
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Antimicrobial and cell-compatible surface-attached polymer networks – how the correlation of chemical structure to physical and biological data leads to a modified mechanism of action†
Peng
Zou
ab,
Dougal
Laird
abc,
Esther K.
Riga
a,
Zhuoling
Deng
ab,
Franziska
Dorner
ab,
Heidi-Rosalia
Perez-Hernandez
ab,
D. Lorena
Guevara-Solarte
ad,
Thorsten
Steinberg
c,
Ali
Al-Ahmad
d and
Karen
Lienkamp
*ab
aDepartment of Microsystems Engineering (IMTEK), Albert-Ludwigs-Universität, Georges-Köhler-Allee 103, 79110 Freiburg, Germany. E-mail: lienkamp@imtek.uni-freiburg.de
bFreiburg Institute for Advanced Studies (FRIAS), Albert-Ludwigs-Universität, Albertstr. 19, 79104 Freiburg, Germany
cOral Biotechnology, University Medical Center of the Albert-Ludwigs-Universität, Hugstetter Str. 55, 79106 Freiburg, Germany
dDepartment of Operative Dentistry and Periodontology, Center for Dental Medicine of the Albert-Ludwigs-Universität, Hugstetter Str. 55, 79106 Freiburg, Germany
First published on 16th June 2015
Abstract
We present a synthetic platform based on photo-induced thiol–ene chemistry, by which surface-attached networks from antimicrobial poly(oxonorbornene) (so-called polymeric synthetic mimics of antimicrobial peptides, SMAMPs) could be easily obtained. By systematically varying hydrophobicity and charge density, surface-attached polymer networks with high antimicrobial activity and excellent cell compatibility were obtained. For the homopolymer networks with constant charge density, antimicrobial activity increased systematically with increasing hydrophobicity (i.e. decreasing swellability and apparent surface energy). Irrespective of charge density, the antimicrobial activity of all networks correlated with the acid constant pK and the isoelectric point (IEP) – the lower pK and IEP, the higher the antimicrobial activity. The cell compatibility of the networks increased with increasing swellability and apparent surface energy, and decreased with increasing charge density. The data corroborates that the mechanism of action of antimicrobial polymer surfaces depends on at least two mechanistic steps, one of which is hydrophobicity-driven and the other charge related. Therefore, we suggest a modified mechanistic model with a charge-driven and a hydrophobicity-driven step. For antimicrobial networks that only varied in hydrophobicity, the antimicrobial activities on surfaces and in solution also correlated – the higher the activity in solution, the higher the activity on surfaces. Thus, the hydrophobicity-driven step for activity on surfaces may be similar to the one in solution. Cell compatibility of SMAMPs in solution and on surfaces also showed a systematic positive correlation for all polymers, therefore this property also depends on the net hydrophobic balance of the polymer.
Introduction
Multi-resistant bacteria are a life-threatening issue worldwide. According to the World Health Organization (WHO), ‘antibiotic resistance is no longer a prediction for the future; it is happening right now, across the world, and is putting at risk the ability to treat common infections in the community and hospitals. Without urgent, coordinated action, the world is heading towards a post-antibiotic era, in which common infections and minor injuries, which have been treatable for decades, can once again kill’.1 Leading global health organizations identified major problems with antimicrobial resistance in S. aureus (hospital-acquired bloodstream, skin and surgical site infections), E. coli (particularly urinary tract infections) and K. pneumoniae (hospital-acquired bloodstream infections, pneumonia).1,2Due to the severe infections caused by these ‘superbugs’, there is an ever-increasing demand for materials that restrain and eradicate them. Multi-resistant bacteria originate from exposure to sub-lethal antibiotic doses; in hospitals, they were found in the ambient atmosphere, on surgical equipment, and on patients' and medical staff's skin or clothing.3 From there, they proliferate and provide a microbe reservoir that defies even the most rigorous hygiene regime.3b,4 As a result, infections with resistant bacteria in hospitals are ever increasing. Patients that have received implants and catheterized patients have a particularly high infection risk,5 as only a few bacteria that contaminate a medical device surface can develop dangerous, antibiotic-resistant biofilms in less than 24 hours.6 Thus, effective medical surfaces that prevent biofilm formation comprise an immediate need and, in addition to strict hygiene regimes, could help contain resistant bacteria.
There are numerous medical applications where contact-killing antimicrobial polymer surface coatings could meet that need. To be truly useful, they must be broad-band active against Gram-negative and Gram-positive bacteria, and non-toxic to the cells of the human organism. Additionally, they should not increase bacterial resistance. Synthetic mimics of antimicrobial peptides (SMAMPs) are polymers that are unlikely to increase bacterial resistance.7 These polymers mimic the structure of natural antimicrobial peptides (AMPs, Fig. 1a).8 Like their natural peptide archetypes, they are facially amphiphilic and act on bacteria through unspecific membrane damage or destruction.7a,c,9 However, because they are cationic, they distinguish between negatively charged bacteria, which are killed, and the cells of the host organism, which are overall charge neutral and therefore much less affected. This feature makes SMAMPs highly cell compatible and therefore ideal candidates for medical applications. Despite the substantial amount of work on cationic antimicrobial polymer surfaces in the literature, the mechanism of action of antimicrobial polymer surfaces and the effect of physical properties on their biological performance is still not fully understood.10 Examples of pioneering work are Klibanov and Tiller's systems,11 and work on poly(diallyl dimethylammonium),12 poly(butyl methacrylate-co-aminoethyl methylacrylate),13 and poly(2-(dimethylamino) ethyl methacrylate)-based surfaces.10b,14 Additionally, recent work by Chan-Park,15 Tiller,16 and Busscher17 made significant contributions towards a mechanistic understanding.
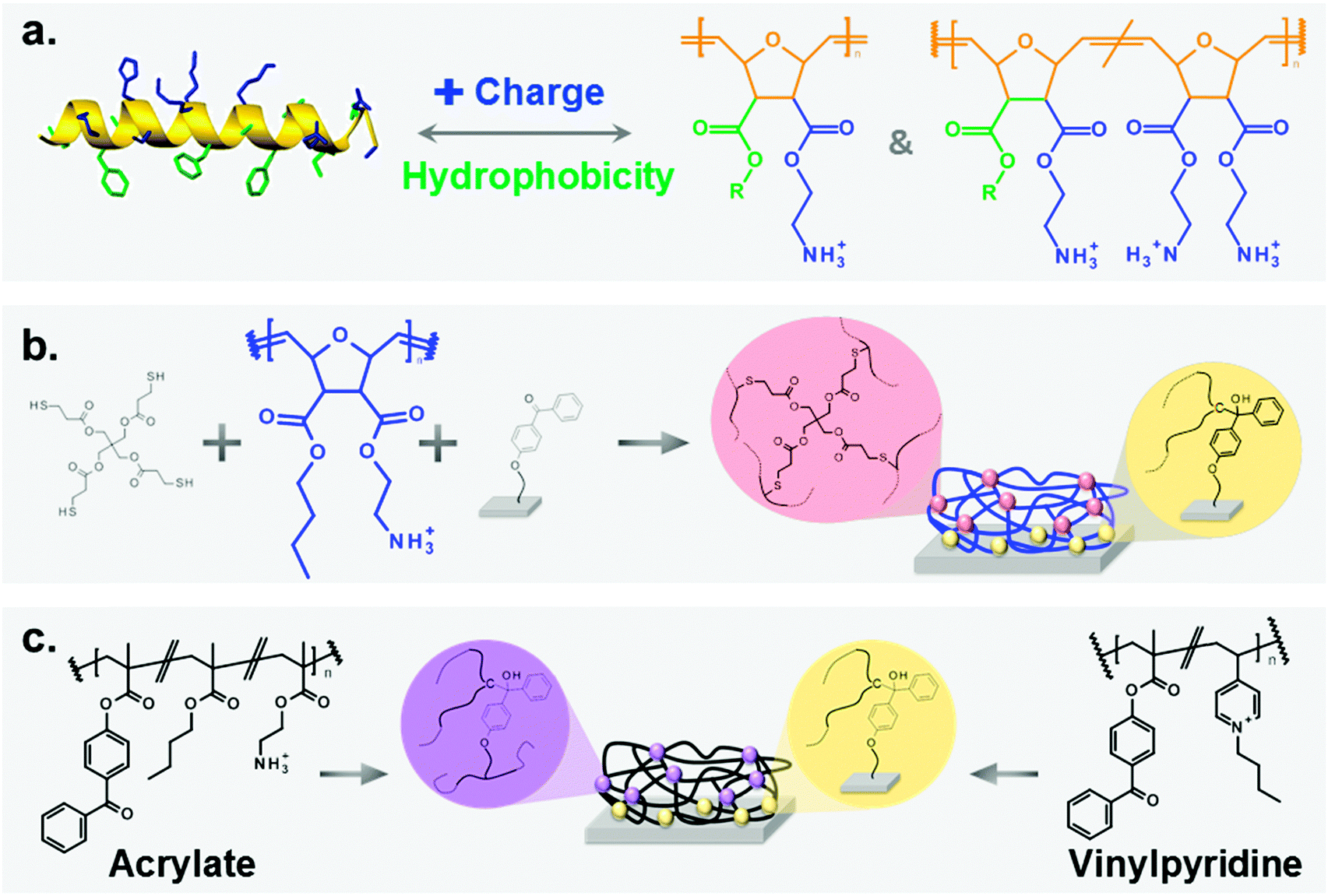 | ||
| Fig. 1 (a) Antimicrobial peptides (AMPs, left) are the natural archetype for poly(oxanorbornene)-based synthetic mimics of antimicrobial peptides (SMAMPs, right). Both consist of hydrophobic (green) and cationic (blue) groups attached to a backbone (orange). Due to their facial amphiphilicity, these molecules are selectively active against bacteria. (b) SMAMPs (blue) were surface immobilized using a benzophenone anchor group, and cross-linked by pentaerithritol-tetrakis(3-mercaptopropionate) (= tetrathiol). As a result, antimicrobial surface-attached polymer networks were obtained. (c) Poly(methacrylate) and poly(vinylpyridine)-based surface-attached networks were synthesized as reference systems. | ||
In this paper, we describe how we translated the SMAMP concept from solution to materials. We synthesized three series of poly(oxonorbornene) SMAMPs with systematically varying hydrophobicity and charge density,7d which are key parameters for antimicrobial activity. We surface-immobilized these polymers by a newly developed, straight-forward process (Fig. 1b) and thus obtained three series of surface-attached antimicrobial polymer networks. We demonstrate that these poly(oxonorbornene)-based SMAMP networks make excellent antimicrobial coatings which, unlike most of the state-of-the-art, are broad-band active against bacteria and highly compatible with primary human fibroblast cells and immortalized human keratinocytes. Additionally, the coating process we describe is so simple that it can be extended onto virtually any 2D or 3D surface that carries hydroxyl groups. We proved this with a model catheter made from medical grade poly(dimethylsiloxane), showing that SMAMPs also work on real-life medical surfaces.
To further advance the mechanistic understanding of antimicrobial surfaces, we fully characterized and quantified the physical properties of our SMAMP networks and correlated these results to their biological properties. This gave additional insight on the relation between molecular design (charge density and hydrophobicity), physical properties (zeta potential, swellability and surface energy) and biological performance (antimicrobial activity and cell compatibility). Moreover, we correlated the biological performance of our surface-attached SMAMP networks to the biological activities of structurally related SMAMPs in solution. In the light of the most recent discussion on the exact mechanism of cationic antimicrobial surfaces, this work may be an important contribution to the way we understand antimicrobial coatings.
