), prepared with a relative concentration of 0.2 nmol per μg rGO. The transmittance spectrum of DMF is shown as a dotted line in (a, b). | |
Table 2 PL lifetime τeff and transfer efficiency η obtained from the time-resolved measurements and eqn (1) (see text) for the PbS QD–silane-f-rGO hybrid materials prepared in DMF compared with the starting PbS QDs in TCE
| Sample |
Solvent |
τ
eff (μs) |
η
|
| OA-capped PbS QDs |
TCE |
1.4 |
— |
| PbS QD–silane-f-rGO |
DMF |
0.066 |
0.95 |
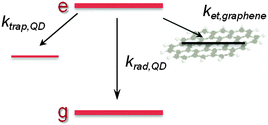 |
| | Scheme 1 Simplified diagram of excited state decay pathways in a QD–rGO hybrid. | |
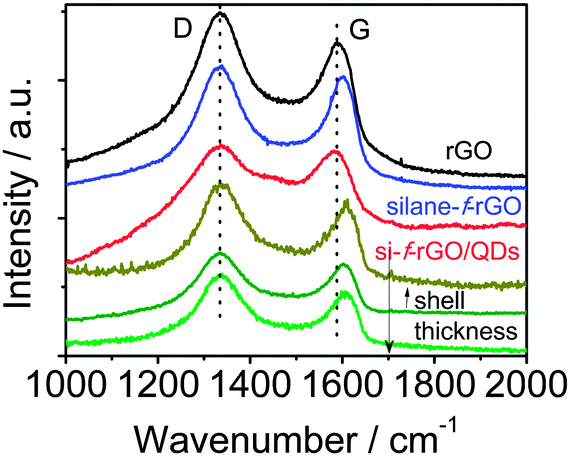
To improve our understanding of the nature of the energy/charge transfer processes in the hybrid system, we carried out complementary studies by means of Raman spectroscopy. This technique allows characterizing graphene-based materials by analyzing the position, relative intensity (ratio) and FWHM of the characteristic D and G bands. Specifically, the G band position shifts with physical and chemical changes in the carbon-network environment. Thus, it is highly sensitive to the presence of excess charges from, for instance, electron-donating molecules, leading to a downshift of the G band position, or electron-acceptor molecules promoting an upshift.15,30,48 The silane functionalization of rGO leads to a G band upshift of 12 ± 2 cm−1 (value determined from the average obtained on three different areas of the sample, see ESI,† Fig. S10), which agrees with a p-doping already observed by the introduction of different functional groups in the graphene network (Fig. 6).38,48 Meanwhile, the attachment of PbS QDs leads to a downshift of 15 ± 1 cm−1 compared with the silane-f-rGO spectrum. In view of the p-doping after silane functionalization, this shift can be ascribed to an electron enrichment of the rGO network in PbS QD–rGO hybrid materials. Therefore, although both energy and electron transfer pathways could still be present, the short distance between the PbS QDs and the rGO sheets induced by the 0.7 nm MPTS linker promotes electron transfer as the main mechanism for the non-radiative relaxation of the QDs.24,29,30,47
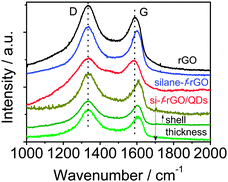 |
| | Fig. 6 Comparison between the representative Raman spectra of the different PbS core and PbS–CdS core-shell QDs and silane-f-rGO hybrid samples. In the lower three spectra (green lines), we systematically varied the shell thickness. The Raman spectrum of the reference material rGO (black line) is also shown. | |
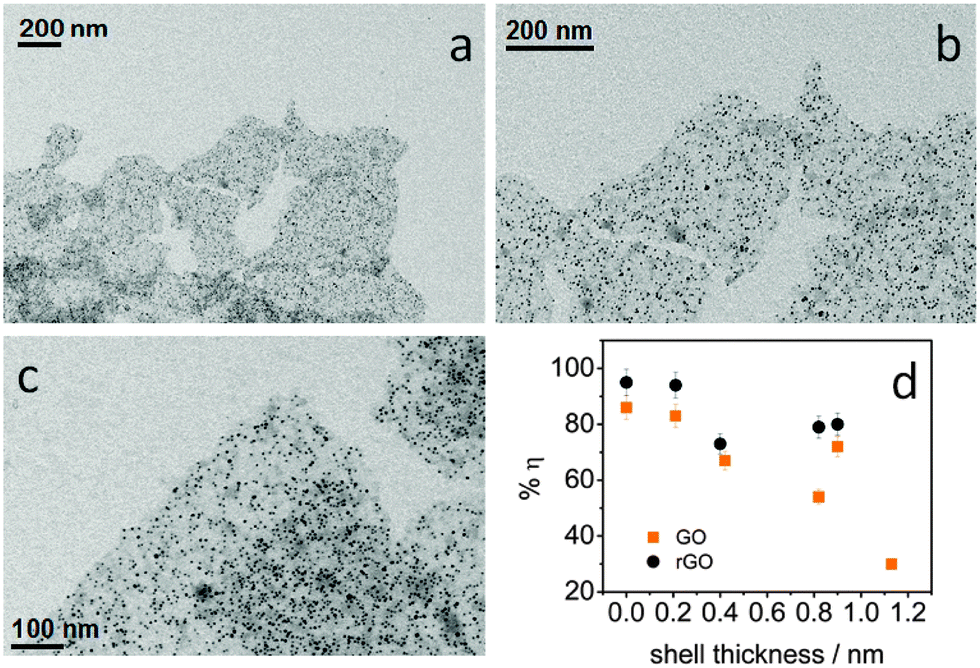