Results
Study design
We chose poly(oxonorbornene) SMAMPs (Fig. 1a) as a synthetic platform that allowed systematic variation of two key parameters affecting antimicrobial activity: hydrophobicity and charge density. These molecules contain a variable substituent R and a positively charged group on each repeat unit. By changing R from methyl to butyl, homopolymers with variable hydrophobicity but constant nominal charge density were obtained. We also made copolymers containing variable amounts of a hydrophilic, doubly charged diamine repeat unit (Fig. 1a, right), and a hydrophobic repeat unit (R = propyl or butyl, respectively).‡ Thus, one polymer series with a hydrophobicity gradient, and two series of copolymers with a simultaneous hydrophobicity and charge density gradient were obtained. This set of polymers enabled us to investigate the effects of gradual molecular changes on the macroscopic physical properties of the resulting polymer surfaces. At the same time, we could optimize the molecular parameters for optimal antimicrobial activity and cell compatibility.The physical properties of these SMAMP networks were carefully characterized. We determined the apparent surface energy, the zeta potential, the swellability, and the surface morphology. The biological characterization included antimicrobial activity tests against 2 to 4 clinically relevant bacterial strains, and two toxicity tests. With this method pool, the present study probably comprises the most comprehensive characterization of antimicrobial polymer surfaces in literature.
Because it is notoriously difficult to compare antimicrobial data from different laboratories, we also included two antimicrobial systems from other groups as reference points. One is a poly(vinylpyrinine based) quaternary ammonium polymer,18 the other a poly(butyl methacrylate-co-ethanolamine methacrylate) system.19 In previous publications, these surfaces had brush-like architectures. For better comparability to our system, we also prepared them as surface-attached polymer networks (Fig. 1c).
Synthesis
The synthesis of the homopolymers and copolymers shown in Fig. 1a had been previously described for low molecular weights,7b–d and was adjusted to obtain the higher molecular weights needed for the network formation. The surface coating process which we developed is shown in Fig. 1b. It makes use of the beautiful benzophenone chemistry that was introduced into the polymer field by Rühe and coworkers,20 which we combined with thiol–ene chemistry. To obtain surface-attached SMAMP networks, a solution of the SMAMP precursor polymer (with an N-Boc protective group) was mixed with the tetrathiol cross-linker shown in Fig. 1b, and spin-coated onto a benzophenone-functionalized substrate. For most experiments, the substrate was a silicon wafer or glass slide functionalized with triethoxysilyl-benzophenone (3EBP, Fig. 2a). For the surface plasmon resonance experiments, we used a gold surface functionalized with 4-hydroxy-benzophenone-lipoic acid ester (DS, Fig. 2b). The benzophenone moiety in 3EBP and DS enabled the covalent link between the surface and adjacent polymer chains (yellow dots in Fig. 1b). The polymer–polymer cross-links were formed by the tetra-functional thiol cross-linker (pink dots in Fig. 1b). These two reactions were simultaneously activated by UV-irradiation, which initiated the C–H-insertion of the benzophenone, and the thiol–ene-reaction between the thiol cross-linker and the poly(oxonorbornene) double bond, respectively. The ratio of double bonds to SH groups was constant for all SMAMP networks to ensure an equal degree of cross-linking. The coatings also had approximately the same thickness (see Table 1 below). Keeping these two parameters constant allowed us to focus on the effect of structural changes in the repeat units on physical and biological properties. We activated the antimicrobial function of the SMAMP networks by converting the tert-butyl carbamate group of the precursor polymers into a primary ammonium group. The full process is described in the ESI† (pages 4–11, Schemes S1–S4).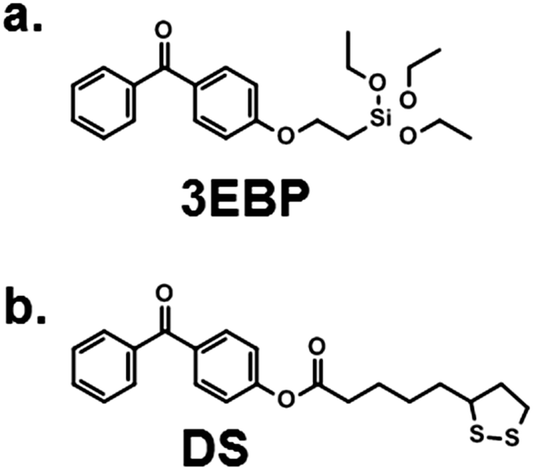 | ||
| Fig. 2 Surface cross-linkers used in this study. (a) 3EBP forms self-assembled layers on any substrate that carries an OH group though Si–O bonds;20 (b) DS attaches to gold via S–Au bonds to form a monolayer.21 | ||
| Dry layer thickness/nm | Apparent surface energy/mN m−1 | Swellability ratio/H2O | ζ max/mV | Iso-electric point | ζ phys/mV | pK | |
|---|---|---|---|---|---|---|---|
| Diamine | 157 ± 4 | 61.0 | 3.2 | 62 ± 3 | 7.5 ± 0.2 | 0 | 7.4 |
| Methyl | 147 ± 3 | 56.8 | 1.9 | 77 ± 5 | 7.6 ± 0.2 | 7 | 7.4 |
| Ethyl | 143 ± 3 | 53.0 | 1.7 | 73 ± 4 | 7.9 ± 0.2 | 23 | 7.7 |
| Propyl | 149 ± 3 | 51.5 | 1.4 | 85 ± 2 | 7.8 ± 0.2 | 11 | 7.5 |
| Butyl | 153 ± 4 | 48.8 | 1.2 | 84 ± 1 | 7.3 ± 0.2 | −2 | 7.2 |
P![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :D = 1:9 :D = 1:9 |
142 ± 4 | 58.0 | |||||
| P:D = 5:5 |
155 ± 3 | 56.3 | |||||
| P:D = 9:1 |
147 ± 3 | 53.5 | |||||
| B:D = 1:9 |
148 ± 3 | 58.0 | 2.3 | 50 ± 3 | 7.5 ± 0.2 | 1 | 7.2 |
| B:D = 5:5 |
158 ± 3 | 55.3 | 1.7 | 74 ± 3 | 7.6 ± 0.2 | 2 | 7.2 |
| B:D = 9:1 |
152 ± 4 | 53.5 | 1.5 | 88 ±3 | 7.5 ± 0.2 | −6 | 7.2 |
A typical 1H-NMR spectrum and a representative GPC curve of the propyl SMAMP precursor are shown in Fig. 3a and b, respectively. The NMR spectrum of the propyl SMAMP precursor demonstrates the high purity of the polymer; the GPC data reveals a monomodal molecular weight distribution, with a molecular weight of 240000 g mol−1 and a polydispersity index of 1.1. The NMR and GPC data for all polymers are given in the ESI† (pages S8–S10). The chemical structure of the polymer networks was characterized by Attenuated Total Reflection Fourier-transform Infrared Spectroscopy (ATR-FTIR, see ESI,† pages S12–S15). The ATR-FTIR spectrum of the propyl SMAMP precursor network and the deprotected, activated propyl SMAMP network are shown in Fig. 3c. The most prominent feature of the spectrum is the characteristic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O band at around 1750 cm−1, which is present in both compounds. After activation, the adsorption band at around 3400 cm−1 in the spectrum of the SMAMP precursor vanishes; this confirms successful removal of the N-Boc protective group. The formation of surface-attached polymer networks from the two antimicrobial reference polymers is illustrated in Fig. 1c. Covalent polymer–polymer cross-links in these reference samples were obtained through reactive methacryloxy benzophenone repeat units22 (purple dots in Fig. 1c), and polymer-surface cross-links through the 3EBP linker (yellow dots in Fig. 1c). The full synthetic procedure is given in the ESI† (page S29, Fig. S17).
O band at around 1750 cm−1, which is present in both compounds. After activation, the adsorption band at around 3400 cm−1 in the spectrum of the SMAMP precursor vanishes; this confirms successful removal of the N-Boc protective group. The formation of surface-attached polymer networks from the two antimicrobial reference polymers is illustrated in Fig. 1c. Covalent polymer–polymer cross-links in these reference samples were obtained through reactive methacryloxy benzophenone repeat units22 (purple dots in Fig. 1c), and polymer-surface cross-links through the 3EBP linker (yellow dots in Fig. 1c). The full synthetic procedure is given in the ESI† (page S29, Fig. S17).
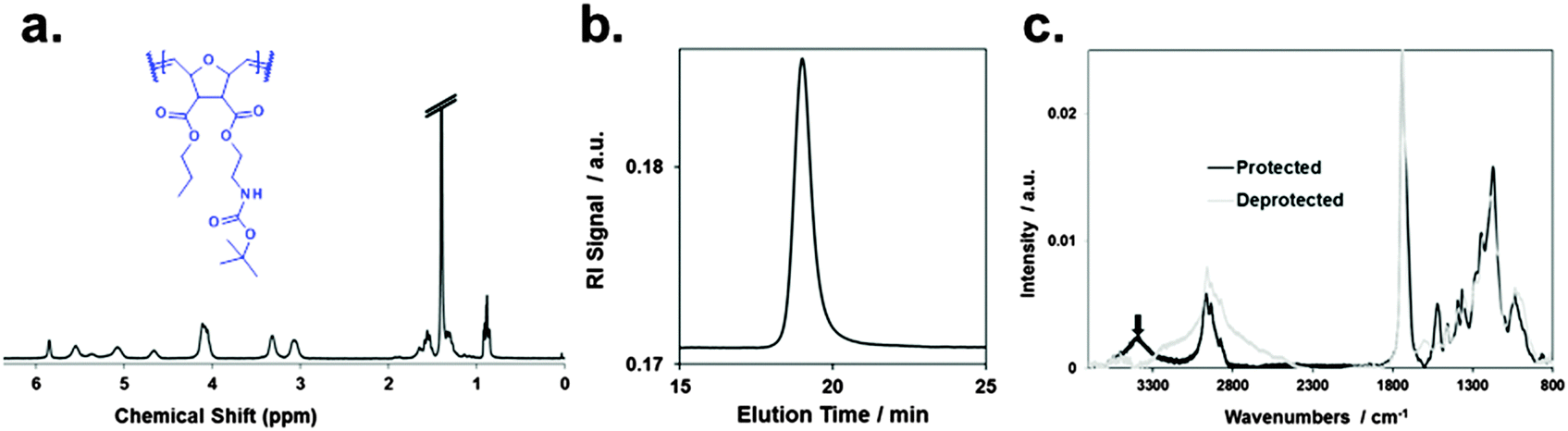 | ||
| Fig. 3 Characterization of the propyl SMAMP precursor polymer and network: (a) 1H-NMR spectrum of the polymer, (b) gel permeation chromatography elugram of the polymer; (c) ATR-FTIR spectrum of the surface-attached precursor polymer network (protected) and activated polymer network (deprotected). | ||
Physical characterization of the networks
The physical characterization of the activated SMAMP networks is summarized in Table 1. All layers had a thickness in the range of 142 to 158 nm. To quantify the relative hydrophobicity of each network, we measured the swelling ratio§ in water using surface plasmon resonance/optical waveguide spectroscopy (SPR/OWG, Table 1).¶ The SPR/OWG spectra of the networks are given in the ESI† (page S15, Table S6); the swelling ratios in water are given in Table 1. In the homopolymer series, the most hydrophilic network was the one with two diamine groups, with a swellability ratio of 3.2, followed by the methyl network with 1.9. The higher alkyl homologous had the expected lower swelling ratios. For the copolymer networks, the swellability also diminished systematically with increasing alkyl repeat unit content. The static and dynamic contact angles of all networks are given in the ESI† (page S18, Tables S7–S12). We quantified the surface energies using the Zisman method.||23 There are some limitations to this method – it is preferentially used for surfaces with low surface energies which do not swell in the solvents used. In our case, the polymer surfaces did swell, but slowly (>30 min). We therefore determined the static contact angle quickly after a defined time interval (1 min). This is a non-equilibrium measurement which only gives relative data; we therefore called these results apparent surface energies. The Zisman plots are given in the ESI† (page S20, Fig. S13–S15), the resulting apparent surface energies are included in Table 1.To check the validity of these measurements, we plotted the swelling ratio in water versus the apparent surface energy (Fig. 4a). This gave an almost linear correlation. The only data point that significantly deviated from linearity was the one corresponding to the highly swollen diamine network. Thus, despite potential limitations of the Zisman method, apparent surface energies and swelling ratios in water were proportional, and therefore either parameter can be used to describe SMAMP hydrophobicity.