Considering the strong distance-dependence of the electron transfer, we were able to modulate the transfer efficiency by controlling the QD–rGO coupling length. We synthesized a thin CdS shell of 0.2–1.1 nm on the PbS QDs by cation exchange. This increases the effective QD–rGO distance, while leaving the assembly itself unaffected, again confirming the universal nature of the approach here developed (Fig. 7a–c). The resulting transfer efficiency, determined again from the PL lifetime, is displayed in Fig. 7d. We observed a notable decrease of the transfer efficiency upon increasing the shell thickness (also see the ESI,† Table S3). As a control, the trend observed for rGO is confirmed by GO. However, we obtained consistently lower transfer efficiencies (this also agrees with a smaller relative shift of the Raman G band in GO-based hybrids, see ESI,† Fig. S11), demonstrating the importance of achieving a sufficiently high degree of reduction for rGO, not only to improve the assembly process, but also to increase the transfer efficiency. The difference between GO and rGO likely lies in the modification of the graphene electronic structure.24,49 More specifically, the larger fraction of C sp2 domains, increasing from 42% in the GO to 56% in the rGO (see ESI†) leads to an increased electronic density of the states50 promoting more efficient charge transfer.
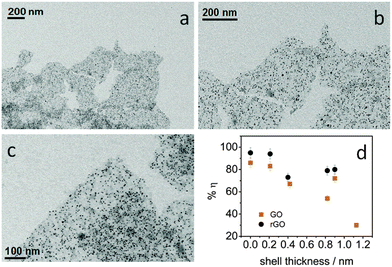 |
| | Fig. 7 (a–c) The representative TEM images at different scales of core–shell PbS–CdS QDs (core 4.2 nm diameter, shell 0.4 nm thickness) coupled with silane-f-rGO. (d) The influence of the CdS shell thickness on the transfer efficiency η between the PbS QDs and the silane-f-(r)GO. The samples were prepared with a ratio of 0.2 nmol QDs per μg (r)GO. | |
By evaluating the electronic interaction between core–shell PbS–CdS QDs and rGO by means of Raman spectroscopy (Fig. 6), we confirmed the role of the shell in inhibiting the electron transfer. When a thick shell is present, the original G band position corresponding to the silane-f-rGO is recovered, indicating that the electron transfer between both materials is suppressed. Most importantly, both PL and Raman spectroscopy data demonstrate that it is possible to maintain a high electron transfer efficiency in PbS–CdS QDs for thin shells of about 0.2 nm. This implies that such core–shell QDs can be for instance used in the implementation of solar cells or other applications operating under ambient conditions, as the CdS shell improves the QD chemical and environmental stability.45,51
Experimental
Materials
PbCl2 (99.999%) and sulfur (99.999%) were purchased from Alfa-Aesar; CdO (≥99.99%) and trioctylphosphine oxide (TOPO, ≥90%) from Strem Chemicals; oleic acid (OA) (90%), 1-octadecene (ODE, 90%), octadecylphosphonic acid (ODPA, 97%), hexylphosphonic acid (HPA, 95%), toluene (≥99.7%), ethanol (≥99.8%, without additive), tetrachloroethylene (TCE, anhydrous, ≥99%) and dimethylformamide (DMF, anhydrous, ≥99.8%) from Sigma Aldrich; oleylamine (OlAm, 80%) from Acros Organics. Graphite flakes were supplied by Graphene Supermarket; H2SO4 (95–98%), NaNO3 (≥99.5%), KMnO4 (≥99%, low in mercury), 4-aminobenzoic acid (ABA, ≥99%), 4-aminothiophenol (ATP, 97%), 3-(mercaptopropyl)trimethoxysilane (MPTS, 95%), caffeic acid (CA, ≥95%) and ammonium hydroxide solution (28%w NH3 in H2O, ≥99.99%) were supplied by Sigma Aldrich. The water used in the experiments was obtained using a MilliQ® system.
Nanocrystal synthesis
OA-capped PbS QDs were prepared following a slight modification of the method described by Moreels et al.52 Briefly, for the synthesis, 1 g of PbCl2 and 7.5 mL of OlAm were degassed for 30 min at 125 °C in a three-neck flack under Argon. Then, the temperature was adjusted to 120–160 °C, depending on the desired QD size, and 2.25 mL of a 0.3 M OlAm–sulfur stock solution was added. After synthesis the OlAm ligands were replaced by adding OA to the QD suspension in toluene, followed by precipitation by EtOH and resuspension in toluene. The diameter was determined from absorption.52 The CdS coating was carried out by cation exchange.53 An OA-capped PbS QD solution of about 10 μM in toluene/ODE (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 vol) was degassed for 1 h under Argon at 80 °C. Then, the solution was heated up to 100 °C and 0.5 mL of a 0.3 M Cd–oleate stock solution was added to form the CdS shell. The reaction continued for 1 to 180 min depending on the desired shell thickness (0.2–1.1 nm). The shell thickness was determined from the blue shift of the first absorption peak after cation exchange.53
:1 vol) was degassed for 1 h under Argon at 80 °C. Then, the solution was heated up to 100 °C and 0.5 mL of a 0.3 M Cd–oleate stock solution was added to form the CdS shell. The reaction continued for 1 to 180 min depending on the desired shell thickness (0.2–1.1 nm). The shell thickness was determined from the blue shift of the first absorption peak after cation exchange.53
CdSe QDs and CdSe–CdS DIRs were prepared according to the procedure reported by Carbone et al.54 Briefly, the CdSe QD synthesis was carried out by mixing a clear solution formed by TOPO–ODPA–CdO with the injection of a Se:TOP solution at 370 °C under nitrogen. The CdSe diameter and concentration were determined from the first absorption peak and absorbance at 350 nm.55 The CdSe–CdS DIRs were prepared via seeded growth by adding the CdSe seeds together with the sulfur precursor to a flask that contains a TOPO–ODPA–HPA–CdO solution at 350 °C. The rod dimensions and concentration were determined using TEM and elemental analysis by inductively coupled plasma optical emission spectroscopy (ICP-OES) of digested solutions, respectively.
Preparation of graphene oxide and reduced graphene oxide
We obtained the GO by a slight modification of the Hummers' method56 from graphite flakes. A paste was formed by mixing graphite–H2SO4–NaNO3–KMnO4, after which the graphite was oxidized for 15 h for an improved exfoliation and oxidation of the material. To chemically reduce the GO, we used CA and introduced several modifications to the reaction in aqueous medium at 95 °C proposed by Bo et al.14 Briefly, in the reduction of a 0.1 mg mL−1 GO aqueous dispersion, we achieved a similar degree of reduction with a smaller amount of CA (CA:GO weight ratio of 1:1) and a shorter reaction time of 3 h, by modifying the pH of the reaction medium (pH = 10 adjusted by NH3 addition). This is based on the literature related to the reduction of GO with endiol structure (HO–A–OH) molecules, such as vitamin C,57 in which the basic conditions favor the hydroxyl dissociation and GO stability, thereby improving the efficiency of the reducing agent.