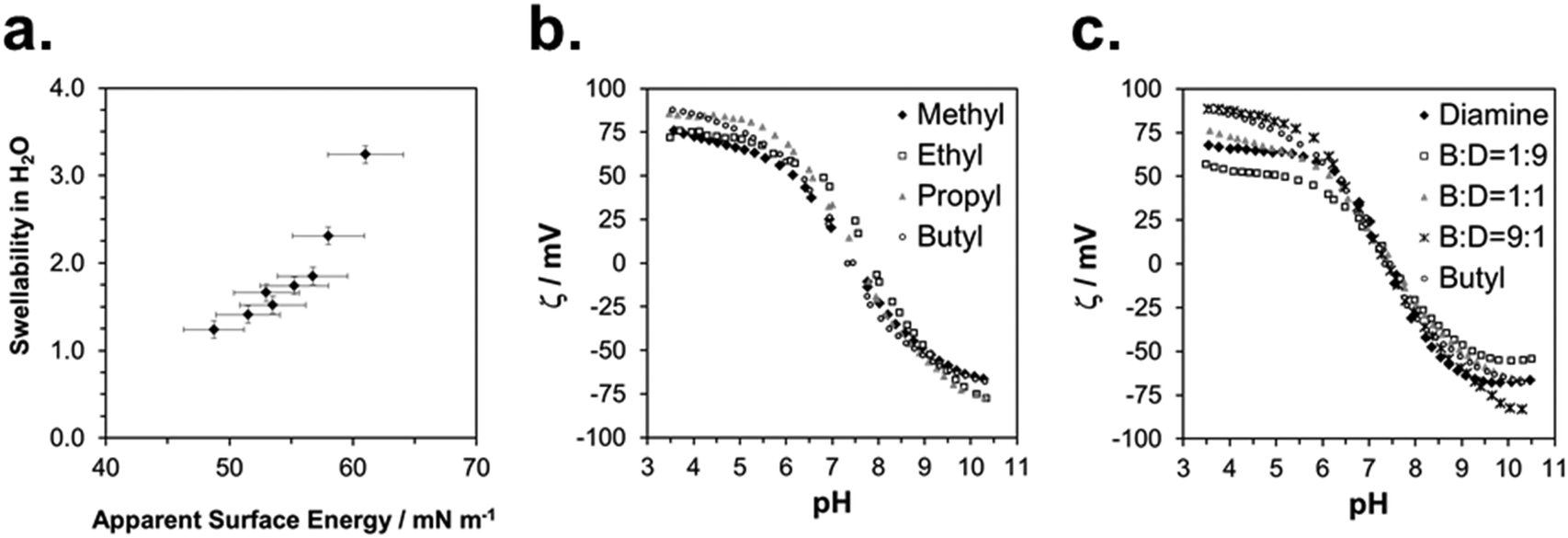 | ||
| Fig. 4 Physical characterization of the SMAMP networks: (a) correlation between swellability in water and apparent surface energy; (b) representative ζ potential titration curves of the SMAMP homopolymer networks; (c) representative ζ potential titration curves of the butyl SMAMP copolymer networks. | ||
We measured the surface charge of the SMAMP networks using elektrokinetic measurements.**24 During these measurements, the samples were titrated in situ from pH 2.5 to pH 10.5. The titration curves of ζ vs. pH (Fig. 4b and c, respectively) conformed to the typical behavior of an amphiphilic polymer surface. At low pH, the zeta potential had a positive plateau and went through a point of inflection. It then leveled off into a negative plateau at high pH. In Fig. 4b, the titration curves for the homopolymers with a hydrophobicity gradient from R = methyl to butyl are shown. The positive plateau values ζmax range from about 85 mV for propyl and butyl, to about 75 mV for methyl and ethyl, the two more hydrophilic polymers. We fitted this data using the Hill equation, a standard fit for sigmoidal curve shapes.†† We also determined the isoelectric point (IEP) and the acid constant (pK value) of the surface bound acid–base pair of the polymer. The latter was calculated from 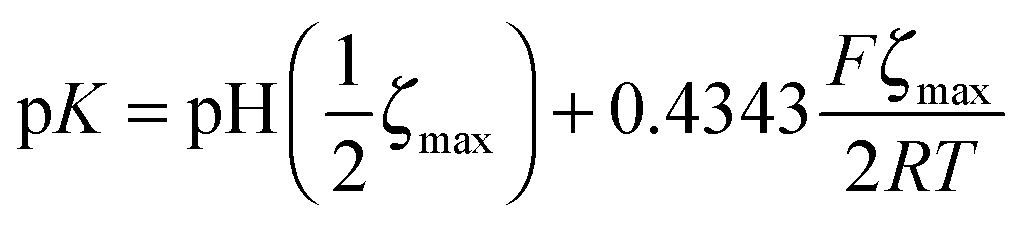 .‡‡25 The data obtained is summarized in Table 1. The isoelectric points and pK values of the homopolymer networks differed about 0.5 pH units, from 7.3 to 7.9 and 7.2 to 7.7, respectively. The titration curves of the butyl copolymer series is shown in Fig. 4c. Here, the differences in ζmax ranged from 50 to 88 mV. Surprisingly, the polymers with higher nominal charge (more diamine content per unit length) had the lowest ζmax values. The reasons for this will be discussed below. The isoelectric points and pK values of the copolymers were much closer than in the homopolymer series, differing only by 0.2 pH units.
.‡‡25 The data obtained is summarized in Table 1. The isoelectric points and pK values of the homopolymer networks differed about 0.5 pH units, from 7.3 to 7.9 and 7.2 to 7.7, respectively. The titration curves of the butyl copolymer series is shown in Fig. 4c. Here, the differences in ζmax ranged from 50 to 88 mV. Surprisingly, the polymers with higher nominal charge (more diamine content per unit length) had the lowest ζmax values. The reasons for this will be discussed below. The isoelectric points and pK values of the copolymers were much closer than in the homopolymer series, differing only by 0.2 pH units.
The exact contribution of each chemical feature of a complicated polymer coating to the zeta potential is not yet fully understood, in particular if the sample can swell. It is clear that the acid–base pair NH3+/NH2 plays a significant role, as does the adsorption of electrolyte ions and aqueous counter-ions.24–26 Swelling influences the zeta potential significantly, and is typically observed by an additional minimum in the negative plateau at alkaline pH.§§ We used the Hill equation to calculate ζ under pseudo-physiological conditions by setting pH = 7.4.¶¶ Thus, we obtained apparent data which we could use to compare the surface properties at pH 7.4. It should also be mentioned that it was difficult to get consistent zeta potential data due to edge effects on the sample slides originating from the coating process. The Helmholtz–Smoluchowski equation relates streaming current and zeta potential. It contains the geometric factor 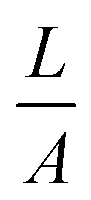 (which depends on surface area A) and is thus sensitive to such effects. Still, several semi-quantitative observations could be derived, which can be used as a guideline to understand the biological effects. This is further discussed below.
(which depends on surface area A) and is thus sensitive to such effects. Still, several semi-quantitative observations could be derived, which can be used as a guideline to understand the biological effects. This is further discussed below.
The morphology of the SMAMP networks was characterized using atomic force microscopy (AFM). Representative AFM height and phase images are given in the ESI† (page S21, Tables S13 and S14). It is evident from these pictures that each SMAMP network homogeneously covered the wafer surface and only had distinct pores in the range of a few tens of nanometers. These originate from phase separation of the polymer and excess tetrathiol cross-linker. The depth of these pores was significantly less than the overall thickness of the network. We found previously that for the given network thickness of about 150 nm, these pores do not impact the antimicrobial activity.27
Antimicrobial activity of the SMAMP networks
The antimicrobial activity of the SMAMP networks was determined using the so-called airborne antimicrobial assay. This assay simulates airborne bacterial contamination, e.g. through sneezing or coughing. We and others recently developed a standardized procedure for this assay.||||27,28 Details are given in the ESI† (page S34). The results of the airborne assay are given in Fig. 5a–f. As this data shows, the SMAMP networks were highly efficient after two hours contact time – many of them killed close to 100% of the bacteria (Fig. 5d–f). In particular, the P:D = 1:9, butyl, B:D = 9:1, B:D = 5:5, and B:D = 1:9 had a reduction of at least 3log units (Fig. 5e and f). The most promising networks were P:D = 1:9 and B:D = 1:9, which quantitatively killed 3 out of 4 tested bacteria, and reduced the fourth by at least 3log units. While this is an exciting result from an applications point of view, it is not possible to correlate structural differences of the SMAMPs when the killing is near-quantitative. We therefore repeated the assay with only 30 min contact time to get a higher amount of surviving colony forming units (CFUs). This data is shown in Fig. 5a–c. For the Gram positive S. aureus bacteria, we observed a correlation between CFU reduction and increasing alkyl chain length in the homopolymer series (Fig. 5a). For Gram negative E. coli bacteria, there was an unexpected minimum for the ethyl networks, which had 100% killing efficiency (Fig. 5a). In the two copolymers series, there was a minimum in the CFU percentage (for P:D = 5:5 and B:D = 5:5, respectively) in the activities against Gram positive S. aureus and E. faecalis at t = 30 min (Fig. 5b and c). This is also seen at 2 h contact time, where these two networks are the only ones in their series that quantitatively kill the two Gram positive bacteria (Fig. 5b and c). For the Gram negative E. coli and P. aeruginosa, the trend is less clear. At 30 min, E. coli was reduced to about 0.5% remaining CFUs for propyl and all its copolymers. In the butyl copolymer series, the number of CFUs was one order of magnitude less, with a quantitative killing of E. coli for B:D = 9:1. P. aeruginosa had a minimum for P:D = 9:1 and P:D = 5:5 in the propyl series at t = 30 min, and was killed quantitatively by all networks at t = 2 h. In general, the Gram negative E. coli and P. aeruginosa were significantly more sensitive to the antimicrobial networks than the Gram positive S. aureus and E. faecalis, in many cases by two log units.