Graphene oxide derivates functionalization
The functionalization of (r)GO with the amino-benzene derivatives was carried out using the methods reported for ABA34 and ATP.35 In the case of the MPTS functionalization, we established the reaction conditions based on the reported literature with GO36 and colloidal nanocrystals.58,59 A GO or rGO dispersion (0.5 mg mL−1) in EtOH was sonicated for 30 min and subsequently functionalized by reflux at 60 °C during 15 h, with 250 μL of MPTS added per mg of (r)GO. The final product was obtained by washing with EtOH to remove the unreacted silane and centrifugation, and finally dispersing the (r)GO in EtOH by sonication (30 min).
Hybrid material fabrication in solution
An aliquot of the QD solution in toluene was mixed with a corresponding amount of the silane-f-(r)GO dispersion in EtOH at room temperature. The mixture was stirred vigorously for 2 minutes, after which the dispersion was precipitated twice with EtOH to remove the toluene and collect the hybrid material formed. The final product was redispersed in DMF by vortexing for at least 12 min.
Structural characterization
The transmission electron microscopy (TEM) images were acquired using a 100 kV JEOL JEM-1011 microscope. The samples were drop cast onto carbon-coated copper grids for the measurements. X-ray photoelectron spectroscopy (XPS) was carried out on a Kratos Axis Ultra DLD spectrometer, using a monochromatic Al Kα source (15 kV, 20 mA). The samples were prepared by drop casting the materials onto 50 nm-gold-coated silicon wafers. High-resolution scans were performed at a constant pass energy of 10 eV and steps of 0.1 eV. The photoelectrons were detected at a take-off angle Φ = 0° with respect to the surface normal. The pressure in the analysis chamber was kept below 7 × 10−9 Torr for data acquisition. The binding energy scale was internally referenced to the Au 4f7/2 peak at 84 eV. The spectra were analysed using the CasaXPS software (version 2.3.16). Fitting of S 2p profiles was done assuming Voigt profiles and Shirley-type background. For each S 2p doublet, we fixed a 1.2 eV energy splitting and a 2:1 intensity ratio between the 3/2 and 1/2 components. Each doublet was identified in the text by the position of the 2p3/2 component.
Optical characterization
The absorption spectra were recorded using a Varian Cary 5000 UV-vis-NIR spectrophotometer. The steady-state and time-resolved photoluminescence (PL) emissions were measured using an Edinburgh Instruments FLS920 spectrofluorometer. The steady-state PL was collected by exciting the samples at 400 nm. The PL decay traces were recorded by exciting the samples at 405 nm using a 50 ps laser diode at a repetition rate of 0.05–1 MHz, to ensure complete decay of the emission between the excitation pulses. The data were collected at the PL peak position with an emission bandwidth of 23 nm.
Raman spectroscopy
The Raman data were collected on silicon wafers using a micro-Raman spectrometer Horiba Jobin-Yvon LabRAM HR800UV at a 632.9 nm laser excitation wavelength, using a 50× objective and neutral filter with an O.D. of 0.6 to reduce the laser intensity. The spectrometer resolution equals 2 cm−1. The spectra were recorded after energy scale calibration by checking the Rayleigh and Si bands at 0 and 520.7 cm−1. The acquisition time was a few minutes at each point, while the laser excitation power was kept below 1 mW to avoid heating and damage of the sample.
Conclusions
In this work, we developed a general up-scalable assembly approach for preparing solution-processed colloidal QD–rGO dispersions as all-in-one hybrid materials, which include both the absorber and the electron transporting material. The results show the formation of well-defined hybrids with controlled QD coverage on the rGO sheets. To demonstrate the potential of the QD–rGO system for photovoltaic or -detector applications, we have investigated the charge transfer from PbS QDs to rGO by means of fluorescence and Raman spectroscopy. The short MPTS linker leads to an efficient electron transfer, as evidenced by a significant shortening of the QD lifetime and downshift of the rGO Raman G band. Further studies with core–shell PbS–CdS QDs indicate a barrier effect of the shell on the QD–rGO interactions, which allowed us to modulate the charge transfer by increasing the shell thickness. Therefore, a trade-off between the shell thickness and transfer efficiency exists for the device design and performance. Nevertheless, we maintained an efficient transfer for thin shells of 0.2 nm, allowing us to combine the charge transfer with an improved inorganic QD surface passivation to enhance the stability of future QD–rGO hybrid-based devices under ambient conditions.
Acknowledgements
We acknowledge support from the European Union 7th Framework Programme under grant agreement no. 604391 Graphene Flagship. Francesco De Donato is acknowledged for providing the DIR samples. Riccardo Carzino is acknowledged for technical assistance with the Raman measurements.
References
- Z. Ning, H. Dong, Q. Zhang, O. Voznyy and E. H. Sargent, ACS Nano, 2014, 8, 10321–10327 CrossRef CAS PubMed.
- C.-H. M. Chuang, P. R. Brown, V. Bulović and M. G. Bawendi, Nat. Mater., 2014, 13, 796–801 CrossRef CAS PubMed.
- F. Bonaccorso, Z. Sun, T. Hasan and A. C. Ferrari, Nat. Photonics, 2010, 4, 611–622 CrossRef CAS PubMed.
- K. S. Novoselov, V. I. Falko, L. Colombo, P. R. Gellert, M. G. Schwab and K. Kim, Nature, 2012, 490, 192–200 CrossRef CAS PubMed.
- F. Bonaccorso and Z. Sun, Opt. Mater. Express, 2014, 4, 63–78 CrossRef.
- G. Konstantatos, I. Howard, A. Fischer, S. Hoogland, J. Clifford, E. Klem, L. Levina and E. H. Sargent, Nature, 2006, 442, 180–183 CrossRef CAS PubMed.
- Y. Shirasaki, G. J. Supran, M. G. Bawendi and V. Bulovic, Nat. Photonics, 2013, 7, 13–23 CrossRef CAS PubMed.
- J. Q. Grim, S. Christodoulou, F. Di Stasio, R. Krahne, R. Cingolani, L. Manna and I. Moreels, Nat. Nanotechnol., 2014, 9, 891–895 CrossRef CAS PubMed.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666–669 CrossRef CAS PubMed.
- J. D. Roy-Mayhew and I. A. Aksay, Chem. Rev., 2014, 114, 6323–6348 CrossRef CAS PubMed.
- I. V. Lightcap and P. V. Kamat, Acc. Chem. Res., 2012, 46, 2235–2243 CrossRef PubMed.
- A. Yeltik, G. Kucukayan-Dogu, B. Guzelturk, S. Fardindoost, Y. Kelestemur and H. V. Demir, J. Phys. Chem. C, 2013, 117, 25298–25304 CAS.
- D. R. Dreyer, S. Park, C. W. Bielawski and R. S. Ruoff, Chem. Soc. Rev., 2010, 39, 228–240 RSC.
- Z. Bo, X. Shuai, S. Mao, H. Yang, J. Qian, J. Chen, J. Yan and K. Cen, Sci. Rep., 2014, 4684 Search PubMed.
- G. Konstantatos, M. Badioli, L. Gaudreau, J. Osmond, M. Bernechea, F. P. G. de Arquer, F. Gatti and F. H. L. Koppens, Nat. Nanotechnol., 2012, 7, 363–368 CrossRef CAS PubMed.
- M. Eck, C. V. Pham, S. Zufle, M. Neukom, M. Sessler, D. Scheunemann, E. Erdem, S. Weber, H. Borchert, B. Ruhstaller and M. Kruger, Phys. Chem. Chem. Phys., 2014, 16, 12251–12260 RSC.
- M.-H. Jung and M.-J. Chu, Nanoscale, 2014, 6, 9241–9249 RSC.
- F.-X. Xiao, J. Miao and B. Liu, J. Am. Chem. Soc., 2014, 136, 1559–1569 CrossRef CAS PubMed.
- Y. Gao, O. Roslyak, E. Dervishi, N. S. Karan, Y. Ghosh, C. J. Sheehan, F. Wang, G. Gupta, A. Mohite, A. M. Dattelbaum, S. K. Doorn, J. A. Hollingsworth, A. Piryatinski and H. Htoon, Adv. Opt. Mater., 2015, 3, 39–43 CrossRef CAS PubMed.