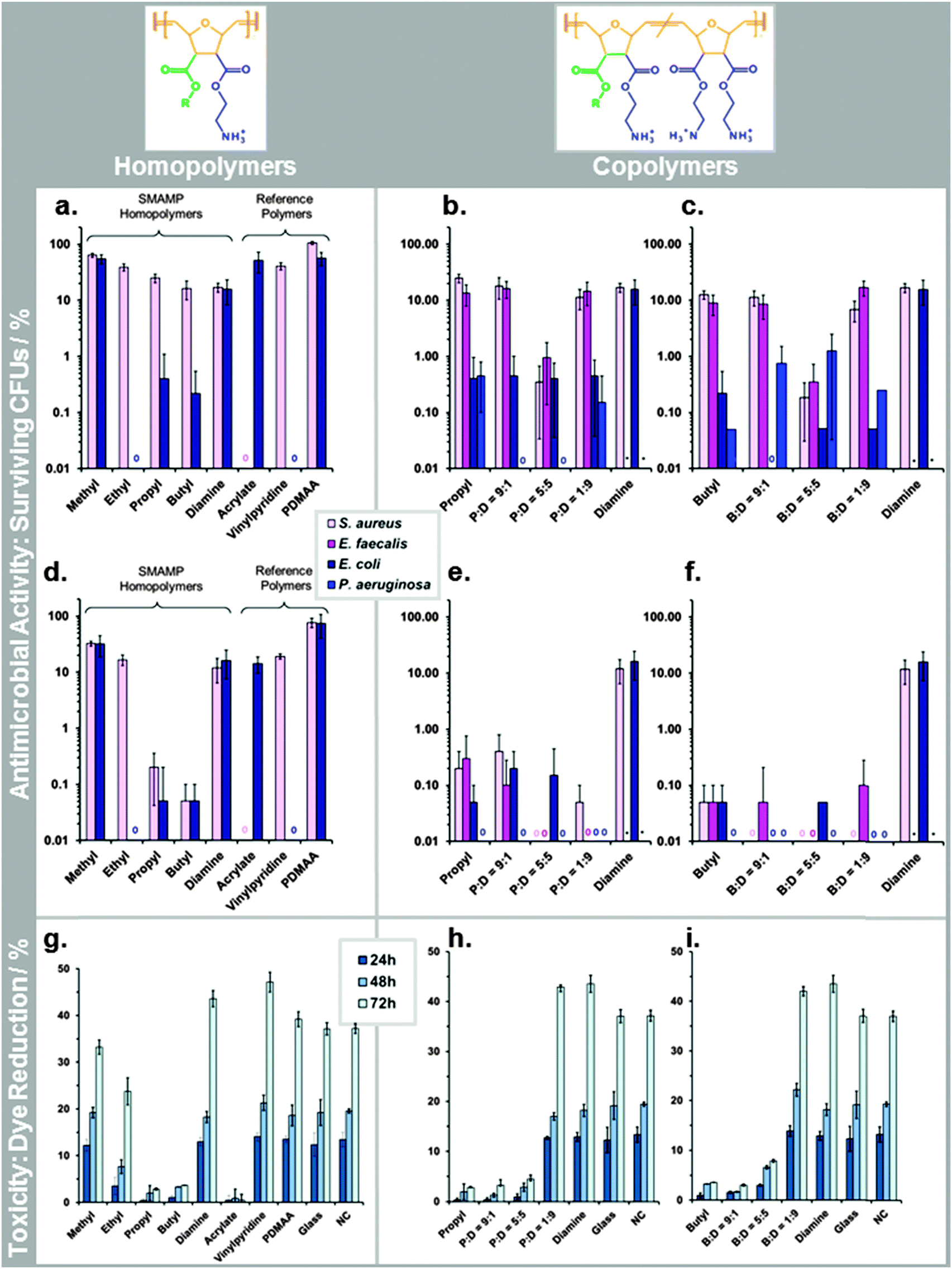 | ||
| Fig. 5 Biological characterization of the SMAMP networks and the reference polymer networks (acrylate, vinylpyridine, PDMAA). (a–f) Antimicrobial activity (reported as percentage of surviving colony forming units, log(%CFUs)) against various bacteria: (a) homopolymers, t = 30 min; (b) and (c): propyl and butyl copolymers, t = 30 min; (d) homopolymers, t = 2 h; (e) and (f) propyl and butyl copolymers, t = 2 h; (g–i) cell compatibility with primary human keratinocytes, reported as % of Alamar Blue dye reduction after 24, 48 and 72 h. (g) Homopolymers, (h) and (i) propyl and butyl copolymers. | ||
To correlate antimicrobial activity with physical properties, we plotted the percentage of surviving CFUs (log(%CFUs) for E. coli an S. aureus at t = 30 min,***) versus swellability, apparent surface energy, zeta potential, pK and the isoelectric point (Fig. 6a–f). The aim of these correlations was to differentiate between the effects of surface hydrophobicity and surface charge on antimicrobial activity. It is important to note that for most of the physical parameters we measured, the effect of changes in surface hydrophobicity and surface charge are coupled. For example, a higher surface charge will make a surface also more hydrophilic and cause more swelling. Likewise, a strong increase in hydrophobicity will also change the zeta potential. Thus, it is very difficult to experimentally separate the two parameters. The best parameter separation was achieved for the homopolymer series with R = methyl to butyl. Here, the nominal charge density per repeat unit was about the same, even in the swollen state, because the swelling ratios of these networks were not too different.
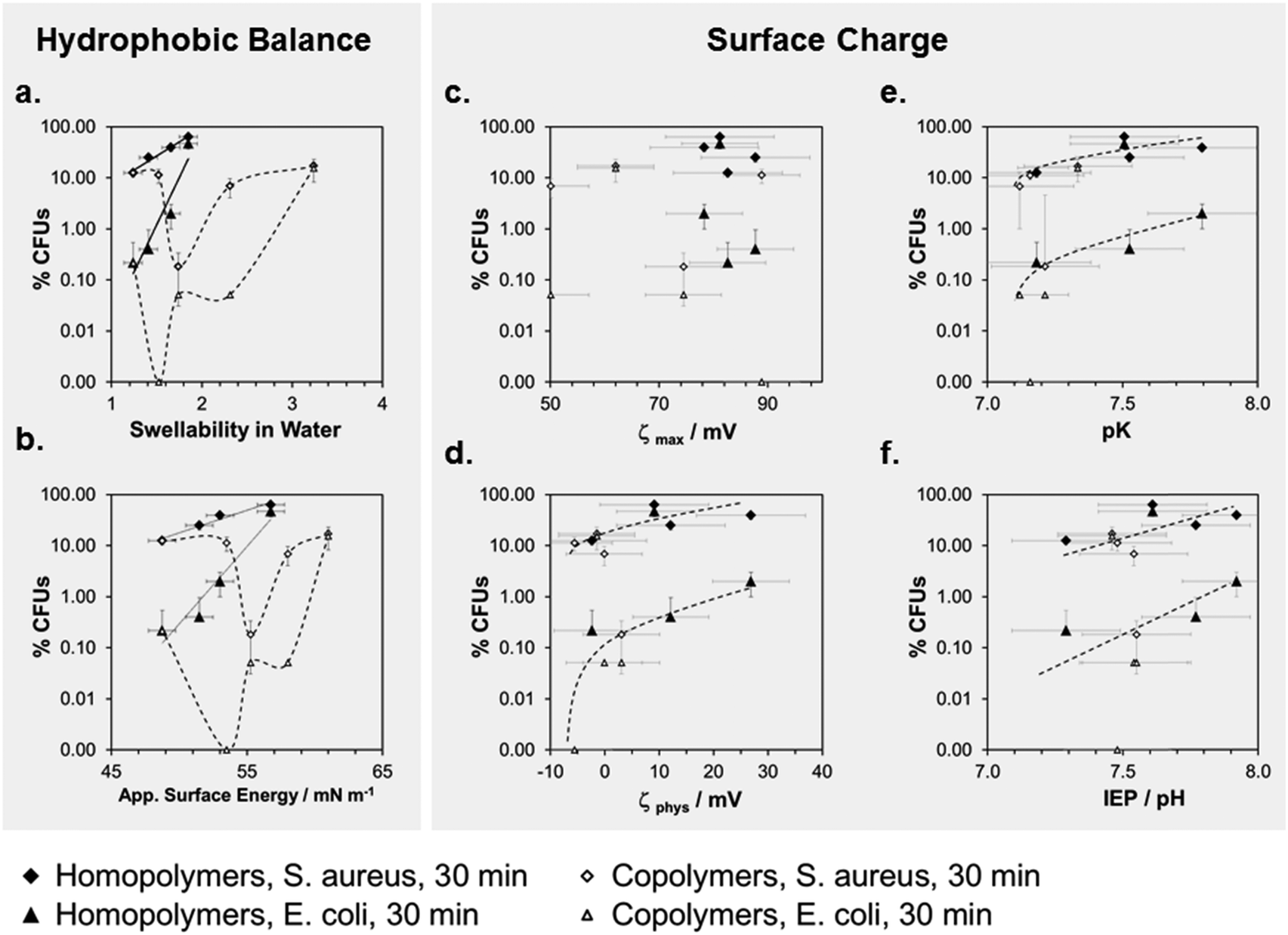 | ||
| Fig. 6 Correlation of physical properties and antimicrobial activities of the surface-attached SMAMP networks: the percentage of surviving colony forming units, log%CFUs, is plotted versus (a) swellability in water; (b) apparent surface energy; (c) maximum zeta potential; (d) zeta potential under pseudo-physiological conditions; (e) acid constant of the polymer network; and (f) isoelectric point of the polymer networks. All lines are guides to the eye only. | ||
We compared these results to the butyl copolymer series with a simultaneous hydrophobicity and charge density gradient. For the homopolymers, we found a linear correlation in the log(%CFUs) vs. swellability plot for both bacteria (Fig. 6a). For both copolymer series, there was a minimum in the log(%CFUs) vs. swellability correlation for both bacteria (Fig. 6a). The same was found in the log(%CFUs) vs. apparent surface energy plot (Fig. 6b): linear correlations were obtained for the homopolymers, minima for the copolymers.
The plots of parameters related to the zeta potential (ζmax, ζphys, pK and IEP) vs. log(%CFUs) are shown in Fig. 6c–f. Overall, these data points scatter more than the data in Fig. 6a and b, probably due to the mentioned edge effects. Still, interesting findings emerge: with few outliners, pK and IEP correlate with log(%CFU) (Fig. 6e and f), no matter whether the data came from the homopolymer or copolymer series. Furthermore, the data for E. coli and S. aureus are well separated, indicating different sensitivities of these two bacteria to the polymer IEP and pK. Thus, pK or IEP, at least in this series of structurally similar polymers samples, may be useful predictors for the antimicrobial activity of a surface against certain bacterial species. This is reasonable because the pK and IEP are sensitive to both the hydrophobic balance of the polymer and its charge density. In Fig. 6d, we also included a plot of the antimicrobial activity vs. the apparent physiological zeta potential. Even though the data scatters more widely, we also see a trend for this parameter.
Cell compatibility of the SMAMP networks
The cell toxicity of the polymer networks was quantified by two methods. First, primary human fibroblasts and immortalized human keratinocytes were grown on the SMAMP networks for 6 hours. Their RNA was then extracted and analyzed for up-regulation of toxicity markers using polymerase chain reaction (PCR). This data is shown in the ESI† (page S36, Fig. S24–S26). For the fibroblasts, the two apoptosis markers (ANXA5, CASP3), the inflammation marker IL6, and the cell proliferation marker MKi67 were on the same level as the control for all SMAMPs (Fig. S25, ESI†). This is an indication that these networks were fully cell-compatible. For the keratinocytes, IL6 was significantly up-regulated (Fig. S26, ESI†), indicating an inflammatory response to the surface. To find out whether this was a significant sign for toxicity, we also performed the Alamar Blue assay on this cell type (ESI,† page 40).††† The results are given in Fig. 5g–i. They nicely complement the PRC data. For the samples where IL6 was most up-regulated (butyl, propyl P:D = 9:1 and P:D = 5:5), Alamar Blue dye reduction was also low, indicating low cell viability. For the samples with low IL6 up-regulation (B:D = 1:9 and P:D = 1:9), the viability was close to that of the control, demonstrating good cell compatibility. For the homopolymer series, the cell toxicity increased with increasing alkyl chain length. The amount of chargeable units did not have an adverse effect on viability: the diamine homopolymer, P:D = 1:9 and B:D = 1:9 had an even higher dye reduction than the negative control, presumably because they swell strongly. For both copolymer series, cell toxicity was systematically reduced as hydrophobicity decreased.
We correlated these cell compatibility results with the physical properties of the surfaces by plotting the percentage of Alamar Blue dye reduction versus swellability, apparent surface energy, and parameters related to the zeta potential (Fig. 7). The 24 h and the 48 h time-point of the cell compatibility data were the most useful ones for these correlations, since the cells had substantially recovered from toxicological effects at t = 72 hours. When plotting log(%Dye reduction) vs. swellability (Fig. 7a), we observed a steep increase of the curve for low swellability, which then leveled off into a plateau. Thus, when a swellability ratio of about 2 was reached, making the surface more hydrophilic did not further improve the cell compatibility. Both copolymers and homopolymers conform to this curve. There is an inverse trend for the plot of dye reduction vs. ζmax, where the amount of dye reduction decreases with increasing surface charge. The other surface charge parameters do not correlate with the cell compatibility data. This will be discussed below.
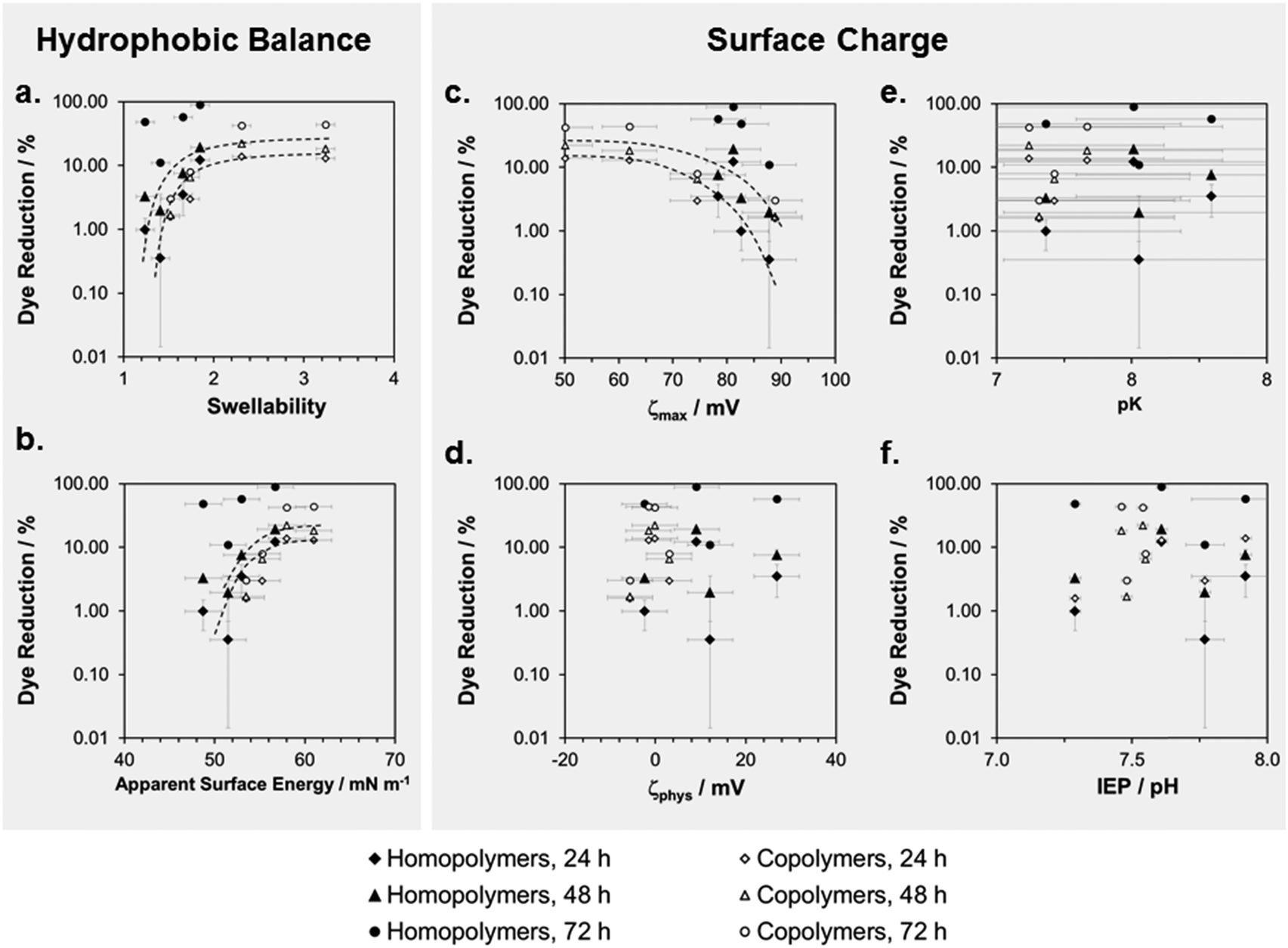 | ||
| Fig. 7 Correlation of physical properties and cell compatibility of surface-attached SMAMP networks: the percentage of Alamar Blue dye reduction is plotted versus (a) swellability in water; (b) apparent surface energy; (c) maximum zeta potential; (d) zeta potential under pseudo-physiological conditions; (e) acid constant of the polymer network; and (f) isoelectric point of the polymer networks. | ||
Possible applications – coating of a model catheter
To obtain proof-of-concept for a real-life application of our surface-attached SMAMP networks, we also coated medical-grade PDMS tubing with propyl SMAMP networks. Since the standard process of network formation compromised the mechanical integrity of the tubing, we modified the process and developed parameters to directly apply the activated SMAMP coating to the PDMS tubing (ESI,† pages S27–S29). Two coatings of 100 nm thickness each were necessary to obtain an antibacterial efficacy of >99.9% (Fig. S16, ESI†). With this new process, which avoids corrosive chemicals and aggressive solvents in direct contact with the tubing, fully bactericidal SMAMP coatings on PDMS tubing were obtained.Discussion
Optimization of biological performance
The first aim of the study was to use the modular chemistry of poly(oxonorbornenes) SMAMPs to obtain surface-attached polymer networks with systematically varying chemical structure. This allowed us to identify the SMAMP surface that had optimal broad band activity against Gram negative and Gram positive bacteria, and excellent compatibility towards human cells. These are crucial properties for all applications that are in contact with the human body for an extended period of time. By gradual variation of hydrophobicity and charge density, we found that P:D = 1:9, butyl, B:D = 9:1, B:D = 5:5, and B:D = 1:9 had excellent antimicrobial properties. Out of these, P:D = 1:9 and B:D = 1:9 also had outstanding cell compatibility, while the others were more toxic. We had also synthesized antimicrobial networks based on the poly(methacrylates) reported by Kuroda and DeGrado,29 and the poly(vinylpyridines) by Tiller and Klibanov as a benchmark for our SMAMP networks.18,30 The data for these samples (included in Fig. 5a and d) showed that the acrylate only killed S. aureus, while the vinylpyridine was only active against E. coli. In the Alamar Blue assay, the acrylate was highly toxic, while the vinylpyridine had excellent cell compatibility. We further tested a surface-attached network made from poly(dimethylacrylamide) (PDMAA),22 which is known for its excellent cell compatibility,31 but is not antimicrobial (Fig. 5a and d). Comparing these three references to our SMAMPs, we find that the vinylpyridine was very similar to the ethyl homopolymer network. Further, none of the SMAMP networks was as toxic as the acrylate. P:D = 1:9 and B:D = 1:9, our most active SMAMPs, were as cell-friendly as PDMAA (Fig. 5g–i), but much more active. In the light of this data, we conclude that the P:D = 1:9 and B:D = 1:9 copolymers, with broadband antimicrobial activity and excellent cell compatibility, are meeting all the requirements for biomedical applications, and may be even setting a new standard for the field.
Correlation of physical properties and antimicrobial activity
The second lead question of our study was – how do the variations in chemical structure correlate with measurable physical quantities, and can we relate these to biological activity? In literature, structural changes in hydrophobicity (e.g. length of alkyl chain) or charge density (e.g. number of charged repeat units) had been previously compared with antimicrobial properties, but the effort typically remained on a qualitative level; physical parameters like zeta potential or surface energy were rarely measured.32 For the series of networks with constant charge density, our results show that swellability and apparent surface energy correlate linearly with antimicrobial activity– the higher the hydrophobicity, the higher the antimicrobial activity. For the series of networks where charge density is not constant, the situation is more complicated. Here, swellability and surface energy do not display any systematic correlation with antimicrobial activity. Thus, swellability and surface energy alone do not sufficiently to describe these systems. On the other hand, we found that pK and IEP correlated systematically with antimicrobial activity for all polymer networks. When scanning the literature for similar findings, we found only scarce reports on ζ measurements of antimicrobial polymer surfaces.32,33 In particular, pH-dependent measurements were rarely performed. Thus, our result that pK and IEP quantitatively correlated with antimicrobial activity, even when correlations to ζmax are not clear (which was the case for our strongly swelling samples) is an important extension to the previously found qualitative correlations between ζmax and antimicrobial activity.33e,fHow can we explain these findings? SMAMP networks are weak polyelectrolyte networks – their theoretical description is complicated, and surface-attached polyelectrolyte networks in particular are not yet fully understood. For example, it is not yet possible to predict the net surface charge of surface-attached weak polyelectrolyte networks. However, the following pieces of theory are useful to understand their behavior: when an unconstrained polyelectrolyte network swells, its chains are extended in three dimensions until there is equilibrium between the osmotic pressure inside the network and the surrounding solvent. Under these conditions, the swelling pressure Π that drives solvent into the network is zero.34 In terms of free energy, ΔG = ΔGmix + ΔGelastic + ΔGion = 0. This equation contains three terms. The one for the contribution of polymer–solvent mixing, ΔGmix, can be described by the Flory–Huggins-theory.35 The elastic energy ΔGelastic is caused by stretching of the polymer chains out of their equilibrium conformation, and ΔGion describes the repulsive electrostatic interactions of the charged polyelectrolyte.34 Thus, ΔGmix and ΔGion cause network extension, while ΔGelastic is the restoring energy that limits swelling. This equation is also valid for surface-attached polyelectrolyte networks, with the exception that these can only swell normal to the surface.22 The problem to theoretically describe swollen surface-attached polyelectrolyte networks is that ΔGion and ΔGelastic become mutually dependent through counter-ion condensation.36 The number of actual charges per unit volume in a polyelectrolyte network will depend on two factors – the distance between available chargeable units d (which is dictated by the molecular structure) and the Bjerrum length l.‡‡‡ Swelling increases d, and that may change the extent of counter-ion condensation if d ≤ l.
For our surface-attached SMAMP homopolymer network series, with a constant number of chargeable sites per unit volume, we estimate that d is on the order of magnitude of 0.75 nm (about 5 carbon–carbon bonds between each NH3+ group). This compares to l = 0.7 nm in water at room temperature. Since SMAMPs are weak polyelectrolytes, we have to consider the acid–base equilibrium of the NH3+/NH2 acid–base pair. With a pK of 7.3–7.9 in the homopolymer series, these groups dissociate 50% or less under physiological conditions (pH = 7.4). Consequently, d > l even without swelling effects. Thus, within this series, the actual surface charge of the homopolymers is dominated by the acid–base equilibrium and not by counter ion condensation, and ΔGion is roughly constant for all homopolymer networks. This is consistent with a difference in ζmax of only 11 mV for the homopolymer series (Fig. 4b and Table 1). Under these conditions, changes in ΔGmix and ΔGelastic through the homopolymer series must compensate each other. Swelling only occurs if ΔGmix is negative. The modulus of ΔGmix should be larger for the more hydrophilic networks, |ΔGmix,methyl| > |ΔGmix,ethyl| > |ΔGmix,Proyl| > |ΔGmix,butyl|. Consequently, for the absolute values of the elastic energy stored in the swollen networks, ΔGelastic,methyl > ΔGelastic,ethyl > ΔGelastic,Proyl > ΔGelastic,butyl. This is consistent with the experimentally found decreasing swelling ratio of 1.9 to 1.2 from methyl to butyl. Thus, what we see when we measure swellability of the SMAMP homopolymer networks are differences in ΔGmix, and consequently in the Flory–Huggins-parameter χ of these polymers. Therefore, for these equally charged, equally cross-linked, equally thick SMAMP networks, χ correlates with antimicrobial activity.
In contrast to the above discussed homopolymers, our copolymers have two chargeable amine groups per repeat unit, and a distance d of about 0.4 nm. Additionally, they have a slightly lower pK (7.2) and are thus slightly more dissociated (60% at pH 7.4). Therefore, for the non-swollen copolymer networks, d ≤ l, and counter-ion condensation becomes significant for the series members with high diamine content. When these networks swell, the extent of counter-ion condensation changes, and we are crossing into the regime where d ≥ e. Thus, swelling affects both ΔGelastic and ΔGion. Experimentally, we observe a systematic increase of swelling with increasing diamine content. This is consistent with an increase of |ΔGmix|, and also with an increasing absolute value of ΔGelastic. The question is – how exactly is ΔGion affected? This should be mirrored in ζmax. We do not see a systematic trend for ζmax, which in fact goes through a minimum for B:D = 1:9 and is smaller for diamine than for butyl, because diamine swells much more strongly. This non-linear behavior, and the resulting non-linear behavior of antimicrobial activity in the copolymer series, may be explained by the overlay of the steadily increasing parameter ΔGmix, which causes swelling, and the non-linear changes in the extent of counter-ion condensation, which affect ΔGion.
In the absence of a more quantitative theory for surface-attached weak polyelectrolyte networks, further correlations are difficult. It is very interesting however, that the parameter pK obtained from the ζ potential titrations correlates with antimicrobial activity for all polymers that were tested. Since the pK data for the homopolymer series shows that at equal charge density, pK changes with hydrophobicity, we find that it is a parameter that is not only affected by the acid–base equilibrium, but also by the overall hydrophobicity of the surface-attached network. This is also well-known for proteins. pK may therefore be the most sensitive parameter in our data set that takes into account both effects – charge and hydrophobicity changes – and could be a useful predictor for antimicrobial activity in a given polymer series. It should be noted however that pK primarily depends on the chemical nature of the cationic group. Thus we can explain a shift in pK for structurally similar polymers, but would not yet dare to propose a general correlation of pK with antimicrobial activity for different cations.
Correlation of physical properties and cell compatibility
We observed an unambiguous correlation between swellability and cell compatibility for all homopolymer and copolymer networks (Fig. 7a). The curve of log(%Dye reduction) vs. swellability first had a steep slope and then leveled into a plateau. Since simultaneous antimicrobial activity and cell compatibility is a subtle balance, the plateau in this curve is an important finding, as it gives a design rule to the polymer chemist. The optimal coating will be found slightly to the left of the plateau. Since swellability and apparent surface energy correlate (Fig. 4a), the same trend is found in the log(Dye reduction) vs. apparent surface energy plot (Fig. 7b). Thus, swellability and apparent surface energy are both suitable to optimize coatings for biocompatibility by physical methods. We found a similar curve shape in the log(Dye reduction) vs. ζmax plots (Fig. 7c). This is consistent with literature reports on the toxicity of too highly charged cationic surfaces.Correlation of biological activity on surfaces and in solution
Lastly, we wanted to see if it was possible to quantitatively correlate the biological activity of SMAMPs on surfaces and in solution. The solution properties of some of our SMAMPs, particularly antimicrobial activity and hemolytic activity, have been carefully characterized before.7b,d,9a We plotted the antimicrobial data of the here reported SMAMP homopolymer networks (for S. aureus and E. coli, respectively) vs. the MIC§§§ data for these homopolymers in solution (at a molecular weight of 3000 g mol−1, data from7d). This is shown in Fig. 8a. The number of available data-points was scarce, nevertheless there seems to be a positive correlation for S. aureus and E. coli – the higher the solution activity, the higher the surface activity (Fig. 8a). For the copolymers, with simultaneous changes in charge density and hydrophobicity, no such correlation could be found (Fig. 8b).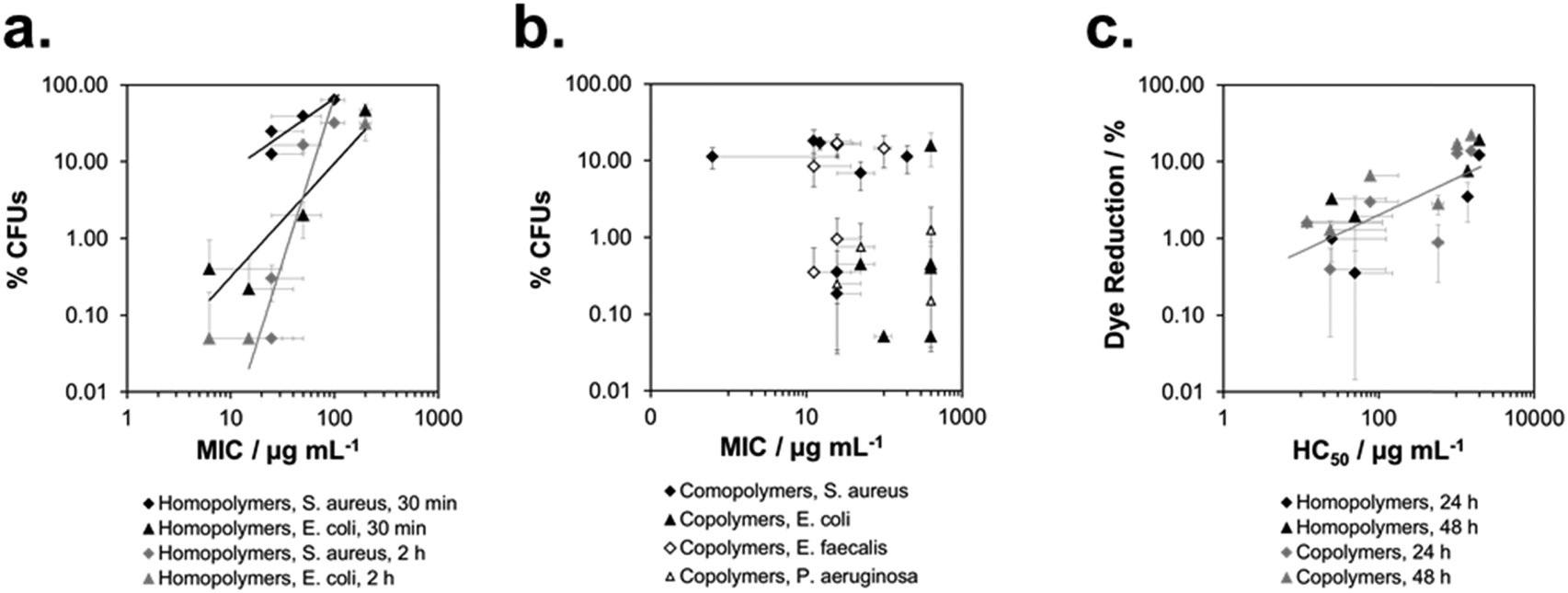 | ||
| Fig. 8 Correlation of biological activity in solution and on surfaces: percentage of surviving colony forming units vs. minimum inhibitory concentration for the homopolymer series (a) and the copolymer series (b); percentage of Alamar Blue dye reduction of human gingiva keratinocytes vs. hemolytic concentration (human red blood cells, c). | ||
To correlate the cell compatibility of SMAMPs on surfaces and in solution, we plotted the amount of Alamar Blue dye reduction on surfaces vs. the hemolytic concentration in solution. Although the data also scatters, there is a clear correlation in the log(%Dye reduction) vs. log(HC50)¶¶¶ plot for homopolymers and copolymers. This indicates that the same parameters govern cell compatibility on surfaces and in solution, and that HC50 is a useful method to also predict the biocompatibility of surface-attached polymers.
Mechanistic considerations
While the mechanism of action of natural antimicrobial peptides (AMPs) and of antimicrobial polymers in solution is meanwhile partially understood,37 the mechanism by which surface-attached polymers kill pathogens is still under debate. Early papers describe a ‘hole-poking mechanism’,11a,d,38 which is probably more of a cell membrane penetration and perturbation by not too densely grafted, surface-attached polyelectrolyte brushes, since such brushes are collapsed and not spike-like under physiological, high salt conditions. The impact of charge density on antimicrobial activity was discussed by Ober,39 Russell and Matyjaszewski,10a,b and Kügler et al., who even found a species-dependent charge threshold for activity.10c As to mechanisms, it was suggested that the exchange of the divalent membrane counter-ions against the polyelectrolyte surface was lethal to bacteria.10c,40 In a recent paper, Chan-Park suggests that highly charged surfaces can suck anionic components out of cell membranes, like an ‘anion sponge’.15 Tiller independently also proposed such a mechanism (‘lipid sponge effect’).16 Busscher recently postulated that there is a ‘lethal regime’, in which the adhesion between bacteria and the cationic substrate is so strong that bacteria are unable to detach and grow, and eventually die.17,41 The existence of such a charge-dependent lethal regime would explain Kügler's species-dependent charge thresholds10c and match the observations of Tiller and Busscher,17 Chan-Park,15 Matyjaszewski and Russell.10a,b We also found a species-dependent correlation between charge and antimicrobial activity (Fig. 6d–f).Such a charge based-mechanism alone, however, does not explain all the observations in this work. It does not explain differences in antimicrobial activity of equally charged polymers with different hydrophobicity, which was observed in our well-defined model system (Fig. 6a and b), and also reported by others.42 Charge density therefore cannot be the whole answer to explain antimicrobial activity on surfaces – the role of hydrophobicity needs to be included into any proposed mechanism. Busscher suggested that the mechanism of action of antimicrobial polymers in solution may be fundamentally different to the mechanism of action of antimicrobial polymers on surfaces.17 In contrast to this, we observed a correlation of antimicrobial activity in solution and on surface for equally charged polymers (Fig. 8a). This would be impossible if the two mechanisms were fundamentally different in all aspects. However, we see no such correlation when we compare the solution and surface activity of polymers with a simultaneous hydrophobicity and charge gradient (Fig. 8b). To understand this, let us look at the mechanism of action of antimicrobial peptides and polymers in solution. The currently accepted model is a two-stage process. Long range electrostatic forces first enable attachment of the cationic antimicrobial molecules to the bacterial surface. In the second step, the hydrophobic groups interact with the bacterial membrane and cause disruption or destabilization. To us, the here reported findings are a strong indication that the mechanism of contact-killing antimicrobial surfaces is also a multi-step process. The step dominated by hydrophobicity (ΔGmix) should be similar to the one found in solution: once in contact with the bacterial surface, the flexible segments of the antimicrobial polymer insert into the bacterial membrane and cause disruption. Thus, the higher the hydrophobicity at constant charge density, the higher the antimicrobial activity. Such an insertion is possible even for surface-attached polymers because they contain swollen, flexible chains or chain segments, and are not rigid bodies. The other mechanistic feature must be charge-dominated, as described by Busscher,17 Tiller16 and Chan-Park.15 The body of data from literature seems to indicate that, unlike in solution, the role of charge is not just to attract the antimicrobial species to the bacterial membrane, and then let hydrophobicity operate. Because the charge density near a cationic surface is much higher than the charge density of low molecular weight antimicrobial polymers in solution, the cationic surface also impacts the viability of bacteria, which cannot fulfil vital functions because they are pinned to the surface by charge. The considerations can be summed up in the following mechanism cartoon (Fig. 9): first, bacteria that have approached the surface are flattened out in the contact zone due to the high local charge density of the antimicrobial surfaces. This flattening will enable close contact between the bacterial envelope and the surface, so that the polymer chain segments at the interface can insert into the bacterial membrane.
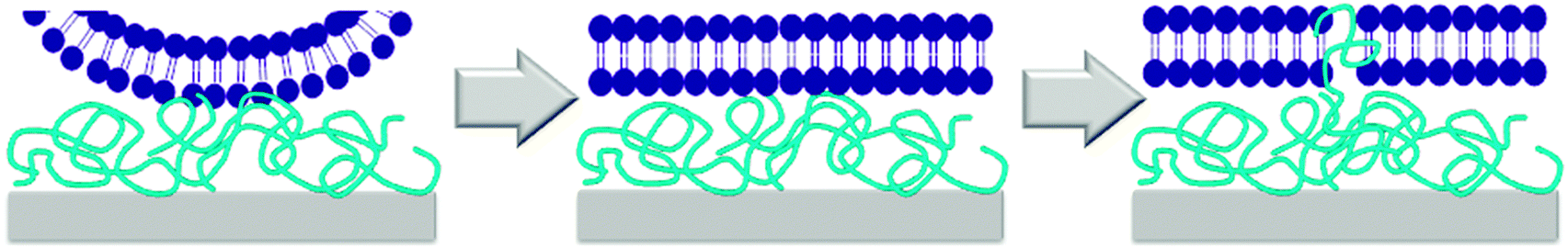 | ||
| Fig. 9 Cartoon illustration of the modified mechanism of action of cationic antimicrobial surfaces: first, the bacterially are locally flattened by the high charge density of the surface. This enables interaction with the polymer segments on the surface, which insert into the membrane and cause damage that may lead to cell death. | ||
Depending on the relative changes in charge density, Δ(ΔGion), or hydrophobicity, Δ(ΔGmix), within a polymer series, one mechanistic feature may dominate the trends in that series, and there may even be opposite effects. For our homopolymer series, we could clearly show the effects of the hydrophobicity-driven mode of action, but we could not yet demonstrate the workings of changes in charge density, because we could not yet design a system where charge changes without affecting hydrophobicity.
Conclusion
Antimicrobial polymer surfaces are attractive materials to fend off the propagation of bacterial pathogens. In this paper, we presented an easy method to obtain surface-attached networks made from poly(oxonorbornene)-based synthetic mimics of antimicrobial peptides (SMAMPs). We systematically varied the structural properties of these networks, and correlated them to physical and biological properties. We could show for surface-attached SMAMP networks with similar thickness and cross-linker density that• by carefully balancing hydrophobicity and charge density, broad-band active antimicrobial surface-attached networks with excellent cell compatibility were obtained, which can also be used in real-life applications such as catheter coatings.
• at constant charge density, swellability and apparent surface energy decreased with increasing antimicrobial activity. This indicates a hydrophobicity-related step in the mechanism of action of antimicrobial polymer networks.
• the acid constant pK correlates decreased with increasing antimicrobial activity for all polymer networks investigated, and thus may be a useful property to predict the relative antimicrobial activity of a series of structurally related polymer networks.
• swellability and apparent surface energy increased with increasing cell compatibility.
• cell compatibility decrease with increasing ζmax. This is consistent with reports that highly charged surfaces are not cell-friendly.
• for equally charged surface-attached antimicrobial networks, antimicrobial activities on surfaces and in solution correlate. This is an indication that the hydrophobic component in the mechanism that describes antimicrobial action in solution and on surfaces is related.
• for networks where hydrophobicity and charge density vary simultaneously, no such correlation was found. This indicates that the step in the mechanism of action that is charge driven differs significantly in solution and on surfaces.
• for all networks, cell compatibility in solution and on surfaces correlate, indicating that the principle that governs these phenomena is similar in both cases.
To our knowledge, this is the first work where such extensive correlations become apparent. However, further studies are needed to derive equations that describe the relations between biological activity and the here investigated physical parameters. We also have to stress that so far, these correlations hold only for structurally closely related compounds. We invite other researchers to complement their studies with these types of measurements, so that a broader data basis for more universal correlations between physical parameters and biological is obtained, which would be of great benefit for the whole field.
Additionally, it has to be noted that the parameters measured here were determined on the macroscopic scale; however, bacteria and cells are micro-objects which may react very sensitive to local effects. For example, they respond to hydrophobic excess in a not-quite randomly-distributed polymer chain,43 or to nanometer-sized surface features. These are effects which may impact the biological activities so strongly that the above observed trends (which we obtained from structurally regular, well-defined, facially amphiphilic SMAMP networks) may not be observable in less homogeneous systems.
Finally, it is well known that cationic polymer surfaces tend to have reduced activity in vivo due to protein adsorption and contamination with biofilms. Thus, to be efficient in medical applications, antimicrobial activity alone is not sufficient. Rather, the ideal material for clinical applications should be also antifouling or self-cleaning. Some progress in this direction has been made: for example, it was shown that some surfaces can be regenerated easily by washing with a cationic surfactant, which is a promising step towards long-term sterile antimicrobial coatings.11c We also reported combined surfaces of antimicrobial SMAMPs and antifouling polyzwitterions that were protein-resistant.21 These are first steps towards long-term active surfaces that are dearly needed for hospital applications.
Experimental
All experimental procedures are given in the ESI.†Acknowledgements
Funding of this project by an Emmy-Noether Grant of the German Research Foundation (DFG, LI1714/5-1) to K.L. is gratefully acknowledged. The catheter coating part of the project has been funded by OphthaSwiss, Herisau. Jürgen Rühe is gratefully acknowledged for very helpful discussions and sharing of many analytical instruments. Experiments were conducted by P.Z., D.L., E.K.R., Z.D., F.D., H.-R.P.-H., and D.L.G.S.. T.S. and A.A.-A. designed the biological experiments, K.L. designed and directed the project, evaluated the data for this manuscript and her habilitation thesis, made the figures, and wrote the manuscript. P.Z. compiled the data and made most of the figures for the ESI,† and independently evaluated the data for his doctoral thesis.References
- World Health Organization, Report on global surveillance of antimicrobial resistance, 2014.
- (a) Center for Disease Control, http://www.cdc.gov/mrsa; (b) European Antimicrobial Resistance Surveillance Network (EARS-Net), Antimicrobial resistance surveillance in Europe 2013, http://www.ecdc.europe.eu/publications.
- (a) O. A. Awoyinka, I. O. Balogun and A. A. Ogunnowo, J. Med. Plants Res., 2007, 1, 63–65 Search PubMed; (b) K. Page, M. Wilson and I. P. Parkin, J. Mater. Chem., 2009, 19, 3819–3831 RSC.
- (a) S. J. Dancer, J. Hosp. Infect., 2004, 56, 10–15 CrossRef CAS PubMed; (b) B. Dietze, A. Rath, C. Wendt and H. Martiny, J. Hosp. Infect., 2001, 49, 255–261 CrossRef CAS PubMed; (c) J. M. Boyce, G. Potter-Bynoe, C. Chenevert and T. King, Infect. Control Hosp. Epidemiol., 1997, 18, 622–627 CrossRef CAS PubMed.
- R. M. Donlan and J. W. Costerton, Clin. Microbiol. Rev., 2002, 15, 167–193 CrossRef CAS.
- (a) C. von Eiff, B. Jansen, W. Kohnen and K. Becker, Drugs, 2005, 65, 179–214 CrossRef CAS PubMed; (b) E. M. Hetrick and M. H. Schoenfisch, Chem. Soc. Rev., 2006, 35, 780–789 RSC.
- (a) K. Lienkamp and G. N. Tew, Chem. – Eur. J., 2009, 15, 11784–11800 CrossRef CAS PubMed; (b) K. Lienkamp, A. E. Madkour, K.-N. Kumar, K. Nuesslein and G. N. Tew, Chem. – Eur. J., 2009, 15, 11715–11722 CrossRef CAS PubMed; (c) K. Lienkamp, K.-N. Kumar, A. Som, K. Nuesslein and G. N. Tew, Chem. – Eur. J., 2009, 15, 11710–11714 CrossRef CAS PubMed; (d) K. Lienkamp, A. E. Madkour, A. Musante, C. F. Nelson, K. Nusslein and G. N. Tew, J. Am. Chem. Soc., 2008, 130, 9836–9843 CrossRef CAS PubMed.
- (a) M. Zasloff, Nature, 2002, 415, 389–395 CrossRef CAS PubMed; (b) K. A. Brogden, M. Ackermann, P. B. McCray and B. F. Tack, Int. J. Antimicrob. Agents, 2003, 22, 465–478 CrossRef CAS PubMed; (c) K. A. Brogden, Nat. Rev. Microbiol., 2005, 3, 238–250 CrossRef CAS PubMed.
- (a) A. Al-Ahmad, D. Laird, P. Zou, P. Tomakidi, T. Steinberg and K. Lienkamp, PLoS One, 2013, 8, e73812 CAS; (b) A. E. Madkour, A. H. R. Koch, K. Lienkamp and G. N. Tew, Macromolecules, 2010, 43, 4557–4561 CrossRef CAS PubMed.
- (a) J. Y. Huang, R. R. Koepsel, H. Murata, W. Wu, S. B. Lee, T. Kowalewski, A. J. Russell and K. Matyjaszewski, Langmuir, 2008, 24, 6785–6795 CrossRef CAS PubMed; (b) H. Murata, R. R. Koepsel, K. Matyjaszewski and A. J. Russell, Biomaterials, 2007, 28, 4870–4879 CrossRef CAS PubMed; (c) R. Kügler, O. Bouloussa and F. Rondelez, Microbiology, 2005, 151, 1341–1348 CrossRef PubMed.
- (a) J. C. Tiller, C. J. Liao, K. Lewis and A. M. Klibanov, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 5981–5985 CrossRef CAS PubMed; (b) J. C. Tiller, S. B. Lee, K. Lewis and A. M. Klibanov, Biotechnol. Bioeng., 2002, 79, 465–471 CrossRef CAS PubMed; (c) J. Lin, J. C. Tiller, S. B. Lee, K. Lewis and A. M. Klibanov, Biotechnol. Lett., 2002, 24, 801–805 CrossRef CAS; (d) J. Lin, S. Y. Qiu, K. Lewis and A. M. Klibanov, Biotechnol. Bioeng., 2003, 83, 168–172 CrossRef CAS PubMed; (e) J. Lin, S. Y. Qiu, K. Lewis and A. M. Klibanov, Biotechnol. Prog., 2002, 18, 1082–1086 CrossRef CAS PubMed; (f) D. Park, J. Wang and A. M. Klibanov, Biotechnol. Prog., 2006, 22, 584–589 CrossRef CAS PubMed; (g) N. M. Milovic, J. Wang, K. Lewis and A. M. Klibanov, Biotechnol. Bioeng., 2005, 90, 715–722 CrossRef CAS PubMed.
- J. Thome, A. Hollander, W. Jaeger, I. Trick and C. Oehr, Surf. Coat. Technol., 2003, 174, 584–587 CrossRef.
- A. E. Madkour, J. A. Dabkowski, K. Nusslein and G. N. Tew, Langmuir, 2009, 25, 1060–1067 CrossRef CAS PubMed.
- (a) S. B. Lee, R. R. Koepsel, S. W. Morley, K. Matyjaszewski, Y. J. Sun and A. J. Russell, Biomacromolecules, 2004, 5, 877–882 CrossRef CAS PubMed; (b) J. Y. Huang, H. Murata, R. R. Koepsel, A. J. Russell and K. Matyjaszewski, Biomacromolecules, 2007, 8, 1396–1399 CrossRef CAS PubMed.
- P. Li, Y. F. Poon, W. Li, H.-Y. Zhu, S. H. Yeap, Y. Cao, X. Qi, C. Zhou, M. Lamrani, R. W. Beuerman, E.-T. Kang, Y. Mu, C. M. Li, M. W. Chang, S. S. J. Leong and M. B. Chan-Park, Nat. Mater., 2011, 10, 149–156 CrossRef CAS PubMed.
- A. M. Bieser and J. C. Tiller, Macromol. Biosci., 2011, 11, 526–534 CrossRef CAS PubMed.
- L. A. T. W. Asri, M. Crismaru, S. Roest, Y. Chen, O. Ivashenko, P. Rudolf, J. C. Tiller, H. C. van der Mei, T. J. A. Loontjens and H. J. Busscher, Adv. Funct. Mater., 2014, 24, 346–355 CrossRef CAS.
- J. C. Tiller, C.-J. Liao, K. Lewis and A. M. Klibanov, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 5981–5985 CrossRef CAS PubMed.
- K. Kuroda, G. A. Caputo and W. F. DeGrado, Chem. – Eur. J., 2009, 15, 1123–1133 CrossRef CAS PubMed.
- O. Prucker, C. A. Naumann, J. Rühe, W. Knoll and C. W. Frank, J. Am. Chem. Soc., 1999, 121, 8766–8770 CrossRef CAS.
- P. Zou, W. Hartleb and K. Lienkamp, J. Mater. Chem., 2012, 22, 19579–19589 RSC.
- R. Toomey, D. Freidank and J. Rühe, Macromolecules, 2004, 37, 882–887 CrossRef CAS.
- R. J. Good, J. Adhes. Sci. Technol., 1992, 6, 1269–1302 CrossRef CAS.
- C. Werner, H. Körber, R. Zimmermann, S. Dukhin and H.-J. Jacobasch, J. Colloid Interface Sci., 1998, 208, 329–346 CrossRef CAS PubMed.
- H.-J. Jacobasch, Prog. Org. Coat., 1989, 17, 115–133 CrossRef CAS.
- (a) C. Werner, U. Konig, A. Augsburg, C. Arnhold, H. Korber, R. Zimmermann and H. J. Jacobasch, Colloids Surf., A, 1999, 159, 519–529 CrossRef CAS; (b) H.-J. Jacobasch, F. Simon, C. Werner and C. Bellmann, Tech. Mess., 1996, 63, 447 CAS.
- A. Al-Ahmad, P. Zou, D. L. Guevara-Solarte, E. Hellwig, T. Steinberg and K. Lienkamp, PLoS One, 2014, e111357 Search PubMed.
- J. Haldar, A. K. Weight and A. M. Klibanov, Nat. Protoc., 2007, 2, 2412–2417 CrossRef CAS PubMed.
- (a) E. F. Palermo, I. Sovadinova and K. Kuroda, Biomacromolecules, 2009, 10, 3098–3107 CrossRef CAS PubMed; (b) K. Kuroda and W. F. DeGrado, J. Am. Chem. Soc., 2005, 127, 4128–4129 CrossRef CAS PubMed.
- J. C. Tiller, S. B. Lee, K. Lewis and A. M. Klibanov, Biotechnol. Bioeng., 2002, 79, 465–471 CrossRef CAS PubMed.
- C. K. Pandiyarajan, O. Prucker, B. Zieger and J. Rühe, Macromol. Biosci., 2013, 13, 873–884 CrossRef CAS PubMed.
- A. M. Kelly, V. Kaltenhauser, I. Muehlbacher, K. Rametsteiner, H. Kren, C. Slugovc, F. Stelzer and F. Wiesbrock, Macromol. Biosci., 2013, 13, 116–125 CrossRef CAS PubMed.
- (a) J. A. Lichter and M. F. Rubner, Langmuir, 2009, 25, 7686–7694 CrossRef CAS PubMed; (b) B. Gottenbos, D. W. Grijpma, H. C. Van der Mei, J. Feijen and H. J. Busscher, J. Antimicrob. Chemother., 2001, 48, 7–13 CrossRef CAS PubMed; (c) D. Iarikov Dmitri, M. Kargar, A. Sahari, L. Russel, T. Gause Katelyn, B. Behkam and A. Ducker William, Biomacromolecules, 2014, 15, 169–176 CrossRef CAS PubMed; (d) S. Y. Wong, L. Han, K. Timachova, J. Veselinovic, M. N. Hyder, C. Ortiz, A. M. Klibanov and P. T. Hammond, Biomacromolecules, 2012, 13, 719–726 CrossRef CAS PubMed; (e) G. Seyfriedsberger, K. Rametsteiner and W. Kern, Eur. Polym. J., 2006, 42, 3383–3389 CrossRef CAS; (f) M. L. Gupta, K. Brunson, A. Chakravorty, P. Kurt, J. C. Alvarez, F. Luna-Vera and K. J. Wynne, Langmuir, 2010, 26, 9032–9039 CrossRef CAS PubMed.
- H. H. Hooper, J. P. Baker, H. W. Blanch and J. M. Prausnitz, Macromolecules, 1990, 23, 1096–1104 CrossRef CAS.
- P. J. Flory, J. Chem. Phys., 1942, 10, 51–61 CrossRef CAS.
- G. S. Manning and J. Ray, J. Biomol. Struct. Dyn., 1998, 16, 461–476 CAS.
- (a) E. A. Porter, X. F. Wang, H. S. Lee, B. Weisblum and S. H. Gellman, Nature, 2000, 404, 565 CrossRef CAS PubMed; (b) D. H. Liu and W. F. DeGrado, J. Am. Chem. Soc., 2001, 123, 7553–7559 CrossRef CAS PubMed; (c) T. L. Raguse, E. A. Porter, B. Weisblum and S. H. Gellman, J. Am. Chem. Soc., 2002, 124, 12774–12785 CrossRef CAS PubMed; (d) G. N. Tew, D. H. Liu, B. Chen, R. J. Doerksen, J. Kaplan, P. J. Carroll, M. L. Klein and W. F. DeGrado, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 5110–5114 CrossRef CAS PubMed; (e) J. A. Patch and A. E. Barron, Curr. Opin. Chem. Biol., 2002, 6, 872–877 CrossRef CAS PubMed; (f) D. H. Liu, S. Choi, B. Chen, R. J. Doerksen, D. J. Clements, J. D. Winkler, M. L. Klein and W. F. DeGrado, Angew. Chem., Int. Ed., 2004, 43, 1158–1162 CrossRef CAS PubMed; (g) K. Kuroda and W. F. DeGrado, J. Am. Chem. Soc., 2005, 127, 4128–4129 CrossRef CAS PubMed; (h) H. Tang, R. J. Doerksen, T. V. Jones, M. L. Klein and G. N. Tew, Chem. Biol., 2006, 13, 427–435 CrossRef CAS PubMed; (i) I. Ivanov, S. Vemparala, V. Pophristic, K. Kuroda, W. F. DeGrado, J. A. McCammon and M. L. Klein, J. Am. Chem. Soc., 2006, 128, 1778–1779 CrossRef CAS PubMed; (j) K. Lienkamp, A. E. Madkour, A. Musante, C. F. Nelson, K. Nusslein and G. N. Tew, J. Am. Chem. Soc., 2008, 130, 9836–9843 CrossRef CAS PubMed; (k) G. J. Gabriel, J. G. Pool, A. Som, J. M. Dabkowski, E. B. Coughlin, M. Muthukumar and G. N. Tew, Langmuir, 2008, 24, 12489–12495 CrossRef CAS PubMed; (l) G. J. Gabriel, J. A. Maegerlein, C. E. Nelson, J. M. Dabkowski, T. Eren, K. Nusslein and G. N. Tew, Chem. – Eur. J., 2009, 15, 433–439 CrossRef CAS PubMed; (m) K. Lienkamp, A. E. Madkour, K. N. Kumar, K. Nusslein and G. N. Tew, Chem. – Eur. J., 2009, 15, 11715–11722 CrossRef CAS PubMed; (n) K. Lienkamp and G. N. Tew, Chem. – Eur. J., 2009, 15, 11784–11800 CrossRef CAS PubMed; (o) B. P. Mowery, A. H. Lindner, B. Weisblum, S. S. Stahl and S. H. Gellman, J. Am. Chem. Soc., 2009, 131, 9735–9745 CrossRef CAS PubMed; (p) K. Kuroda, G. A. Caputo and W. F. DeGrado, Chem. – Eur. J., 2009, 15, 1123–1133 CrossRef CAS PubMed; (q) G. N. Tew, R. W. Scott, M. L. Klein and W. F. Degrado, Acc. Chem. Res., 2010, 43, 30–39 CrossRef CAS PubMed.
- (a) J. Lin, S. Qiu, K. Lewis and A. M. Klibanov, Biotechnol. Bioeng., 2003, 83, 168–172 CrossRef CAS PubMed; (b) A. M. Klibanov, J. Mater. Chem., 2007, 17, 2479–2482 RSC.
- S. Krishnan, R. J. Ward, A. Hexemer, K. E. Sohn, K. L. Lee, E. R. Angert, D. A. Fischer, E. J. Kramer and C. K. Ober, Langmuir, 2006, 22, 11255–11266 CrossRef CAS PubMed.
- H. Nikaido and T. Nakae, Adv. Microb. Physiol., 1979, 20, 163–250 CrossRef CAS.
- H. J. Busscher and H. C. van der Mei, PLoS Pathog., 2012, 8, e1002440 CAS.
- S. Ye, P. Majumdar, B. Chisholm, S. Stafslien and Z. Chen, Langmuir, 2010, 26, 16455–16462 CrossRef CAS PubMed.
- G. J. Gabriel, J. A. Maegerlein, C. F. Nelson, J. M. Dabkowski, T. Eren, K. Nusslein and G. N. Tew, Chem. – Eur. J., 2009, 15, 433–439 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5tb00906e |
| ‡ Our sample nomenclature indicates the repeat unit content of the copolymers. For example, P:D = 1:9 designates a polymer with 10 mol% propyl and 90 mol% diamine repeat units. |
| § Swelling ratio = swollen layer thickness divided by dry layer thickness. |
| ¶ In SPR/OWG, p-polarized light excites surface oscillations (plasmon and waveguide modes) in the gold layer, which result in light adsorption under certain angles of incidence. Thus, when measuring light intensity as a function of observation angle, several intensity minima (the plasmon and the waveguide modes) are obtained. These can be fitted to obtain the thickness and dielectric constants of the polymer layer on the gold surface. When the layer swells, the thickness and the dielectric constants change, which results in a shift of the plasmon and wave guide modes. |
| || In this approach, the static contact angle (CA) of a test surface in contact with a series of solvent mixtures is measured. Plotting cos(CA) against the known surface energies of these solvent mixtures and extrapolation to cos(CA) = 0 gives the surface energy of a liquid that would fully wet the test surface, which is set equal to the surface energy of the test surface itself. |
** In the set-up used (SurPass, AntonPaar, Graz), the zeta potential ζ was obtained by measuring the streaming current through a sample chamber holding two planar, parallel sample slides. ζ was calculated from the Helmholtz–Smoluchowski equation 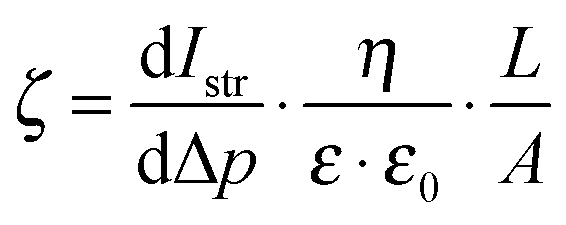 , where dIstr is the streaming current and Δp the pressure difference during the measurement; η, ε and ε0 are parameters that describe the measurement electrolyte (viscosity, relative and absolute dielectric constant); L and A describe the sample geometry (distance between the sample slides and sample area, respectively). , where dIstr is the streaming current and Δp the pressure difference during the measurement; η, ε and ε0 are parameters that describe the measurement electrolyte (viscosity, relative and absolute dielectric constant); L and A describe the sample geometry (distance between the sample slides and sample area, respectively). |
†† 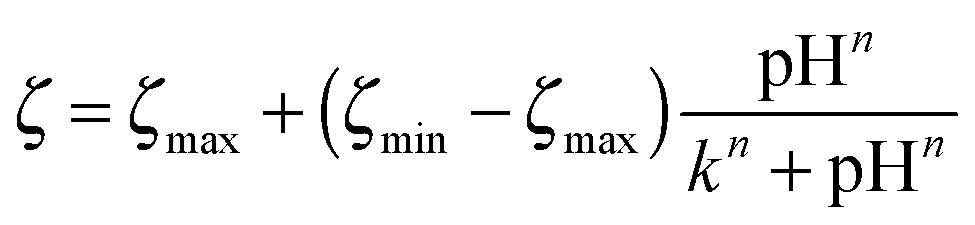 . The fitting parameters were: ζmax, the extrapolated start value of the fitted positive plateau; ζmin, the extrapolated end value of the fitted negative plateau; k, the point of inflection and n, a measure of the width and steepness of the sigmoidal curve. . The fitting parameters were: ζmax, the extrapolated start value of the fitted positive plateau; ζmin, the extrapolated end value of the fitted negative plateau; k, the point of inflection and n, a measure of the width and steepness of the sigmoidal curve. |
| ‡‡ pH(½ζmax) is the pH value where the half maximum of ζ is measured. The second term (containing Faraday's constant, the universal gas constant and the absolute temperature) is a term that corrects for the ionic strength. |
| §§ For B:D = 1:9 and the diamine polymer, the most strongly swelling polymers in this sample set, the titration curves at high pH bent slightly upwards, indicating that swelling effects were present. |
| ¶¶ This is not exactly correct, because ζ is sensitive to ionic strength and was measured at 0.001 mol L−1 in KCl (=0.002 osmol L−1), and not under physiological conditions of about 0.3 osmol L−1. However, this high salt concentration could not be used because it is outside the range in which the ζ can be measured with our set-up. ζ typically decreases with increasing ionic strength. |
| |||| In short, bacteria are sprayed on a test surface and remain in contact with this surface for two hours. They are then removed and transferred onto agar plates. After incubation, the number of colony forming units (CFUs) is counted, and the antimicrobial activity is reported as the percentage of surviving CFUs (%CFU) relative to a negative control. |
| *** For the other antimicrobial data sets, the percentage of CFUs was so low – close to 100% killing – that we were in the limit of accuracy of the assay, where no meaningful correlations could be made. |
| ††† In this assay, keratinocytes were cultivated on the SMAMP networks in the presence of Alamar Blue, a dye that is metabolized by viable cells. Thus, the amount of dye reduction relative to a negative control can be used to quantify cell viability. |
| ‡‡‡ The Bjerrum length is the distance where the thermal energy of two charges is equal to their electrostatic repulsion, i.e. the minimum distance of two charges. |
| §§§ The MIC (= minimum inhibitory concentration) is the smallest concentration that inhibits bacterial growth. |
| ¶¶¶ The HC50 (= hemolytic concentration) is the concentration where 50% of red blood cells were damaged. |
| This journal is © The Royal Society of Chemistry 2015 |