-
P. Reiss, Synthesis of Semiconductor Nanocrystals in Organic Solvents, in Semiconductor Nanocrystal Quantum Dots: Synthesis, Assembly, Spectroscopy and Applications, ed. A. L. Rogach, Springer-Verlag/Wien, Austria, 2008, pp. 35–73 Search PubMed.
- T. Avellini, C. Lincheneau, M. La Rosa, A. Pertegas, H. J. Bolink, I. A. Wright, E. C. Constable, S. Silvi and A. Credi, Chem. Commun., 2014, 50, 11020–11022 RSC.
- Z. Zheng, W. Xie, Z. S. Lim, L. You and J. Wang, Sci. Rep., 2014, 5721 CAS.
- A. A. Bakulin, S. Neutzner, H. J. Bakker, L. Ottaviani, D. Barakel and Z. Chen, ACS Nano, 2013, 7, 8771–8779 CrossRef CAS PubMed.
- S. Guo, D. Bao, S. Upadhyayula, W. Wang, A. B. Guvenc, J. R. Kyle, H. Hosseinibay, K. N. Bozhilov, V. I. Vullev, C. S. Ozkan and M. Ozkan, Adv. Funct. Mater., 2013, 23, 5199–5211 CrossRef CAS PubMed.
- Y. Zhu, X. Meng, H. Cui, S. Jia, J. Dong, J. Zheng, J. Zhao, Z. Wang, L. Li, L. Zhang and Z. Zhu, ACS Appl. Mater. Interfaces, 2014, 6, 13833–13840 CAS.
- S. Wang, J. Li, X. Zhou, C. Zheng, J. Ning, Y. Zhong and Y. Hu, J. Mater. Chem. A, 2014, 2, 19815–19821 CAS.
- B. H. Juárez, M. Meyns, A. Chanaewa, Y. Cai, C. Klinke and H. Weller, J. Am. Chem. Soc., 2008, 130, 15282–15284 CrossRef PubMed.
- F. Iacono, C. Palencia, L. de la Cueva, M. Meyns, L. Terracciano, A. Vollmer, M. J. de la Mata, C. Klinke, J. M. Gallego, B. H. Juarez and R. Otero, ACS Nano, 2013, 7, 2559–2565 CrossRef CAS PubMed.
- I. V. Lightcap and P. V. Kamat, J. Am. Chem. Soc., 2012, 134, 7109–7116 CrossRef CAS PubMed.
- S. Kundu, S. Sadhu, R. Bera, B. Paramanik and A. Patra, J. Phys. Chem. C, 2013, 117, 23987–23995 CAS.
- Z. Chen, S. Berciaud, C. Nuckolls, T. F. Heinz and L. E. Brus, ACS Nano, 2010, 4, 2964–2968 CrossRef CAS PubMed.
- B. Rogez, H. Yang, E. Le Moal, S. Lévêque-Fort, E. Boer-Duchemin, F. Yao, Y.-H. Lee, Y. Zhang, K. D. Wegner, N. Hildebrandt, A. Mayne and G. Dujardin, J. Phys. Chem. C, 2014, 118, 18445–18452 CAS.
- X. Gao, J. Jang and S. Nagase, J. Phys. Chem. C, 2010, 114, 832–842 CAS.
- M. Jahan, Q. Bao, J.-X. Yang and K. P. Loh, J. Am. Chem. Soc., 2010, 132, 14487–14495 CrossRef CAS PubMed.
- J. Debgupta and V. K. Pillai, Nanoscale, 2013, 5, 3615–3619 RSC.
- H. Yang, F. Li, C. Shan, D. Han, Q. Zhang, L. Niu and A. Ivaska, J. Mater. Chem., 2009, 19, 4632–4638 RSC.
- Y. Lin, J. Jin and M. Song, J. Mater. Chem., 2011, 21, 3455–3461 RSC.
- R. Wang, S. Wang, D. Zhang, Z. Li, Y. Fang and X. Qiu, ACS Nano, 2011, 5, 408–412 CrossRef CAS PubMed.
- L. Newton, T. Slater, N. Clark and A. Vijayaraghavan, J. Mater. Chem. C, 2013, 1, 376–393 RSC.
- Y. An, M. Chen, Q. Xue and W. Liu, J. Colloid Interface Sci., 2007, 311, 507–513 CrossRef CAS PubMed.
- W. Ai, L. Xie, Z. Du, Z. Zeng, J. Liu, H. Zhang, Y. Huang, W. Huang and T. Yu, Sci. Rep., 2014, 2341 Search PubMed.
- H. Hamoudi, M. Prato, C. Dablemont, O. Cavalleri, M. Canepa and V. A. Esaulov, Langmuir, 2010, 26, 7242–7247 CrossRef CAS PubMed.
-
C. Reichardt, Empirical Parameters of Solvent Polarity, Solvents and Solvent Effects in Organic Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, 2004, pp. 389–469 Search PubMed.
- Y. Q. Huang, R. J. Zhu, N. Kang, J. Du and H. Q. Xu, Appl. Phys. Lett., 2013, 103, 143119 CrossRef PubMed.
- L.-H. Lai, L. Protesescu, M. V. Kovalenko and M. A. Loi, Phys. Chem. Chem. Phys., 2014, 16, 736–742 RSC.
- F. C. J. M. van Veggel, Chem. Mater., 2014, 26, 111–122 CrossRef CAS.
- L. Gaudreau, K. J. Tielrooij, G. E. D. K. Prawiroatmodjo, J. Osmond, F. J. G. de Abajo and F. H. L. Koppens, Nano Lett., 2013, 13, 2030–2035 CrossRef CAS PubMed.
- H. Liu, Y. Liu and D. Zhu, J. Mater. Chem., 2011, 21, 3335–3345 RSC.
- B. Rajbanshi, S. Sarkar and P. Sarkar, J. Mater. Chem. C, 2014, 2, 8967–8975 RSC.
- Y. Dong and D. H. Son, J. Phys. Chem. Lett., 2015, 6, 44–47 CrossRef CAS.
- D. C. J. Neo, C. Cheng, S. D. Stranks, S. M. Fairclough, J. S. Kim, A. I. Kirkland, J. M. Smith, H. J. Snaith, H. E. Assender and A. A. R. Watt, Chem. Mater., 2014, 26, 4004–4013 CrossRef CAS.
- I. Moreels, Y. Justo, B. De Geyter, K. Haustraete, J. C. Martins and Z. Hens, ACS Nano, 2011, 5, 2004–2012 CrossRef CAS PubMed.
- Y. Justo, P. Geiregat, K. V. Hoecke, F. Vanhaecke, C. De Mello Donega and Z. Hens, J. Phys. Chem. C, 2013, 117, 20171–20177 CAS.
- L. Carbone, C. Nobile, M. De Giorgi, F. D. Sala, G. Morello, P. Pompa, M. Hytch, E. Snoeck, A. Fiore, I. R. Franchini, M. Nadasan, A. F. Silvestre, L. Chiodo, S. Kudera, R. Cingolani, R. Krahne and L. Manna, Nano Lett., 2007, 7, 2942–2950 CrossRef CAS PubMed.
- J. Jasieniak, L. Smith, J. V. Embden, P. Mulvaney and M. Califano, J. Phys. Chem. C, 2009, 113, 19468–19474 CAS.
- W. S. Hummers and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef CAS.
- J. Gao, F. Liu, Y. Liu, N. Ma, Z. Wang and X. Zhang, Chem. Mater., 2010, 22, 2213–2218 CrossRef CAS.
- K. Luo, S. Zhou, L. Wu and G. Gu, Langmuir, 2008, 24, 11497–11505 CrossRef CAS PubMed.
- M.-Q. Zhu, E. Chang, J. Sun and R. A. Drezek, J. Mater. Chem., 2007, 17, 800–805 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: XPS analysis of GO and rGO; functionalization of the (r)GO; XPS analysis of ATP and ABA functionalized (r)GO and TEM images of the QD coupling results; estimation of the density of PbS QDs on the (r)GO sheets; complementary control experiments: fluorescence spectroscopy characterization of MUA-capped PbS QDs and the effect of EtOH washing; Raman characterization; and PL dynamics in core–shell PbS–CdS QD and silane-f-(r)GO hybrid materials. See DOI: 10.1039/c5tc01137j |
|
| This journal is © The Royal Society of Chemistry 2015 |

 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence